
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time monitoring of the sialic acid biosynthesis pathway by NMR†
Jacob L.
Gorenflos López
 ab,
Peter
Schmieder
a,
Kristin
Kemnitz-Hassanin
a,
Hatice Ceyda
Asikoglu
ac,
Arif
Celik
ab,
Christian E.
Stieger
ab,
Dorothea
Fiedler
ab,
Stephan
Hinderlich
*c and
Christian P. R.
Hackenberger
*ab
ab,
Peter
Schmieder
a,
Kristin
Kemnitz-Hassanin
a,
Hatice Ceyda
Asikoglu
ac,
Arif
Celik
ab,
Christian E.
Stieger
ab,
Dorothea
Fiedler
ab,
Stephan
Hinderlich
*c and
Christian P. R.
Hackenberger
*ab
aLeibniz-Institut für Molekulare Pharmakologie, Robert-Roessle-Strasse 10, 13125 Berlin, Germany. E-mail: hackenbe@fmp-berlin.de
bHumboldt Universität zu Berlin, Department Chemie, Brook-Taylor-Strasse 2, 12489, Germany
cBerliner Hochschule für Technik, Department Life Sciences & Technology, Seestrasse 64, 13347 Berlin, Germany
First published on 21st February 2023
Abstract
Sialic acids are part of the outermost component of the glycocalyx of all vertebrates; as such, they are fundamental markers in physiological and pathological processes. In this study, we introduce a real-time assay to monitor individual enzymatic steps of sialic acid biosynthesis, either with recombinant enzymes, in particular using UDP-N-acetylglucosamine 2-epimerase (GNE) or N-acetylmannosamine kinase (MNK), or in cytosolic rat liver extract. Using state-of-the-art NMR techniques, we are able to follow the characteristic signal of the N-acetyl methyl group, which displays different chemical shifts for the biosynthesis intermediates UDP-N-acetylglucosamine, N-acetylmannosamine (and its 6-phosphate) and N-acetylneuraminic acid (and its 9-phosphate). Pseudo 2- and 3-D NMR demonstrated that in rat liver cytosolic extract, the phosphorylation reaction of MNK is exclusive for N-acetylmannosamine generated by GNE. Thus, we speculate that phosphorylation of this sugar from other sources (e.g. external application to cells) or N-acetylmannosamine derivatives often applied in metabolic glycoengineering is not conducted by MNK but by a yet unknown sugar kinase. Competition experiments with the most prevalent neutral carbohydrates demonstrated that of these, only N-acetylglucosamine slowed N-acetylmannosamine phosphorylation kinetics, suggesting an N-acetylglucosamine-preferring kinase as the acting enzyme.
Introduction
Sialic acid is the umbrella term for a class of nonose carbohydrates.1 The individual carbohydrates are called neuraminic acids with their most prevalent form N-acetylneuraminic acid (Neu5Ac). Usually, sialic acids are found on glycans' termini, making them a critical component in many cell–cell and host–pathogen interactions.2In vertebrates, de novo sialic acid biosynthesis proceeds in four consecutive steps in the cytosol. First, uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), produced from fructose 6-phosphate by the hexosamine biosynthetic pathway,3 is epimerised to produce N-acetylmannosamine (ManNAc). Subsequently, using ATP, it is phosphorylated to ManNAc-6-phosphate (ManNAc-6P). Both steps are catalysed by the bifunctional UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE/MNK).4 Then, sialic acid synthase (SAS) catalyses the condensation reaction of phosphoenolpyruvic acid (PEP) and ManNAc-6P to Neu5Ac 9-phosphate (Neu5Ac-9P). Neu5Ac-9P is dephosphorylated by sialic acid phosphatase (SAP), the only non-essential enzyme of this pathway.5 Neu5Ac is activated to CMP-Neu5Ac by CMP-Neu5Ac synthetase (CMP-SA) in the nucleus. Afterwards, it is transported by the specific nucleotide sugar transporter solute carrier 35A1 (SLC35A1) into the Golgi,6 where it serves as a substrate for sialyltransferases (Scheme 1).
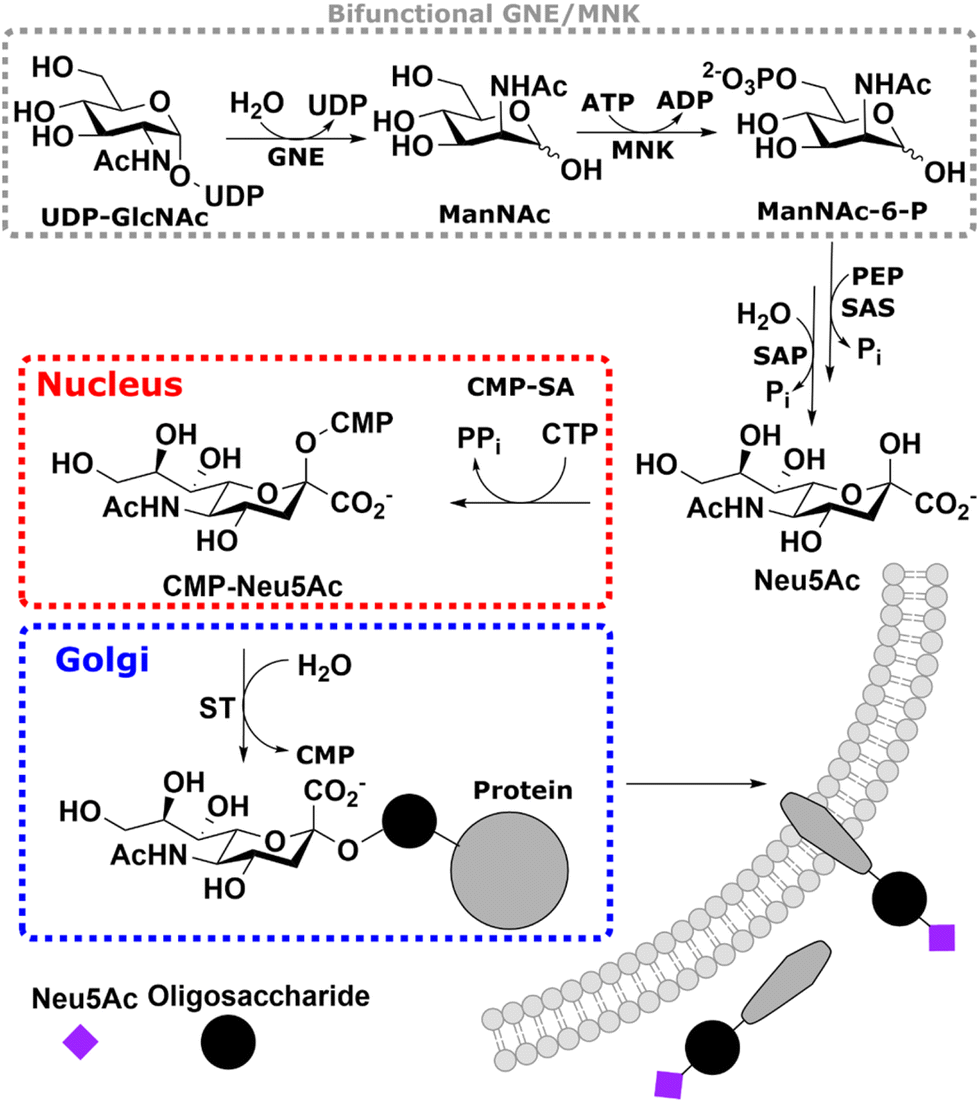 | ||
| Scheme 1 – Overview of the sialic acid biosynthetic pathway. The bifunctional UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE/MNK) catalyses the first two steps of the de novo synthesis of Neu5Ac in cells. The produced ManNAc-6P is then condensed with PEP to Neu5Ac-9P, followed by dephosphorylation by SAP. Neu5Ac is finally activated to CMP-sialic acid in the nucleus to be used to synthesise sialylated glycans in the Golgi. | ||
Metabolic glycan engineering (MGE) is a methodology used to introduce specific chemical entities into cells via glycobiological pathways.7 Because of the terminal position of sialic acids and the promiscuity of the involved enzymes, many MGE approaches use ManNAc derivatives, which are directly metabolised to sialic acids.7,8 The most widely used of these is N-azidoacetylmannosamine (ManNAz), usually in its per-acetylated form to ensure cell permeability, which is metabolised to N-azidoacetylneuraminic acid (Neu5Az) and can be used as an easily accessible bioorthogonal handle targeting the azide on cell surface glycoconjugates9,10 and soluble glycoproteins.11,12 The first metabolic step of this and related N-acyl-modified ManNAc derivatives, after their acetyl esters have been cleaved off by unspecific esterases, is the phosphorylation to ManNAc-6P derivatives. First, it was speculated that MNK catalyses this step. Still, low conversion efficiencies of the recombinant kinase13 and studies with GNE/MNK knock-down cells14 indicated that another kinase might be responsible for this enzymatic step. Furthermore, experiments with external ManNAc (we differentiate between biosynthetic ManNAc produced in cells and external ManNAc, which is added artificially) rescues the cell surface sialic acid content of lymphoma BJA-B cells with a GNE/MNK knock-out.15
Analysis of sialic acid biosynthesis metabolites is a challenging approach. Recombinant and purified enzymes often allow colourimetric assays or optical tests for the characterisation of single steps of the pathway, including recording of enzyme kinetics.16,17 Analysis of the metabolites in a cellular context was initially performed by radioactive compounds, which were separated by paper or thin-layer chromatography.18 Improved methods allowed high-performance liquid chromatography (HPLC) assays for labelled Neu5Ac and its derivatives19 or HPLC-mass spectrometry (MS), thereby often employing 13C or 1H labelled compounds.20,21 However, all of these methods are endpoint assays, which so far did not allow real-time monitoring of one metabolite or even several compounds in parallel. Therefore, our aim in the current paper was to develop a real-time assay to monitor key steps in sialic acid biosynthesis in a cell-free cytosolic system.
