
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the mechanism of synergetic photoredox/copper(I)-catalyzed carbocyanation of 1,3-dienes: a DFT study†
Yanhong
Liu
 ab,
Aili
Feng
a,
Rongxiu
Zhu
a and
Dongju
Zhang
*a
ab,
Aili
Feng
a,
Rongxiu
Zhu
a and
Dongju
Zhang
*a
aKey Lab of Colloid and Interface Chemistry, Ministry of Education, Institute of Theoretical Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: zhangdj@sdu.edu.cn
bSchool of Radiology, Shandong First Medical University & Shandong Academy of Medical Sciences, Taian 271016, P. R. China
First published on 29th March 2023
Abstract
This work presents a DFT-based computational study to understand the mechanism, and regio- and enantioselectivities in the synergetic photoredox/copper(I)-catalyzed carbocyanation of 1,3-dienes with alkyl redox-active esters. The calculated results show an unprecedented copper catalytic mechanism, where the reaction follows a catalytic cycle involving CuI-only catalysis, instead of a Cu(I)/Cu(II)/Cu(III)/Cu(I) cycle as proposed in the experimental study. Moreover, it is found that the critical step involves the reaction of the cyanocopper(I) species with an allyl cation rather than the cyanocopper(II) species reacting with an allyl radical as proposed in the experiment, and that the photocatalyst is regenerated via single electron transfer from the allyl radical to the oxidized photocatalyst. In the newly proposed photoredox/copper(I) catalysis, the reaction consists of four stages: (i) generation of the copper(I) active catalyst, (ii) formation of an allyl radical with oxidative quenching of the photoexcited species, (iii) generation of an allylcopper complex accompanied by the regeneration of the photocatalyst, and (iv) formation of the allyl cyanide product with the regeneration of the copper(I) active catalyst. The cyanation of the allyl cation is calculated to be the regio- and enantioselectivity-determining step. The excellent regio- and stereoselectivities are attributed to the favorable CH–π interaction between the substrate and catalyst as well as the small distortion of the substrate.
Introduction
The selective difunctionalization of 1,3-dienes, in particular, their asymmetric difunctionalization, which provides various value-added chemicals, has attracted considerable attention from both industrial and academic communities.1,2 Over the last few decades, synthetic chemists have developed an efficient and practical protocol for the asymmetrical and selective difunctionalization of 1,3-dienes by using transition metal catalysts, such as Ni,3 Pd,4 Rh,5,6 and Cu.7,8 Recently, the merger of photoredox catalysis with chiral organometallic catalysis has found widespread application in a variety of catalytic asymmetric difunctionalizations of 1,3-dienes, including their 1,2-dialkylations,9 1,2-alkylesterification,10,11 and 1,2- or 1,4-aminoalkylation.12,13Nitriles are an important class of organic compounds and the cyano group is a versatile building block which can be easily converted into various derivatives such as carboxylic acids, amines, amides, and ketones.14,15 Introducing the cyano group into organic molecules is an important strategy in synthetic chemistry.16–18 In recent years, the cyanations of alkenes via using trimethylsilyl cyanide (TMSCN) as the provider of the cyano group have been well developed.19–25
Quite recently, Xiao and co-workers reported the first example of the visible-light-induced enantioselective carbocyanation of 1,3-dienes through synergetic photoredox/copper catalysis.26 As shown by a representative reaction in Scheme 1, the carbocyanation of 1-phenyl-1,3-diene (1) is achieved by using a cyclohexyl (Cy)-substituted carboxylic acid derived N-hydroxyphthalimide (NHP) ester (2) and TMSCN (3) in the presence of an organic photocatalyst (PC), Cu(MeCN)4BF4, and chiral bisoxazoline ligand (L). The reaction selectively affords the desired 1,2-difunctionalization product (4) in good yield (71%) and excellent enantioselectivity (92% ee) without the observation of the 1,4-regioisomer (5).
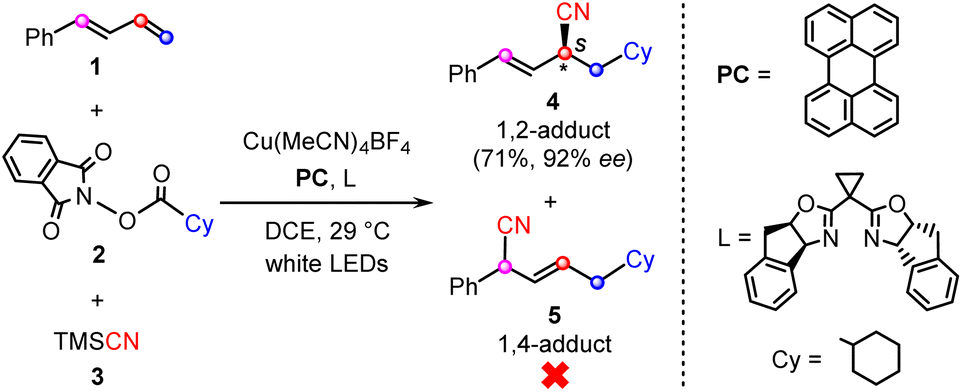 | ||
| Scheme 1 Representative reaction of photoredox/copper-catalyzed carbocyanations of 1,3-dienes, reported by Xiao et al.26 | ||
Scheme 2 summarizes the proposed reaction mechanism of synergetic photoredox/copper catalysis by Xiao et al.26 Initially, under visible light irradiation, the photocatalyst (PC) is excited to its triplet excited-state PC*, from which a single electron transfer (SET) to redox-active NHP ester 2 occurs to give the reduced ester 2˙− with the oxidized photocatalyst PC˙+. The resulting 2˙− carries out decarboxylation with the release of CO2 and the NHP− anion to afford cyclohexyl radical Cy˙, which is then captured by 1,3-diene 1 to yield allyl radical A. Meanwhile, the chiral copper(I) catalyst B combines a cyanide anion (CN−) generated in situ from the reaction between TMSCN (3) with the anion NHP− to give the neutral CuI–CN complex C, which is subsequently oxidized by PC˙+ to CuII–CN complex Dvia SET, closing the photocatalytic cycle. Next, allyl radical A adds to the CuII center to form π-allylcopper(III) complexes E. Finally, reductive elimination from the Cu(III) species leads to product 4 with the regeneration of CuI catalyst B.
