
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAminals as powerful XAT-reagents: activation of fluorinated alkyl chlorides†
Vladislav S.
Kostromitin
ab,
Artem O.
Sorokin
ab,
Vitalij V.
Levin
 a and
Alexander D.
Dilman
*a
a and
Alexander D.
Dilman
*a
aN. D. Zelinsky Institute of Organic Chemistry, Leninsky prosp. 47, 119991 Moscow, Russian Federation. E-mail: adil25@mail.ru
bLomonosov Moscow State University, Department of Chemistry, Leninskie Gory 1-3, 119991 Moscow, Russian Federation
First published on 21st February 2023
Abstract
Readily available 1,3,5-trimethyl-1,3,5-triazinane serves as an efficient reagent for halogen atom transfer. Under photocatalytic conditions, the triazinane generates an α-aminoalkyl radical, which can activate the C–Cl bond of fluorinated alkyl chlorides. The hydrofluoroalkylation reaction between fluorinated alkyl chlorides and alkenes is described. The efficiency of the diamino-substituted radical derived from the triazinane is associated with stereoelectronic effects defined by a six-membered cycle forcing the anti-periplanar arrangement of the radical orbital and lone pairs of adjacent nitrogen atoms.
Introduction
The activation of the carbon–halogen bond is a widely used pathway for the generation of organic radicals.1 The advent of photocatalysis2–5 has made a notable breakthrough in this area leading to the development of more efficient approaches to the application of organic halides, which allowed to escape from classic organotin based reagents.6 In a series of conventional alkyl halides, iodides are activated most easily, while chlorides are the least reactive, though methods for the activation of the carbon–chlorine bond have been described.7 Thus, organometallic and related reagents capable of direct abstraction of the chlorine by virtue of strong a chlorine-element bond8–10 or transition metals11–13 may be used. Another approach is based on photoredox promoted single electron reduction. However, this step requires catalysts having strongly reducing excited states,14–18 which in some cases may be accessed via a two-photon absorption mechanism.19,20Recently, a concept involving halogen atom transfer (XAT)21,22 by using α-aminoalkyl radicals was introduced by Leonori23 (Scheme 1). These radicals are generated from readily available tertiary amines by single electron oxidation followed by proton loss. These α-aminoalkyl radicals effectively activate alkyl iodides23,24 and fluorinated bromides,25 while reactions with chlorides are