Real-time NMR (RT-NMR) monitoring of metabolic pathways in physiological environments has received considerable attention in recent years. Due to the non-invasive, non-disruptive and quantitative nature of RT-NMR, with NMR signal intensities reflecting the absolute concentrations of components,22–26 we considered RT-NMR to be the ideal method to follow different biosynthetic intermediates of the sialic acid biosynthesis pathway, such as UDP-GlcNAc, ManNAc, ManNAc-6P and Neu5Ac, even in the complex environment of a tissue homogenate. In particular, we envisioned using this approach to obtain more information on the metabolic pathway flux and on the phosphorylation of biosynthetic vs. external ManNAc, due to its importance for MGE.
Results and discussion
Chemical shifts of carbohydrates involved in the sialic acid biosynthesis pathway in 1H NMR
We started our investigation by identifying suitable NMR peaks of carbohydrates involved in the sialic acid biosynthesis pathway. We focused on 1H-NMR because it does not require labelled compounds. The N-Ac methyl of UDP-GlcNAc, ManNAc, ManNAc-6P and Neu5Ac displayed characteristic chemical shifts around 2.1–1.9 ppm (Fig. 1A and ESI† Fig. 1). Whereas ManNAc and ManNAc-6P showed two peaks, one for each anomer, UDP-GlcNAc and Neu5Ac only presented one signal. Furthermore, Neu5Ac has C3 ring proton signals between 2.15–2.05 ppm (equatorial) and 1.75–1.65 ppm (axial) with characteristic signals (ESI† Fig. 2),27,28 which helped to validate its presence in complex mixtures.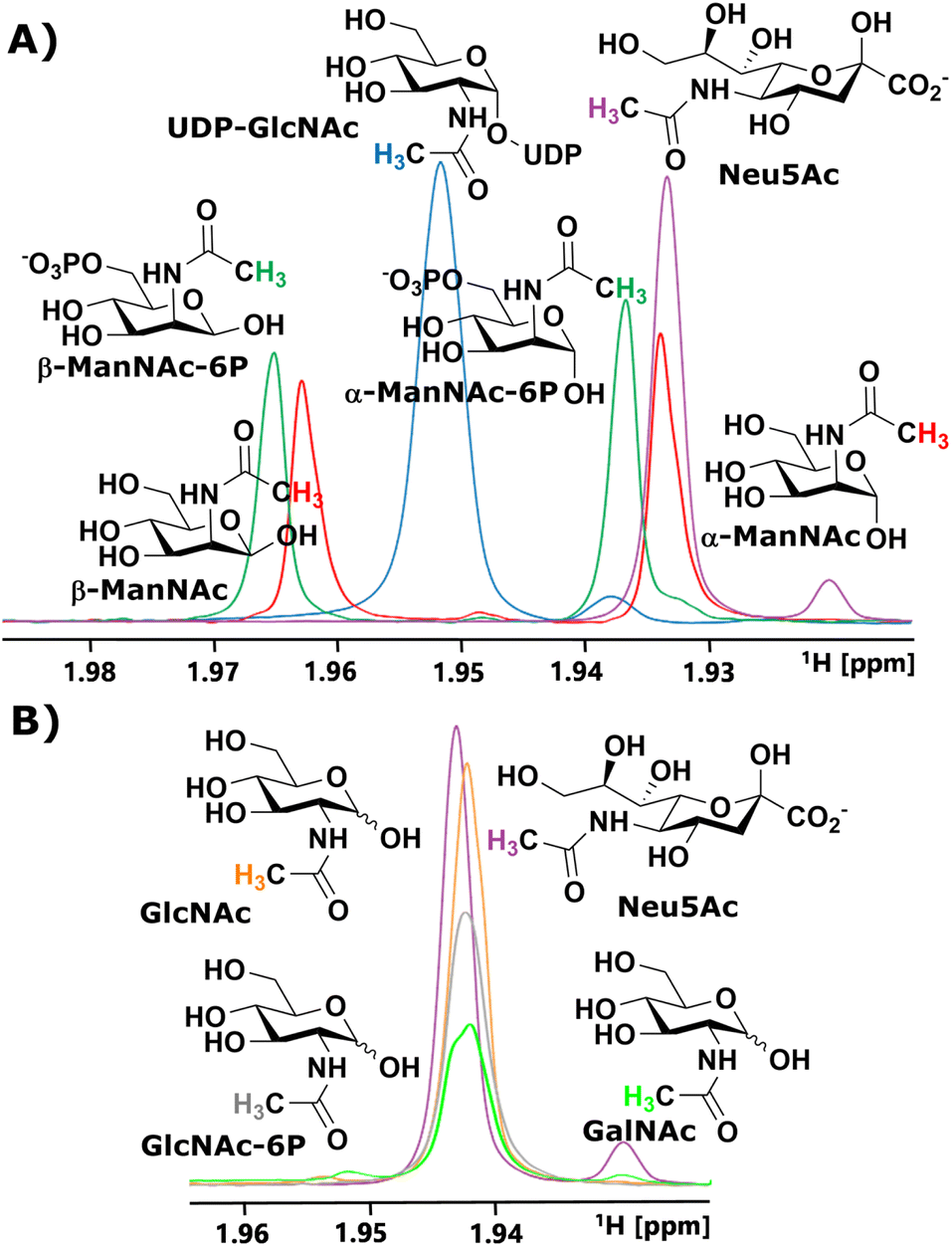 | ||
| Fig. 1 (A) Characteristic chemical shifts in 1H-NMR of the N-acetyl groups of the intermediates in the sialic acid biosynthesis pathway. (B) Characteristic chemical shifts in 1H-NMR of the N-acetyl groups of GlcNAc, GlcNAc-6P, GalNAc and Neu5Ac. | ||
The α to β ratios of ManNAc and ManNAc-6P were determined by integrating the peaks to roughly be 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI† Fig. 3). A combination of HMBC and HMQC revealed, in tune with the literature, the downfield peak of ManNAc to be the β-anomer (ESI† Fig. 4).29 The methyl groups of GlcNAc and GalNAc only present one peak for both anomers. Surprisingly, both anomers of ManNAc-6P show the same chemical shift difference of 0.03 ppm for the N-Ac methyl in 1H-NMR compared to ManNAc. This was not the case for the GlcNAc and GlcNAc-6P pair (Fig. 1B). We speculated that the axial orientation of the N-Ac methyl in ManNAc and ManNAc-6P positions the methyl group to interact with the anomeric hydroxyl, thus causing peak separation between α- and β-anomer. In the case of the chemical shift between the phosphorylated and the unphosphorylated ManNAc, we considered that the phosphate interacts with the axial amide either sterically, through a salt bridge or a hydrogen bond (ESI† Fig. 5).
:1 (ESI† Fig. 3). A combination of HMBC and HMQC revealed, in tune with the literature, the downfield peak of ManNAc to be the β-anomer (ESI† Fig. 4).29 The methyl groups of GlcNAc and GalNAc only present one peak for both anomers. Surprisingly, both anomers of ManNAc-6P show the same chemical shift difference of 0.03 ppm for the N-Ac methyl in 1H-NMR compared to ManNAc. This was not the case for the GlcNAc and GlcNAc-6P pair (Fig. 1B). We speculated that the axial orientation of the N-Ac methyl in ManNAc and ManNAc-6P positions the methyl group to interact with the anomeric hydroxyl, thus causing peak separation between α- and β-anomer. In the case of the chemical shift between the phosphorylated and the unphosphorylated ManNAc, we considered that the phosphate interacts with the axial amide either sterically, through a salt bridge or a hydrogen bond (ESI† Fig. 5).
In parallel to measuring the chemical shifts of the methyl group of N-acetyl hexosamines, 1H-NMR also enabled the monitoring of many other proton-containing molecules, such as ATP, ADP, AMP, by the aromatic protons of the nucleotides, and PEP and pyruvate by their vinyl and methyl protons, respectively. With the help of standards, characteristic peaks for these molecules could be attributed to their origin, which provided further highly valuable information on coenzymes and substrates from our assay (ESI† Fig. 6).
