
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free reductive desulfurization of C-sp3-substituted thiols using phosphite catalysis†
Rana M. I.
Morsy
a,
Ganesh
Samala
a,
Ankur
Jalan
 b,
Michael E.
Kopach
b,
Naresh M.
Venneti
*a and
Jennifer L.
Stockdill
*a
b,
Michael E.
Kopach
b,
Naresh M.
Venneti
*a and
Jennifer L.
Stockdill
*a
aDepartment of Chemistry, Wayne State University, Detroit, MI 48202, USA. E-mail: gu0132@wayne.edu; stockdill@wayne.edu
bEli Lilly and Company, Indianapolis, IN 46285, USA
First published on 13th July 2023
Abstract
Phosphines and phosphites are critical tools for non-metal desulfurative methodologies due to the strength of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond. An overarching premise in these methods has been that stoichiometric (or excess) P(III) reagent is required for reactivity. Despite decades of research, a desulfurative process that is catalytic in phosphine/phosphite has not been reported. Here, we report the successful merging of two thermal radical processes: the desulfurization of unactivated and activated alkyl thiols and the reduction of P(V) = S to P(III) by reaction with a silyl radical species. We employ catalytic trimethyl phosphite, catalytic azo-bis(cyclohexyl)nitrile, and two equivalents of tris(trimethylsilyl)silane as the stoichiometric reductant and sulfur atom scavenger. This method is tolerant of common organic functional groups and affords products in good to excellent yields.
S bond. An overarching premise in these methods has been that stoichiometric (or excess) P(III) reagent is required for reactivity. Despite decades of research, a desulfurative process that is catalytic in phosphine/phosphite has not been reported. Here, we report the successful merging of two thermal radical processes: the desulfurization of unactivated and activated alkyl thiols and the reduction of P(V) = S to P(III) by reaction with a silyl radical species. We employ catalytic trimethyl phosphite, catalytic azo-bis(cyclohexyl)nitrile, and two equivalents of tris(trimethylsilyl)silane as the stoichiometric reductant and sulfur atom scavenger. This method is tolerant of common organic functional groups and affords products in good to excellent yields.
Introduction
Desulfurization is an important process1 for diverse applications from organic synthesis2 and chemical protein synthesis2 to crude oil processing and refinement.3 Numerous reports describe transition metal-based reduction reactions, including Ni,4 Co,5 W,6 Mo,7 and others.8 Unfortunately, critical limitations remain, which include pyrophoricity,4,5 toxic H2S gas evolution,4 reactivity with other S-functional groups,4,6 and the need for specialized reaction setups.5 Additionally, when the desulfurized products are deployed in living systems, trace metal contamination can be problematic.9 Photoredox strategies10a,b avoid functional group incompatibility10a and may enable further functionalization of the substrate; however, they do require the use of expensive rare earth transition metal complexes, which may limit industrial scale applications. With only two exceptions,10d,e modern and non-metal strategies for thiol desulfurization10,11 employ excess phosphine/phosphite reagents, whose aquatic toxicity9 has limited their utility in >kg-scale processes. The last several years have seen a surge of interest in the potential of P(III)/P(V) catalysis,12 largely focused on strategies to facilitate polar P(III)/P(V) = O cycles.13 We were intrigued by the potential of employing P(III)/P(V) = S catalysis for desulfurative radical transformations. Here, we present the first step along this journey, a metal-free desulfurization of unactivated thiols by P(III)/P(V) = S catalysis. We achieve this reactivity by leveraging the affinity of silyl radicals toward sulfur14 to achieve in situ regeneration of phosphites by reduction of the PS bond while sequestering the S atom to avoid H2S formation.
Results and discussion
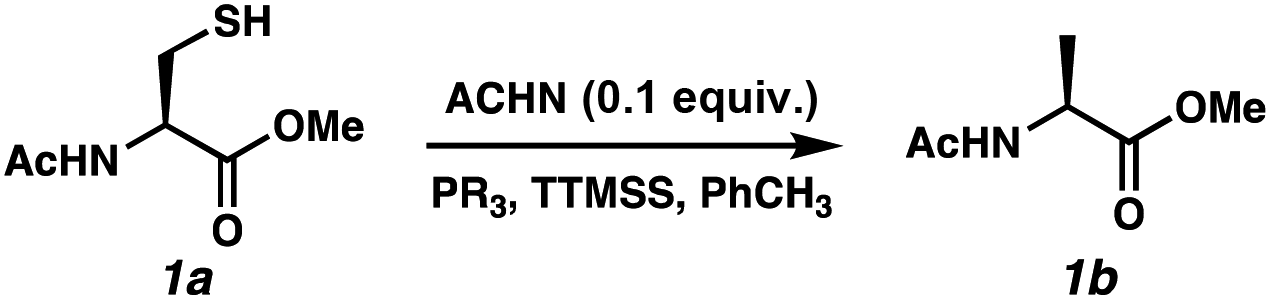
Reports as recent as 2021 have asserted that stoichiometric phosphine is a requirement for radical desulfurization reactions.12b Both phosphines and phosphites are well established reagents for desulfurization reactions,11,15 with the electronics of the R group on PR3 being key to reactivity.16 Phosphine sulfides react with super silyl hydride (TTMSS)17 under free radical conditions to produce phosphines in good yields.14 Unlike P(III)/P(V) = O redox cycles, which occur via polar pathways, the P(III)/P(V) = S reduction occurs via a radical mechanism.14 Thus, we envisioned that this cycle would merge seamlessly with a radical desulfurization mechanism. We employed Ac-N-Cys-OMe18 (1a) as a model substrate to investigate the feasibility of the catalytic transformation in the presence of various phosphines/phosphites with TTMSS as a phosphine sulfide reductant14 and/or terminal hydrogen atom donor19 (Table 1). We selected