
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective construction of triaryl-substituted all-carbon quaternary stereocenters via organocatalytic arylation of oxindoles with azonaphthalenes†
Pengquan
Chen
ab,
Mei-Jun
Lv
b,
Jun Kee
Cheng
b,
Shao-Hua
Xiang
 b,
Xiang-Zhong
Ren
a,
Junmin
Zhang
*a and
Bin
Tan
*b
b,
Xiang-Zhong
Ren
a,
Junmin
Zhang
*a and
Bin
Tan
*b
aInternational Joint Research Center for Molecular Science, College of Chemistry and Environmental Engineering, College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: zhangjm@szu.edu.cn
bShenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: tanb@sustech.edu.cn
First published on 6th February 2023
Abstract
Azonaphthalenes have been verified as a class of effective arylation reagents in a variety of asymmetric transformations. Here a highly efficient approach to construct triaryl-substituted all-carbon quaternary stereocenters through chiral phosphoric acid-catalyzed enantioselective arylation of 3-aryl-2-oxindoles with azonaphthalenes is disclosed. This chemistry is scalable and displays excellent functional group tolerance, furnishing a series of 3,3-disubstituted 2-oxindole derivatives in good yields with excellent enantiocontrol. Preliminary mechanistic data suggest that the initially formed direct addition intermediate undergoes intramolecular annulation under acidic reaction conditions.
Introduction
Structures that contain all-carbon quaternary centers are widely found in natural products and pharmaceutical molecules.1,2 The development of highly efficient and enantioselective approaches to construct congested centers has been a long-standing challenge due to the steric repulsion encountered in installing the carbon appendages.3 Although diverse synthetic strategies have been elegantly devised in the last decade,4 the realization of enantioselective versions remains difficult. A more challenging scenario arises when the stereocenters contain triaryl-substitution, where the subtle difference between the three arene substituents could hinder stereodifferentiation.5,6 Therefore, straightforward routes to prepare specifically functionalized frameworks with triaryl-substituted all-carbon quaternary stereocenters are always desirable.5,6 The unique electronic nature of ortho- or para-quinone methides7 renders a convenient platform to forge such carbon centers by means of bifunctional chiral phosphoric acid (CPA) catalysis.8 A series of pioneering