rare.26 We proposed that switching from amines to aminals may provide more effective XAT reagents due to the additional donor effect of the second nitrogen on the radical center. Herein, we report that 1,3,5-triazinane N1 exhibits very high reactivity in XAT processes owing to its cyclic structure. The observed phenomenon is based on stereoelectronic effects originating from the fixation of the nitrogen lone pairs in a proper spatial arrangement with respect to the singly occupied orbital.27,28 We demonstrate the application of cyclic amine N1 for the activation of the carbon–chlorine bond of dichlorodifluoromethane (CF2Cl2, Freon R-12) and other fluorinated chlorides. In fact, Freon R-12 is one of the most readily available commodity chemicals, and it was used as a refrigerant, aerosol spray propellant, and fire retardant.29,30 Apparently, it would be attractive to use this relatively unreactive chemical for the synthesis of valuable organofluorine compounds,31,32 while there are only a few limited radical based methods affording CF2Cl-compounds.33,34
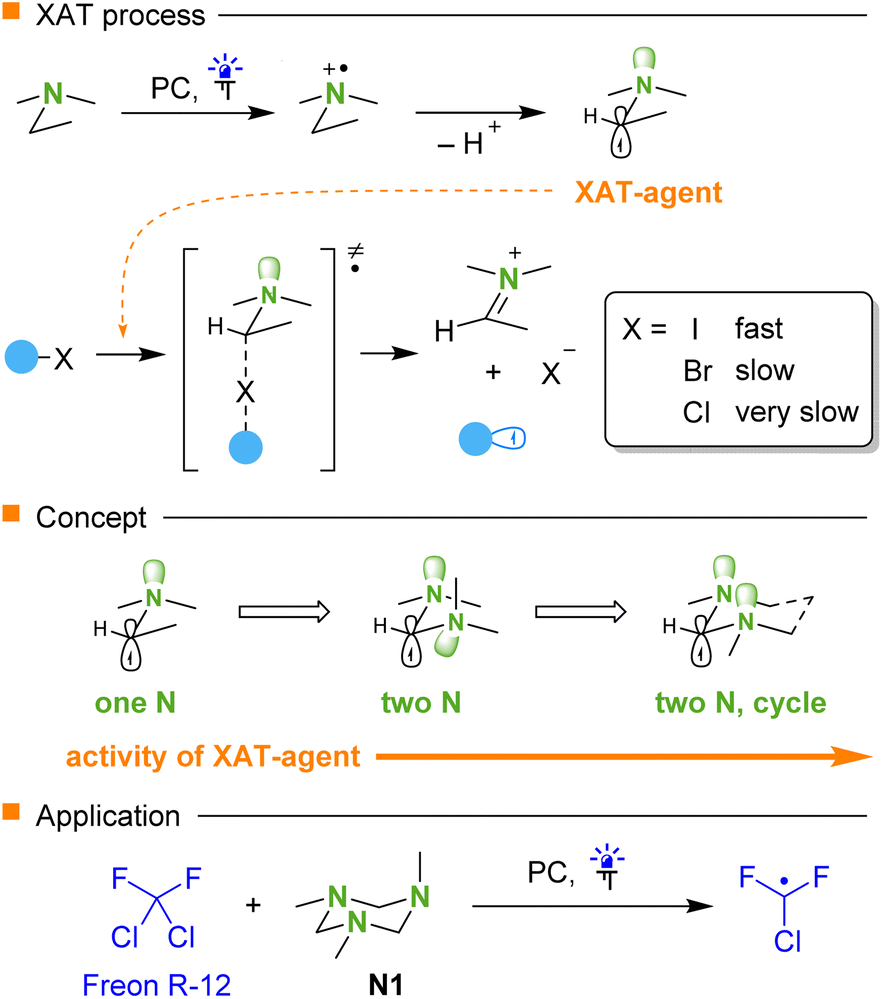 | ||
| Scheme 1 Reactions of α-aminoalkyl radicals. | ||
Results and discussion
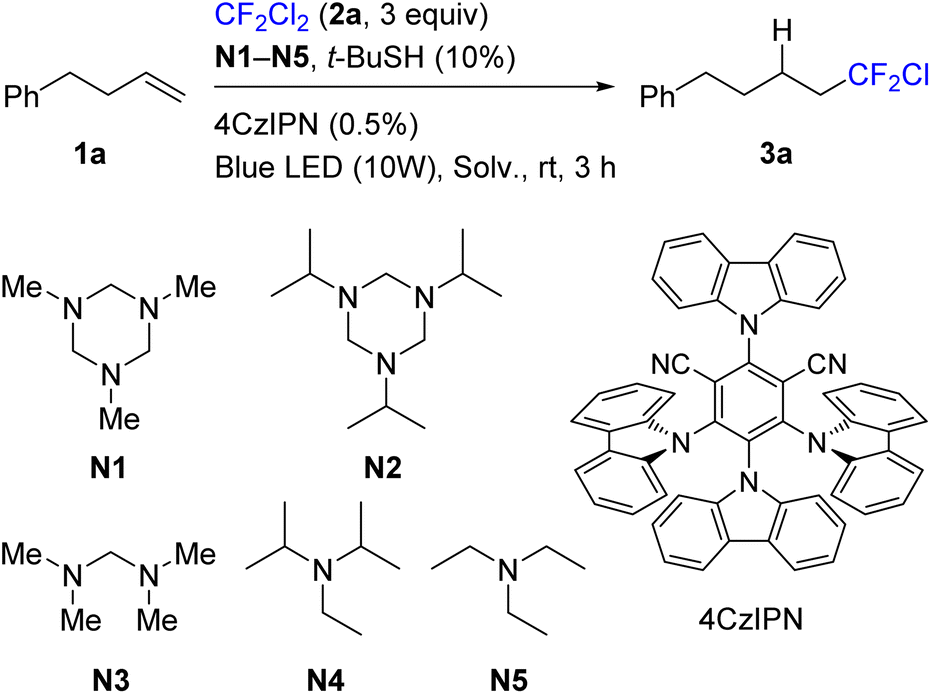
4-Phenylbut-1-ene (1a) was selected as a model substrate and its reaction with Freon R-12 (2a) was evaluated (Table 1). A typical carbazole-type photocatalyst (4CzIPN)35 was used under blue light irradiation. tert-Butyl thiol in catalytic amounts (10%) was added as a mediator of hydrogen atom transfer.36–39 A series of amines N1–N5, which can serve both as XAT-type activating agents and as a source of the hydrogen atom, were evaluated. Triazinanes N1 and N2 were found to be optimal (entries 1 and 5), while acyclic aminal N3 was less efficient, and conventional tertiary amines were notably less reactive (entries 8 and 9). Acetonitrile was routinely used as a solvent in this process, though dimethylsulfoxide and dichloromethane performed similarly. Without tert-butyl thiol the reaction was slower, and by-products were formed (entry 4). Gaseous 2a was used in excess (3 equiv.), while the decrease of its amount twice led to some decrease of the product yield (entry 10). Interestingly, the reaction slowly proceeded even without the photocatalyst (entry 11). Finally, the use of 2 equiv. of triazinane N1 gave higher conversion, and hydrofluoroalkylation product 3a was isolated in 92% yield (entry 7). Between triazinanes N1 and N2, we selected the former for further experiments because of the atom efficiency issue. It should also be noted that reagent N1 is commercially available or can be easily obtained from cheap one-carbon precursors – methylamine and paraform.|