azo-bis(cyclohexyl)nitrile (ACHN) as the initiator because of its 10 h half-life20 and to avoid the formation of oxygenated by-products commonly observed in the presence of organic peroxide initiators.21 Because of the electrophilic nature of thiyl radicals, 22 we began our studies with 10 mol% of the electron-rich tris(dimethylanion)phosphine ((Me2N)3P).23 Only a single turnover was observed, with the desired product (1b) formed in 21% conversion (entry 1). We limited reaction time to 24 h for the purposes of this screen. The less basic tri-tert-butylphosphine was markedly better with 65% conversion (entry 2), while the less hindered and less electron-rich tri(n-butyl)phosphine led to 54% conversion (entry 3). Interestingly, tri-cyclohexylphosphine, which bridges these steric and electronic properties, was even less effective (38% conversion, entry 4). Trimethyl phosphite balanced these effects, improving the conversion to 80% (entry 5). Increasing the phosphite loading to 20 mol% led to complete conversion by 16 h (entry 6).24 Control experiments were consistent with the radical pathway we envisioned for this chemistry. Omission of the radical initiator (entry 7) or the phosphite (entry 8) abolished reactivity. In the absence of TTMSS, slow conversion was observed and did not reach 20% because the substrate thiol must serve as the H-atom donor for C–H bond formation (entry 9). Finally, heating is required for the reduction of the PS bond by TTMSS, as evidenced by entry 10.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | PR3 (equiv.) | TTMSS (equiv.) | ACHN (equiv.) | Temp. (°C) | Time (h) | Conversionb (%) |
| a Reactions were performed with 1 equiv. 1a (0.05 M) in degassed PhMe. b Conversion = consumption of 1a; based on 1H-NMR integration. | ||||||
| 1 | (Me2N)3P (0.1) | 1 | 0.1 | 80 | 24 | 21 |
| 2 | t Bu3P (0.1) | 1 | 0.1 | 80 | 24 | 65 |
| 3 | n Bu3P (0.1) | 1 | 0.1 | 80 | 24 | 54 |
| 4 | P(Cy)3 (0.1) | 1 | 0.1 | 80 | 24 | 38 |
| 5 | P(OMe)3 (0.1) | 1 | 0.1 | 80 | 24 | 80 |
| 6 | P(OMe) 3 (0.2) | 2 | 0.1 | 88 | 16 | >99 |
| 7 | P(OMe)3 (0.2) | 2 | — | 88 | 16 | 0 |
| 8 | — | 2 | 0.1 | 88 | 16 | 0 |
| 9 | P(OMe)3 (0.2) | — | 0.1 | 88 | 16 | 7 |
| 10 | P(OMe)3 (0.2) | 2 | 0.1 | 25 | 16 | 0 |
We proceeded with trimethyl phosphite at 88 °C for 16 h while screening different reaction parameters such as initiator, reductant, and solvent (Table 2). Using different initiators like VA-044 or Luperox A98 led to large decreases in conversion (entries 1–2). Dicumyl peroxide led to a 3-fold increase in conversion percentage relative to VA-044 at the same temperature (entry 3), but it was still inferior to ACHN.
|
|
||||
|---|---|---|---|---|
| Entry | Initiator | Reductant | Solvent | Conversionb (%) |
| a Reactions were performed with 1 equiv. 1a (0.05 M) in degassed PhMe. b Conversion = consumption of 1a; based on 1H-NMR integration. | ||||
| 1 | VA-044 | TTMSS | H2O/MeCN | 44 |
| 2 | Luperox A98 | TTMSS | PhMe | 22 |
| 3 | Dicumyl peroxide | TTMSS | PhMe | 65 |
| 4 | ACHN | TIPS | PhMe | 7 |
| 5 | ACHN | Et3GeH | PhMe | 43 |
| 6 | ACHN | n Bu3SnH | PhMe | 63 |
| 7 | ACHN | TTMSS | MeCN | 62 |
| 8 | ACHN | TTMSS | THF | 82 |
| 9 | ACHN | TTMSS | 1,4-Dioxane | >99 |

Alternative hydrogen atom/silyl radical donors had a deleterious effect on conversion. When tri-isopropylsilane (TIPS) was used, the reaction takes place, but the phosphite is not reduced (Table 2, entry 4). This apparent limited efficiency of TIPS compared to TTMSS is logical given the difference in their Si–H bond-dissociation energies (BDEs) with trialkylsilanes ranging from (90–96 kcal mol−1)25 compared to the Si–H in TTMSS at 79 kcal mol−1.26 The instability of the TIPS˙ interferes with the turnover of the phosphine sulfide, presumably because H-atom abstraction from the substrate or solvent occurs more rapidly than Si–S bond formation. Alternatively, the i-Pr group is slow to migrate after α-scission. Consistent with this reasoning, and with the similar properties of Ge and Si,27 Et3GeH (86 kcal mol−1) was able to effect ∼1 phosphine sulfide turnover in 16 h, proceeding with 43% conversion (entry 5). The best alternate PS reductant was n-Bu3SnH (74 kcal mol−1), which still reduced the phosphine sulfide more slowly than TTMSS (entry 6).28 The respective BDEs of TTMSS and n-Bu3SnH indicate that the Si˙ and Sn˙ species are similarly stable and the rate difference likely arises from differences in affinity to sulfur or the ability of TTMSS to irreversibly sequester the S-atom via an additional α-fragmentation and 1,2-group shift (see Scheme 2).
In terms of solvent, THF and MeCN, each of which were performed at 88 °C using a sealed microwave tube, were less competent than toluene (entries 7–8). Interestingly, the reaction proceeded to complete conversion with 1,4-dioxane as solvent (entry 9). Indeed, the success of 1,4-dioxane increases the scope of this method to include polar substrates.