contributions have been made by the Sun group in utilizing racemic tertiary alcohols or 1,1-diarylethylenes as the precursors for ortho- or para-quinone methides in various heteroarylation reactions (Scheme 1a).5 In Liu's protocol, racemic 2,2-diarylacetonitriles were used for enantioselective oxidative cross-coupling with (hetero)arenes (Scheme 1b).6 Despite the robustness of these routes, an ortho- or para-hydroxyl group should be installed at one of the aromatic rings in the diaryl substrates for in situ generation of the reactive quinone methide intermediates. In this context, the exploration of novel strategies which could accommodate substrates with diverse structures remains meaningful and necessary.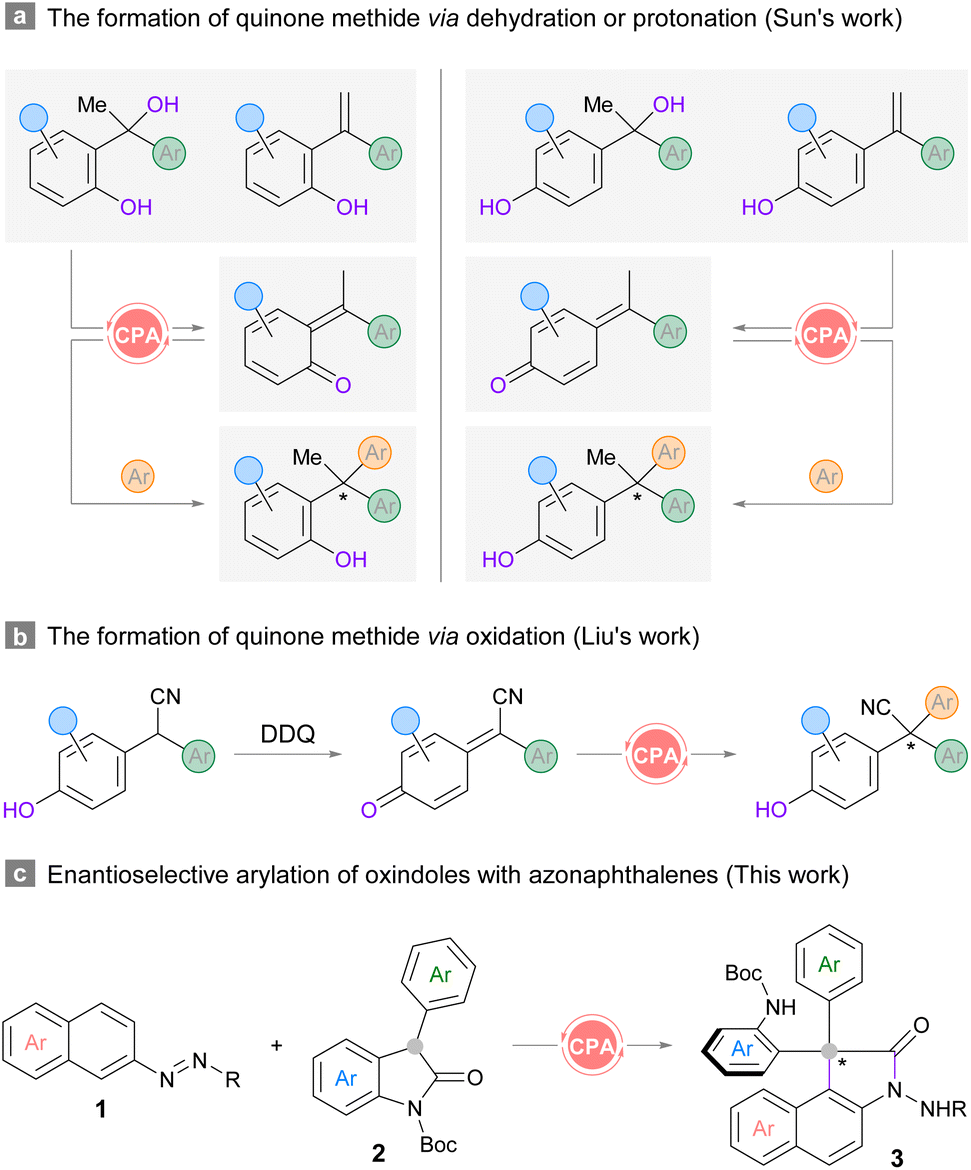 | ||
| Scheme 1 Representative catalytic asymmetric construction triaryl-substituted all-carbon quaternary centers and our design. | ||
C3-substituted oxindoles exist extensively in biologically active compounds,2,9 and they are also appreciated as fundamental building blocks10 and reaction intermediates11 due to the wide range of synthetic opportunities. Accordingly, the synthesis and transformation of C3-substituted oxindoles as well as their derivatives receive intense research focus, especially in the total synthesis of complex molecules. We contemplated a scheme to construct sterically congested triaryl-substituted quaternary stereocenters by harnessing the verified nucleophilicity of 3-aryl-2-oxindoles, which would necessitate suitable electrophilic arenes as the arylation reagents. Our previous finding proved that the umpolung of the nucleophilic aromatic rings could be realized by installing an azo substituent at the C2-position of naphthalene as the directing and activating group in the presence of a CPA catalyst.12a This strategy has also been applied in the construction of diverse axially and centrally chiral structures by our and other groups.12,13 This success led us to consider the union of azonaphthalenes and C3-aryl-2-oxindoles for the enantioselective construction of triaryl-substituted all-carbon quaternary stereocenters. The recruitment of a bifunctional CPA catalyst should be able to control the configuration of the stereogenic carbon center when both reacting partners are brought together. However, the regioselectivity might be complicated by the steric size of oxindole nucleophiles, directing arylation at more accessible sites on azo-arene substrates.