Monitoring the conversions of the sialic acid biosynthesis pathway by recombinant enzymes with 1H NMR
To obtain a model system of the first two reactions of the sialic acid biosynthesis pathway, we expressed GNE30 and MNK31 recombinantly and purified them based on previously published protocols (ESI† Fig. 7 and 8). Because 1H-NMR experiments with UDP-GlcNAc, ManNAc and ManNAc-6P showed separate peaks for their methyl groups in the range around 2.1–1.9 ppm (Fig. 1A), we were able to monitor their conversions by the two enzymes in a RT-NMR approach. The key to applying RT-NMR spectroscopy for us was adjusting the pre-measurement calibration to be as quick and efficient as possible, primarily by using pre-optimized parameters. Specific numbers of experiments in regularly interspaced intervals with defined starting time were set up. The specified starting time allowed to pre-mix the reaction components at a predefined time point, ensuring consistent reactions. In this study, all reactions were started 2 min before the time point zero measurements, which gave us enough time to mix the samples, transfer them to an NMR vial and place them into the sample holder (ESI† Fig. 9). To ensure that all experiments fit in their allotted time frame, the speed at which samples were processed once placed in the sample holder was optimised to 5 min.At 10 μg mL−1 (215 nM) GNE converted 400 μM UDP-GlcNAc in about 5 h to ManNAc (half-life 1.1 h, ESI† Table 1). 1H-NMR spectra were recorded in hourly intervals. The stacking of these spectra enabled the quantification of the epimerization of UDP-GlcNAc to ManNAc. The UDP-GlcNAc signal was normalised to the first measurement. The ManNAc signals were normalised to the averaged signal of the plateau after 7 h (Fig. 2A). This and all following reactions were analysed using a one-phase decay model (ESI† Fig. 10).
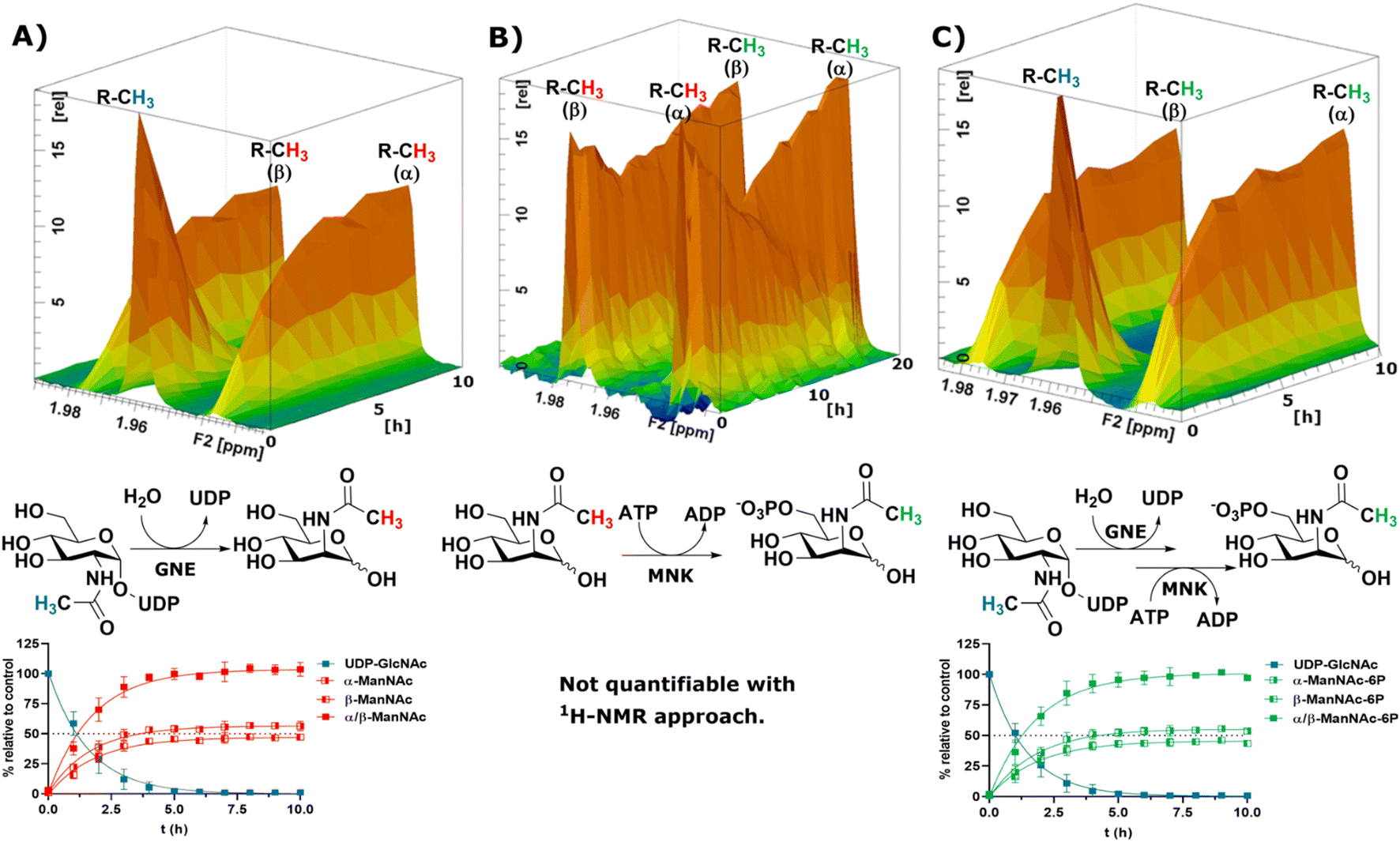 | ||
| Fig. 2 Topographic view (oblique mode) of the pseudo-2D NMR; the monitored reactions and the graphs resulting from the integration of the raw data are displayed here in triplicates. (A) Real-time 1H-NMR of the epimerisation of UDP-GlcNAc (400 μM) to ManNAc by recombinant GNE (10 μg mL−1) – nonlinear fit data in ESI† Table 1. (B) Real-time 1H-NMR of the phosphorylation of ManNAc to ManNAc-6p by recombinant MNK (1 μg mL−1) with ATP (5 mM). (C) Real-time 1H-NMR of the conversion of UDP-GlcNAc (400 μM) to ManNAc-6p by recombinant GNE/MNK (10 μg mL−1) with ATP (5 mM) – nonlinear fit data in ESI† Table 2. | ||
Recombinant MNK proved to be significantly more active than recombinant GNE, 1 μg mL−1 (29 nM) MNK was determined to be a more suitable concentration for the 1 h measuring intervals (ESI† Fig. 11). Real-time 1H-NMR spectroscopy enabled monitoring of the phosphorylation of ManNAc to ManNAc-6P by MNK, but due to poor peak separation the ManNAc and ManNAc-6P peaks could not be integrated separately, which made separate quantification of educt and product unreliable (Fig. 2B).
To demonstrate that we could monitor epimerisation and phosphorylation a cocktail of both enzymes at 10 μg mL−1 of each, GNE (215 nM) and MNK (290 nM), was employed to convert UDP-GlcNAc to ManNAc-6P (Fig. 2C). To mimic the production of Neu5Ac by the sialic acid synthase, commercially available sialic acid aldolase from E. coli (1 mg mL−1; 29 μM) and pyruvate (200 mM) were used. ManNAc was generated in situ using GNE and UDP-GlcNAc. Real-time 1H-NMR spectroscopy proved to be efficient at monitoring the conversion reactions (ESI† Fig. 12A). Neu5Ac production was corroborated by the characteristic signal of the axial ring protons at C3. Part of the ring proton signal overlapped with unidentified compounds (ESI† Fig. 12B).
SOFAST-HMQC NMR to monitor the phosphorylation of isotopically labelled ManNAc to ManNAc-6P
As mentioned before, the phosphorylation of ManNAc by recombinant MNK could not be monitored separately because the methyl peaks of educt and product overlapped. The topographic view (oblique mode) of the pseudo-2D NMR spectrum appears to show good separation, but a look at an individual plane (contour mode) still revealed substantial overlap of the peaks (Fig. 3A). An HSQC spectrum of 1:1 ManNAc and ManNAc-6P revealed peak separation of the methyl group in 1H and 13C (Fig. 3B and ESI† Fig. 13). Based on this finding, we hypothesized that a SOFAST-HMQC experiment with minimal spectral width in 13C could be designed to be used for real-time resolution of this conversion. As such, we synthesized ManNAc-2-13C by activating acetic acid-2-13C to an NHS ester and coupling it to mannosamine (ESI† Fig. 14).
