DOI:
10.1039/D3SC00118K
(Edge Article)
Chem. Sci., 2023,
14, 4134-4142
Enantio- and diastereodivergent synthesis of fused indolizines enabled by synergistic Cu/Ir catalysis†
Received
7th January 2023
, Accepted 13th March 2023
First published on 14th March 2023
Abstract
Highly diastereo-/enantioselective assembly of 2,3-fused indolizine derivatives could be easily available through a cascade allylation/Friedel–Crafts type reaction enabled by a synergistic Cu/Ir catalysis. This designed protocol provides an unprecedented and facile route to enantioenriched indolizines bearing three stereogenic centers in moderate to high yields with excellent stereoselective control, which also featured broad substrate generality. Remarkably, four stereoisomers of the 2,3-fused indolizine products could be efficiently constructed in a predictable manner through the pairwise combination of copper and iridium catalysts. The synthetic utility of this method was readily elaborated by a gram-scale reaction, and synthetic transformations to other important chiral indolizine derivatives. Quantum mechanical explorations constructed a plausible synergetic catalytic cycle, revealed the origins of stereodivergence, and rationalized the protonation-stimulated stereoselective Friedel–Crafts type cyclization to form the indolizine products.
Introduction
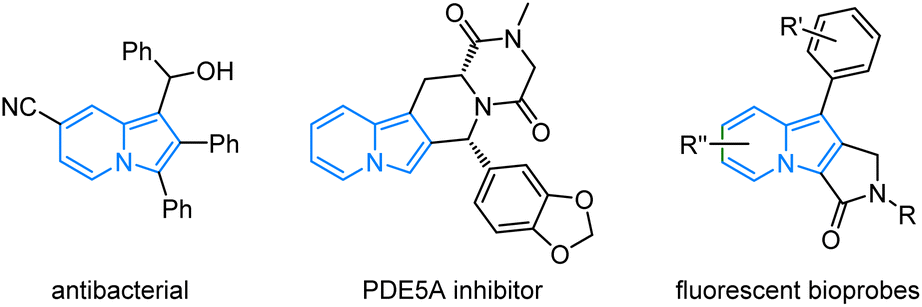
The incorporation of stereogenic centers into privileged scaffolds to enhance the three-dimensional complexity represents a significant objective of chemical synthesis in support of pharmaceutical and materials science.1 Indolizines, as one kind of important N-heterocycles, display an impressive range of bioactivities,2 such as anticancer, antioxidant, antifungal, antiviral, and photophysical properties (Fig. 1).3 The partially or fully reduced indolizine (mostly in enantiopure form) constitutes the core skeleton of over thousands of alkaloids.4 The broad utilities of indolizine have long motivated the development of efficient methods for their preparation.
 |
| | Fig. 1 Examples of bioactive compounds and organic fluorophores bearing indolizine heterocycles. | |
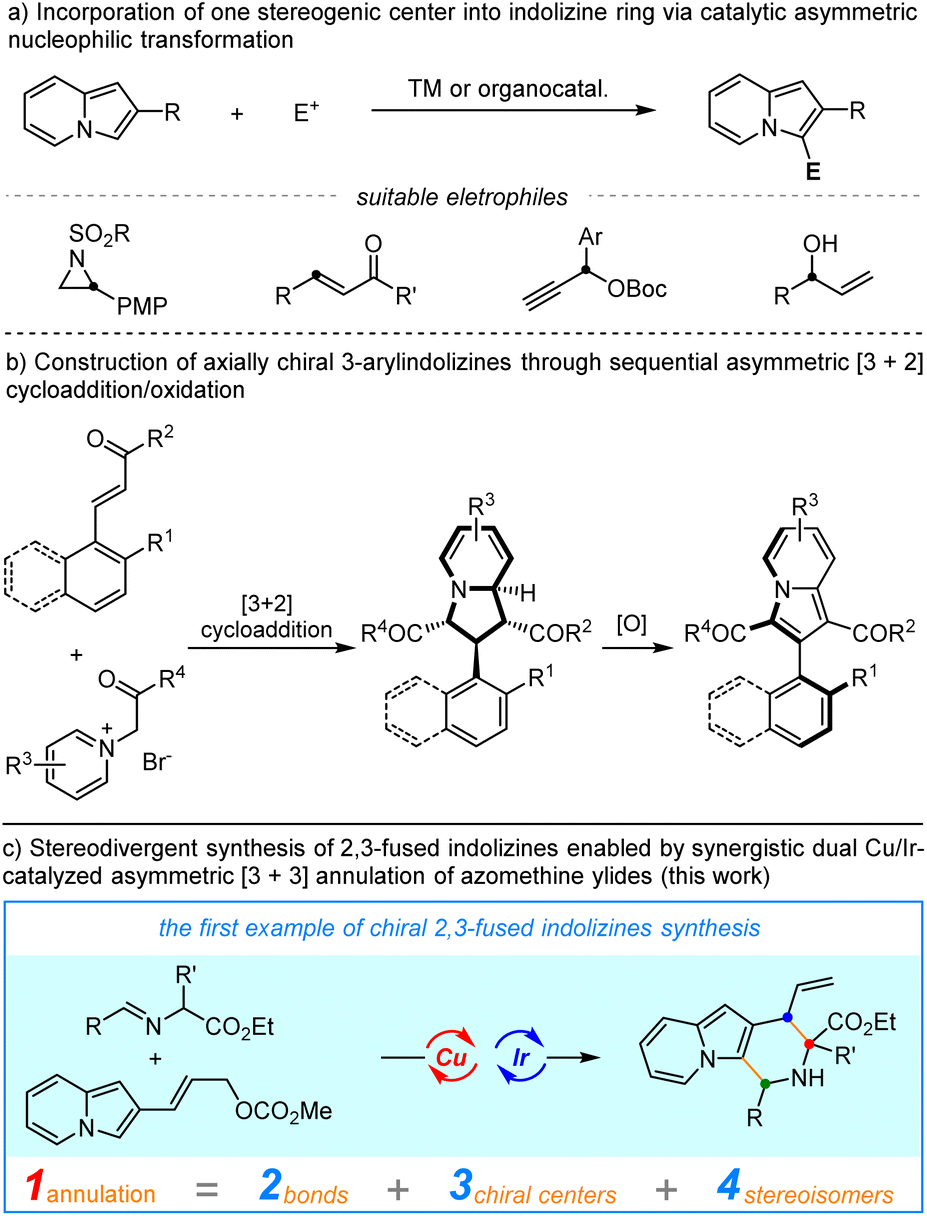