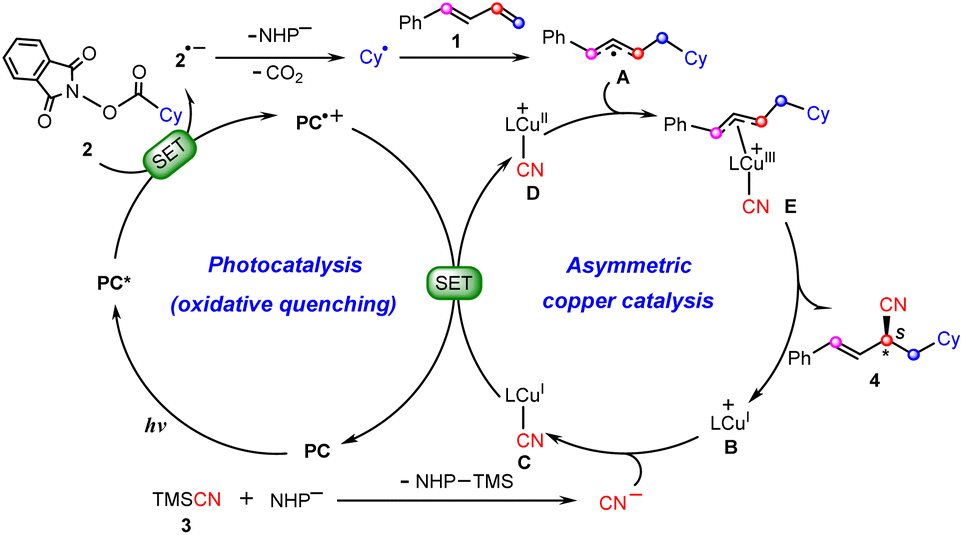 | ||
| Scheme 2 Proposed mechanisms of dual photoredox and copper-catalyzed carbocyanation of 1,3-diene reactions. | ||
Obviously, the mechanism described above involves a Cu(I)/Cu(II)/Cu(III)/Cu(I) cycle, which is similar to those found recently by Guan,10,27,28 Lin,29,30 and Lan20via density functional theory (DFT) calculations on the synergetic photoredox/copper(I)-catalyzed cyanations of alkenes and enynes. However, our calculations show that such a copper and photoredox catalytic cycle is not operative. Alternatively, we propose an only CuI-involving mechanism, where the allyl cyanide product is obtained by direct coupling between the copper(I) species and allyl cation through a unique outer-sphere mechanism. In the photoredox cycle, the oxidized photocatalyst is reduced by the allyl radical. With this new mechanism, the observed regio- and enantioselectivities can be rationalized well. The present study is expected to provide insights into designing future copper-based catalytic systems.
Computational details
The reaction shown in Scheme 1 was chosen as the prototype system of photoredox/copper(I)-catalyzed carbocyanation of 1,3-dienes reported by Xiao et al.26 All DFT calculations were carried out using the Gaussian 16 program.31 Geometry optimizations of stationary points were performed using the M06 functional32 with the LANL2DZ effective core potential (ECP) basis set33 augmented by an f-type polarization function (ζf = 3.525)34 for the copper atom and 6-31G(d,p)35 for other atoms. Our benchmark calculations, utilizing both the SDD36 and Def2-TZVP37 basis sets for the Cu atom, provide support for the utilization of the LANL2DZ ECP basis set for the current Cu-containing system (Table S1†). The M06 suite of density functionals has been evaluated by Truhlar et al.,32 who recommend it for the thermochemistry study of organometallic and inorganometallic systems and for the study of noncovalent interactions. The extensive subsequent uses of this functional attest to its effectiveness in describing structures and calculating thermochemistry in transition metal systems.38,39 The solvent effect of 1,2-dichloroethane (DCE) was taken into account using the polarizable continuum model (PCM)40 in the geometry optimization calculations. Frequency analyses were performed at the same level of theory to confirm the optimized structures to be local minima (with zero imaginary frequency) or first-order saddle points (with only one imaginary frequency). Intrinsic reaction coordinate (IRC)41 calculations were also performed to ensure that the transition states connected the reactant or product properly. The relative Gibbs free energy of each structure was further refined by performing single-point energy calculations with the larger 6-311+G(d,p) basis set for all atoms except for the copper atom. All reported energies of intermediates and transition states are solvated Gibbs free energies. The energy calibration calculations (Table S2†) were performed using the domain-based local pair natural orbital coupled cluster method,42 DLPNO-CCSD(T), with the cc-PVTZ basis set,43 in conjunction with the SMD solvation model,44 as implemented in the Orca 5.0.3 program.45 The input file for the DLPNO-CCSD(T)/cc-PVTZ calculations was prepared using Multiwfn 3.8 software.46 Natural population analyses (NPA) were carried out using the NBO 6.0 program.47 The 3D images of key transition states were obtained using the CYLview program,48 and the non-covalent interactions (NCIs) were visualized using the visual molecular dynamics (VMD) software.49Results and discussion
It is anticipated that with the presence of cyclopropyl-substituted bisoxazoline ligand L, the precatalyst Cu(MeCN)4+ can be transformed into several different copper(I) species by performing ligand exchange, as shown by several representative examples in Fig. 1. In terms of the calculated Gibbs free energy changes, LCu(MeCN)+ is identified as the most stable structure and is therefore chosen as the energy reference point.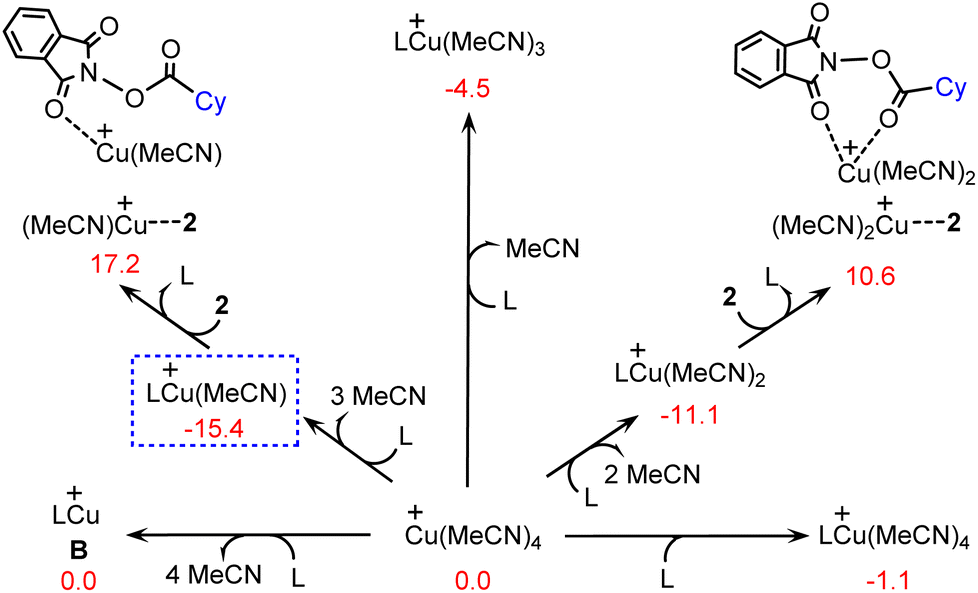 | ||
| Fig. 1 Calculated Gibbs free energy changes (kcal mol−1) for the formation of several copper(I) species. | ||
According to Scheme 2, the reaction involves three key processes: generation of the allyl radical, formation of the allylcopper complex, and formation of the allyl cyanide product. The following subsections discuss these processes one by one.