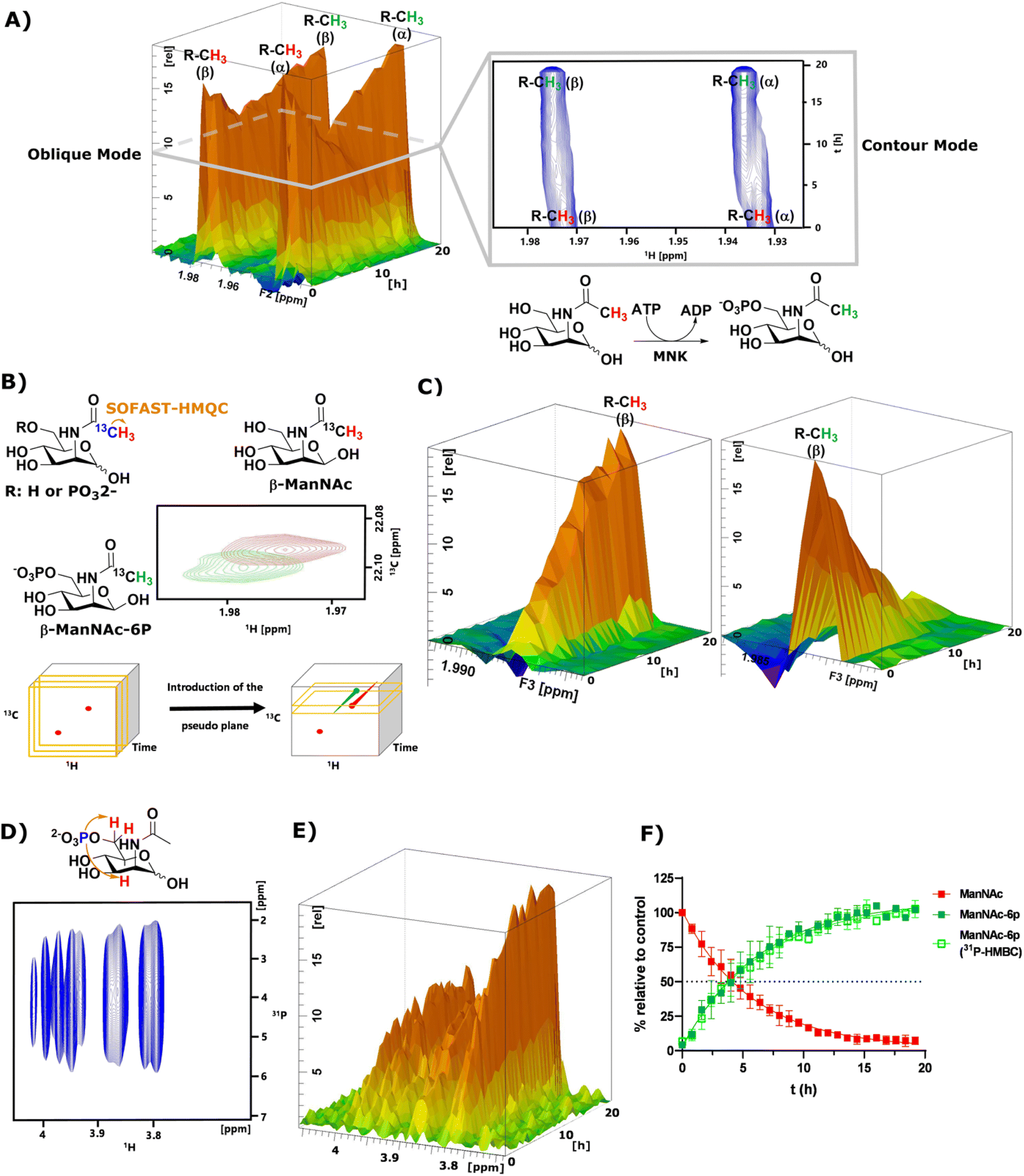 | ||
| Fig. 3 (A) Topographic view (oblique mode) of the pseudo 2D NMR spectrum of the phosphorylation of ManNAc (400 μM) to ManNAc-6p by recombinant MNK (1 μg mL−1) with ATP (5 mM). The individual plane (contour mode) demonstrates shows the lack of peak separation of ManNAc and ManNAc-6P in 1H-NMR. (B) Schematic representation of SOFAST-HMQC coupling of ManNAc/-6P-2-13C and the resulting 2D spectra. These 2D spectra can be stacked into a cube, introducing two pseudo planes, each containing separate peaks; (C) pseudo planes of β-ManNAc/-6P described in (B). The ManNAc and ManNAc-6P peaks can be integrated separately. (D) 31P-HMBC spectrum of ManNAc-6P. (E) 31P-HMBC spectra can be stacked into cubes, allowing the introduction of pseudo planes displayed here. (F) Integration of the pseudo planes of the SOFAST-HMQC and 31P-HMBC – non-linear fit data in ESI† Table 4. | ||
The phosphorylation of ManNAc-2-13C by MNK was monitored using SOFAST-HMQC,32 which was run using non uniform sampling (NUS)33 measuring only 25% of all data points and reducing the spectral width in the 13C-dimension to 0.6 ppm around the ManNAc/-6P signals, which differed on the two utilized spectrometers (22.0 ± 0.3 ppm). The resulting spectra were stacked in a cube, and the peak separation was achieved through the introduction of pseudo-planes (Fig. 3B), which enabled the separation of the β-ManNAc and the β-ManNAc-6P peaks and the integration (Fig. 3C). The same was possible with the α-ManNAc signal (ESI† Fig. 18). All following experiments were analysed using the β-ManNAc and the β-ManNAc-6P peaks because the α signals in part overlapped with other N-acetyl methyl signals, like that of GlcNAc (Fig. 1A/B). To further validate this method, a 31P-HMBC approach was designed, which was also facilitated by NUS of 25% of the data points. ManNAc-6P has a characteristic 31P-HMBC signal (Fig. 3D) that can be stacked like the SOFAST-HMQC spectra. Again time-resolved analysis became possible by introducing a pseudo plane and the integration (Fig. 3E). Multiple measurements were run in sequence. First, the 1H-NMR was run, then the SOFAST-HMQC and last, the 31P-HMBC. All three experiments in a row had been optimised to take less than 5 min, which included moving the probe in the autosampler. While the 1H-NMR provided an initial overview of the data (Fig. 3A), which can also be used to monitor parallel reactions of ATP and PEP, the SOFAST-HMQC offered a powerful method to monitor ManNAc phosphorylation by recombinant enzymes. For recombinant MNK, the 31P-HMBC method could validate the rate constants from the SOFAST-HMQC experiments (Fig. 3F). 31P-HMBC therefore proved to be a robust method to monitor the phosphorylation of monosaccharides by recombinant enzymes.
Monitoring the conversions of the sialic acid biosynthesis pathway in cytosolic extract
After the RT-NMR method had been developed on recombinant enzymes, we applied it to study cytosolic liver extracts. For two reasons liver extracts have long been used as sialic acid biosynthesis pathway model systems.4 First, the tissue is available in homogenous quality by laboratory animals. Second, and more important, rat liver displays the highest expression of sialic acid biosynthesis enzymes of all tissues.34 Liver extracts were obtained by freezing the livers in liquid nitrogen immediately after removing. The tissue was then thawed in buffer, shredded and centrifuged. The supernatant was taken as rat liver cytosolic extract (RLCE), aliquoted and frozen in liquid nitrogen. Although liver is the organ with the highest number of expressed proteins, we performed semi-quantitative proteomics on the enzymes of the sialic acid biosynthesis in our prepared RLCE. GNE/MNK and SAS were found at high expression levels (ESI† Fig. 20). To avoid slight changes in enzyme expression of different RLCE preparations, most presented experiments were conducted using the same extract obtained from one adult female Sprague Dawley rat.First, we performed a GNE assay with RLCE. In the absence of ATP and PEP, UDP-GlcNAc was converted in roughly equal parts to ManNAc and GlcNAc. This is likely due to the activity of GlcNAc 2-epimerase.35,36 However, we could not rule out that UDP-GlcNAc is converted to GlcNAc by a non-physiological activity of GNE/MNK in the absence of ATP, or an independent UDP-GlcNAc hydrolysing enzyme. Because of overlapping signals with GlcNAc, we assumed an α to β ratio of 1.2:1 for ManNAc based on our initial experiments (ESI† Table 5). 27% of the initial UDP-GlcNAc signal was not regained by the sum of ManNAc and GlcNAc signals (Fig. 4A). It is likely, that both amino sugars were further converted by catabolic pathways, including release of the N-acetyl group. This cleaved group is not detected by our NMR assay, leading to loss of signal.