However, in comparison with the well-established non-asymmetric approaches, access to enantioenriched indolizines remains rare.5 Successful catalytic asymmetric synthesis of chiral indolizine derivatives heavily relies on the Friedel–Crafts-type transformations with electrophiles to incorporate one stereogenic center on the C3-position of indolizine (Scheme 1a), including asymmetric conjugate addition, asymmetric propargylation and asymmetric allylic alkylation.6 Recently, Feng, Liu, and coworkers developed an elegant diastereo-/enantioselective [3 + 2]-cycloaddition between pyridinium ylides and enones to deliver chiral tetrahydroindolizines, which could be easily oxidized to access high-value enantioenriched 3-arylindolizines with axial chirality.7 Synthetic methods for other types of chiral indolizines, in particular, the privileged fused indolizine motifs have been rarely exploited. Therefore, a conceptually different strategy to achieve catalytic enantioselective functionalization of indolizine is urgently needed. On the other hand, the efficient generation of compound libraries with a broad stereochemical diversity is of great significance in both synthetic and medicinal chemistry.8 In this aspect, the stereodivergent synthesis which involves the concurrent activation of two reactants via a dual organo-/metal, metal/metal or organo-/organo-catalysis is among the most effective methodologies.9–11 In continuation of our longstanding interest in stereodivergent synthesis of α-amino acids,9b,9d we were intrigued by the possibility of π-allyl-Ir species derived from 2-indolizine allyl carbonates serving as 3-atom amphiphilic species in a cascade annulation with 1,3-dipolar-type metallated azomethine ylides. We anticipated that the addition of a nucleophilic Cu-azomethine ylides,12 to such electrophilic π-allyl-Ir species followed by an intramolecular Friedel–Crafts-type annulation would provide a straightforward entry to chiral indolizines bearing three stereogenic centers. In addition, the high stereocontrol imparted by Ir catalysis on allylic alkylation13 and Cu catalysis on α-functionalization of azomethine ylides might be able to control the generated stereogenic centers, thus allowing the precise construction of different stereoisomers of the 2,3-fused indolizines. Herein, we describe the first example of enantio-/diastereoselective synthesis of chiral indolizines via Cu/Ir-catalyzed asymmetric annulation through a cascade allylic alkylation/Friedel–Crafts reaction in high yields with excellent stereoselective control.