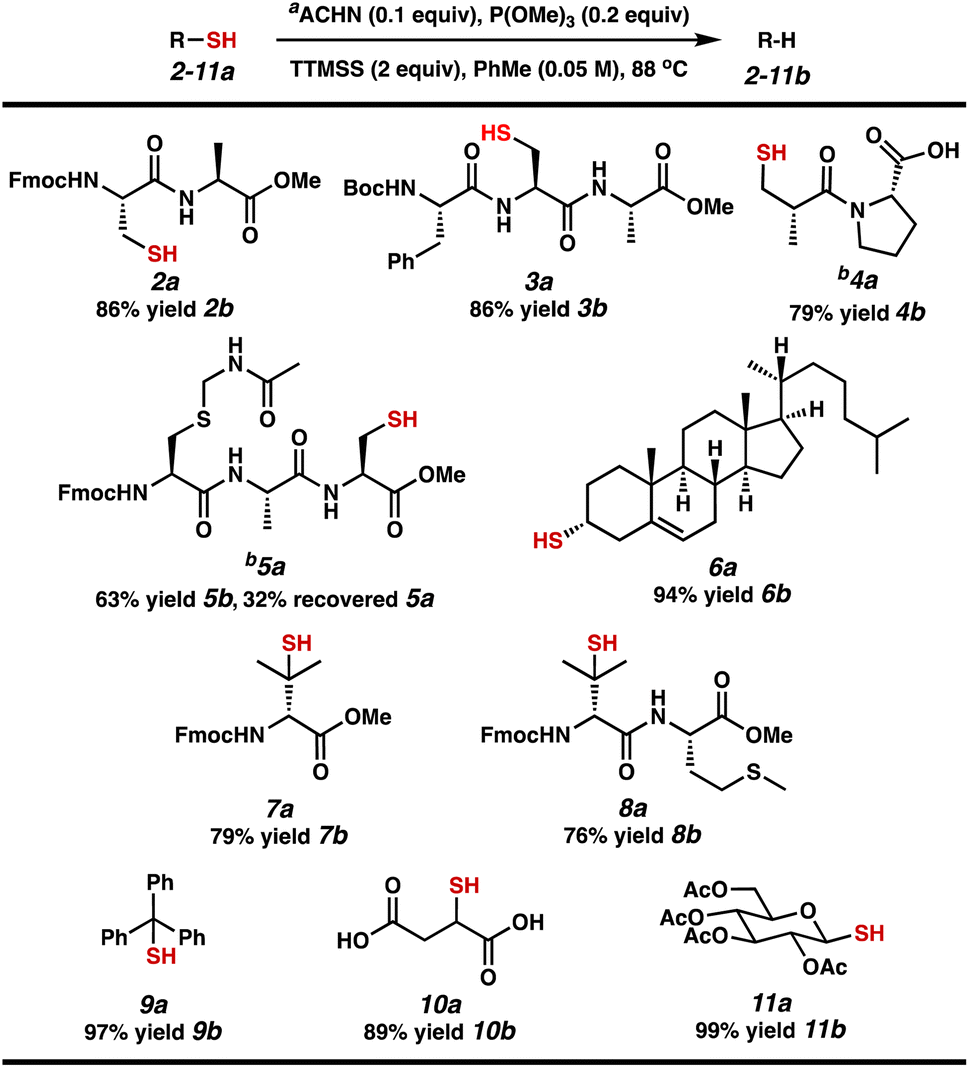
With two sets of established conditions in hand, we evaluated the scope of our efficient reductive desulfurization reaction on different relevant substrates, including short peptides and small molecule substrates that would form intermediate C-radicals with varying stability (Table 3). Both protected dipeptide 2a and tripeptide 3a – each containing a 1° alkyl thiol – were desulfurized in 86% yield. Carboxylic acid-containing captopril (4a) was desulfurized in 79% yield. Tripeptide 5a performed more sluggishly, producing 5b in 63% yield with 32% recovered 5a. We next investigated 2° and 3° alkyl thiol substrates. Cholesterol derivative 6a was converted to cholest-5-ene 6b in 94% yield. The B-ring olefin was unaffected. Penicillamine derivatives 7a and 8a proceed through tertiary C-radicals to afford valine-containing peptides 7b (79% yield) and 8b (76% yield). Notably, the thioether of methionine was stable to these conditions. Finally, we tested substrates that proceed through α-stabilized radicals. Trityl thiol 9a was desulfurized to form triphenylmethane (9b) in 97% yield. The thiols in diacid 10a and glucose derivative 11a were readily converted to the corresponding C–H bonds to provide 10b in 89% yield and a chiral pyran derivative (11b) in 99% yield.
Efforts to extend these results to longer and unprotected peptides were not fruitful due to lack of solubility of the starting materials. A brief co-solvent screen of a dipeptide acid (Fmoc-Cys-Ala-OH, SI-13†) was conducted to probe the conversion efficiency in the presence of various amounts of water, which would be needed to increase the efficiency of the desulfurization process for peptide substrates. Water is tolerated in the reaction, with the caveat that the reactivity is significantly slowed and by-product formation occurs (disulfide, thioether, and C–C bond formation were observed).23 The maximum conversion obtained for dipeptide SI-13† was 62%, requiring 2 additions of ACHN and P(OMe)3. We suspect that the hydrophobicity of TTMSS is limiting in these reactions. Presently, a water-soluble analog of TTMSS is not available, and this chemistry is therefore best suited to organic-soluble substrates. Importantly, our interest was not in achieving an ideal peptide desulfurization, 10e but rather in demonstrating P(III)/P(V) = S catalysis and learning about its behavior.