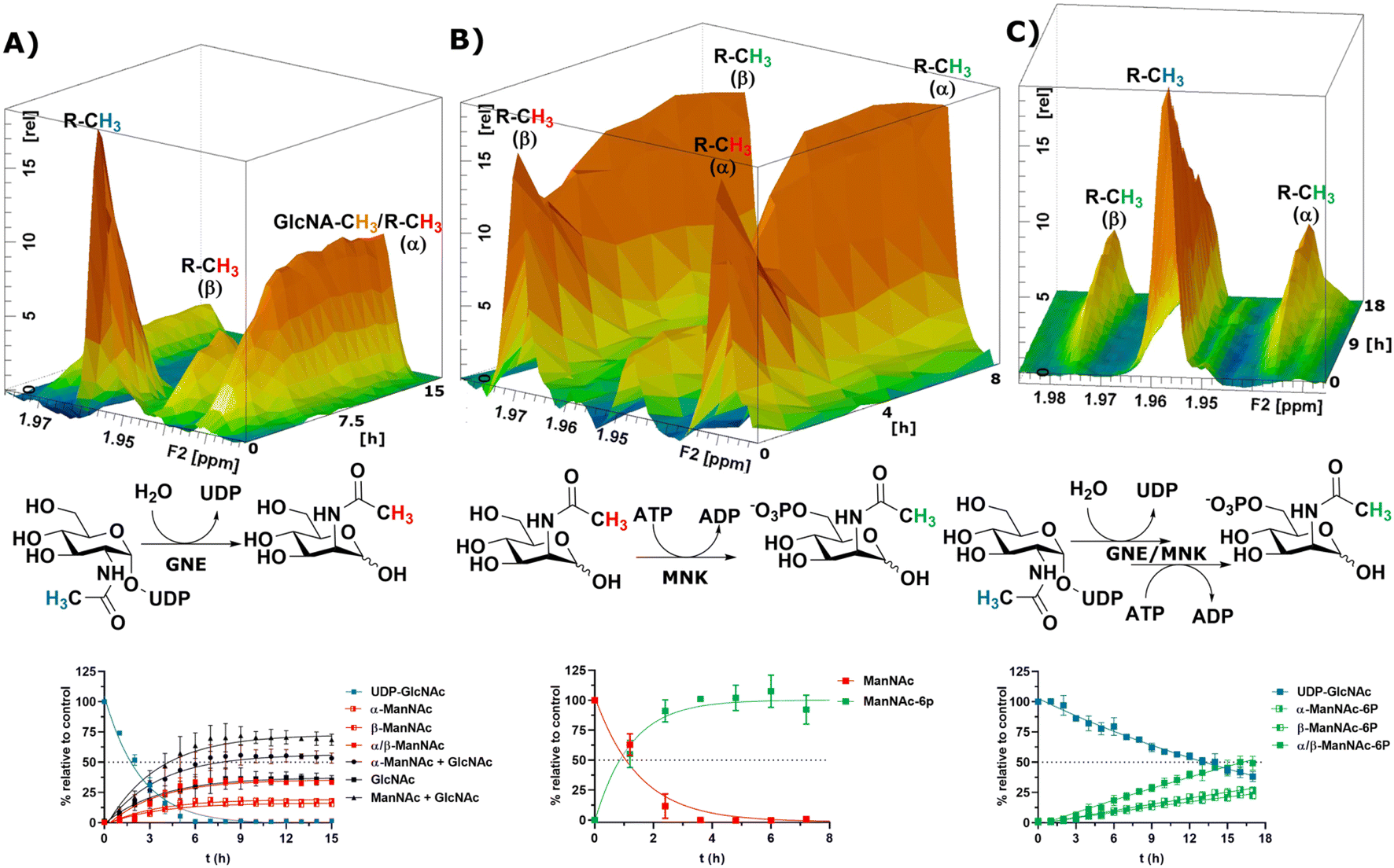 | ||
| Fig. 4 Topographic view (oblique mode) of the pseudo-2D NMR; the monitored reactions and the graphs resulting from the integration of the raw data are displayed here in triplicates. (A) Real-time 1H-NMR of the conversion of UDP-GlcNAc (400 μM) in RLCE (1.6 mg mL−1) without further cofactors. The percentages are given relative to the initial UDP-GlcNAc signal – nonlinear fit data in ESI† Table 5. (B) Real-time 1H-NMR of the phosphorylation of ManNAc-2-13C (400 μM) in RLCE (1.6 mg mL−1) – nonlinear fit data in ESI† Table 6. (C) Real-time 1H-NMR of the conversion of UDP-GlcNAc (400 μM) to ManNAc-6P with ATP (5 mM) and PEP (25 mM) in RLCE (1.6 mg mL−1) – nonlinear fit data in ESI† Table 7. | ||
External ManNAc (400 μM) was phosphorylated to ManNAc-6P in RLCE (1.6 mg mL−1). The half-life of the reaction was 0.84 h (ESI† Table 6). After 3 h of reaction, ManNAc was completely converted to ManNAc-6P (Fig. 4B). Although significant amounts of PEP were present, ManNAc-6P was not converted to Neu5Ac. It is notable that under the same conditions the half-life of the phosphorylation of external ManNAc to ManNAc-6P (0.84 h) was more than an order of magnitude higher than the conversion of UDP-GlcNAc to ManNAc-6P 18.83 h (ESI† Table 7), indicating the activity of another kinase than MNK phosphorylating ManNAc.
In the presence of ATP and PEP, RLCE converts UDP-GlcNAc to ManNAc-6P (Fig. 4C). Though, the half-life of UDP-GlcNAc increases by an order of magnitude from 1.66 h (ESI† Table 5) to 18.83 h (ESI† Table 7). Under these conditions, the production of ManNAc was faster than the production of ManNAc-6P (ESI† Fig. 22).
Commercially available ManNAc-6P was converted with PEP to Neu5Ac/-9P in RLCE (6 mg mL−1). Due to the unavailability of Neu5Ac-9P standards, we could not differentiate whether the detected signal stemmed from the phosphorylated or the dephosphorylated Neu5Ac. Because of overlapping signals with Neu5Ac, we again could not measure the α-anomer signals of ManNAc-6P. Therefore, we calculated Neu5Ac concentrations assuming an α to β ratio of 1.2:1 (ESI† Fig. 23 and Table 8). Of all the recorded reactions of the sialic acid biosynthesis pathway in RLCE, the synthase reaction was the slowest – RLCE concentration had to be increased almost 4-fold to obtain a system in which significant concentrations of substrate were converted to product. Under these conditions, the half-life of ManNAc-6P was 6.46 h (ESI† Table 8). GNE/MNK and SAS are expressed in comparable levels in RLCE (ESI† Fig. 20). Therefore, specific activity of SAS must be significantly lower than the ones of GNE/MNK. Former studies revealed that in RLCE both specific activities of GNE/MNK4 are more than one order of magnitude higher than the specific activity of SAS.37 In conclusion, SAS could be suggested as the bottleneck for sialic acid biosynthesis in RLCE. This is in agreement with the essential role of SAS in sialic acid biosynthesis,5 which is also true for glycoengineering approaches to establish the pathway in sialic acid-deficient cells from insects38 or plants.39
Taken together, we could demonstrate that our RT-NMR approach using 1H-NMR and SOFAST-HMQC experiments could be employed to measure substrate conversions of GNE/MNK and SAS in RLCE in the complex environment of RLCE. Furthermore, the flexibility of the assay to measure unpredicted side products (e.g. GlcNAc) was demonstrated.
Substrate competition for ManNAc phosphorylation in rat liver cytosolic extract
In light of the different reaction velocities in RLCE of UDP-GlcNAc to ManNAc-6P vs. the phosphorylation of ManNAc to ManNAc-6P, we hypothesized that a further kinase in addition to MNK is able to phosphorylate external ManNAc in RLCE. To validate the impact of different kinases on the phosphorylation of external ManNAc in RLCE, UDP-GlcNAc and ManNAc were combined in a reaction cocktail at equimolar concentrations (400 μM) with ATP (5 mM) and PEP (25 mM). The two substrates were monitored independently by 1H-NMR for the decreasing UDP-GlcNAc signal and with HMBC-NMR for the phosphorylation of external ManNAc to ManNAc-6P. The presence of both substrates in the reaction mixture did not significantly alter the conversion velocities of either of them, validating the hypothesis that another kinase than MNK might phosphorylate ManNAc (Fig. 5A). It is important to note, that this experiment was conducted with a different RLCE than the one used in all other experiments, and conversion velocities are not directly comparable.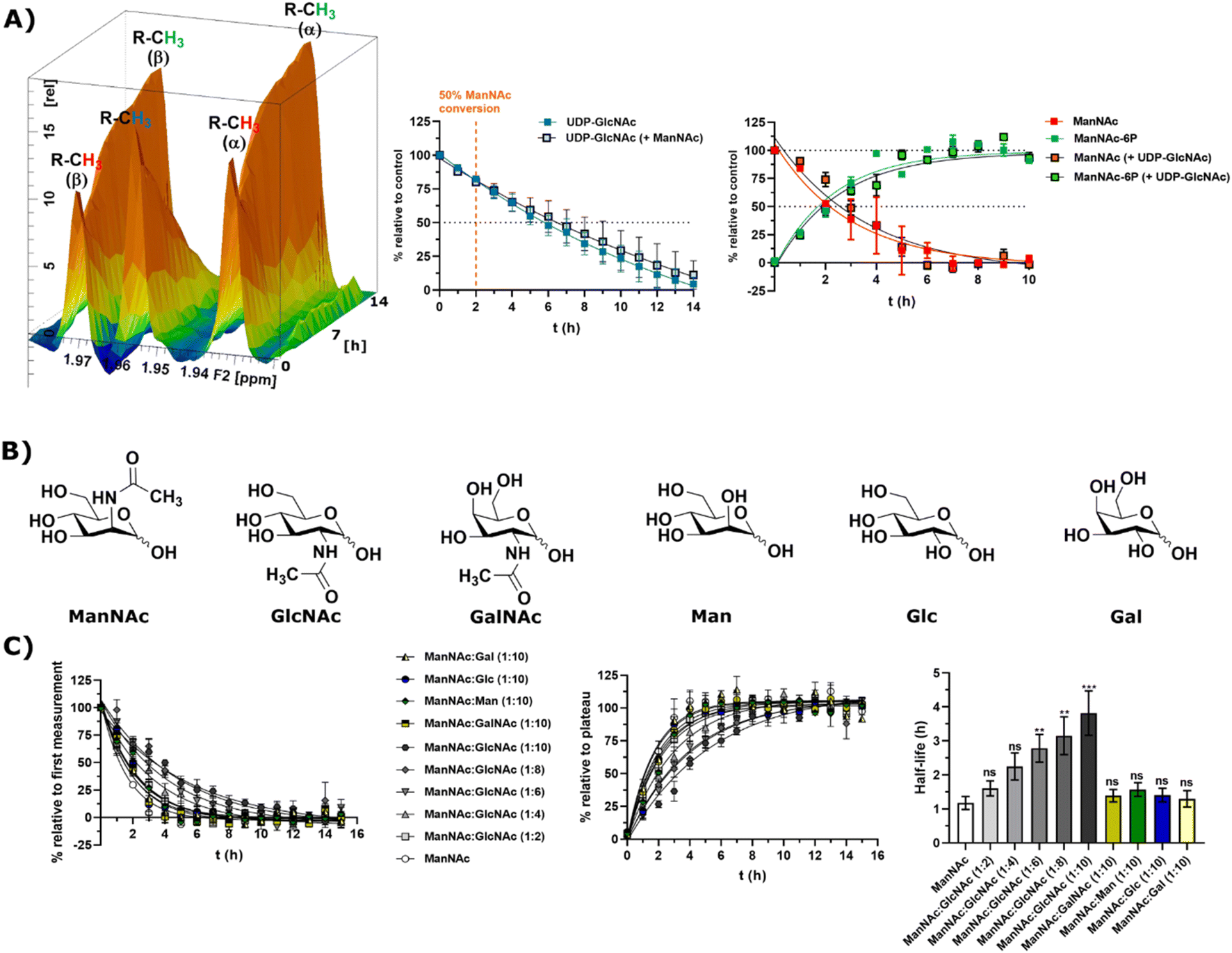 | ||
| Fig. 5 (A) Simultaneous conversion of UDP-GlcNAc (400 μM) and ManNAc (400 μM) in RLCE (1.6 mg mL−1). The decreasing UDP-GlcNAc signals were monitored and the conversion of ManNAc to ManNAc-6P. The respective systems with either only UDP-GlcNAc or only ManNAc were used as control experiments, to reference the conversion velocities – nonlinear fit data in and ESI† Table 10. This experiment was conducted with a different RLCE than all other experiments. (B) Highly abundant monosaccharides ordered by structural similarity to ManNAc. (C) Competition experiments of the conversion of ManNAc (200 μM) to ManNAc-6P with other carbohydrates. The influence of GlcNAc was assessed at increasing ratios (1:2 to 1:10). Other relevant carbohydrates were only used at the ratio of 1:10. The decreasing ManNAc signal and the increasing ManNAc-6P signals were modelled with a one-phase decay model (nonlinear fit data in ESI† Table 11) and the resulting half-life times were plotted. The significance is given relative to the experiment without competing carbohydrate; data are represented as means ± SEM (n = 45; 15 time points per experiment conducted in triplicates) analysed by multiple t-test (P-value: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***) and <0.0001). | ||