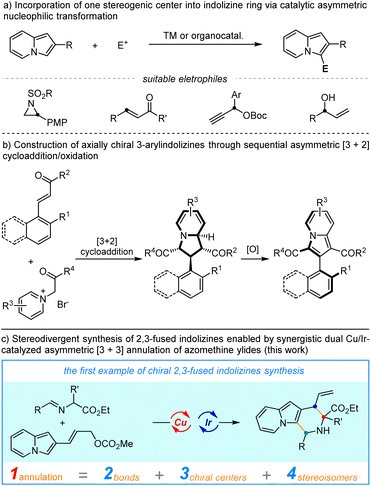 |
| | Scheme 1 Catalytic asymmetric synthesis of chiral indolizine derivatives. | |
Results and discussion
Optimization of reaction conditions
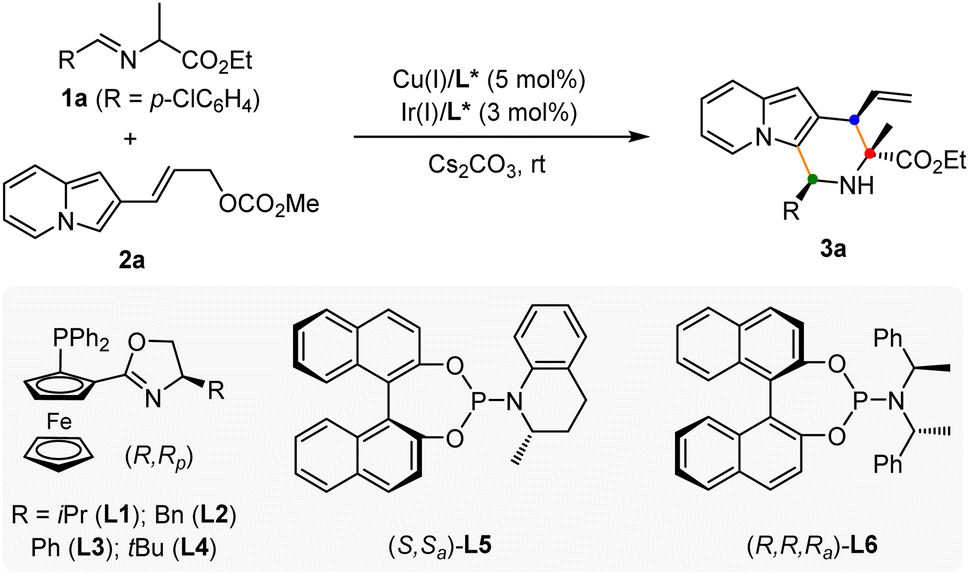
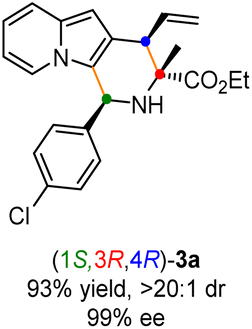
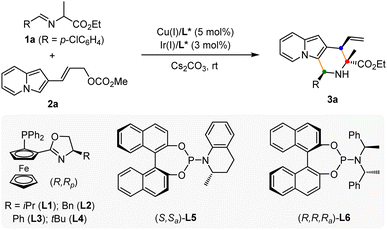
To verify our hypothesis, a model reaction was conducted using rac-alanine ethyl ester derived aldimine ester 1a and 2-indolizinyl allyl carbonate 2a as the substrates, Cs2CO3 as the base, and dichloromethane as the solvent. As shown in Table 1, we first tested the catalytic efficiency of the combined [Cu(I)/(R,Rp)-L1 + Ir(I)/(S,Sa)-L5] system which was used for the previous reactions with 2-indolyl allyl carbonates.11q,14 The reaction proceeded smoothly via the designed cascade allylation/Friedel–Crafts annulation pathway, giving (1S,3R,4R)-3a in 73% yield albeit with unsatisfactory diastereoselectivity and enantioselectivity (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 87% ee for the major isomer) (Table 1, entry 1). Although chiral phosphoramidite L5 is privileged in our previous work,11q however, this reactivity and enantioselectivity on the allylic stereogenic center did not translate to 2-indolizinyl allyl carbonate. Encouragingly, using chiral phosphoramidite ligand (R,R,Ra)-L6 (ref. 15) for the iridium catalyst instead of L5, the catalytic reactivity and stereoselective control were significantly enhanced, affording the desired product 3a in 93% yield with >20:1 dr and 99% ee (Table 1, entry 2). No better results were achieved through the screening of a series of Phosferrox ligands16L2–L4 for the copper catalyst (Table 1, entries 3–5). The effect of solvent was then tested, and dichloromethane provided the best results with a 93% yield among these solvents. No reaction was observed in the absence of either copper or iridium catalysts, which showed that both two chiral metal complexes are critical for this cascade allylation/Friedel–Crafts annulation to deliver the [3 + 3] annulation product (Table 1, entries 10–11).