Herein we report an organocatalytic enantioselective arylation of C3-aryl-2-oxindoles with azonaphthalenes. The reaction develops through sequential nucleophilic addition at the ortho-carbon and skeletal rearrangement. The latter is initiated by the electrophilic interception at the hydrazine nitrogen under acidic conditions. The in situ polarity inversion of the nitrogen on the azo-group enables an overall intermolecular annulation between the two substrates to provide oxindoles harbouring a triaryl-substituted all-carbon quaternary stereocenter (Scheme 1c). This protocol also enriches the current synthetic toolbox of 3,3-diaryl-2-oxindoles, which mostly generates racemic analogues through substrate-specific reactions, including Friedel–Crafts type arylations, cyclizations and rearrangement reactions,11c,14 or from pre-functionalized substrates.15
Results and discussion
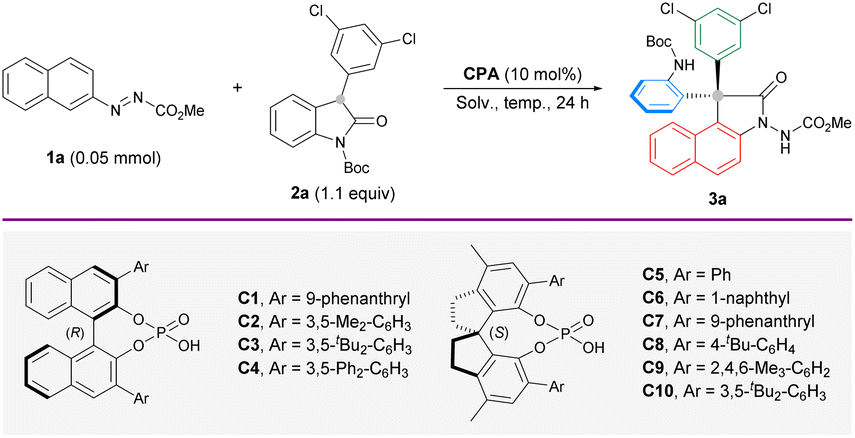
Our initial attempt of enantioselective arylation was performed with azonaphthalene 1a and 3-aryl-2-oxindole 2a as the model substrates in the presence of catalyst (R)-C1. Delightfully, the reaction performed in toluene at 25 °C gave an annulated product 3a in 31% yield and 35% ee (Table 1, entry 1), which features a change in the original connectivity of the substrates. This interesting outcome encouraged us to screen different CPA catalysts. Having observed that BINOL-derived catalysts could not effectively improve the yield and enantioselectivity (entries 2–4), another subclass of CPAs based on a methyl-SPINOL skeleton (entries 5–10) was investigated. By focusing on the influence of the steric environment, (S)-C10 with a bulky 3,5-di-butyl-phenyl group was found to afford the best enantiocontrol (70% ee) in 44% yield (entry 10). The much lower ee achieved with the BINOL-derived counterpart highlights the importance of both the backbone and sidearm components in enhancing reaction efficiency and stereoselectivity (entry 3). Subsequent optimization was conducted with the best-performing (S)-C10. Compared to toluene, other solvents dramatically hampered the enantioselectivity (entries 11–14). This remarkable solvent effect indicates the positive influence of a nonpolar solvent on the stereocontrol. It was noted that the yield remained very low at room temperature throughout the optimization process. As some unstable intermediates could be traced from thin-layer chromatography (TLC) analysis, we sought to drive better conversion by elevating the reaction temperature. A temperature gradient from 50 °C to 90 °C was explored (entries 15–19). As expected, the chemical yield saw substantial improvement. Surprisingly, the enantioselectivity increased significantly. From these attempts, 80 °C was identified as the most optimal temperature (entry 18) to furnish the desired arylation product 3a in 89% yield and 96% ee. A slightly better yield (93%) was obtained when the reaction was performed on the 0.2 mmol scale (entry 20).|