Generation of the allyl radical
According to Scheme 2, this process starts from the oxidative quenching of photoexcited high-energy species PC*via a SET to the redox-active ester 2. Our calculations indicate that this process is endergonic by 20.8 kcal mol−1 and involves a barrier of 20.9 kcal mol−1 (Fig. S1†), which is estimated from Marcus–Hush theory.50,51 Interestingly, we located an energetically more favourable pathway for the oxidative quenching of PC* (Fig. 2), in which the quencher used is the complex LCu+⋯2 of NHP ester 2 with LCu+. This complex is generated through a ligand exchange reaction, where MeCN in the catalytically active species LCu(MeCN)+ is replaced by NHP ester 2.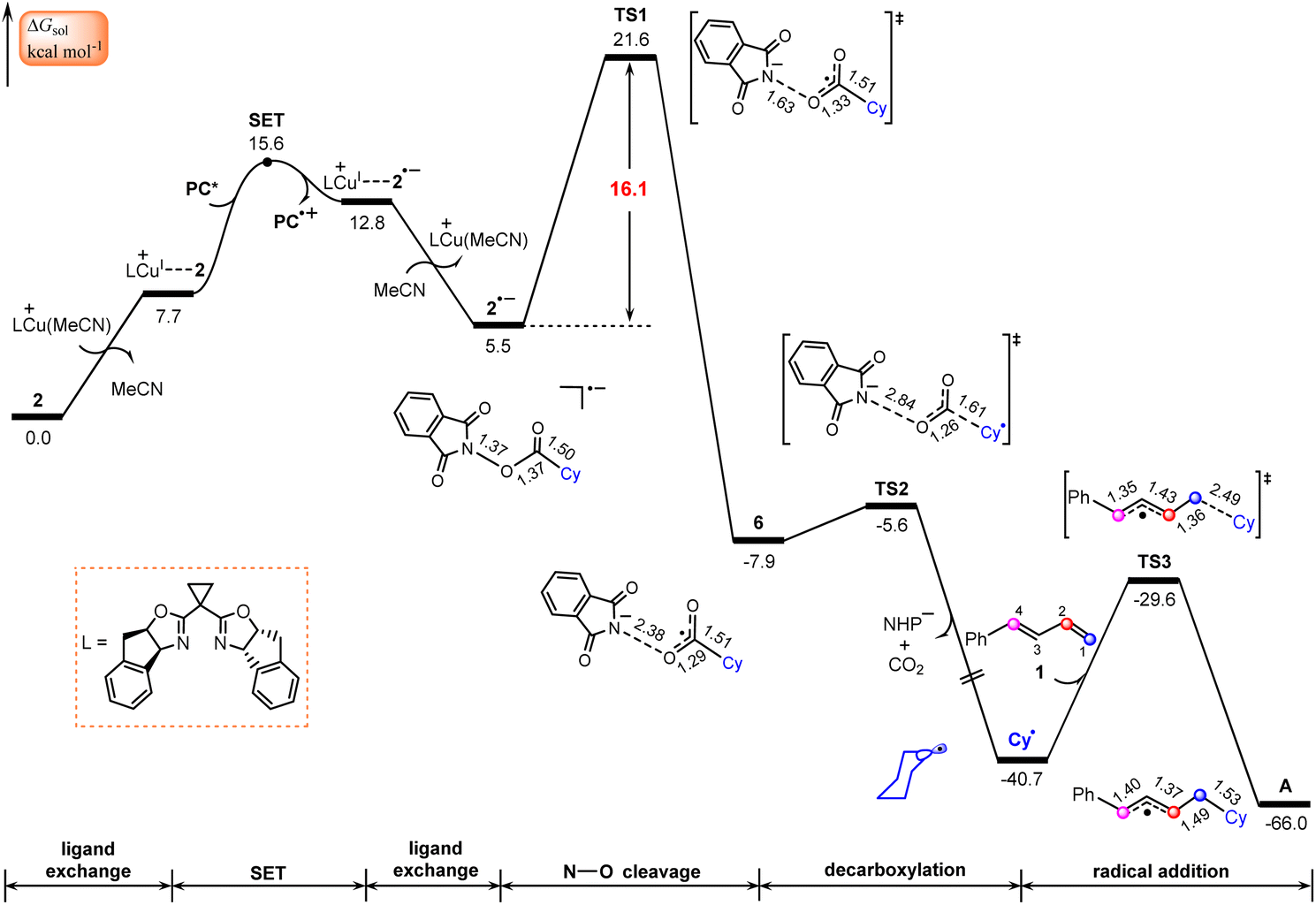 | ||
| Fig. 2 Calculated Gibbs free energy profile for generation of the allyl radical. Bond distances are in Å. | ||
The SET from PC* to LCu+⋯2, leading to the oxidized photocatalyst PC˙+ and LCu+⋯2˙−, an ion-pair complex between 2˙− and LCu+, is found to be endergonic by 5.1 kcal mol−1 with a barrier of 7.9 kcal mol−1, which is energetically more favourable than that using 2 as quencher (Fig. S1†). This fact indicates that the complex LCu+⋯2 of NHP ester 2 with LCu+ rather than by itself plays the role of quencher in the oxidative quenching of photoexcited high-energy species PC*. In other words, the LCu+ species assists the oxidative quenching of photoexcited species PC*. The lower LUMO energy level of complex LCu+⋯2 in comparison with NHP ester 2 is mainly responsible for the relatively easier SET process (−3.32 vs. −2.68 eV in Fig. S2†). Note that the radical species LCu(I)+⋯2˙− resulting from the photocatalytic cycle may also release the radical species 2˙−. This raises the question of whether these two radicals might add to 1-phenyl-1,3-diene (1) and thus interrupt the catalytic cycle. To address this question, we calculated the thermodynamics of adding these two species to 1, as shown in Fig. S3.† The calculated results indicate that both processes were endergonic, suggesting that they are unlikely to occur and would not interrupt the catalytic cycle.
Once formed, the ion-pair complex LCu+⋯2˙− undergoes ligand exchange with MeCN to give the reduced NHP ester 2˙− and release LCu(MeCN)+ (Fig. 2). NPA for 2˙− (Fig. S4†) show that the spin is located on the five-membered 2,3-dicarboxylimide heterocycle. Thus, the subsequent N–O bond cleavage of 2˙−viaTS1 would lead to intermediate 6, a complex between the NHP− anion and the cyclohexyl acyl radical. Next, 6 undergoes decarboxylation viaTS2 to generate the cyclohexyl radical Cy˙ and NHP− anion with release of CO2. The overall barrier for the formation of Cy˙ is calculated to be 21.6 kcal mol−1, implying that the Cy˙ radical is easily formed from the NHP esters. This is consistent with literature reports in which NHP esters are suggested to be the precursors of the alkyl radicals under visible light irradiation.26,52,53
It is known that 1,3-dienes are competent radical trapping reagents.54–56 Therefore, the generated Cy˙ radical can be captured by 1-phenyl-1,3-diene (1), leading to generation of allyl radical complex A. It is found that the addition of the Cy˙ radical on C1 of 1viaTS3 with a barrier of 11.1 kcal mol−1 is both kinetically and thermodynamically the most favorable pathway in comparison with three other pathways where the addition of the Cy˙ radical occurs on C2, C3, and C4 atoms, respectively (Fig. S5†).