:1 dr, 87% ee for the major isomer) (Table 1, entry 1). Although chiral phosphoramidite L5 is privileged in our previous work,11q however, this reactivity and enantioselectivity on the allylic stereogenic center did not translate to 2-indolizinyl allyl carbonate. Encouragingly, using chiral phosphoramidite ligand (R,R,Ra)-L6 (ref. 15) for the iridium catalyst instead of L5, the catalytic reactivity and stereoselective control were significantly enhanced, affording the desired product 3a in 93% yield with >20:1 dr and 99% ee (Table 1, entry 2). No better results were achieved through the screening of a series of Phosferrox ligands16L2–L4 for the copper catalyst (Table 1, entries 3–5). The effect of solvent was then tested, and dichloromethane provided the best results with a 93% yield among these solvents. No reaction was observed in the absence of either copper or iridium catalysts, which showed that both two chiral metal complexes are critical for this cascade allylation/Friedel–Crafts annulation to deliver the [3 + 3] annulation product (Table 1, entries 10–11).
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
L* for Cu |
L* for Ir |
Solvent |
drb |
Yieldc (%) |
eed (%) |
|
All reactions were carried out with 0.3 mmol 1a and 0.2 mmol 2a in 2 mL of solvent within 2–12 h.
dr was determined by the crude 1H NMR analysis.
Yields refer to the isolated products after chromatographic purification.
ee was determined by chiral HPLC analysis.
|
| 1 |
(R,Rp)-L1 |
(Sa,S)-L5 |
CH2Cl2 |
3:1 |
73 |
87 |
| 2 |
(R,Rp)-L1 |
(Ra,R,R)-L6 |
CH2Cl2 |
>20:1 |
93 |
99 |
| 3 |
(R,Rp)-L2 |
(Ra,R,R)-L6 |
CH2Cl2 |
19:1 |
83 |
97 |
| 4 |
(R,Rp)-L3 |
(Ra,R,R)-L6 |
CH2Cl2 |
>20:1 |
85 |
99 |
| 5 |
(S,Sp)-L4 |
(Sa,S,S)-L6 |
CH2Cl2 |
>20:1 |
87 |
99 |
| 6 |
(R,Rp)-L1 |
(Ra,R,R)-L6 |
THF |
>20:1 |
85 |
99 |
| 7 |
(R,Rp)-L1 |
(Ra,R,R)-L6 |
Toluene |
>20:1 |
87 |
99 |
| 8 |
(R,Rp)-L1 |
(Ra,R,R)-L6 |
EtOAc |
>20:1 |
84 |
99 |
| 9 |
(R,Rp)-L1 |
(Ra,R,R)-L6 |
MeCN |
19:1 |
80 |
97 |
| 10 |
(R,Rp)-L1 |
No Ir complex |
CH2Cl2 |
— |
NA |
NA |
| 11 |
No Cu complex |
(Ra,R,R)-L6 |
CH2Cl2 |
— |
NA |
NA |
Substrate scope study
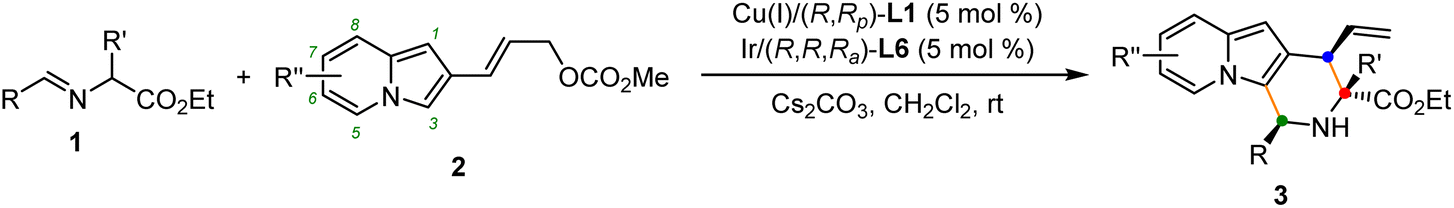
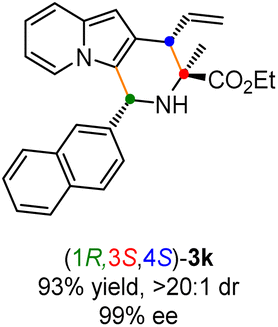
The generality of the current protocol with respect to the azomethine ylide precursors was first examined under the established reaction conditions. As shown in Table 2, the scope of the substituted aldimine esters for this synergetic Cu/Ir-catalyzed asymmetric allylation/Friedel–Crafts type reaction was remarkably broad. A wide range of natural and unnatural amino acids derived aldimine esters underwent this cascade reaction with 2-indolizinyl allyl carbonates efficiently, giving the corresponding annulation products in good to high yields with excellent stereoselective control. The aromatic aldehyde derived α-methyl substituted aldimine esters (1a–1i) bearing substituents of