|
|||||
|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.05 mmol), 2a (1.1 equiv.) and CPA (10 mol%) in solvent (1 mL) for 24 h unless noted otherwise. b Isolated yield. c The ee values were determined by chiral HPLC analysis. d 1a (0.20 mmol) and 2a (0.22 mmol) in solvent (4 mL). | |||||
| Entry | CPA | Solv. | T (°C) | Yieldb (%) | eec (%) |
| 1 | (R)-C1 | Toluene | 25 | 31 | 35 |
| 2 | (R)-C2 | Toluene | 25 | 29 | 20 |
| 3 | (R)-C3 | Toluene | 25 | 30 | 20 |
| 4 | (R)-C4 | Toluene | 25 | 27 | 8 |
| 5 | (S)-C5 | Toluene | 25 | 40 | 9 |
| 6 | (S)-C6 | Toluene | 25 | 38 | 17 |
| 7 | (S)-C7 | Toluene | 25 | 43 | 60 |
| 8 | (S)-C8 | Toluene | 25 | 39 | 14 |
| 9 | (S)-C9 | Toluene | 25 | 36 | 31 |
| 10 | (S)-C10 | Toluene | 25 | 44 | 70 |
| 11 | (S)-C10 | CH2Cl2 | 25 | 44 | 35 |
| 12 | (S)-C10 | CHCl3 | 25 | 51 | 18 |
| 13 | (S)-C10 | MeCN | 25 | 37 | 55 |
| 14 | (S)-C10 | Dioxane | 25 | 17 | 50 |
| 15 | (S)-C10 | Toluene | 50 | 76 | 89 |
| 16 | (S)-C10 | Toluene | 60 | 88 | 85 |
| 17 | (S)-C10 | Toluene | 70 | 88 | 91 |
| 18 | (S)-C10 | Toluene | 80 | 89 | 96 |
| 19 | (S)-C10 | Toluene | 90 | 91 | 90 |
| 20d | (S)-C10 | Toluene | 80 | 93 | 96 |
Having established the optimal reaction conditions, we turned to investigate the substrate scope of this enantioselective arylation reaction with respect to the azonaphthalene component. Aliphatic esters other than methyl ester were also compatible to deliver products 3b–g in 75–92% yield and 84–95% ee (Scheme 2a). However, the yield and enantioselectivity decreased gradually as steric hindrance intensified from ethyl to tert-butyl groups (3b–e). In general, the position (C6 or C7) and electronic nature of the aryl substituents on azonaphthalene had limited influence on the enantioselectivities of arylation products (3h–o). It is worth pointing out that the reaction with C6-ester-substituted azonaphthalene (3k) proceeded in poorer yield (74%) and the formation of unidentified byproducts could be detected on the TLC. Remarkably, 3-bromoazonaphthalene was well-suited to this reaction system to deliver product 3p in 89% yield and 92% ee (Scheme 2b). This result suggests that the substrate–catalyst interaction is not interfered with by the presence of a bulky substituent adjacent to the azo group at the naphthalene ring.