|
|||
|---|---|---|---|
| Entry | XAT reagent (equiv.) | Solv. | Ratio 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1aa :1aa |
| a Determined by GC-MS. b Without t-BuSH. c Isolated yield of 3a. d 1.5 equiv. of CF2Cl2. e Without 4CzIPN. | |||
| 1 | N1 (1.5) | MeCN | 97:3 |
| 2 | N1 (1.5) | DMSO | 96:4 |
| 3 | N1 (1.5) | DCM | 97:3 |
| 4b | N1 (1.5) | MeCN | 28:72 |
| 5 | N2 (1.5) | MeCN | 95:5 |
| 6 | N3 (1.5) | MeCN | 71:29 |
| 7 | N1 (2) | MeCN | > 99:1 (92c) |
| 8 | N4 (2) | MeCN | 5:95 |
| 9 | N5 (2) | MeCN | 26:74 |
| 10d | N1 (2) | MeCN | 76:24 |
| 11e | N1 (2) | MeCN | 14:86 |
Under the optimized conditions, a series of alkenes were fluoroalkylated using Freon R-12 and other fluorinated chlorides (Scheme 2). Typically, hydrofluoroalkylation products 3–5 were obtained in high yields. The reaction tolerates a wide variety of functional groups in the alkene component including keto (4d), cyano (4k), ester (3b, g, k, m, and 4m), free hydroxyl group (3f), free carboxylic acid (5d), and diverse protecting groups (silyl, benzyl, PMP, and MEM), N-Boc protected amine (4p), phosphonate (4g), boryl (4h), acetal (4o) and sulfide (3j and l) fragments. Besides terminal alkenes, 1,1-, 1,2-disubstituted and trisubstituted alkenes were also successfully involved (products 4i, 4j, and 4s, respectively). Even a fully substituted alkene gave the desired product (4t) though in a decreased yield. In some cases (products 3l, n, and 4t), an increase of the tert-butyl thiol additive to 20% was needed to suppress the formation of by-products, which according to NMR and GC-MS analysis was tentatively ascribed to the alkenes (m/z values less by 2 relative to that of expected products). It is worth noting that reagent N1 is very selective and activates only Freon R-12 and tolerates the C–Cl bond of compounds 3, as no by-products arising from the interaction of 3 with alkenes 1 were observed. Other fluorinated chlorides were found to be good reaction partners such as ethyl 4-chloro-2,2,3,3-tetrafluoropropionate (products 4), methyl chlorodifluoroacetate (products 5a and b), and higher polyfluorinated chlorides (products 5c–e). We also evaluated chloroform as a non-fluorinated chloride, which afforded the desired product 5g, though the reaction required higher loadings of triazinane and the photocatalyst.