different electronic properties on different positions participated in this reaction smoothly, affording the desired products (3a–3i) in 85–95% yields and generally >20:1 dr and 99% ee (Table 2, entries 1–9). In addition, the naphthyl substituted aldimine esters with steric hindrance also worked well to deliver the desired products 3j and 3k with excellent results (86% yield, >20:1 dr, 99% ee and 95% yield, >20:1 dr, 99%, respectively, entries 10 and 11). It's worth noting that the heteroaromatic substituted substrates containing 2-thenyl, 2-furanyl, and N-Ts 3-indolyl groups, were well compatible to generate the corresponding products 3l–3n in 82–90% yields, >20:1 dr and 99% ee (entries 12–14). To our delight, the more challenging aliphatic aldehyde-derived aldimine esters 1o and 1p also can undergo this transformation smoothly, leading to the products 3o and 3p with excellent results (59% yield, 10:1 dr, 96% ee for 3o, and 60% yield, >20:1 dr, 99% ee for 3p, entries 15 and 16). Promoted by the success of α-methyl substituted aldimine esters in this cascade reaction, we then turned our attention to further investigating other α-substituted aldimine esters. When the methyl group was replaced by n-butyl (1q), i-butyl (1r), benzyl (1s), and phenylethyl (1t) groups, the corresponding aldimine esters worked well to give the desired products (3q–3t) with excellent results (85–96% yields, >20:1 dr, generally 99% ee, entries 17–20). Furthermore, we found that a series of aldimine esters containing α-functionalized substituted groups, such as allyl (1u), cinnamyl (1v), CH2CH2SMe (1w), CH2CH2CO2Et (1x), (CH2)4NHCbz (1y) groups, could function as compatible substrates to provide products (3u–3y) in 91–97% yields with >20:1 dr and all with 99% ee (entries 21–25). Tryptophan-derived aldimine ester bearing α-3-indolymethyl (1z) with high steric hindrance worked well to give the corresponding cycloadduct 3z in 55% yield with exclusive diastereoselectivity and excellent enantioselectivity (entry 26). Aldimine esters bearing sterically hindered α-substituted groups, such as isopropyl or cyclohexyl groups, did not work in this catalytic system. Remarkably, glycine-derived aldimine ester 1A, which posed an additional challenge to control the stereoselectivity since the initially formed allylation intermediate still contains a readily-racemized proton, also reacted smoothly with 2a affording desired product 3A with satisfactory results (entry 27). To further expand the scope generality of this protocol, a variety of indolizine-derived allyl carbonates bearing varied substituents with different electronic properties were all well-tolerated in this synergistic Cu/Ir catalytic system, providing the highly functionalized indolizine derivatives (3B–3E) in moderate to good yields with excellent stereoselectivities (75–83% yields, all with >20:1 dr and 99% ee, entries 28–31).