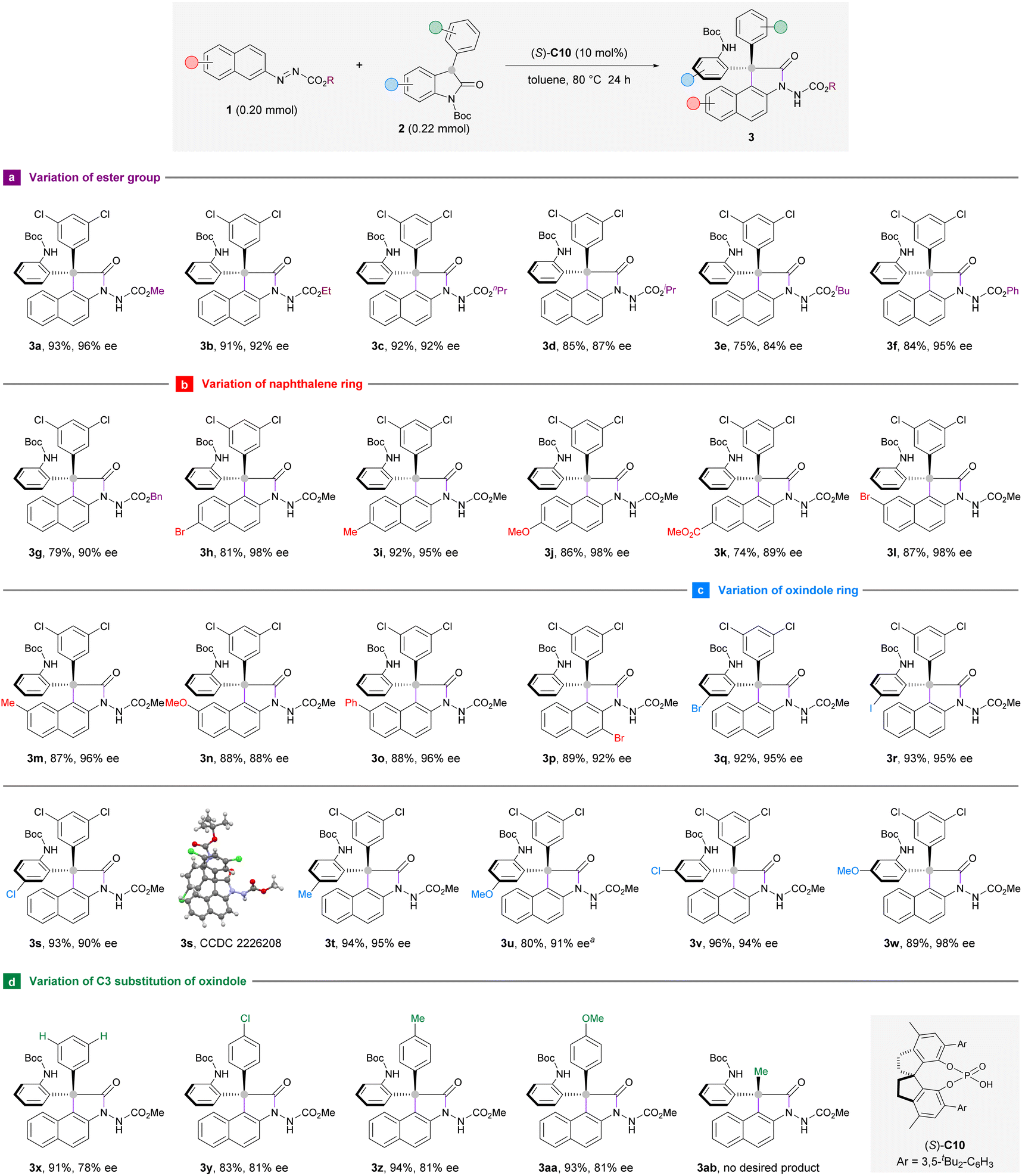 | ||
| Scheme 2 Substrate generality of the CPA catalyzed enantioselective arylation reaction. Reaction conditions: 1 (0.20 mmol), 2 (0.22 mmol), (S)-C10 (10 mol%) in toluene (4 mL) at 80 °C for 24 h under argon, unless noted otherwise. Yield of the isolated product is given. The ee values were determined by HPLC analysis using a chiral stationary phase. aReaction was carried out at 25 °C for 48 h. | ||
To further define the scope of our developed protocol, the optimal conditions were then applied to different oxindole derivatives with azonaphthalene 1a as the standard arylation partner (Scheme 2c). The formation of triaryl-substituted stereocenters occurred smoothly, regardless of the identity of C5- or C6-substituents of the aromatic ring. Oxindoles that bear an electron-withdrawing (3q–s and 3v) or electron-donating (3t, 3u, and 3w) group could afford the products with high yields and excellent stereoselectivities. Considering that the Boc-protecting group of 3u would be labile at 80 °C, the reaction to prepare 3u was carried out at 25 °C with an extended time (48 hours). Furthermore, the C3 position of oxindole could be occupied by a phenyl group (3x) or its 4-substituted analogues with electron-withdrawing (3y) or electron-donating (3z and 3aa) functionalities. However, these reactions have afforded moderate enantioselectivities (78–81% ee). The introduction of a methyl group to this C3-position resulted in a messy reaction system and the target product 3ab bearing a diaryl-substituted stereocenter could not be isolated (Scheme 2d). Product 3s carrying a chloride substituent was crystalline, which allows unequivocal determination of the stereochemistry to be (S) at the all-carbon quaternary stereocenter by means of X-ray crystallographic analysis. The configurations of the other products displayed in this scheme were assigned by analogy.
We next examined the practicality and reliability of the present synthetic method by implementing a gram-scale reaction with 1a (1.07 g) and 2a (2.08 g) under standard reaction conditions. The isolation of 2.6 g of 3a (88% yield) in 96% ee showed that the outcomes were largely preserved (Scheme 3a). To gain more mechanistic insights into the reaction, the coupling of 1a and 2i was conducted at 25 °C instead of 80 °C used in the substrate survey. An unstable arylation compound 4 and (rac)-3x could be separated in 26% and 33% yields, respectively. On subjection to 10 mol% diphenyl phosphate, 4 converted completely into 3x after 4 hours at 25 °C (Scheme 3b). The application of standard conditions afforded 3x with 70% ee instead, after 30 minutes. These results suggested the intermediacy of 4 in this intermolecular annulation reaction, and a secondary kinetic resolution could be operative during the transamidation step to improve the enantiopurity of the products.