 | ||
| Scheme 2 Fluoroalkylation of alkenes.a | ||
While reactions were typically performed on a 0.5 mmol scale, they can be readily scaled-up, as was demonstrated by the synthesis of gram quantities of products 3a and 5a starting from 20 and 10 mmol of 1a, respectively (Scheme 3). Importantly, these products were isolated without chromatography by distillation of the crude material. In these experiments, only 0.25 mol% of the photocatalyst, that is half of the standard amount, was used. Compound 5a was further converted into alcohol 6 and amide 7 by reactions typical for the ester group.
 | ||
| Scheme 3 Gram-scale experiments and reactions of 5a. | ||
The fluoroalkylation was successfully applied to difluorinated bromide 8 affording product 5g in good yield (Scheme 4). At the same time, difluorinated iodide 9 gave a complex mixture containing traces of the desired product 5h. We may propose that for the iodide, after the radical addition step, the secondary alkyl radical undergoes fast iodine atom transfer to give alkyl iodide followed by dehydroiodination (a series of alkenes were observed by GC-MS analysis). While products 3 are inactive under typical conditions (see above), isolated compound 3a could be coupled with another alkene by using a different photocatalyst and more powerful purple irradiation (400 nm). As a result, product 10 was assembled from two alkenes and Freon R-12 as a source of the difluoromethylene fragment (Scheme 4, bottom equation).
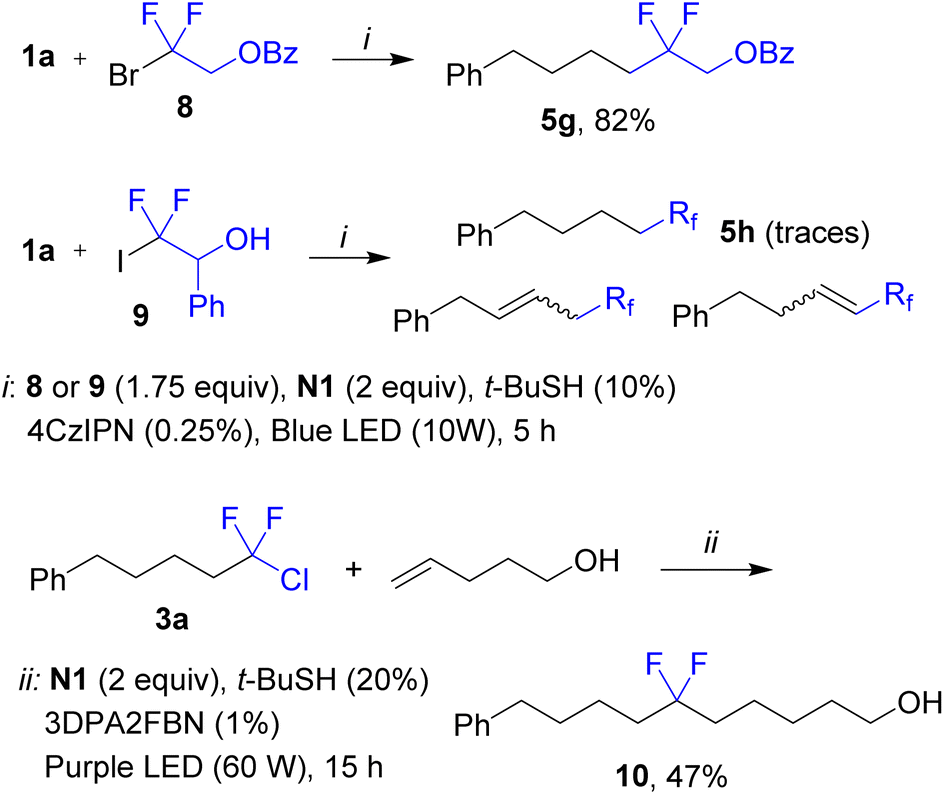 | ||
| Scheme 4 Scope of difluorinated halides. | ||
The radical character of the fluoroalkylation was supported by a radical clock experiment involving diallylmalonate, which afforded product 11, likely via rapid 5-exo cyclization of the intermediate addition radical (Scheme 5A). Reactions performed in acetonitrile are accompanied by precipitation of a white solid. This precipitate was isolated and identified as previously known iminium salt 12, which is formed from triazinane N1 almost quantitatively (Scheme 5B). Performing Stern–Volmer analysis suggested strong quenching of the fluorescence of photoexcited 4CzIPN by triazinane N1, while there is virtually no quenching by ethyl 4-chloro-2,2,3,3-tetrafluoropropionate and other components. Taking into account that the redox potential of triazinane N1 of +0.94 V (vs. SCE)40 matches well the potential of the excited state of 4CzIPN (+1.43 V vs. SCE),35 the single electron oxidation of N1 by the photocatalyst seems likely. The reaction quantum yield was found to be 0.44, thereby suggesting that the process is either non-chain or has a chain character with short propagating efficiency.