Formation of the allylcopper complex
The calculated results according to Scheme 2 are shown in Fig. 3. Initially, LCu(MeCN)+ undergoes ligand exchange with the NHP− anion generated in situ from the NHP ester 2, and subsequently TMSCN (3) coordinates to the copper(I) center to afford an ion-pair intermediate 7, which then performs desilylation with the assistance of the NHP− anion viaTS4 with a relative Gibbs free energy of 14.6 kcal mol−1 to generate the isocyanocopper(I) intermediate 8 with release of the NHP–TMS species. Once formed, 8 can evolve into the more stable cyanocopper(I) intermediate Cvia a three-membered transition state, TS5. This process is calculated to be exergonic by 10.0 kcal mol−1 with a barrier of 5.3 kcal mol−1. Next, the cyanocopper(I) intermediate C reduces PC˙+ through the second SET to provide a cyanocopper(II) species D and regenerate the photocatalyst PC. The barrier of this SET process is estimated to be 19.0 kcal mol−1. We also calculated an alternative pathway leading to the cyanocopper(II) species D, in which the oxidation of isocyanocopper(I) intermediate 8 by PC˙+ occurs prior to the isomerization from isocyanocopper to cyanocopper. However, such a pathway is significantly less favorable than that discussed above, as indicated by the red energy profile in Fig. 3.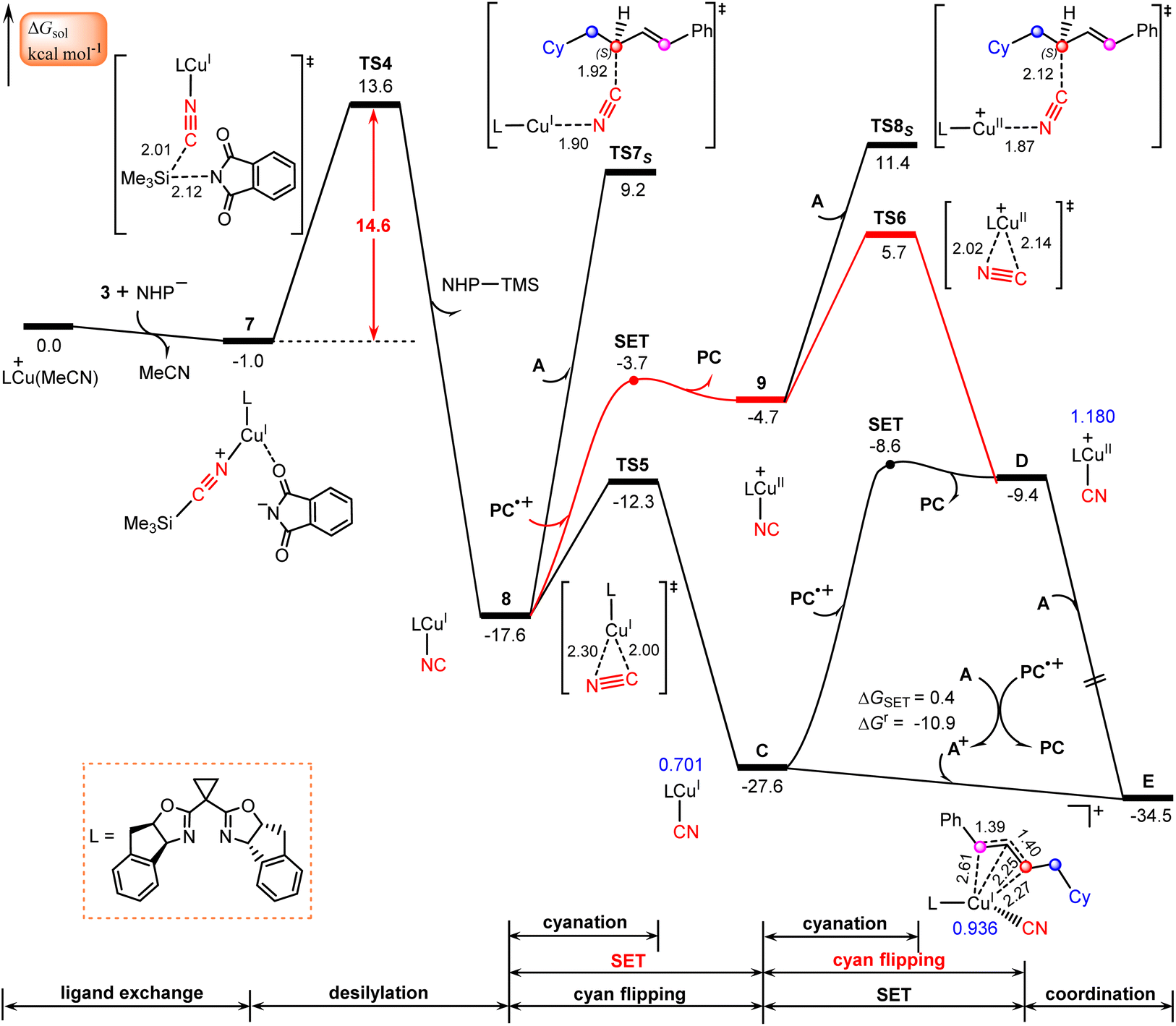 | ||
| Fig. 3 Calculated Gibbs free energy profiles for generation of the allylcopper complex accompanied by the regeneration of photocatalyst PC. Bond distances (black font) and NPA charges (blue font) are in Å and e, respectively. | ||
According to the mechanism proposed in Scheme 2, the cyanocopper(II) species D captures allyl radical A, leading to the allylcopper complex E. Interestingly, based on the following facts we confirm that E is an allylcopper(I) complex rather than an allylcopper(III) complex as proposed in Scheme 2. Firstly, as indicated by the blue values in Fig. 3, for structure E, the calculated NPA charge of the copper center is 0.936 e, which is comparable to that of the cyanocopper(I) species C at 0.701 e, but significantly lower than that of the cyanocopper(II) species D at 1.180 e. Secondly, Fig. 4 shows the calculated barrier of SET, redox potentials and HOMO-LUMO energy gaps of the electron donor–acceptor pairs, the cyanocopper(II) species D and allyl radical A, and all these results consistently rule out the electron transfer process from D to A. As indicated by calculated data in Fig. 4a, if there is a cyanocopper(II) species like D, the electron transfer between D and A must be from A to D, resulting in D returning to the cyanocopper(I) intermediate C. These results clearly indicate that E is actually a complex of C with allyl cation A+, which can be easily formed via an almost barrierless SET process from A to PC˙+ (Fig. 3). This inspired us to propose a new pathway, in which the allylcopper complex E is directly formed via the interaction between C and A+, which is shown to be a thermodynamically favorable process and is calculated to be exergonic by 6.9 kcal mol−1. According to previous studies,57,58 our calculations also located two other potential pathways involving a direct cyanation step from the isocyanocopper(I) intermediate 8viaTS7S (26.8 kcal mol−1) and from the isocyanocopper(II) intermediate 9viaTS8S (29.0 kcal mol−1). It is found that these two pathways are energetically less favorable than that discussed above.