Table 2 Reaction scope of Cu/Ir-catalyzed asymmetric [3 + 3] annulationa
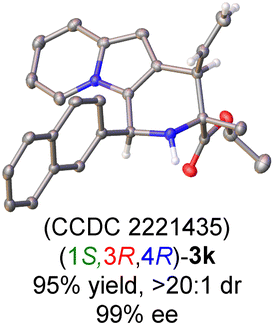
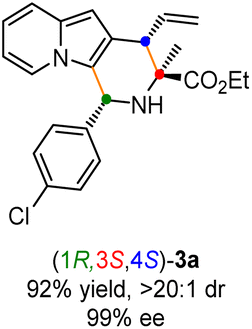
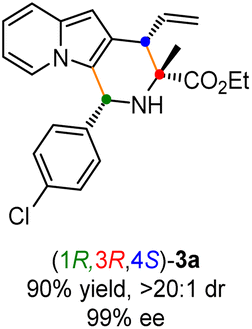
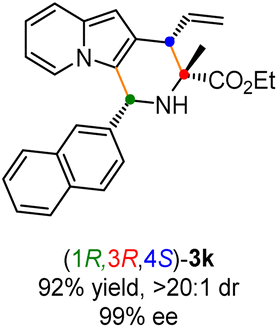
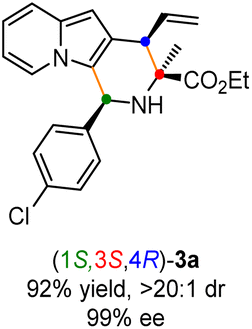
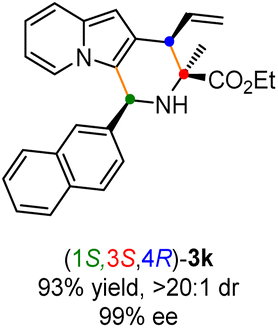
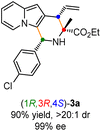
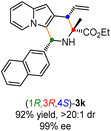
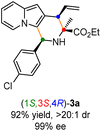
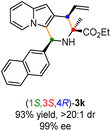
Having established the optimal reaction conditions for the preparation of (1S,3R,4R)-3a, we began to evaluate the feasibility of stereodivergent synthesis via the synergistic Cu/Ir catalysis (Table 3). To our delight, the four stereoisomers of the indolizine derivative 3a could be successfully prepared in high yields (90–93%) with exclusive diastereoselectivities and almost perfect enantioselectivities (all >20:1 dr and 99% ee) by simply switching the configuration combination of two chiral ligands L1 and L6 in the combined two chiral metal complexes. In addition, four stereoisomers of 2-naphthyl fused indolizine 3k could be predictably achieved in the same manner. The absolute configuration of 3k obtained with the combined [Cu(I)/(R,Rp)-L1 + Ir(I)/(R,R,Ra)-L5] was unanimously determined to be (1S,3R,4R) by X-ray diffraction analysis (CCDC 2221435),17 and the absolute configuration of other products was tentatively assigned by structural analogy.
Table 3 Stereodivergent synthesis
| [Cu]/(R,Rp)-L1 + [Ir]/(R,R,Ra)-L6 |
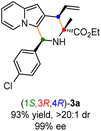
|
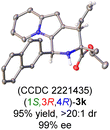
|
| [Cu]/(S,Sp)-L1 + [Ir]/(S,S,Sa)-L6 |
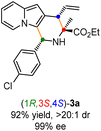