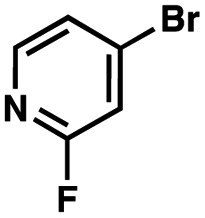
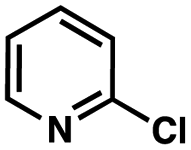
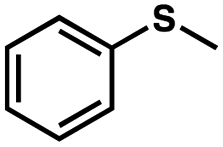
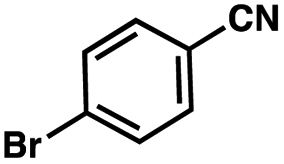
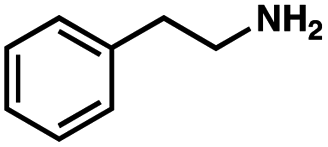
To probe the amenability of the desulfurization to large scale conditions, we performed the reaction on 4-methoxybenzyl mercaptan (12a). On 6.84 mmol scale in 1,4-dioxane, 100% conversion was observed in 3 h, with clean reactivity and no unexpected by-product formation (Scheme SI-01†). To probe the compatibility of various functional groups with the reaction conditions, we again employed 4-methoxybenzyl mercaptan (12a) in toluene along with different additives. The additives shown in Table 4 were compatible with the reaction conditions, showing no impact on the additive. In most cases, good to excellent reactivity of 12a was maintained. In entries 6 and 8, the desulfurization reaction did not occur. Alcohol additives had no negative effect on the desulfurization but were silylated. Aldehyde and ketone additives led to an unknown by-product and interfered somewhat with the desulfurization (see Table SI-02†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
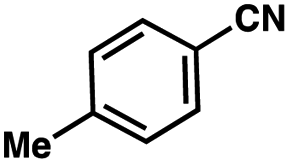
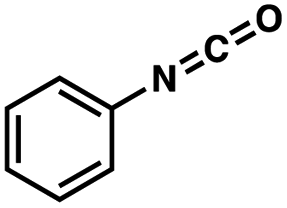
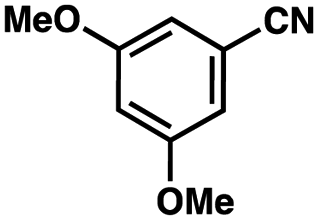
| Entrya | Additive | Unreacted 12ab (%) | GC yield 12bb (%) | Entrya | Additive | Unreacted 12ab (%) | GC yield 12ab (%) |
| a Reaction conditions: (4-methoxyphenyl)methanethiol (0.2 mmol, 1 equiv.), P(OMe)3 (0.04 mmol, 0.2 equiv.), ACHN (0.02 mmol, 0.1 equiv.), TTMSS (0.4 mmol, 2 equiv.), toluene (0.05 M), 88 °C, 3 h. b Calculated via GCMS using 1,3,5-trimethoxybenzene as internal standard. | |||||||
| 1 | None | 0 | 87 | 6 |
|
100 | 0 |
| 2 |
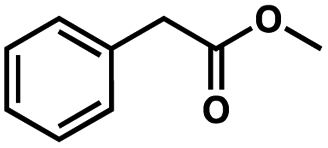
|
0 | 87 | 7 |
|
0 | 57 |
| 3 |
|
7 | 91 | 8 |
|
95 | 4 |
| 4 |
|
0 | 93 | 9 |
|
0 | 81 |
| 5 |
|
0 | 59 | 10 |
|
0 | 55 |

A radical clock experiment (Scheme 1) was performed with cyclopropane-containing thiol, 13a, which underwent ring opening to afford olefin 13b in 55% yield. This indicates that a carbon-centered radical is an intermediate in this reaction. Based on our observations, we hypothesize that reduction of the phosphine sulfide is rate-limiting and proceeds via the radical pathway described by Chatgilialoglu.14 The postulated overall radical chain mechanism for the catalytic reductive desulfurization is illustrated in Scheme 2.
 | ||
| Scheme 1 Cyclopropylcarbinyl radical rearrangement product supports a radical pathway. | ||
 | ||
| Scheme 2 Mechanism of catalytic desulfurization. | ||
Upon heating, ACHN releases N2 to initiate the reaction by forming the C-centered cyclohexyl nitrile radical. H-atom abstraction from TTMSS leads to super silyl radical 14. H-atom abstraction from the sulfhydryl group in 15a forms thiyl radical 15c and regenerates TTMSS.22 Thiyl radical 15c reacts with phosphite2916 to generate phosphoranyl radical intermediate 17.30 Tetravalent P-centered radical 17 undergoes β-scission to produce trimethyl thiophosphate 18 and the carbon centered radical 15d. H-atom transfer from TTMSS to 15d produces the desulfurized product 15b and regenerates TTMSS radical 14. Trimethyl thiophosphate 18 reacts with silyl radical 14 to form phosphoranyl radical intermediate 19. Subsequent α-scission regenerates the catalytically active phosphite (16) to be reused in the propagation step. Notably, this α-scission is facilitated by the thiophilic nature of the silane (Si–S 148.4 versus P–S 105.6 kcal mol−1).25 The resulting S-centered radical 20 induces a 1,2-group shift31 of a TMS group from silicon to sulfur, furnishing silyl radical 21 that reacts with a second equivalent of TTMSS to afford TTMSS radical 14 and silyl sulfide 22.32 This completes the in situ reduction process and permits propagation of the radical cascade. GCMS analysis of the crude reaction mixture indicates the presence of a small amount of phosphine sulfide 18 and multiple TTMSS-related peaks during the reaction.
Conclusions
In summary, we have established reaction conditions for P(III)/P(V) = S catalysis that proceeds through a tetravalent P-centered radical rather than through a pentavalent P(V) intermediate as is observed for PO reduction. The reductive desulfurization reaction sequesters the S atom, avoids the use of rare earth and transition metals, and affords desulfurized products in good yields. The method has been tested across a range of substrate types including thiols that lead to benzylic, primary, secondary, and tertiary alkyl C-centered radicals. Importantly, good functional group compatibility is observed, including free carboxylic acid, ester, carbamate, amide, nitrile, aryl halide, amine, isocyanate, thioether functionalities. Steroid, carbohydrate, amino acid, pyridine, and benzene scaffolds are compatible with the reaction. No special setup is required for this catalytic method, so it can be readily employed and adapted. This study expands the scope of metal-free and thiol-additive-free desulfurization methods while establishing the viability of P(III)/P(V) = S catalysis under radical conditions. Efforts to harness this catalysis strategy to effect other transformations and develop new phosphorus-based catalysts are ongoing.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
All authors have contributed to this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
In Fondest Memory of Prof. Robert H. Grubbs, a world leader in chemistry, humility, and kindness. These studies were supported by the National Science Foundation (CAREER Award to JLS; CHE-1554752) and the Lilly Research Award Program (LRAP) at Eli Lilly and Co. We thank A. Paul and Carol C. Schaap and Wayne State University for a Rumble-Schaap Fellowship to (RMIM).Notes and references
- J. Rentner, M. Kljajic, L. Offner and R. Breinbauer, Recent advances and applications of reductive desulfurization in organic synthesis, Tetrahedron, 2014, 70, 8983–9027, DOI:10.1016/j.tet.2014.06.104.