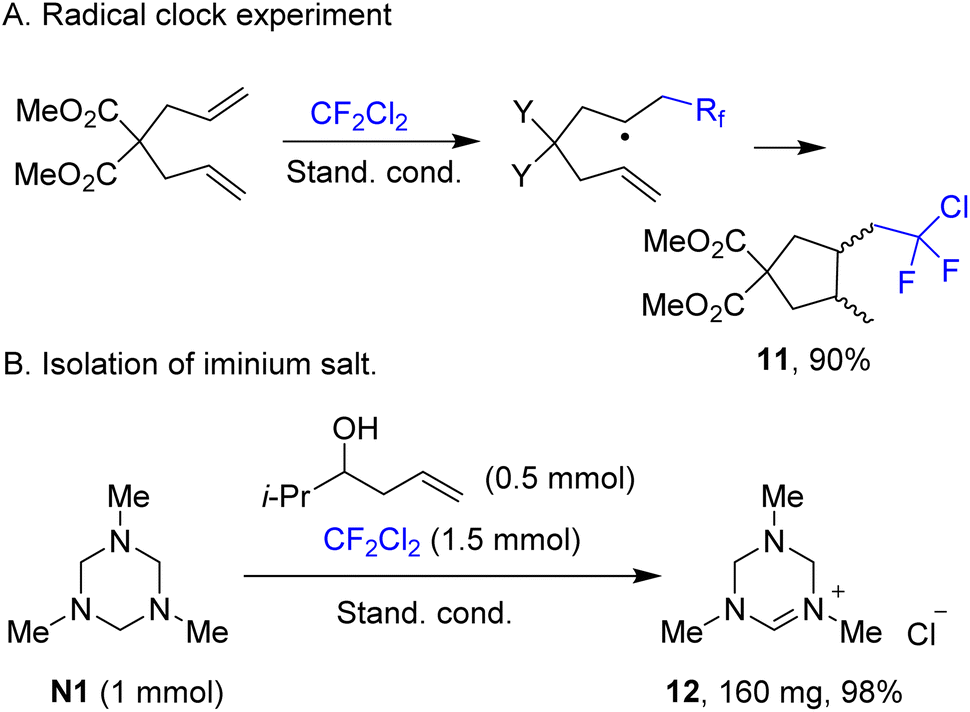 | ||
| Scheme 5 Mechanistic experiments. | ||
The proposed mechanism is shown in Scheme 6. Triazinane N1 is oxidized by an excited photocatalyst followed by loss of a proton with the generation of a triazinanyl radical T1. The XAT event generates the fluorinated radical, which attacks the double bond. Finally, the secondary alkyl radical undergoes hydrogen atom transfer from the thiol. The thiyl radical is either reduced by the photocatalyst (path a) or abstracts the hydrogen atom from triazinane to regenerate an aminoalkyl radical T1 (path b). The latter pathway may contribute to a chain character of the reaction, and it may be responsible for the slow reaction without the photocatalyst (Table 1, entry 11). The interaction of radical T1 with fluoroalkyl halide may be realized via single-electron reduction, but it is difficult to distinguish between these two mechanisms.