We assumed that the responsible kinase for external ManNAc phosphorylation uses highly abundant and structurally related monosaccharides as substrates. These encompass primarily the N-acetyl hexosamines GlcNAc and GalNAc, and the hexoses mannose (Man), glucose (Glc) and galactose (Gal) (Fig. 5B). Only GlcNAc had a significant effect on the half-life of ManNAc-6P production from ManNAc, which increased from 1.20 h to nearly 3.76 h with a 10-fold excess of GlcNAc over ManNAc (Fig. 5C and ESI† Table 12) by use of a decreased RCLE concentration to 1 mg mL−1. This makes a kinase with a primary activity for GlcNAc a prime candidate to phosphorylate external ManNAc, instead of a kinase with a primary activity for ManNAc phosphorylation. Previously, significant MNK activity was already observed for partially purified N-acetylglucosamine kinase (NAGK) from rat liver by Allen and Walker.40 This observation was confirmed by homogenous purification of rat liver NAGK; MNK activity of NAGK could clearly be distinguished from the MNK activity of GNE/MNK by chromatographic separation.41 Our data were further underlined by human cell lines with low or completely absent GNE/MNK expression, where MNK activity could almost completely assigned to NAGK.15 Treatment of GNE myopathy, a hereditary inclusion body myopathy caused by mutations in GNE/MNK,42 with ManNAc, is currently in a phase 2 clinical study. Understanding the nature of the kinase responsible for the first step of metabolisation will enhance the knowledge with regard to failure and success of this trials.43,44
Recombinant MNK and NAGK with different substrates
Next, we decided to test the activity of recombinant human NAGK31 (ESI† Fig. 24) in comparison to the recombinant MNK domain of human GNE/MNK, first assessed in a plate reader-based ATP detection assay (ATP-Glo) with 100 μM of carbohydrates and 100 μM of ATP to adjust a useful enzyme concentration. NAGK phosphorylated GlcNAc efficiently at an enzyme concentration of 0.1 μg mL−1. At the highest tested concentration (10 μg mL−1) NAGK showed minor phosphorylation activity for ManNAc (ESI† Fig. 25B). MNK phosphorylated ManNAc efficiently. However, about 10-fold higher enzyme concentrations are needed compared to NAGK to observe comparable enzyme activities. This is in agreement with the about 10-fold higher specific activity of recombinant NAGK compared to recombinant MNK.31,45 At the highest tested concentration (200 μg mL−1) MNK does not consume ATP in the presence of GlcNAc (ESI† Fig. 25C), suggesting an exclusive substrate specificity for ManNAc.Based on the plate-reader experiments, two concentrations were tested for the GlcNAc phosphorylation by NAGK in NMR experiments. Whereas 10 μg mL−1 NAGK did not allow quantification of enzyme activity because the conversion was finished before the first measurement, 0.1 μg mL−1 NAGK showed complete phosphorylation of GlcNAc after 6 h (Fig. 6A). The phosphorylation of ManNAc (400 μM) by 10 μg mL−1 MNK with ATP (5 mM) was finished after 48 min. At 1 μg mL−1, complete transformation was reached at 16 h (Fig. 6B). These data confirm the activities of the two recombinant kinases observed in the plate-reader assay.
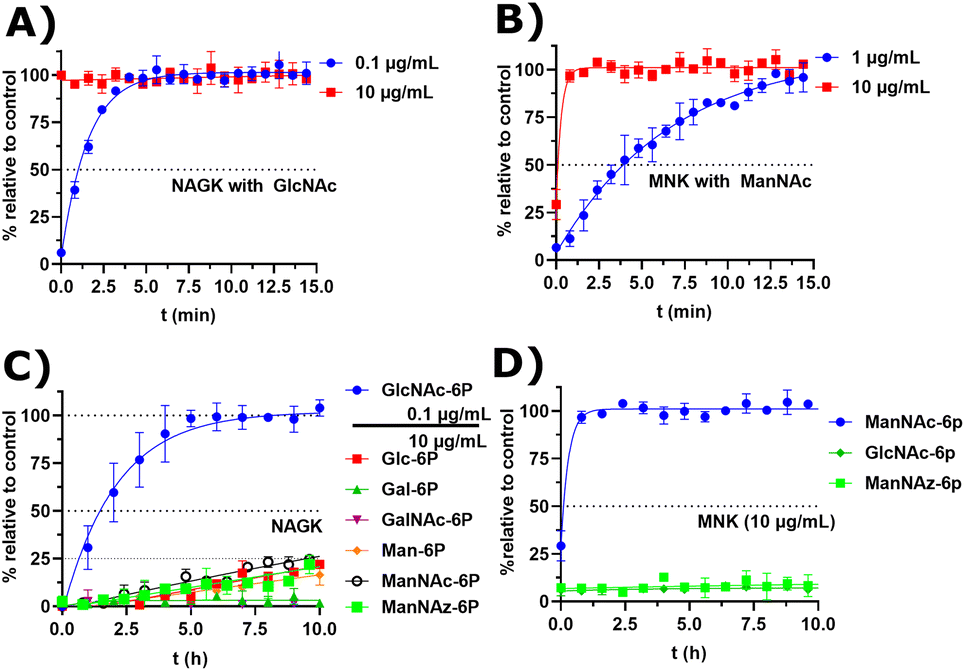 | ||
| Fig. 6 (A and B) Real-time 31P-HMBC-NMR of the phosphorylation (ATP 5 mM) of GlcNAc and ManNAc (400 μM) by NAGK and MNK at different concentrations - nonlinear fit data in ESI† Table 13 and 14. (C and D) Real-time 31P-HMBC-NMR of the phosphorylation of different carbohydrates (400 μM) by NAGK and MNK with ATP (5 mM) - nonlinear fit data in ESI† Tables 15 and 16. | ||
GlcNAc-6P standard revealed a similar signal pattern in a 31P-HMBC as ManNAc-6P (ESI† Fig. 26). From this, we extrapolated that all hexoses phosphorylated at the hydroxy groups at carbon 6 show similar patterns in 31P-HMBC NMR spectra. As such, we used the 31P-HMBC real-time NMR assay to do studies over 18 h on the activity of NAGK and MNK with different substrates. At 10 μg mL−1 NAGK phosphorylates ManNAc, but also Glc and Man with similar rates within the detection limits of this assay. These data are in agreement with the identification of NAGK as “extrahepatic glucokinase”,46 suggesting that NAGK is an enzyme with high substrate promiscuity, at least in in vitro assays, with high Km values for ManNAc (Km = 0.95 mM) and Glc (Km = 600 mM).47 On the other hand, NAGK reveals no activity with Gal and GalNAc (Fig. 6C), revealing a physiologically relevant selection of substrates. MNK does not appear to be promiscuous over longer incubation periods (Fig. 6D), underlining its specificity for ManNAc as a substrate.
In the context of MGE the question is still up to debate, which kinase phosphorylates the ManNAc derivatives. Earlier studies had demonstrated the ability of GNE/MNK knock down cells to still metabolize ManNAz,13 the most commonly used ManNAc derivative for MGE.7 Previous studies had further shown that recombinant rat GNE/MNK could phosphorylate certain ManNAc derivatives with low efficiency in dependence of the length of their N-acyl side chains.13 We therefore investigated both kinases for their ability to phosphorylate ManNAz. Surprisingly, MNK showed no activity for this artificial sugar, whereas NAGK was able to phosphorylate ManNAz with the same activity as shown for the other alternative substrates (Fig. 6C and D), again pointing towards NAGK as a responsible kinase in MGE of N-acyl-mannosamine derivatives.