- (a) J. J. Eisch, L. E. Hallenbeck and K. I. Han, Organic chemistry of subvalent transition metal complexes. 12. Hydrodesulfurization of organosulfur heterocycles by metal hydride-nickel (0) complexes: accelerated single-electron transfer in carbon-sulfur bond cleavage, J. Am. Chem. Soc., 1986, 108, 7763–7767, DOI:10.1021/ja00284a045; (b) H. Rohde and O. Seitz, Invited review ligation—desulfurization: a powerful combination in the synthesis of peptides and glycopeptides, Pept. Sci., 2010, 94, 551–559, DOI:10.1002/bip.21442; (c) K. Jin and X. Li, Advances in native chemical ligation–desulfurization: a powerful strategy for peptide and protein synthesis, Chem.–Eur. J., 2018, 24, 17397–17404, DOI:10.1002/chem.201802067.
- J. Zhao and R. Wang, Research Progress of Phase Transfer Catalysts Used in Oxidative Desulfurization of Fuel Oil, Mini Rev. Org. Chem., 2021, 18, 626–648, DOI:10.2174/1570193X17999200820160851.
- (a) K. M. Ho, C. H. Lam and T. Y. Luh, Transition metal promoted reactions. Part 27. Nickel (II)-lithium aluminum hydride mediated reduction of carbon-sulfur bonds, J. Org. Chem., 1989, 54, 4474–4476, DOI:10.1021/jo00279a046; (b) M. C. Chan, K. M. Cheng, K. M. Ho, C. T. Ng, T. M. Yam, B. S. Wang and T. Y. Luh, Transition-metal-promoted reactions. 19. Nickelocene-lithium aluminum hydride: a versatile desulfurization reagent, J. Org. Chem., 1988, 53, 4466–4471, DOI:10.1021/jo00254a009; (c) M. C. Chan, K. M. Cheng, M. K. Li and T. Y. Luh, Nickelocene–lithium aluminium hydride, a new effective desulphurization reagent, J. Chem. Soc., Chem. Commun., 1985, 1610–1611, 10.1039/C39850001610.
- (a) H. Alper, F. Sibtain and J. Heveling, Phase transfer catalyzed desulfurization reactions, Tetrahedron Lett., 1983, 24, 5329–5332, DOI:10.1016/S0040–4039(00)87860-0; (b) S. C. Shim, S. Antebi and H. Alper, Desulfurization of mercaptans to hydrocarbons by carbon monoxide and water in the presence of cobalt carbonyl, Tetrahedron Lett., 1985, 26, 1935–1938, DOI:10.1016/S0040-4039(00)98345-X.
- C. T. Ng, X. Wang and T. Y. Luh, Transition metal-promoted reactions. Part 21. Tungsten hexacarbonyl-mediated carbon-sulfur bond cleavage reactions, J. Org. Chem., 1988, 53, 2536–2539, DOI:10.1021/jo00246a024.
- (a) H. Alper and C. Blais, Modified molybdenum carbonyl species; excellent reagents for the desulphurization of thiols, J. Chem. Soc., Chem. Commun., 1980, 169–170, 10.1039/C39800000169; (b) T. Y. Luh and C. S. Wong, Transition-metal promoted reactions. 12. Molybdenum hexacarbonyl-promoted reductive cleavage of the carbon-sulfur bond, J. Org. Chem., 1985, 50, 5413–5415, DOI:10.1021/jo00225a091; (c) Z. Wang, Y. Kuninobu and M. Kanai, Molybdenum-mediated desulfurization of thiols and disulfides, Synlett, 2014, 25, 1869–1872, DOI:10.1055/s-0034-1378315.
- (a) H. Alper, S. Ripley and T. L. Prince, Desulfurization of thiols and thioketones by sodium triethylborohydride and iron (II) chloride on alumina, J. Org. Chem., 1983, 48, 250–252, DOI:10.1021/jo00150a020; (b) Z. Yu and J. G. Verkade, Mild and efficient desulfurization of alkyl sulfides with sodium, Tetrahedron Lett., 1998, 39, 2671–2674, DOI:10.1016/S0040-4039(98)00368-2; (c) H. Alper and T. L. Prince, Exceedingly Mild Desulfurization of Thiols by Sodium Triethylhydroborate and Transition Metal Chlorides, Angew. Chem., Int. Ed. Engl., 1980, 19, 315–316, DOI:10.1002/anie.198003151; (d) T. Ganguly, A. Das and A. Majumdar, Iron (II) Mediated Desulfurization of Organosulfur Substrates Produces Nonheme Diiron (II)-hydrosulfides, Inorg. Chem., 2019, 58, 9998–10011, DOI:10.1021/acs.inorgchem.9b01144; (e) E. Gretz, W. M. Vetter, H. A. Stecher and A. Sen, Homogeneous Desulfurization of Thiols and Sulfides by Yb [N (SiMe3)2]2.2 THF. Relevance to Heterogeneous and Biological Desulfurizations and the Synthesis of Lanthanide (II) Sulfides, Isr. J. chem., 1990, 30, 327–329, DOI:10.1002/ijch.199000034.