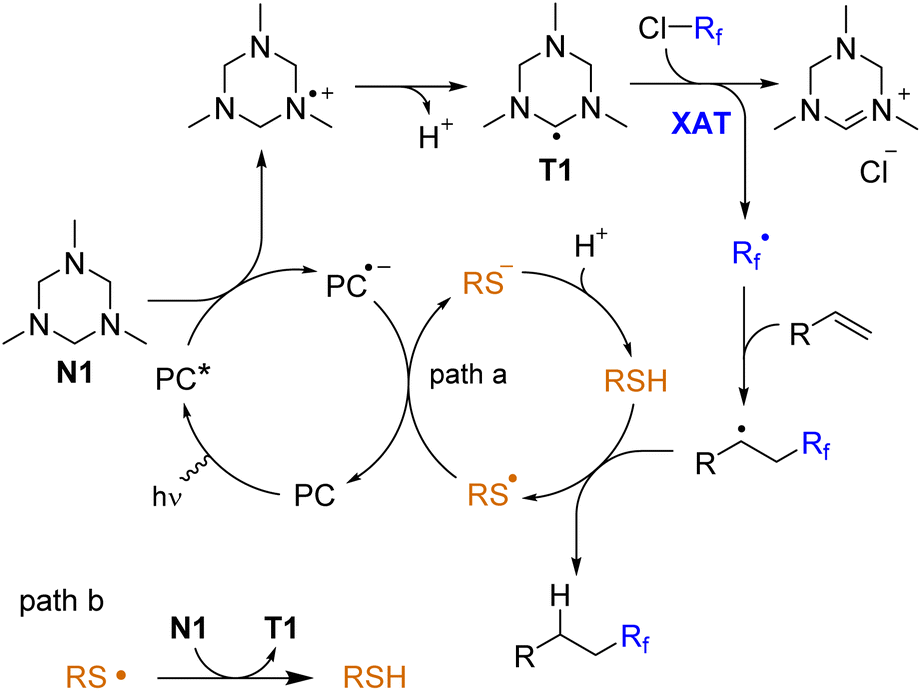 | ||
| Scheme 6 Proposed mechanism. | ||
To explain the efficiency of triazinane N1 compared to other amines in the XAT process, the interaction of various aminoalkyl radicals with CF2Cl2 was evaluated by DFT calculations (Fig. 1). Thus, the activation free energies for the reactions of radicals T5 and T3 derived from monoamine and acyclic aminal differ only by 0.5 kcal mol−1. However, for the triazinane-derived radical T1, the activation energy decreases further by 4.5 kcal mol−1, which corresponds to three orders of magnitude in the rate constant. To rationalize this trend in transition state energies, we analyzed the conformations of aminoalkyl radicals. Thus, for triazinane system T1 we may consider three structures C1–C3 with various orientations of nitrogen lone pairs, based on the well-studied conformational behavior of triazinanes.41–43 The most stable is structure C1 having two lone pairs of adjacent nitrogens arranged in anti-orientation with respect to the radical. For the diamine-type radical T3, the most stable structure has one anti and one gauche arrangement of lone pairs with respect to the radical, which resemble that of structure C2. Interestingly, attempts to locate the T3 structure with a double anti arrangement similar to C1 (shown in parentheses) was unsuccessful, with the optimization leading to the most stable structure via rotation around the C–N bond (see the ESI† for relaxed potential energy scan). As follows from NBO analysis, the interactions of nitrogen lone pairs with the adjacent carbon singly occupied orbital are stronger for the double anti arrangement [net energies are shown in brackets in Fig. 1].