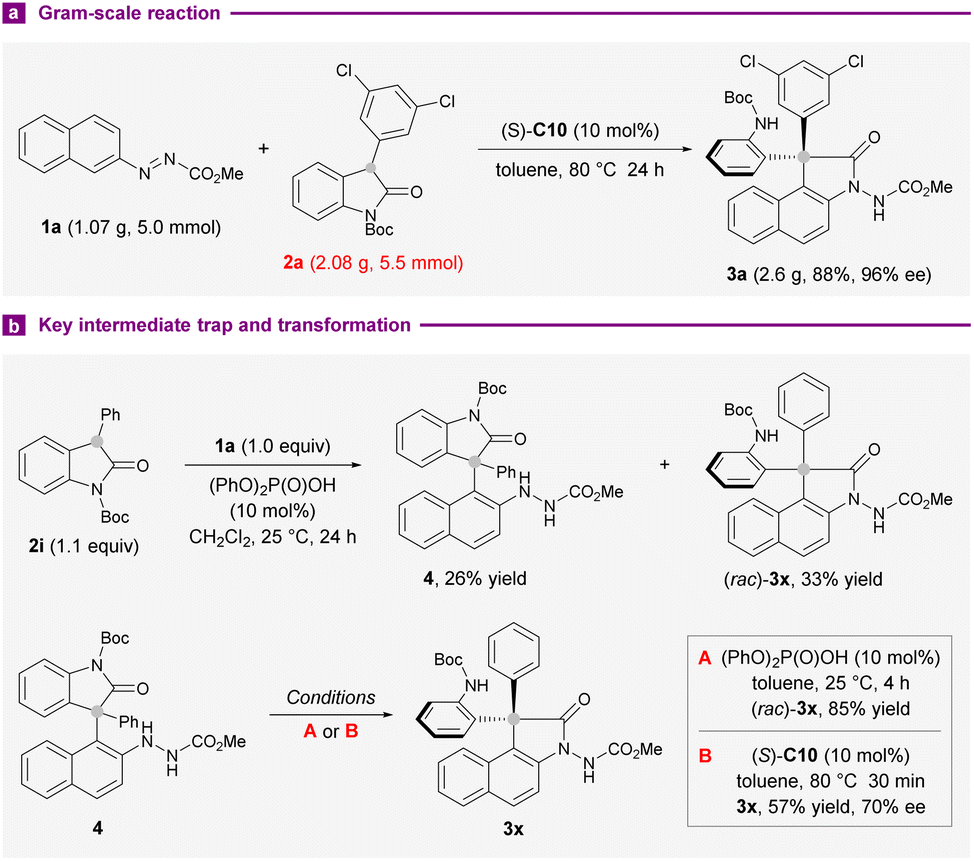 | ||
| Scheme 3 Gram-scale synthesis and mechanistic investigations. | ||
On the basis of these experimental observations and previous literature, we proposed a reaction mechanism as outlined in Scheme 4. The bifunctional CPA catalyst simultaneously activates azonaphthalene 1 and oxindole 2′ which is in the enol form through multiple hydrogen bondings, which leads to a stereoselective nucleophilic attack at the α-carbon of 1 to form intermediate A. By comparing the conversion rate of intermediate 4 and the reaction rate of 1a and 2i under standard conditions, this arylation process is likely to be rate-determining (for more details, see the ESI†). The following rearomatization step reveals key intermediate B, which undergoes an acid-promoted intramolecular addition to a carbonyl group on oxindole. Subsequently, acid mediates the ring opening of Cvia cleavage of the C–N bond to release the observed product 3.
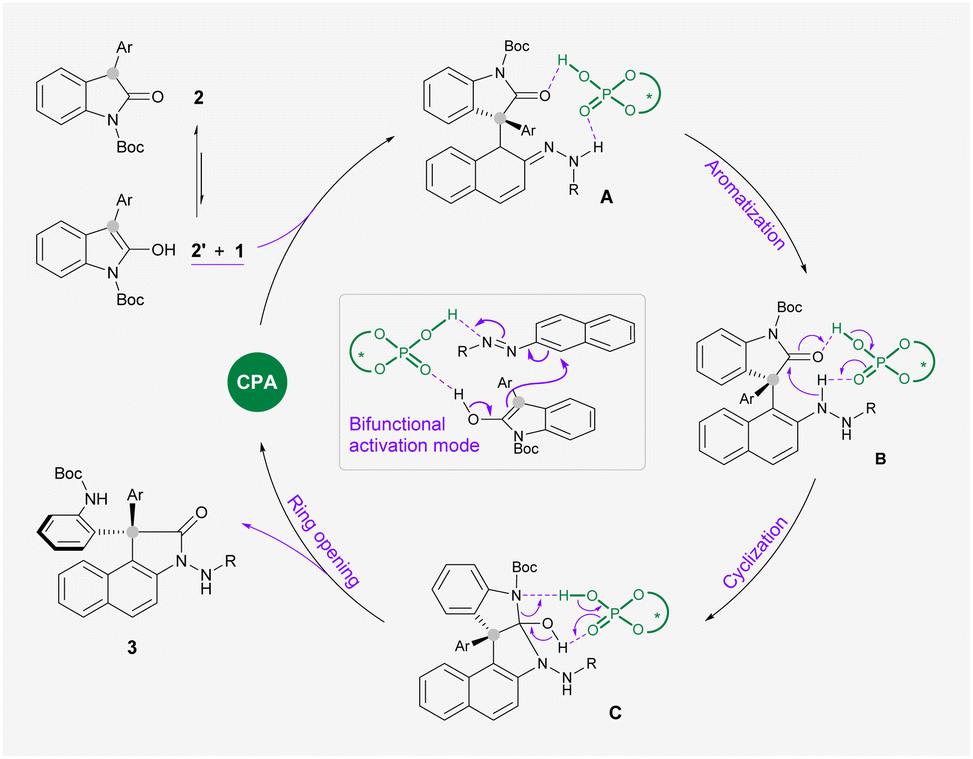 | ||
| Scheme 4 Proposed reaction mechanism and stereocontrol mode. | ||
Conclusions
In summary, we have successfully developed a CPA-catalyzed asymmetric arylation of oxindole derivatives with azonaphthalenes as the electrophilic arylation reagents for efficient construction of triaryl-substituted quaternary stereocenters in good yields with excellent enantioselectivities. This protocol provided a general and practical method to prepare a variety of 3,3-disubstituted oxindole derivatives with fairly good functional group tolerance for both azonaphthalene and oxindole partners. The proposed reaction mechanism is supported by the isolation of a key intermediate and its conversion to the target product. The overall reaction enantioselectivity could be enhanced by a secondary kinetic resolution. Efforts are currently underway in our laboratory to further investigate synthetic application and generality of this strategy.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 3s have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2226208.Author contributions
B. T. & J. Z. conceived and directed the project. P. C. designed and performed the experiments. M.-J. L., J. K. C., S.-H. X. & X.-Z. R. helped with the collection of some new compounds and data analysis. B. T., P. C., J. K. C. & S.-H. X. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21825105, 22231004 and 22271135), National Key R&D Program of China (2021YFF0701604), Guangdong Provincial Key Laboratory of Catalysis (2020B121201002), Guangdong Innovative Program (2019BT02Y335), and Shenzhen Science and Technology Program (KQTD20210811090112004, JCYJ20210324120205016, and JCYJ20210324105005015). The authors appreciate the assistance of SUSTech Core Research Facilities.Notes and references