Conclusions
In this study, we developed an NMR assay to monitor the conversion velocities during sialic acid biosynthesis by recombinant proteins and in cytosolic liver extract in real time. With this assay, we demonstrated that the phosphorylation of external ManNAc is most likely not performed by GNE/MNK. We observed that the phosphorylation of external ManNAc was inhibited by more than equimolar concentrations of GlcNAc, which is a strong indication that the responsible kinase is NAGK. This finding is in line with several studies indicating that NAGK phosphorylates external ManNAc.15,47 Nevertheless, recombinant NAGK did not phosphorylate ManNAc at physiologically relevant velocities, in contrast to GlcNAc. One potential explanation is that specific posttranslational modifications are necessary to give this enzyme the ability to phosphorylate ManNAc.GNE myopathy is presumably caused by a lack of sialic acid production due to a malfunctioning GNE/MNK. As such, our study supports the clinical application of external ManNAc, because external ManNAc phosphorylation was shown functioning independently of GNE/MNK. Still, it also highlights a problem with this treatment: the deregulation of sialic acid biosynthesis. The utilization of external ManNAc circumvents the master regulator of sialic acid biosynthesis: GNE/MNK.48 Without the feedback inhibition of the end-product CMP-Neu5Ac sialic acid production is deregulated and could cause symptoms akin to Sialuria, a hereditary developmental disorder caused by mutations in the CMP-Neu5Ac binding pocket of GNE/MNK.49,50 Furthermore, we think that this deregulation of sialic acid biosynthesis is the reason for why MGE with ManNAc derivatives has been employed so successfully. A recent study showed that azide modifications of carbohydrates were hampering metabolisation rates.51,52 Nonetheless, in a HEK 293T cell model, 65% of the sialome after 24 h of treatment with Ac4ManNAz carried an azide.53 We speculate that the kinase responsible for the phosphorylation of external ManNAc, also phosphorylates ManNAc derivatives.
In summary, with this study we highlight the power of a real-time NMR assay, which, in contrast to other endpoint methods, can differentiate between forming monosaccharide species in situ. As such, we envision this method to be a singularly useful approach to study the conversion velocities of biologically highly relevant carbohydrates in complex environments to evaluate the performance of potential inhibitors and elucidate metabolic pathways.
Data availability
The raw data has been uploaded to Zenodo DOI: http://10.5281/zenodo.7648851. The NMR and the proteomics data were uploaded separately under the same DOI. The NMR data-set contains a read-me file designed to guide the future user.Author contributions
J. L. G. L. and P. S. developed and performed the NMR assay. J. L. G. L., K. K.-H. and A. C. expressed and purified the recombinant proteins. J. L. G. L. and H. C. A. prepared the RCLE and performed experiments with them. C. S. performed the proteomic analysis of the RLCE. J. L. G. L., S. H., P. S. and C. P. R. H. wrote the manuscript. S. H., D. F. and C. P. R. H. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project ID 431232613 – SFB 1449. We would like to thank Minh Nguyen Trung for inspiring the NMR method development and Ellen Avery for providing the rat liver.Notes and references
- F. G. Blix, A. Gottschalk and E. Klenk, Proposed nomenclature in the field of Neuraminic and Sialic acids, Nature, 1957, 179, 1088 CrossRef CAS PubMed.
- A. Varki, Sialic acids in human health and disease, Trends Mol. Med., 2008, 14, 351–360 CrossRef CAS PubMed.
- N. M. Akella, L. Ciraku and M. J. Reginato, Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer, BMC Biol., 2019, 17, 52–66 CrossRef CAS PubMed.
- S. Hinderlich, R. Stäsche, R. Zeitler and W. Reutter, A Bifunctional Enzyme Catalyzes the First Two Steps in N-Acetylneuraminic Acid Biosynthesis of Rat Liver, J. Biol. Chem., 1997, 272, 24313–24318 CrossRef CAS PubMed.
- A. P. Willems, L. Sun, M. A. Schulz, W. Tian, A. Ashikov, M. van Scherpenzeel, E. Hermans, H. Clausen, Z. Yang and D. J. Lefeber, Activity of N-acylneuraminate-9-phosphatase (NANP) is not essential for de novo sialic acid biosynthesis, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1471–1479 CrossRef CAS PubMed.
- S. Ahuja and M. R. Whorton, Structural basis for mammalian nucleotide sugar transport, eLife, 2019, 8, 1–27 CrossRef PubMed.
- P. R. Wratil, R. Horstkorte and W. Reutter, Metabolic Glycoengineering with N-Acyl Side Chain Modified Mannosamines, Angew. Chem., Int. Ed., 2016, 55, 9482–9512 CrossRef CAS PubMed.
- C. Agatemor, M. J. Buettner, R. Ariss, K. Muthiah, C. T. Saeui and K. J. Yarema, Exploiting metabolic glycoengineering to advance healthcare, Nat. Rev. Chem., 2019, 605–620 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Cell Surface Engineering by a Modified Staudinger Reaction, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Copper-free click chemistry for dynamic in vivo imaging, Proc. Natl. Acad. Sci., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- S. J. Luchansky, S. Argade, B. K. Hayes and C. R. Bertozzi, Metabolic Functionalization of Recombinant Glycoproteins, Biochemistry, 2004, 43, 12358–12366 CrossRef CAS PubMed.
- W. Qin, K. Qin, X. Fan, L. Peng, W. Hong, Y. Zhu, P. Lv, Y. Du, R. Huang, M. Han, B. Cheng, Y. Liu, W. Zhou, C. Wang and X. Chen, Artificial Cysteine S-Glycosylation Induced by Per-O-Acetylated Unnatural Monosaccharides during Metabolic Glycan Labeling, Angew. Chem., Int. Ed., 2018, 57, 1817–1820 CrossRef CAS PubMed.
- C. L. Jacobs, S. Goon, K. J. Yarema, S. Hinderlich, H. C. Hang, D. H. Chai and C. R. Bertozzi, Substrate Specificity of the Sialic Acid Biosynthetic Pathway, Biochemistry, 2001, 40, 12864–12874 CrossRef CAS PubMed.
- H. Möller, V. Böhrsch, L. Lucka, C. P. R. Hackenberger and S. Hinderlich, Efficient metabolic oligosaccharide engineering of glycoproteins by UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) knock-down, Mol. BioSyst., 2011, 7, 2245–2251 RSC.
- S. Hinderlich, M. Berger, O. T. Keppler, M. Pawlita and W. Reutter, Biosynthesis of N-Acetylneuraminic Acid in Cells Lacking UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase, Biol. Chem., 2001, 382, 291–297 CAS.
- L. Roden, H. Yu, J. Jin, G. Ekborg, A. Estock, N. R. Krishna and P. Livant, Analysis of the Morgan-Elson chromogens by high-performance liquid chromatography, Anal. Biochem., 1997, 254, 240–248 CrossRef CAS PubMed.
- T. Takahashi, M. Ikegami-Kawai, R. Okuda and K. Suzuki, A fluorimetric Morgan-Elson assay method for hyaluronidase activity, Anal. Biochem., 2003, 322, 257–263 CrossRef CAS PubMed.
- N. Taniguchi, K. Honke, M. Fukuda, H. Narimatsu, Y. Yamaguchi and T. Angata, Handbook of glycosyltransferases and related genes, Springer, 2014 Search PubMed.
- J. E. G. A. Dold and V. Wittmann, Metabolic Glycoengineering with Azide- and Alkene-Modified Hexosamines: Quantification of Sialic Acid Levels, ChemBioChem, 2021, 22, 1243–1251 CrossRef CAS PubMed.
- M. van der Ham, T. J. de Koning, D. Lefeber, A. Fleer, B. H. C. M. T. Prinsen and M. G. M. de Sain-van der Velden, Liquid chromatography–tandem mass spectrometry assay for the quantification of free and total sialic acid in human cerebrospinal fluid, J. Chromatogr. B, 2010, 878, 1098–1102 CrossRef CAS PubMed.
- X.-Y. Wen, M. Tarailo-Graovac, K. Brand-Arzamendi, A. Willems, B. Rakic, K. Huijben, A. Da Silva, X. Pan, S. El-Rass, R. Ng, K. Selby, A. M. Philip, J. Yun, X. C. Ye, C. J. Ross, A. M. Lehman, F. Zijlstra, A. A. Bakar, B. Drögemöller, J. Moreland, W. W. Wasserman, H. Vallance, M. Van Scherpenzeel, F. Karbassi, M. Hoskings, U. Engelke, A. De Brouwer, R. A. Wevers, A. V. Pshezhetsky, C. D. M. Van Karnebeek and D. J. Lefeber, Sialic acid catabolism by N-acetylneuraminate pyruvate lyase is essential for muscle function, JCI Insight, 2018, 3, e122373 CrossRef PubMed.
- M. J. Smith, C. B. Marshall, F.-X. Theillet, A. Binolfi, P. Selenko and M. Ikura, Real-time NMR monitoring of biological activities in complex physiological environments, Curr. Opin. Struct. Biol., 2015, 32, 39–47 CrossRef CAS PubMed.
- J. Eicher, J. Snoep and J. Rohwer, Determining Enzyme Kinetics for Systems Biology with Nuclear Magnetic Resonance Spectroscopy, Metabolites, 2012, 2, 818–843 CrossRef CAS PubMed.