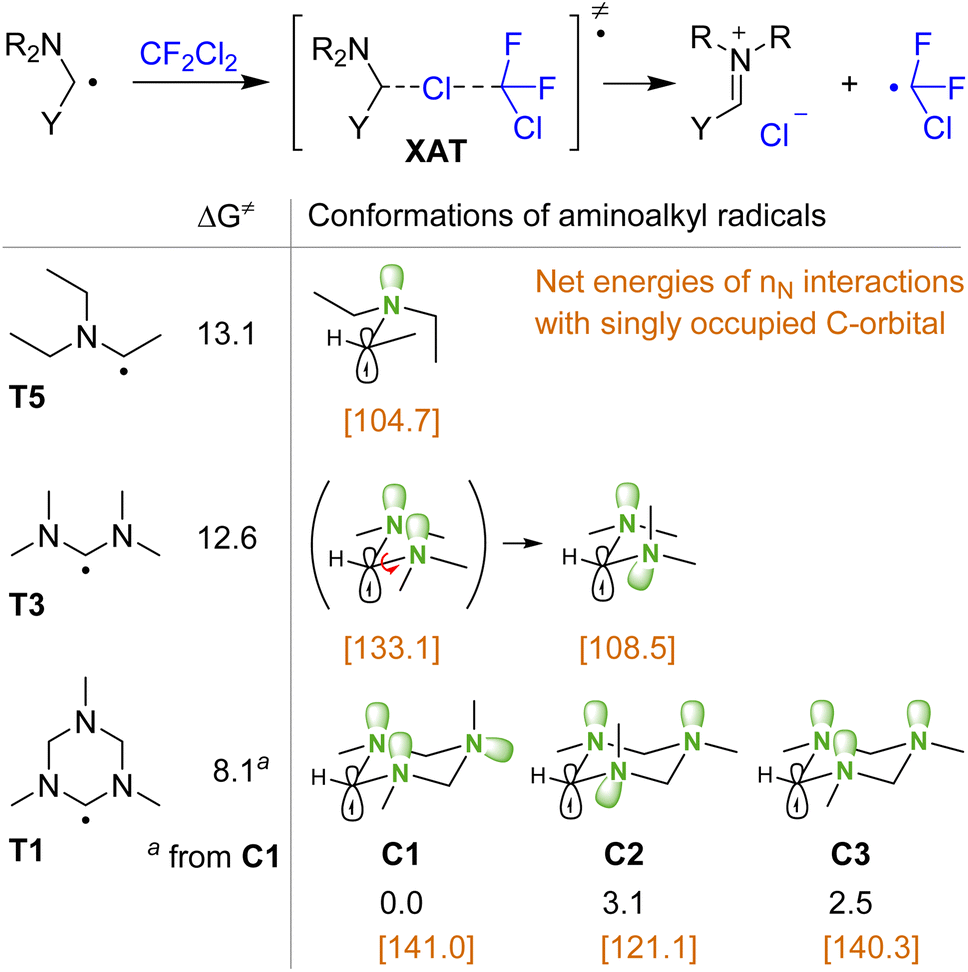 | ||
| Fig. 1 Activation free energies for the XAT process and conformations of aminoalkyl radicals. Method: uwB97xd/def2svp-CPCM(MeCN). In brackets, energies obtained from second order perturbation theory in NBO analysis are shown (for radicals T3 and C1–C3, the sum of interactions for the two lone pairs is given). All energies are in kcal mol−1. | ||
This analysis demonstrates that the ability of the cycle to freeze the rotation and stabilize the structure having two lone pairs anti to the free radical is a key factor responsible for the observed reactivity of the triazinane N1.
Conclusions
In summary, an application of a readily available amine-based reagent for an effecting XAT process under photocatalytic conditions is described. Triazinane serves as a precursor of the α-aminoalkyl radical capable of activating the carbon–chlorine bond of various clorofluoro organic compounds. The enhanced reactivity of the cyclic radical derived from triazinane compared to acyclic mono- and diamino-based analogues is associated with stereoelectronic effects. The cyclic aminals may be considered as promising reagents for the generation of alkyl radicals.Data availability
Procedures, characterization data, NMR spectra, details of DFT calculations, and Stern–Volmer plots are available in the ESI.†Author contributions
V. S. Kostromitin and A. O. Sorokin: investigation; V. V. Levin: investigation, conceptualization, and writing; A. D. Dilman: conceptualization and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr M. Medvedev (Zelinsky Institute) for assistance with NBO analysis.Notes and references
- P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, 2001 Search PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Nat. Catal., 2020, 3, 872–886 CrossRef CAS.
- Z.-Q. Zhang, Y.-Q. Sang, C.-Q. Wang, P. Dai, X.-S. Xue, J. L. Piper, Z.-H. Peng, J.-A. Ma, F.-G. Zhang and J. Wu, J. Am. Chem. Soc., 2022, 144, 14288–14296 CrossRef CAS PubMed.