- (a) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (b) S.-J. Han, F. Vogt, S. Krishnan, J. A. May, M. Gatti, S. C. Virgil and B. M. Stoltz, Org. Lett., 2014, 16, 3316–3319 CrossRef CAS PubMed; (c) S.-J. Han, F. Vogt, J. A. May, S. Krishnan, M. Gatti, S. C. Virgil and B. M. Stoltz, J. Org. Chem., 2015, 80, 528–547 CrossRef CAS PubMed.
- (a) M. K. Uddin, S. G. Reignier, T. Coulter, C. Montalbetti, C. Grånäs, S. Butcher, C. Krog-Jensen and J. Felding, Bioorg. Med. Chem. Lett., 2007, 17, 2854–2857 CrossRef CAS PubMed; (b) P. Paira, A. Hazra, S. Kumar, R. Paira, K. B. Sahu, S. Naskar, P. saha, S. Mondal, A. Maity, S. Banerjee and N. B. Mondal, Bioorg. Med. Chem. Lett., 2009, 19, 4786–4789 CrossRef CAS PubMed.
- (a) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef; (b) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (c) B. M. Trost and C. Jiang, Synthesis, 2006, 3, 369–396 CrossRef; (d) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (e) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC.
- (a) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314–19317 CrossRef CAS PubMed; (b) L. Gao, B. C. Kang and D. H. Ryu, J. Am. Chem. Soc., 2013, 135, 14556–14559 CrossRef CAS PubMed; (c) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (d) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef CAS PubMed; (e) K. Hojoh, Y. Shido, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2014, 53, 4954–4958 CrossRef CAS PubMed; (f) J.-S. Lin, T.-T. Li, J.-R. Liu, G.-Y. Jiao, Q.-S. Gu, J.-T. Cheng, Y.-L. Guo, X. Hong and X.-Y. Liu, J. Am. Chem. Soc., 2019, 141, 1074–1083 CrossRef CAS PubMed; (g) D. Ma, C.-B. Miao and J. Sun, J. Am. Chem. Soc., 2019, 141, 13783–13787 CrossRef CAS PubMed.
- (a) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910–1913 CrossRef CAS PubMed; (b) Z. Wang, Y. F. Wong and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 13711–13714 CrossRef CAS PubMed; (c) Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383–389 CrossRef CAS PubMed; (d) Z. Li, Y. Li, X. Li, M. Wu, M.-L. He and J. Sun, Chem. Sci., 2021, 12, 11793–11798 RSC.
- Z. Wang, Y. Zhu, X. Pan, G. Wang and L. Liu, Angew. Chem., Int. Ed., 2020, 59, 3053–3057 CrossRef CAS PubMed.
- (a) X. Li, Z. Li and J. Sun, Nat. Synth., 2022, 1, 426–438 CrossRef; (b) C. Dorsch and C. Schneider, Synthesis, 2022, 54, 3125–3141 CrossRef CAS.