- (a) A. M. Sciuto, B. J. Wong, M. E. Martens, H. Hoard-Fruchey and M. W. Perkins, Phosphine toxicity: a story of disrupted mitochondrial metabolism, Ann. N. Y. Acad. Sci., 2016, 1374, 41–51, DOI:10.1111/nyas.13081; (b) J. Roels and W. Verstraete, Biological formation of volatile phosphorus compounds, Bioresour. Technol., 2001, 79, 243–250, DOI:10.1016/S0960-8524(01)00032-3; (c) B. Wang, Q. Shen, C. Han, Y. Zheng, Z. Wang, C. Liu, L. Zhang and J. Ren, New insights into the growth response of the macrophyte Vallisneria natans exposed to phosphite, Sci. Total Environ., 2022, 851, 158189, DOI:10.1016/j.scitotenv.2022.158189.
- (a) X. F. Gao, J. J. Du, Z. Liu and J. Guo, Visible-light-induced specific desulfurization of cysteinyl peptide and glycopeptide in aqueous solution, Org. Lett., 2016, 18, 1166–1169, DOI:10.1021/acs.orglett.6b00292; (b) M. Lee, S. Neukirchen, C. Cabrele and O. Reiser, Visible-light photoredox-catalyzed desulfurization of thiol-and disulfide-containing amino acids and small peptides, J. Pept. Sci., 2017, 23, 556–562, DOI:10.1002/psc.3016; (c) E. H. Discekici, S. L. Shankel, A. Anastasaki, B. Oschmann, I. H. Lee, J. Niu, A. J. McGrath, P. G. Clark, D. S. Laitar, J. Read de Alaniz, C. J. Hawker and D. J. Lunn, Dual-pathway chain-end modification of RAFT polymers using visible light and metal-free conditions, Chem. Commun., 2017, 53, 1888–1891, 10.1039/C6CC08370F; (d) S. Kaur, D. P. Luciano, X. Fan, G. Zhao, S. Messier, M. M. Walker, Q. Zhang and T. Wang, Radical functionalization of thioglycosides in aqueous medium, Tetrahedron Lett., 2021, 86, 153499, DOI:10.1016/j.tetlet.2021.153499; (e) N. M. Venneti, G. Samala, R. M. I. Morsy, L. G. Mendoza, A. Isidro-Llobet, J. K. Tom, S. Mukherjee, M. E. Kopach and J. L. Stockdill, Phosphine-dependent photoinitiation of alkyl thiols under near-UV light facilitates user-friendly peptide desulfurization, J. Am. Chem. Soc., 2023, 145, 1053–1061 CrossRef CAS PubMed.
- For selected publications see (a) F. W. Hoffmann, R. J. Ess, T. C. Simmons and R. S. Hanzel, The Desulfurization of Mercaptans with Trialkyl Phosphites, J. Am. Chem. Soc., 1956, 78, 6414, DOI:10.1021/ja01605a033; (b) C. Walling and R. Rabinowitz, The Reaction of Thiyl Radicals with Trialkyl Phosphites, J. Am. Chem. Soc., 1957, 79, 5326, DOI:10.1021/ja01576a077; (c) C. Walling and R. Rabinowitz, The reaction of trialkyl phosphites with thiyl and alkoxy radicals, J. Am. Chem. Soc., 1959, 81, 1243–1249, DOI:10.1021/ja01514a057; (d) C. Walling, O. H. Basedow and E. S. Savas, J. Am. Chem. Soc., 1960, 82, 2181–2184, DOI:10.1021/ja01494a023; (e) C. Walling and M. S. Pearson, Some Radical Reactions of Trivalent Phosphorus Derivatives with Mercaptans, Peroxides, and Olefins. A New Radical Cyclization, J. Am. Chem. Soc., 1964, 86, 2262–2266, DOI:10.1021/ja01065a032; (f) W. G. Bentrude, Phosphoranyl radicals-their structure, formation, and reactions, Acc. Chem. Res., 1982, 15, 117–125, DOI:10.1021/ar00076a004; (g) W. G. Bentrude, E. R. Hansen, W. A. Khan and P. E. Rogers, α vs. β Scission in Reactions of Alkoxy and Thiyl Radicals with Diethyl Alkylphosphonites, J. Am. Chem. Soc., 1972, 94, 2867–2868, DOI:10.1021/ja00763a059; (h) W. G. Bentrude, E. R. Hansen, W. A. Khan, T. B. Min and P. E. Rogers, Free-Radical Chemistry of Organophosphorus Compounds. III. α vs. β Scission in Reactions of Alkoxy and Thiyl Radicals with Trivalent Organophosphorus Derivatives, J. Am. Chem. Soc., 1973, 95, 2286–2293, DOI:10.1021/ja00788a031; (i) W. G. Bentrude, J. H. Hargis, N. A. Johnson, T. B. Min, P. E. Rusek Jr, H. W. Tan and R. A. Wielesek, Free radical studies of organophosphorus compounds. 7. The stereochemistries of alkoxy and thiyl radical oxidations of phosphines and cyclic phosphites. Possible permutational isomerization modes for phosphoranyl radicals, J. Am. Chem. Soc., 1976, 98, 5348–5357, DOI:10.1021/ja00433a049; (j) W. G. Bentrude, J. J. L. Fu and P. E. Rogers, Free-radical chemistry of organophosphorus compounds. IV. Polar, bond strength, and resonance effects on phosphoranyl radical formation, J. Am. Chem. Soc., 1973, 95, 3625–3635, DOI:10.1021/ja00792a027; (k) W. G. Bentrude and P. E. Rogers, Free-radical chemistry of organophosphorus compounds. V. Reactions of thiyl radicals with alkylphosphonites. Effects of activation energies and A values on the. alpha. vs. beta. scission competition, J. Am. Chem. Soc., 1976, 98, 1674–1677, DOI:10.1021/ja00423a006; (l) W. G. Bentrude and F. R. Hartley, The Chemistry of Organophosphorus Compounds, Wiley, Chichester, 1990, vol. 1, pp. 531–566, DOI:10.1002/9780470034439m; (m) S. Marque and P. Tordo, Reactivity of Phosphorus Centered Radicals, Top. Curr. Chem., 2005, 250, 43–76, DOI:10.1007/b100981.