- I. Alshamleh, N. Krause, C. Richter, N. Kurrle, H. Serve, U. L. Günther and H. Schwalbe, Real-Time NMR Spectroscopy for Studying Metabolism, Angew. Chem., Int. Ed., 2020, 59, 2304–2308 CrossRef CAS PubMed.
- R. K. Harmel, R. Puschmann, M. Nguyen Trung, A. Saiardi, P. Schmieder and D. Fiedler, Harnessing13C-labeled myo-inositol to interrogateinositol phosphate messengers by NMR, Chem. Sci., 2019, 10, 5267–5274 RSC.
- A. Canal-Martín, J. Sastre, M. J. Sánchez-Barrena, A. Canales, S. Baldominos, N. Pascual, L. Martínez-González, D. Molero, M. E. Fernández-Valle, E. Sáez, P. Blanco-Gabella, E. Gómez-Rubio, S. Martín-Santamaría, A. Sáiz, A. Mansilla, F. J. Cañada, J. Jiménez-Barbero, A. Martínez and R. Pérez-Fernández, Insights into real-time chemical processes in a calcium sensor protein-directed dynamic library, Nat. Commun., 2019, 10, 2798–2811 CrossRef PubMed.
- E. B. Brown, W. S. Brey and W. Weltner, Cell-surface carbohydrates and their interactions: I. NMR of N-acetyl neuraminic acid, Biochim. Biophys. Acta, Gen. Subj., 1975, 399, 124–130 CrossRef CAS PubMed.
- T. Klepach, I. Carmichael and A. S. Serianni, 13C-Labeled N-Acetyl-neuraminic Acid in Aqueous Solution: Detection and Quantification of Acyclic Keto, Keto Hydrate, and Enol Forms by 13C NMR Spectroscopy, J. Am. Chem. Soc., 2008, 130, 11892–11900 CrossRef CAS PubMed.
- A. J. Benie, A. Blume, R. R. Schmidt, W. Reutter, S. Hinderlich and T. Peters, Characterization of Ligand Binding to the Bifunctional Key Enzyme in the Sialic Acid Biosynthesis by NMR, J. Biol. Chem., 2004, 279, 55722–55727 CrossRef CAS PubMed.
- S. C. Chen, C. H. Huang, S. J. Lai, C. S. Yang, T. H. Hsiao, C. H. Lin, P. K. Fu, T. P. Ko and Y. Chen, Mechanism and inhibition of human UDP-GlcNAc 2-epimerase, the key enzyme in sialic acid biosynthesis, Sci. Rep., 2016, 6, 23274–23284 CrossRef CAS PubMed.
- J. Martinez, L. D. Nguyen, S. Hinderlich, R. Zimmer, E. Tauberger, W. Reutter, W. Saenger, H. Fan and S. Moniot, Crystal structures of N-acetylmannosamine kinase provide insights into enzyme activity and inhibition, J. Biol. Chem., 2012, 287, 13656–13665 CrossRef CAS PubMed.
- P. Schanda, V. Forge and B. Brutscher, Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy, Proc. Natl. Acad. Sci., 2007, 104, 11257–11262 CrossRef CAS PubMed.
- D. Gołowicz, P. Kasprzak, V. Orekhov and K. Kazimierczuk, Fast time-resolved NMR with non-uniform sampling, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 116, 40–55 CrossRef PubMed.
- J. Van Rinsum, W. Van Dijk, G. J. Hooghwinkel and W. Ferwerda, Subcellular localization and tissue distribution of sialic acid precursor-forming enzymes, Biochem. J., 1983, 210, 21–28 CrossRef CAS PubMed.
- S. Ghosh and S. Roseman, The Sialic acids V. N-acyl-D-glucosamine 2-epimerase, J. Biol. Chem., 1965, 240, 1531–1536 CrossRef CAS PubMed.
- S. J. Luchansky, K. J. Yarema, S. Takahashi and C. R. Bertozzi, GlcNAc 2-epimerase can serve a catabolic role in Sialic acid metabolism, J. Biol. Chem., 2003, 278, 8035–8042 CrossRef CAS PubMed.
- H. Chen, A. Blume, M. Zimmermann-Kordmann, W. Reutter and S. Hinderlich, Purification and characterization of N-acetylneuraminic acid-9-phosphate synthase from rat liver, Glycobiology, 2002, 12, 65–71 CrossRef CAS PubMed.
- K. Viswanathan, S. Lawrence, S. Hinderlich, K. J. Yarema, Y. C. Lee and M. J. Betenbaugh, Engineering Sialic Acid Synthetic Ability into Insect Cells: Identifying Metabolic Bottlenecks and Devising Strategies To Overcome Them, Biochemistry, 2003, 42, 15215–15225 CrossRef CAS PubMed.
- A. Castilho, R. Strasser, J. Stadlmann, J. Grass, J. Jez, P. Gattinger, R. Kunert, H. Quendler, M. Pabst, R. Leonard, F. Altmann and H. Steinkellner, In planta protein sialylation through overexpression of the respective mammalian pathway, J. Biol. Chem., 2010, 285, 15923–15930 CrossRef CAS PubMed.
- M. B. Allen and D. G. Walker, The isolation and preliminary characterization of Nacetyl-D-glucosamine kinase from rat kidney and liver, Biochem. J., 1980, 185, 565–575 CrossRef CAS PubMed.
- S. Hinderlich, S. Nohring, C. Weise, P. Franke, R. Stasche and W. Reutter, Purification and characterization of N-acetylglucosamine kinase from rat liver. Comparison with UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase, Eur. J. Biochem., 1998, 252, 133–139 CrossRef CAS PubMed.
- N. Carrillo, M. C. Malicdan and M. Huizing, GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges, Neurotherapeutics, 2018, 15, 900–914 CrossRef PubMed.
- S. Van Wart, D. E. Mager, C. J. Bednasz, M. Huizing and N. Carrillo, Population Pharmacokinetic Model of N-acetylmannosamine (ManNAc) and N-acetylneuraminic acid (Neu5Ac) in Subjects with GNE Myopathy, Drugs R&D, 2021, 21, 189–202 CrossRef CAS PubMed.
- N. Carrillo, M. C. Malicdan, P. Leoyklang, J. A. Shrader, G. Joe, C. Slota, J. Perreault, J. D. Heiss, B. Class, C.-Y. Liu, K. Bradley, C. Jodarski, C. Ciccone, C. Driscoll, R. Parks, S. Van Wart, L. Bayman, C. S. Coffey, M. Quintana, S. M. Berry, M. Huizing and W. A. Gahl, Safety and efficacy of N-acetylmannosamine (ManNAc) in patients with GNE myopathy: an open-label phase 2 study, Genet. Med., 2021, 23, 2067–2075 CrossRef CAS PubMed.
- S. Hinderlich, M. Berger, M. Schwarzkopf, K. Effertz and W. Reutter, Molecular cloning and characterization of murine and human N-acetylglucosamine kinase, Eur. J. Biochem., 2000, 267, 3301–3308 CrossRef CAS PubMed.
- J. Davagnino and T. Ureta, The identification of extrahepatic “glucokinase” as N-acetylglucosamine kinase, J. Biol. Chem., 1980, 255, 2633–2636 CrossRef CAS PubMed.
- M. B. Allen and D. G. Walker, Kinetic characterization of Nacetyl-D-glucosamine kinase from rat liver and kidney, Biochem. J., 1980, 185, 577–582 CrossRef CAS PubMed.
- S. Hinderlich, W. Weidemann, T. Yardeni, R. Horstkorte and M. Huizing, UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A Master Regulator of Sialic Acid Synthesis, Top. Curr. Chem., 2015, 366, 97–137 CrossRef CAS PubMed.
- R. Seppala, V.-P. Lehto and W. A. Gahl, Mutations in the Human UDP-N-Acetylglucosamine 2-Epimerase Gene Define the Disease Sialuria and the Allosteric Site of the Enzyme, Am. J. Hum. Genet., 1999, 64, 1563–1569 CrossRef CAS PubMed.
- H. Ferreira, R. Seppala, R. Pinto, M. Huizing, E. Martins, A. C. Braga, L. Gomes, D. M. Krasnewich, M. C. Sa Miranda and W. A. Gahl, Sialuria in a Portuguese Girl: Clinical, Biochemical, and Molecular Characteristics, Mol. Genet. Metab., 1999, 67, 131–137 CrossRef CAS PubMed.
- F. Liu, H.-M. Chen, Z. Armstrong and S. G. Withers, Azido Groups Hamper Glycan Acceptance by Carbohydrate Processing Enzymes, ACS Cent. Sci., 2022, 8, 656–662 CrossRef CAS PubMed.
- L. A. Walter, A. R. Batt, N. Darabedian, B. W. Zaro and M. R. Pratt, Azide- and Alkyne-Bearing Metabolic Chemical Reporters of Glycosylation Show Structure-Dependent Feedback Inhibition of the Hexosamine Biosynthetic Pathway, ChemBioChem, 2018, 19, 1918–1921 CrossRef CAS PubMed.
- J. E. G. A. Dold and V. Wittmann, Metabolic Glycoengineering with Azide- and Alkene-Modified Hexosamines: Quantification of Sialic Acid Levels, ChemBioChem, 2020, 22, 1243–1251 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06986e |
| This journal is © The Royal Society of Chemistry 2023 |