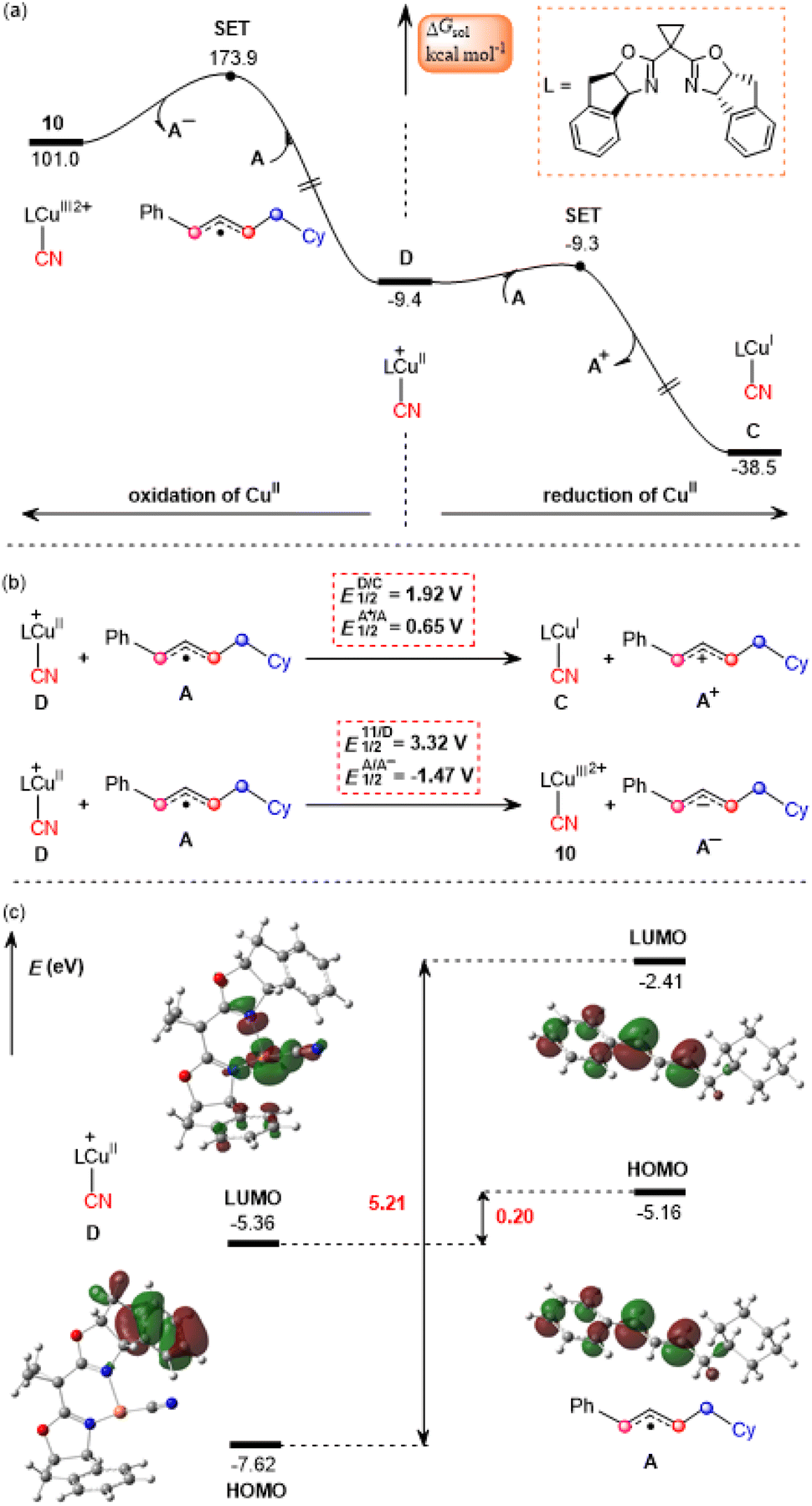 | ||
| Fig. 4 Calculated data of Gibbs free energy profiles for (a) the single electron transfer (SET) process, (b) redox potentials (ESCE = 4.28 V), and (c) HOMO-LUMO energy gaps of the electron donor–acceptor pairs between the cyanocopper(II) species D and allyl radical A. | ||
Formation of allyl cyanide products
Finally, we focus our attention on the generation of the allyl cyanide products. Fig. 5 illustrates the calculated pathways for the formation of 1,2-carbocyanation products 4S and 4R, and their 1,4-regioisomers 5S and 5Rvia the reaction of allylcopper(I) complex E with MeCN. From the above discussion, it is clear that the reductive elimination mechanism is not operative due to the fact that species E is an allylcopper(I) complex rather than an allylcopper(III) complex.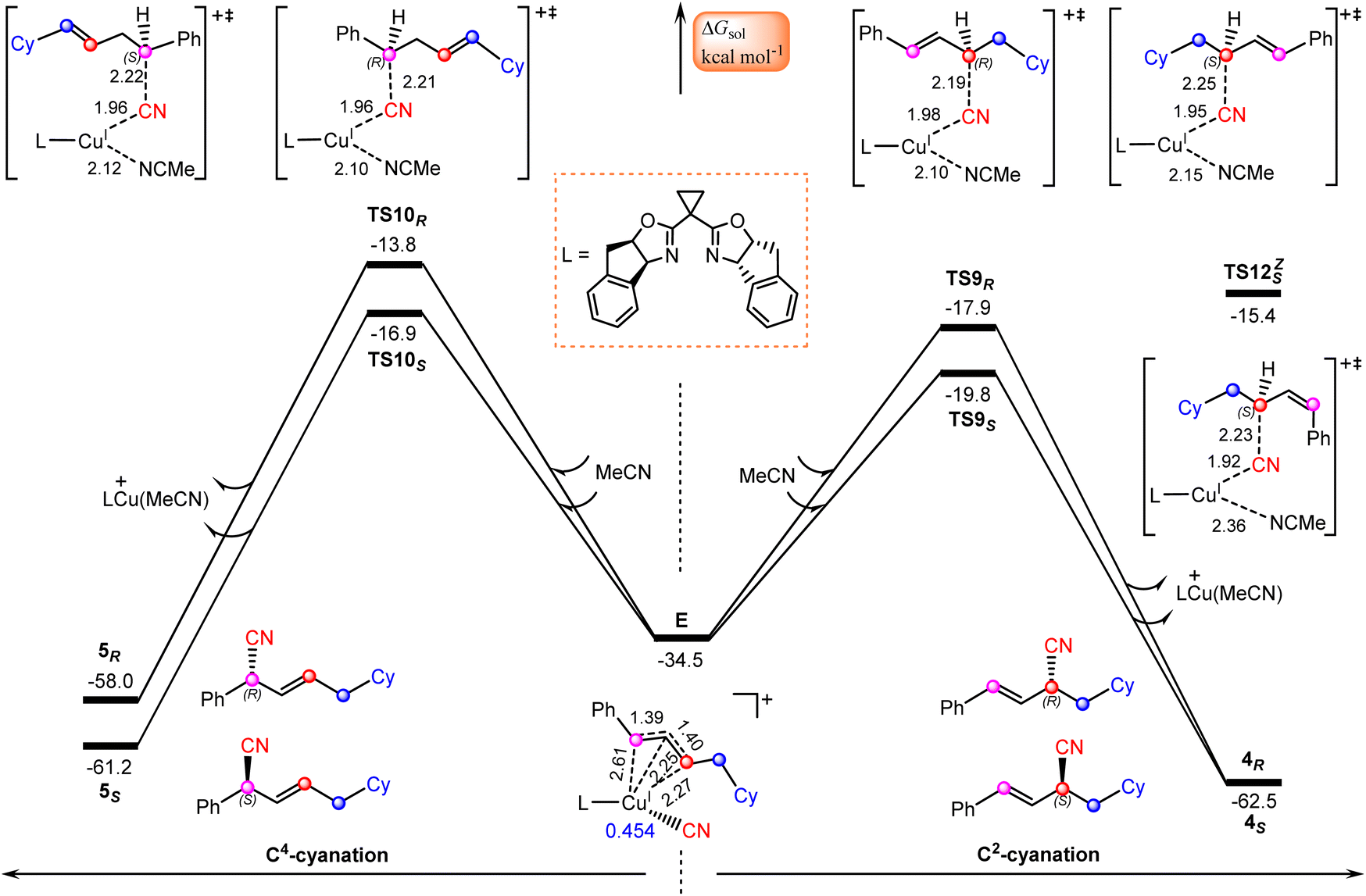 | ||
| Fig. 5 Calculated Gibbs free energy profiles for generation of allyl cyanide products. Bond distances are in Å. | ||
Alternatively, our calculations identified an electrostatic interaction-induced cyanation mechanism for the formation of allyl cyanide products. In this mechanism, the cyanation of the allyl cation is achieved with the assistance of a MeCN molecule. As indicated by the calculated transition state geometries, the carbon atom of cyanide in E attacks the C2 atom of the allyl cation moiety from the Re-face viaTS9R or from the Si-face viaTS9S, leading to formation of an R- or S-1,2-allyl cyanide product, or attacks the C4 atom of an allyl cation moiety from the Re-face viaTS10R or from the Si-face viaTS10S, leading to formation of an R- or S-1,4-allyl cyanide product. It is found that the reaction viaTS9S with a barrier of 14.7 kcal mol−1 is the energetically most favorable, delivering the experimentally observed product 4S.