- (a) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine organocatalysis, Chem. Rev., 2018, 118, 10049–10293, DOI:10.1021/acs.chemrev.8b00081; (b) C. Xie, A. J. Smaligo, X. R. Song and O. Kwon, Phosphorus-based catalysis, ACS Cent. Sci., 2021, 7, 536–558, DOI:10.1021/acscentsci.0c01493; (c) M. C. Fu, J. X. Wang and R. Shang, Triphenylphosphine-Catalyzed Alkylative Iododecarboxylation with Lithium Iodide under Visible Light, Org. Lett., 2020, 22, 8572–8577, DOI:10.1021/acs.orglett.0c03173.
- (a) N. L. Dunn, M. Ha and A. T. Radosevich, Main group redox catalysis: reversible PIII/PV redox cycling at a phosphorus platform, J. Am. Chem. Soc., 2012, 134, 11330–11333, DOI:10.1021/ja302963p; (b) K. D. Reichl, N. L. Dunn, N. J. Fastuca and A. T. Radosevich, Biphilic organophosphorus catalysis: Regioselective reductive transposition of allylic bromides via PIII/PV redox cycling, J. Am. Chem. Soc., 2015, 137, 5292–5295, DOI:10.1021/jacs.5b01899; (c) K. Lee, A. V. Blake, A. Tanushi, S. M. McCarthy, D. Kim, S. M. Loria, C. M. Donahue, K. D. Spielvogel, J. M. Keith, S. R. Daly and A. T. Radosevich, Validating the biphilic hypothesis of nontrigonal phosphorus (III) compounds, Angew. Chem., Int. Ed., 2019, 58, 6993, DOI:10.1002/anie.201901779; (d) G. Li, T. V. Nykaza, J. C. Cooper, A. Ramirez, M. R. Luzung and A. T. Radosevich, AnImprovedPIII/PV = O-Catalyzed Reductive C–N Coupling of Nitroaromatics and Boronic Acids by Mechanistic Differentiation of Rate- and Product- Determining Steps, J. Am. Chem. Soc., 2020, 142, 6786–6799, DOI:10.1021/jacs.0c01666; (e) J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. Taylor, Phosphoranyl radical fragmentation reactions driven by photoredox catalysis, ACS Catal., 2020, 10, 7250–7261, DOI:10.1021/acscatal.0c01923; (f) G. Zeng, S. Maeda, T. Taketsugu and S. Sakaki, Theoretical study of hydrogenation catalysis of phosphorus compound and prediction of catalyst with high activity and wide application scope, ACS Catal., 2016, 6, 4859–4870, DOI:10.1021/acscatal.5b02968.
- R. Romeo, L. A. Wozniak and C. Chatgilialoglu, Radical-based reduction of phosphine sulfides and phosphine selenides by (Me3Si)3SiH, Tetrahedron Lett., 2000, 41, 9899–9902, DOI:10.1016/S0040-4039(00)01759-7.
- J. M. Lopp and V. A. Schmidt, Intermolecular Phosphite-Mediated Radical Desulfurative Alkene Alkylation Using Thiols, Org. Lett., 2019, 21, 8031–8036, DOI:10.1021/acs.orglett.9b03018.
- J. M. Lopp and V. A. Schmidt, Experimental and Computational Investigations into Trivalent Phosphorous-Mediated Radical Thiol Desulfurization, ChemRxiv, Cambridge Open Engage, Cambridge, 2021, preprint, DOI:10.26434/chemrxiv-2021-fgq3c.
- (a) C. Chatgilialoglu, Organosilanes in radical chemistry, John Wiley & Sons, 2004, DOI:10.1002/0470024755; (b) C. Chatgilialoglu, Organosilanes as radical-based reducing agents in synthesis, Acc. Chem. Res., 1992, 25, 188–194, DOI:10.1021/ar00016a003; (c) C. Chatgilialoglu and V. I. Timokhin, Silanes as reducing reagents in radical chemistry, in Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012, DOI:10.1002/9781119953678.rad074; (d) H. Janbazi, C. Schulz, I. Wlokas, H. Wang and S. Peukert, Thermochemistry of organosilane compounds and organosilyl radicals, Proc. Combust. Inst., 2021, 38, 1259–1267, DOI:10.1016/j.proci.2020.06.114; (e) B. Giese, B. Kopping and C. Chatgilialoglu, Tris(trimethylsilyl) silane as mediator in organic synthesis via radicals, Tetrahedron Lett., 1989, 30, 681–684, DOI:10.1016/S0040-4039(01)80281-1; (f) M. Lesage, C. Chatgilialoglu and D. Griller, Tris (trimethylsilyl) silane: A catalyst for radical mediated reduction reactions, Tetrahedron Lett., 1989, 30, 2733–2734, DOI:10.1016/S0040-4039(00)99111-1; (g) M. Ballestri, C. Chatgilialoglu, K. B. Clark, D. Griller, B. Giese and B. Kopping, Tris (trimethylsilyl) silane as a radical-based reducing agent in synthesis, J. Org. Chem., 1991, 56, 678–683, DOI:10.1021/jo00002a035; (h) C. Chatgilialoglu, A. Guerrini and M. Lucarini, The trimethylsilyl substituent effect on the reactivity of silanes. Structural correlations between silyl radicals and their parent silanes, J. Org. Chem., 1992, 57, 3405–3409, DOI:10.1021/jo00038a031; (i) C. Chatgilialoglu, The Tris (trimethylsilyl) silane/Thiol Reducing System: A Tool for Measuring Rate Constants for Reactions of Carbon-Centered Radicals with Thiols, Helv. Chim. Acta, 2006, 89, 2387–2398, DOI:10.1002/hlca.200690221; (j) C. Chatgilialoglu, (Me3Si)3SiH: Twenty Years After Its Discovery as a Radical-Based Reducing Agent, Chem.–Eur. J., 2008, 14, 2310–2320, DOI:10.1002/chem.200701415; (k) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Thirty years of (TMS)3SiH: A milestone in radical-based synthetic chemistry, Chem. Rev., 2018, 118, 6516–6572, DOI:10.1021/acs.chemrev.8b00109; (l) C. Chatgilialoglu and J. Lalevée, Recent applications of the (TMS)3SiH radical-based reagent, Molecules, 2012, 17, 527–555, DOI:10.3390/molecules17010527; (m) C. Chatgilialoglu and V. I. Timokhin, 2 Silyl Radicals in Chemical Synthesis, Adv. Organomet. Chem., 2008, 57, 117–181 CrossRef CAS.