- For pioneering work: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed, For typical reviews: (c) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (d) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- (a) A. A. Raj, R. Raghunathan, M. R. SrideviKumari and N. Raman, Bioorg. Med. Chem. Lett., 2003, 11, 407–419 CrossRef CAS PubMed; (b) A. Kamal, Y. V. V. Srikanth, M. N. A. Khan, T. B. Shaik and M. Ashraf, Bioorg. Med. Chem. Lett., 2010, 20, 5229–5231 CrossRef CAS PubMed; (c) G. Wang, J. Wang, Z. Xie, M. Chen, L. Li, Y. Peng, S. Chen, W. Li and B. Deng, Bioorg. Chem., 2017, 72, 228–233 CrossRef CAS PubMed.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS; (c) A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed; (d) X. Tang, H. Ni and Y. Lu, Org. Chem. Front., 2021, 8, 4485–4489 RSC; (e) J. Lin, M. Jia and S. Ma, Chin. J. Chem., 2021, 39, 3044–3050 CrossRef CAS; (f) M. Gao, Y. Li, X. Li and L. Hu, Org. Lett., 2022, 24, 875–880 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, M. Bella, D. Y.-K. Chen, X. Huang, T. Ling and S. A. Snyder, Angew. Chem., Int. Ed., 2002, 41, 3495–3499 CrossRef CAS; (b) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS; (c) K. C. Nicolaou, D. Y.-K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, J. Am. Chem. Soc., 2004, 126, 12888–12896 CrossRef CAS PubMed; (d) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (e) C.-K. Mai, M. F. Sammons and T. Sammakia, Angew. Chem., Int. Ed., 2010, 49, 2397–2400 CrossRef CAS PubMed; (f) J. Xu, L.-D. Shao, D. Li, X. Deng, Y.-C. Liu, Q.-S. Zhao and C. Xia, J. Am. Chem. Soc., 2014, 136, 17962–17965 CrossRef CAS PubMed; (g) J. Park, A. Jean and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2017, 56, 14237–14240 CrossRef CAS PubMed.
- (a) L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58–64 CrossRef CAS PubMed; (b) L.-W. Qi, S. Li, S.-H. Xiang, J. Wang and B. Tan, Nat. Catal., 2019, 2, 314–323 CrossRef CAS; (c) W. Xia, Q.-J. An, S.-H. Xiang, S. Li, Y.-B. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775–6779 CrossRef CAS PubMed.
- (a) C. Ma, J.-Y. Zhou, Y.-Z. Zhang, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5398–5402 CrossRef CAS PubMed; (b) Z. Wu and J. Wang, Chem. Sci., 2019, 10, 2501–2506 RSC; (c) Y.-L. Hu, Z. Wang, H. Yang, J. Chen, Z.-B. Wu, Y. Lei and L. Zhou, Chem. Sci., 2019, 10, 6777–6784 RSC; (d) H. Zhao, H. Yuan, Y. Zhang, R. Li and W. Li, Org. Lett., 2019, 21, 6557–6561 CrossRef CAS PubMed; (e) H. Yuan, Y. Li, H. Zhao, Z. Yang, X. Li and W. Li, Chem. Commun., 2019, 55, 12715–12718 RSC; (f) S. Cen, N. Huang, D. Lian, A. Shen, M.-X. Zhao and Z. Zhang, Nat. Commun., 2022, 13, 4735 CrossRef CAS PubMed.
- (a) A. Swetha, G. S. Kumar, A. S. Kumar and H. M. Meshram, Tetrahedron Lett., 2014, 55, 4705–4710 CrossRef CAS; (b) K. K. S. Sai, P. M. Esteves, E. T. da Penha and D. A. Klumpp, J. Org. Chem., 2008, 73, 6506–6512 CrossRef CAS PubMed; (c) D. A. Klumpp, K. Y. Yeung, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1998, 63, 4481–4484 CrossRef CAS; (d) P. Magnus and R. Turnbull, Org. Lett., 2006, 8, 3497–3499 CrossRef CAS PubMed.
- (a) S. Shirakawa, K. Koga, T. Tokuda, K. Yamamoto and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 6220–6223 CrossRef CAS PubMed; (b) C.-K. Mai, M. F. Sammons and T. Sammakia, Org. Lett., 2010, 12, 2306–2309 CrossRef CAS PubMed; (c) R. Samineni, C. R. C. Bandi, P. Srihari and G. Mehta, Org. Lett., 2016, 18, 6184–6187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2226208. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc07103g |
| This journal is © The Royal Society of Chemistry 2023 |