To confirm this new mechanism, we carried out IRC calculations for TS9S and the results are shown in Fig. 6, where several key bond distances along the reaction coordinate are given. Clearly, at the transition state region the C2–CN distance remarkably decreases and once the C2–CN bond is formed, both the Cu–CN and Cu–C2 distances increase rapidly. Furthermore, it is found that the Cu–NCMe distance monotonically decreases along the reaction coordinate. The electrostatic interaction between the allyl cation and the nitrile anion in allylcopper(I) complex E is believed to be the main driving force for the formation of the allyl cyanide product.
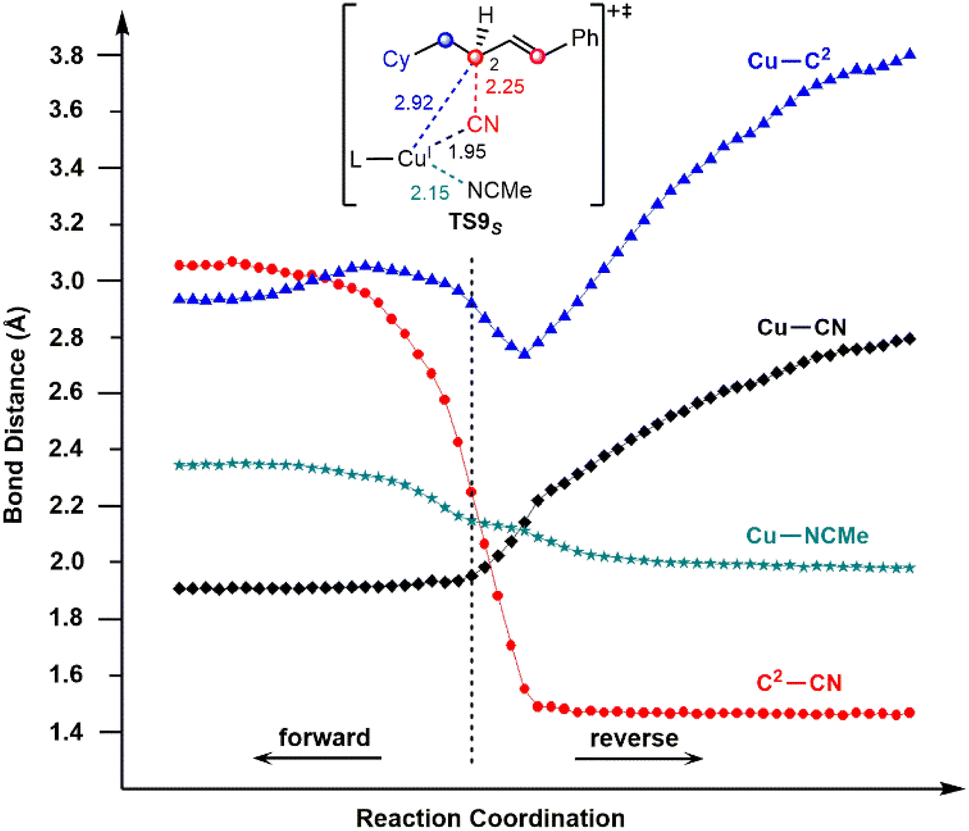 | ||
| Fig. 6 IRC calculation results for transition state TS9S. | ||
Note that all four transition state geometries are in their E-conformations, leading to E-allyl cyanides. Taking TS9S as an example, we also optimized its Z-isomer, which is found to be energetically less favorable by 4.4 kcal mol−1. This is consistent with the experimental observation that no Z-allyl cyanide product was detected.26
In comparison with TS9S (Si-face coupling) delivering the S-allyl cyanide product 4S, TS9R (Re-face coupling) leading to the R-allyl cyanide product 4R is energetically less favorable by 1.9 kcal mol−1. A Gibbs free energy barrier difference of 1.9 kcal mol−1 corresponds to an ee value of ∼93% for the 4S product, which is in good agreement with the experimentally observed enantioselectivity, 92% ee value (Scheme 1). The distortion–interaction analyses,59–62 using the allyl cation and [LCu(CN)(MeCN)] as fragments, show that the stronger electronic interaction in TS9S is mainly responsible for its higher stability than TS9R, as indicated by the calculated interaction energies between the two fragments (15.5 vs. 13.3 kcal mol−1 in Table S3†). Furthermore, the non-covalent interactions (NCIs) analyses indicate that in TS9S there are significant CH–π interactions between the two fragments, while in TS9R these CH–π interactions are absent and only a weak π–π interaction is observed (Fig. S6†).
Furthermore, the formation of 1,4-carbocyanation products 5S and 5R is also found to be energetically less than that for 4S, as indicated by the calculated relatively higher Gibbs free energies of TS10S and TS10R, −16.9 and −13.8 kcal mol−1, which are less stable by 2.9 and 6.0 kcal mol−1 than TS9S, respectively. These results are also consistent with the experimental observation that only the desired 1,2-allyl cyanide product 4S is observed (Scheme 1).
Other pathways for the attack of the carbon atom of cyanide in E on the C1 or C3 atom of the allyl cation moiety were also considered, however, the designed geometries were consistently observed to collapse into those at the C2 or C4 position.