- Ac-N-Cys-OMe was prepared according to reported procedure: Y. A. Lin, J. M. Chalker, N. Floyd, G. J. Bernardes and B. G. Davis, Allyl sulfides are privileged substrates in aqueous cross-metathesis: application to site-selective protein modification, J. Am. Chem. Soc., 2008, 130, 9642–9643, DOI:10.1021/ja8026168.
- C. Chatgilialoglu, D. Griller and M. Lesage, Tris(trimethylsilyl) silane. A new reducing agent, J. Org. Chem., 1988, 53, 3641–3642, DOI:10.1021/jo00250a051.
- https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/411/888/thermal_initiators.pdf, accessed 28 November 2022.
- M. B. Cole Jr, An “azo” substitute for benzoyl peroxide in polymerizing glycol methacrylate (GMA), Micron, 1979, 10, 135–137, DOI:10.1016/0047-7206(79)90005-0.
- (a) H. Subramanian, R. Moorthy and M. P. Sibi, Thiyl Radicals: From Simple Radical Additions to Asymmetric Catalysis, Angew. Chem., Int. Ed., 2014, 53, 13660–13662, DOI:10.1002/anie.201408781; (b) F. Denes, M. Pichowicz, G. Povie and P. Renaud, Thiyl radicals in organic synthesis, Chem. Rev., 2014, 114, 2587–2693, DOI:10.1021/cr400441m.
- See the ESI† for details.
- Use of an aromatic phosphine (PPh3) under the optimized conditions afforded 98% (toluene) and 92% (1,4-dioxane) conversions.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 1st edn, 2007, DOI:10.1201/9781420007282, ISBN 9780429128684.
- (a) J. F. Brazeau, Tris(trimethylsilyl) silane (TTMSS), Synlett, 2007, 1972–1973, DOI:10.1055/s-2007-984873; (b) L. J. Laarhoven, P. Mulder and D. D. Wayner, Determination of bond dissociation enthalpies in solution by photoacoustic calorimetry, Acc. Chem. Res., 1999, 32, 342–349, DOI:10.1021/ar9703443.
- J. Lalevée, N. Blanchard, B. Graff, X. Allonas and J. P. Fouassier, Tris (trimethylsilyl) silyl versus tris (trimethylsilyl) germyl: radical reactivity and oxidation ability, J. Organomet. Chem., 2008, 693, 3643–3649, DOI:10.1016/j.jorganchem.2008.08.039.
- G. Avar and W. P. Neumann, J. Organomet. Chem., 1977, 131, 207–214, DOI:10.1016/S0022-328X(00)93403-X.
- (a) C. Walling and R. Rabinowitz, The reaction of thiyl radicals with trialkyl phosphites1, J. Am. Chem. Soc., 1957, 79, 5326, DOI:10.1021/ja01576a077; (b) F. W. Hoffmann, R. J. Ess, T. C. Simmons and R. S. Hanzel, The desulfurization of mercaptans with trialkyl phosphites, J. Am. Chem. Soc., 1956, 78, 6414, DOI:10.1021/ja01605a034; (c) C. Walling and M. S. Pearson, Some radical reactions of trivalent phosphorus derivatives with mercaptans, peroxides, and olefins. A new radical cyclization, J. Am. Chem. Soc., 1964, 86, 2262–2266, DOI:10.1021/ja01065a032.
- (a) D. Leca, L. Fensterbank, E. Lacôte and M. Malacria, Recent advances in the use of phosphorus-centered radicals in organic chemistry, Chem. Soc. Rev., 2005, 34, 858–865, 10.1039/B500511F; (b) W. G. Bentrude, Phosphoranyl radicals-their structure, formation, and reactions, Acc. Chem. Res., 1982, 15, 117–125, DOI:10.1021/ar00076a004; (c) K. Luo, W. C. Yang and L. Wu, Photoredox catalysis in organophosphorus chemistry, Asian J. Org. Chem., 2017, 6, 350–367, DOI:10.1002/ajoc.201600512; (d) C. Lopin, G. Gouhier, A. Gautier and S. R. Piettre, Phosphonyl, Phosphonothioyl, Phosphonodithioyl, and Phosphonotrithioyl Radicals: Generation and Study of Their Addition onto Alkenes, J. Org. Chem., 2003, 68, 9916–9923, DOI:10.1021/jo0348064.
- (a) M. Ballestri, C. Chatgilialoglu and G. Seconi, (Me3Si)3SiS. H.: a new radical-based reducing agent, J. Organomet. Chem., 1991, 408, C1–C4, DOI:10.1016/0022-328X(91)80107-U; (b) C. Chatgilialoglu, Structural and chemical properties of silyl radicals, Chem. Rev., 1995, 95, 1229–1251, DOI:10.1021/cr00037a005.
- C. Chatgilialoglu and K. D. Asmus, Sulfur-centered reactive intermediates in chemistry and biology, Springer Science & Business Media, 2013, vol. 197, DOI:10.1007/978-1-4684-5874-9.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, tabulated spectroscopic data, and images of spectra. See DOI: https://doi.org/10.1039/d3sc00045a |
| This journal is © The Royal Society of Chemistry 2023 |