- T. Kawamoto and I. Ryu, Org. Biomol. Chem., 2014, 12, 9733–9742 RSC.
- X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836–14843 CrossRef CAS PubMed.
- M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2019, 58, 4869–4874 CrossRef CAS PubMed.
- J. Aragón, S. Sun, D. Pascual, S. Jaworski and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2022, 61, e202114365 Search PubMed.
- V. S. Kostromitin, A. A. Zemtsov, V. V. Levin and A. D. Dilman, Adv. Synth. Catal., 2021, 363, 5336–5340 CrossRef CAS.
- N. Noto and S. Saito, ACS Catal., 2022, 12, 15400–15415 CrossRef CAS.
- D. Wei, X. Li, L. Shen, Y. Ding, K. Liang and C. Xia, Org. Chem. Front., 2021, 8, 6364–6370 RSC.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS PubMed.
- M. Giedyk, R. Narobe, S. Weiss, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- T. Constantin, B. Górski, M. J. Tilby, S. Chelli, F. Juliá, J. Llaveria, K. J. Gillen, H. Zipse, S. Lakhdar and D. Leonori, Science, 2022, 377, 1323–1328 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Julia and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- N. Sanosa, B. Peñín, D. Sampedro and I. Funes-Ardoiz, Eur. J. Org. Chem., 2022, 2022, e202200420 CrossRef CAS.
- W.-J. Yue, C. S. Day, A. J. Brenes Rucinski and R. Martin, Org. Lett., 2022, 24, 5109–5114 CrossRef CAS PubMed.
- Y. Cheng, Z. Qu, S. Chen, X. Ji, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2022, 364, 1573–1579 CrossRef CAS.
- I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons, 2016 Search PubMed.
- I. V. Alabugin, L. Kuhn, N. V. Krivoshchapov, P. Mehaffy and M. G. Medvedev, Chem. Soc. Rev., 2021, 50, 10212–10252 RSC.
- A. J. Sicard and R. T. Baker, Chem. Rev., 2020, 120, 9164–9303 CrossRef CAS PubMed.
- A. K. Vuppaladadiyam, E. Antunes, S. S. V. Vuppaladadiyam, Z. T. Baig, A. Subiantoro, G. Lei, S.-Y. Leu, A. K. Sarmah and H. Duan, Sci. Total Environ., 2022, 823, 153670 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- D. Meng, L. Li, A. Brown, J.-N. Desrosiers, S. Duan, C. M. Hayward, Z. He, J. Hu, T. Makowski, M. Maloney, S. Monfette, H. Perfect, J. L. Piper, M. Zhou and D. W. Widlicka, Cell Rep. Phys. Sci., 2021, 2, 100394 CrossRef CAS.
- S. Kawamura, C. J. Henderson, Y. Aoki, D. Sekine, S. Kobayashi and M. Sodeoka, Chem. Commun., 2018, 54, 11276–11279 RSC.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- S. Escoubet, S. Gastaldi, N. Vanthuyne, G. Gil, D. Siri and M. P. Bertrand, J. Org. Chem., 2006, 71, 7288–7292 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
- V. S. Kostromitin, V. V. Levin and A. D. Dilman, J. Org. Chem., 2022 DOI:10.1021/acs.joc.2c00712.
- S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS.
- J. G. Jewett, J. J. Breeyear, J. H. Brown and C. H. Bushweller, J. Am. Chem. Soc., 2000, 122, 308–323 CrossRef CAS.
- C. H. Bushweller, M. Z. Lourandos and J. A. Brunelle, J. Am. Chem. Soc., 1974, 96, 1591–1593 CrossRef CAS.
- V. J. Baker, I. J. Ferguson, A. R. Katritzky, R. Patel and S. Rahimi-Rastgoo, J. Chem. Soc., Perkin Trans. 2, 1978, 377–381 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, compound characterization, NMR spectra, and calculations. See DOI: https://doi.org/10.1039/d3sc00027c |
| This journal is © The Royal Society of Chemistry 2023 |