Proposed mechanism for photoredox/copper catalysis
Based on the above discussion, we summarize the mechanism of the synergetic photoredox/copper catalysis in Scheme 3. This is an unprecedented reaction mechanism, in which both the photoredox catalytic cycle and the copper catalytic cycle are different from those proposed in the experimental study. In the photoredox cycle, the oxidized photocatalyst PC˙+ is reduced by the allyl radical A generated in situ rather than the cyanocopper(I) species C, and this process generates the key allyl cation A+. On the other hand, in the copper catalytic cycle, the allyl cyanide product is obtained by direct coupling between the cyanocopper(I) species C and allyl cation A+ through the unique outer-sphere mechanism rather than the reductive elimination mechanism from a hypothetical Cu(III) species that cannot be located from the present calculations.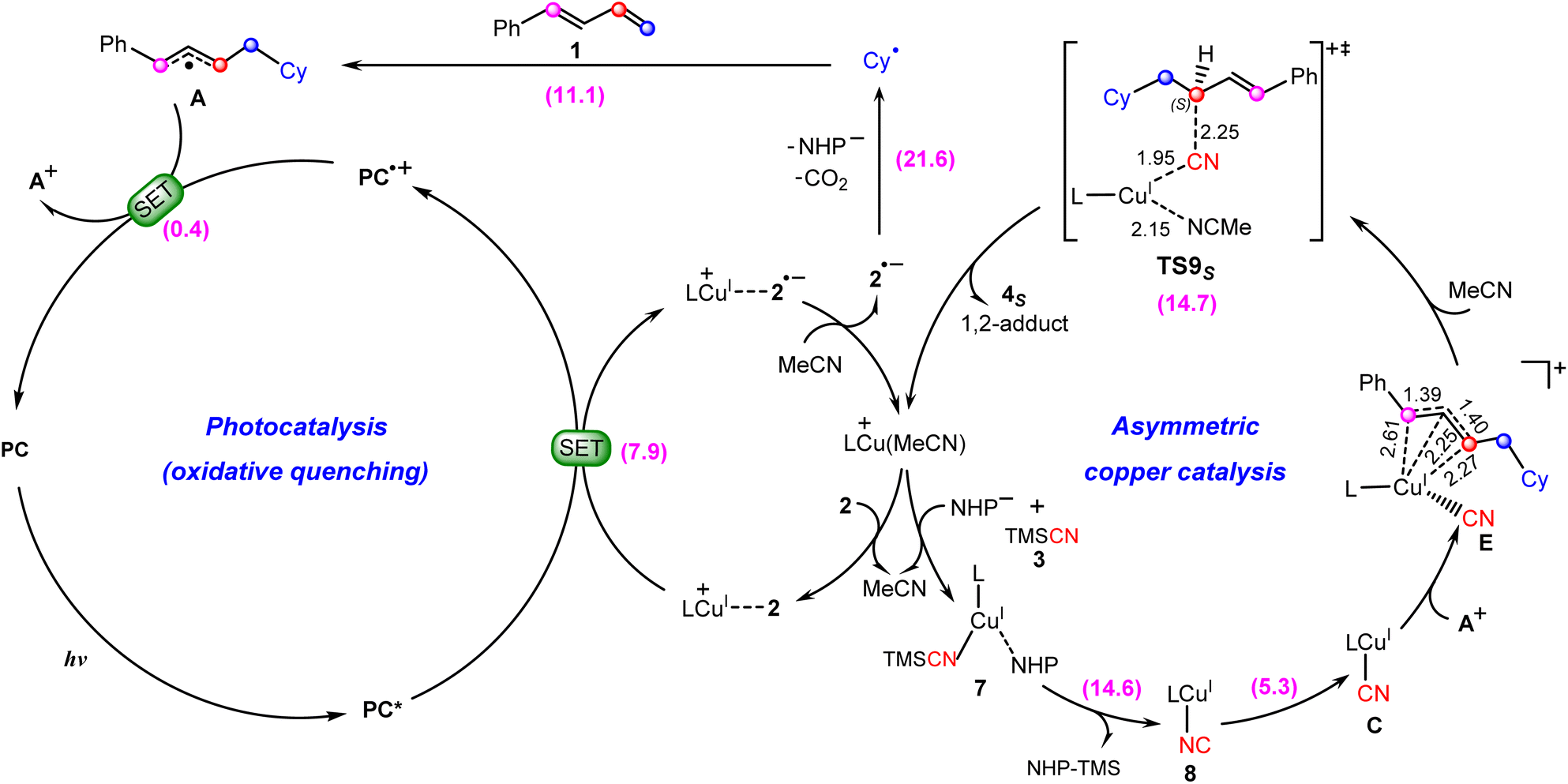 | ||
| Scheme 3 Newly proposed mechanism for photoredox/copper(I)-catalyzed carbocyanation of 1,3-dienes with the calculated Gibbs free energy barriers (in kcal mol−1). | ||
In the newly proposed mechanism, throughout the whole reaction process, only copper(I) species are involved. This is very different from the previously proposed mechanism (Scheme 2) in which the reaction occurs via a CuI/CuII/CuIII/CuI cycle. From our calculations, the allylcopper complex E is identified as a monovalent copper species rather than a trivalent species. Note that in the previous experimental and theoretical studies10,20,21,26,28–30 on the photoredox/copper(I)-catalyzed carbocyanation of alkenes or alkynes using trimethylsilyl cyanide (TMSCN) as the provider of the cyano group, the desilylation process was proposed to proceed via the reaction of a Cu(II) species with TMSCN. However, for the present system our calculations show that the involvement of Cu(II) in the desilylation process is expected to be energetically less favorable due to the higher barrier involved in the formation of the Cu(II) species, which requires an energy input of 31.7 kcal mol−1 (Fig. S7†). In contrast, the reaction of Cu(I) species involves a lower barrier of 14.6 kcal mol−1, as shown in Fig. 3.
In addition, we also investigated the reaction of the n-propyl-substituted N-hydroxyphthalimide (NHP) ester (2′) with the p-phenylmethyl-substituted 1,3-diene (1′), and have included the results of the four transition states involved in the cyanation of the allyl cation (regio- and enantioselectivity-determining step) in Fig. S8.† It is found that the formation of the S-1,2-allyl cyanide product is the energetically most favorable. This is consistent with the results obtained for the reaction of 2 with 1 discussed above. Based on these results, we conclude that our proposed mechanism can explain Xiao's experimental observation that different allyl esters yield the same chiral allyl cyanides.
Energy transfer mechanism
According to previous reports in the literature,63,64 the reaction may be initiated via a triplet–triplet energy transfer (TTET) mechanism in some photocatalytic reactions. The donor is the triplet state of the excited photocatalyst and the acceptor is the singlet state of the transition metal catalyst. However, for the present reaction, the TTET mechanism is not found to be operative due to the incompatible singlet-triplet energy gaps of the donor and the acceptor (active copper catalyst LCu(MeCN)+). Fig. 7 shows the calculated energies of the singlet and triplet states of PC and LCu(MeCN)+. The calculated singlet-triplet energy gap for PC is 1.37 eV, while that for LCu(MeCN)+ is 3.41 eV. Clearly, the singlet-triplet energy gaps of the donor and acceptor differ significantly, which hinders effective TTET between the excited photocatalyst and copper catalyst according to Marcus theory.65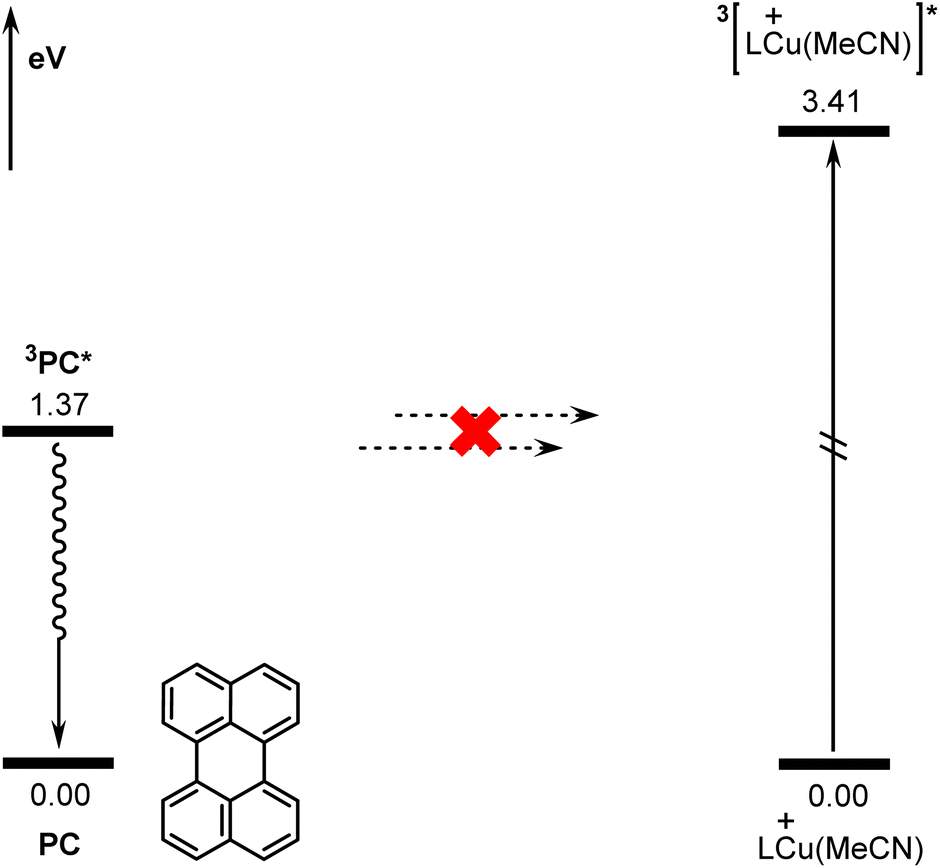 | ||
| Fig. 7 Calculated singlet-triplet energy gaps of PC and copper(I) species LCu(MeCN)+. | ||
Conclusions
In summary, by performing DFT calculations we have shown the mechanistic details of the synergetic photoredox/copper-catalyzed carbocyanation of 1,3-dienes. The calculated results indicated a significantly different photoredox/copper catalytic mechanism from that proposed in the experimental study. New insights on the catalytic mechanism can be summarized in the following three points. Firstly, the copper catalytic cycle involves only CuI species, instead of a Cu(I)/Cu(II)/Cu(III)/Cu(I) cycle as proposed in the experimental study. Secondly, the cyanation of the cyanocopper(I) species is achieved through its reaction with an allyl cation rather than an allyl radical as proposed in the experiment. Thirdly, the photocatalyst is regenerated via single electron transfer from the allyl radical rather than cyanocopper(I) species to the oxidized photocatalyst. The present calculations show that the cyanation of the allyl cation is the regio- and enantioselectivity-determining step of the reaction.Data availability
All the data supporting this work have been included in the main text and the ESI.†Author contributions
Yanhong Liu performed the calculations and wrote the original draft, Aili Feng validated the results, Rongxiu Zhu analyzed the results, and Dongju Zhang wrote and reviewed the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21833004 and 22273051).Notes and references
- Y. Xiong, Y. Sun and G. Zhang, Tetrahedron Lett., 2018, 59, 347–355 CrossRef CAS.
- N. J. Adamson and S. J. Malcolmson, ACS Catal., 2020, 10, 1060–1076 CrossRef CAS.
- C.-G. Wang, Y. Zhang, S. Wang, B. Chen, Y. Li, H.-L. Ni, Y. Gao, P. Hu, B.-Q. Wang and P. Cao, Org. Lett., 2021, 23, 535–541 CrossRef CAS PubMed.
- L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784–5787 CrossRef CAS PubMed.
- Y. Chu, S. Bi, X. Wu, Y. Jiang, Y. Liu, B. Ling and T.-A. Yuan, Mol. Catal., 2020, 497, 111222 CrossRef CAS.
- X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef CAS PubMed.
- Y. Liu, D. Fiorito and C. Mazet, Chem. Sci., 2018, 9, 5284–5288 RSC.
- G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS.
- J. L. Schwarz, H.-M. Huang, T. O. Paulisch and F. Glorius, ACS Catal., 2020, 10, 1621–1627 CrossRef CAS.
- J. Chen, Y.-J. Liang, P.-Z. Wang, G.-Q. Li, B. Zhang, H. Qian, X.-D. Huan, W. Guan, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2021, 143, 13382–13392 CrossRef CAS PubMed.
- P.-Z. Wang, X. Wu, Y. Cheng, M. Jiang, W.-J. Xiao and J.-R. Chen, Angew. Chem., Int. Ed., 2021, 60, 22956–22962 CrossRef CAS PubMed.
- K. P. S. Cheung, D. Kurandina, T. Yata and V. Gevorgyan, J. Am. Chem. Soc., 2020, 142, 9932–9937 CrossRef PubMed.
- C. Gosset, A. Moncomble, C. Dumont, S. Pellegrini, T. Bousquet, M. Sauthier and L. Pélinski, Adv. Synth. Catal., 2020, 362, 3100–3104 CrossRef CAS.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed.
- S. Tong, K. Li, X. Ouyang, R. Song and J. Li, Green Synth. Catal., 2021, 2, 145–155 CrossRef.
- D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359–373 RSC.
- B. Ghosh, A. Banerjee, L. Roy, R. N. Manna, R. Nath and A. Paul, Angew. Chem., Int. Ed., 2022, 61, e202116868 CAS.
- J. Liu, X.-P. Liu, H. Wu, Y. Wei, F.-D. Lu, K.-R. Guo, Y. Cheng and W.-J. Xiao, Chem. Commun., 2020, 56, 11508–11511 RSC.
- F.-D. Lu, D. Liu, L. Zhu, L.-Q. Lu, Q. Yang, Q.-Q. Zhou, Y. Wei, Y. Lan and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167–6172 CrossRef CAS PubMed.
- P.-Z. Wang, Y. Gao, J. Chen, X.-D. Huan, W.-J. Xiao and J.-R. Chen, Nat. Commun., 2021, 12, 1815 CrossRef CAS PubMed.
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed.
- Q. Guo, M. Wang, Y. Wang, Z. Xu and R. Wang, Chem. Commun., 2017, 53, 12317–12320 RSC.
- L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio Jr. and S. Lin, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed.
- M. Zheng, K. Gao, H. Qin, G. Li and H. Lu, Angew. Chem., Int. Ed., 2021, 60, 22892–22899 CrossRef CAS PubMed.
- F.-D. Lu, L.-Q. Lu, G.-F. He, J.-C. Bai and W.-J. Xiao, J. Am. Chem. Soc., 2021, 143, 4168–4173 CrossRef CAS PubMed.
- Y. Liang, G. Sun, Z. Su and W. Guan, Dalton Trans., 2020, 49, 15276–15286 RSC.
- P.-Z. Wang, Y.-J. Liang, X. Wu, W. Guan, W.-J. Xiao and J.-R. Chen, ACS Catal., 2022, 12, 10925–10937 CrossRef CAS.
- F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140–7145 CrossRef CAS PubMed.
- R. Lu, T. Yang, X. Chen, W. Fan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2021, 143, 14451–14457 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- S. Huzinaga, Comput. Phys. Rep., 1985, 2, 281–339 CrossRef.
- D. Andrae, U. Häuβermann, M. Dolg, H. Stoll and H. Preuβ, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. Koshikawa, J. Nogami, Y. Nagashima and K. Tanaka, ACS Catal., 2022, 12, 14330–14336 CrossRef CAS.
- L. Wu, F. K. Sheong and Z. Lin, ACS Catal., 2020, 10, 9585–9593 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke: Sherbrooke, Quebec, Canada, 2009 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. A. Marcus, Can. J. Chem., 1959, 37, 155–163 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- Y. Liu, Y. Yang, R. Zhu and D. Zhang, ACS Catal., 2020, 10, 5030–5041 CrossRef CAS.
- A. Feng, Y. Yang, Y. Liu, C. Geng, R. Zhu and D. Zhang, J. Org. Chem., 2020, 85, 7207–7217 CrossRef CAS PubMed.
- H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC.
- F. Li, S. Lin, Y. Chen, C. Shi, H. Yan, C. Li, C. Wu, L. Lin, C. Duan and L. Shi, Angew. Chem., Int. Ed., 2021, 60, 1561–1566 CrossRef CAS PubMed.
- S. Ma, F. Li, G. Zhang, L. Shi and X. Wang, ACS Catal., 2021, 11, 14848–14853 CrossRef CAS.
- X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS PubMed.
- Y. Jiao, M.-F. Chiou, Y. Li and H. Bao, ACS Catal., 2019, 9, 5191–5197 CrossRef CAS.
- I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS PubMed.
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated Gibbs free energy profiles, thermodynamic data, and cartesian coordinates of all structures (PDF). See DOI: https://doi.org/10.1039/d3sc00002h |
| This journal is © The Royal Society of Chemistry 2023 |