|
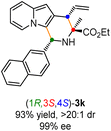
|
| [Cu]/(R,Rp)-L1 + [Ir]/(S,S,Sa)-L6 |
|
|
| [Cu]/(S,Sp)-L1 + [Ir]/(R,R,Ra)-L6 |
|
|
Scale-up experiments and synthetic application
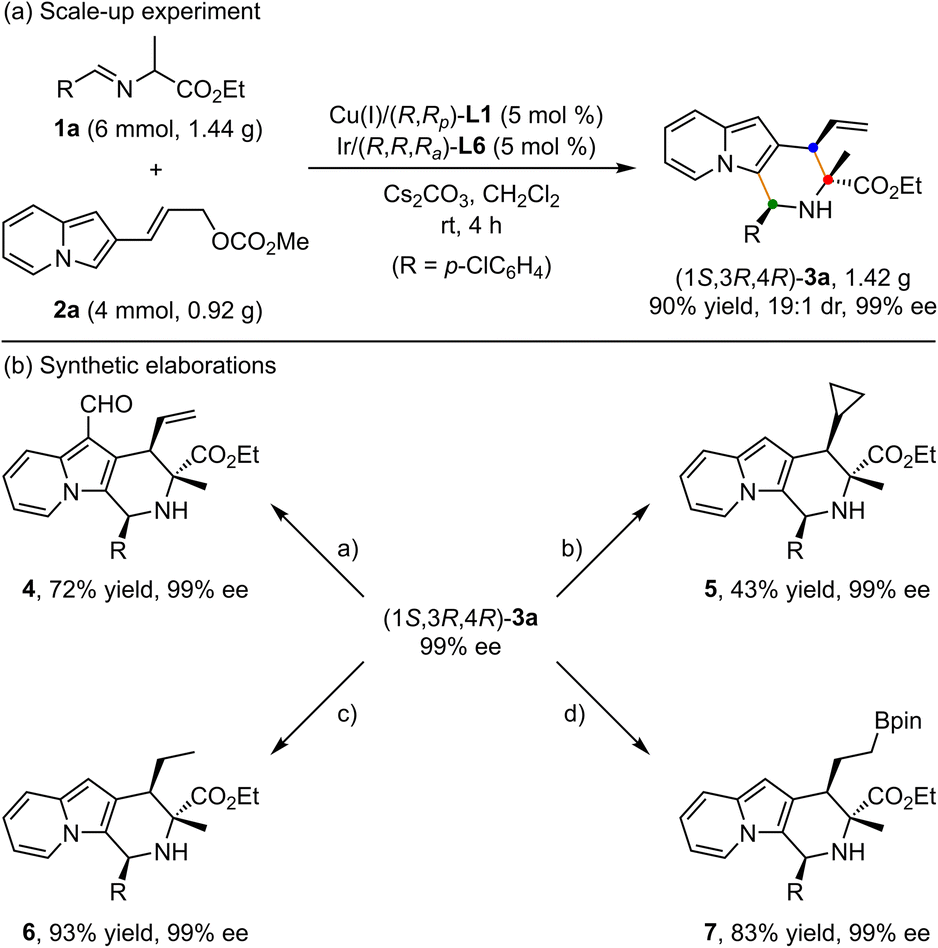
To showcase the synthetic utility of this methodology, first, a gram-scale of synergistic Cu/Ir-catalyzed asymmetric [3 + 3] annulation of alanine ethyl ester derived aldimine ester 1a and 2-indolizinyl allyl carbonate 2a was conducted (Scheme 2a), and the enantiopure 2,3-fused indolizine (1S,3R,4R)-3a could be smoothly achieved in 90% yield with 19:1 dr and 99% ee. As outlined in Scheme 2b, some synthetic transformations of (1S,3R,4R)-3a were further surveyed. It went through the Vilsmeier–Haack reaction efficiently to install a formyl group at the C1-position, providing the corresponding aldehyde compound 4 in 72% yield without any loss of enantioselectivity. The terminal alkenyl motif of product 3a could react with CH2N2 catalyzed by Pd(OAc)2 to generate product 5 containing the cyclopropane unit in moderate yield with maintained diastereoselectivity and enantioselectivity. In addition, Pd/C-catalyzed direct hydrogenation afforded product 6 in 93% yield without loss of diastereoselectivity. Moreover, the Ir/DPPM-catalyzed hydroboration of compound 3a smoothly provided product 7 in 83% yield and without erosion of enantioselectivity.
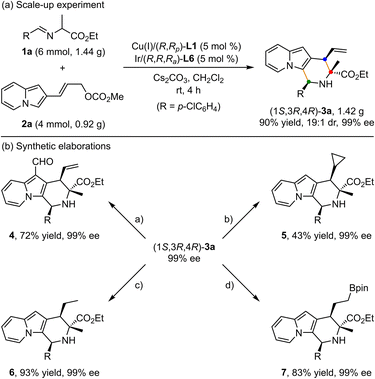 |
| | Scheme 2 Scale-up experiments and synthetic elaborations. Reaction conditions: (a) POCl3, DMF, 0 °C; (b) Pd(OAc)2 (1 mol%), CH2N2 Et2O, −20 °C; (c) Pd/C, H2, rt; (d) [Ir(COD)Cl]2 (5 mol%), dppm (10 mol%), HBpin, DCM, rt. | |
Mechanistic studies
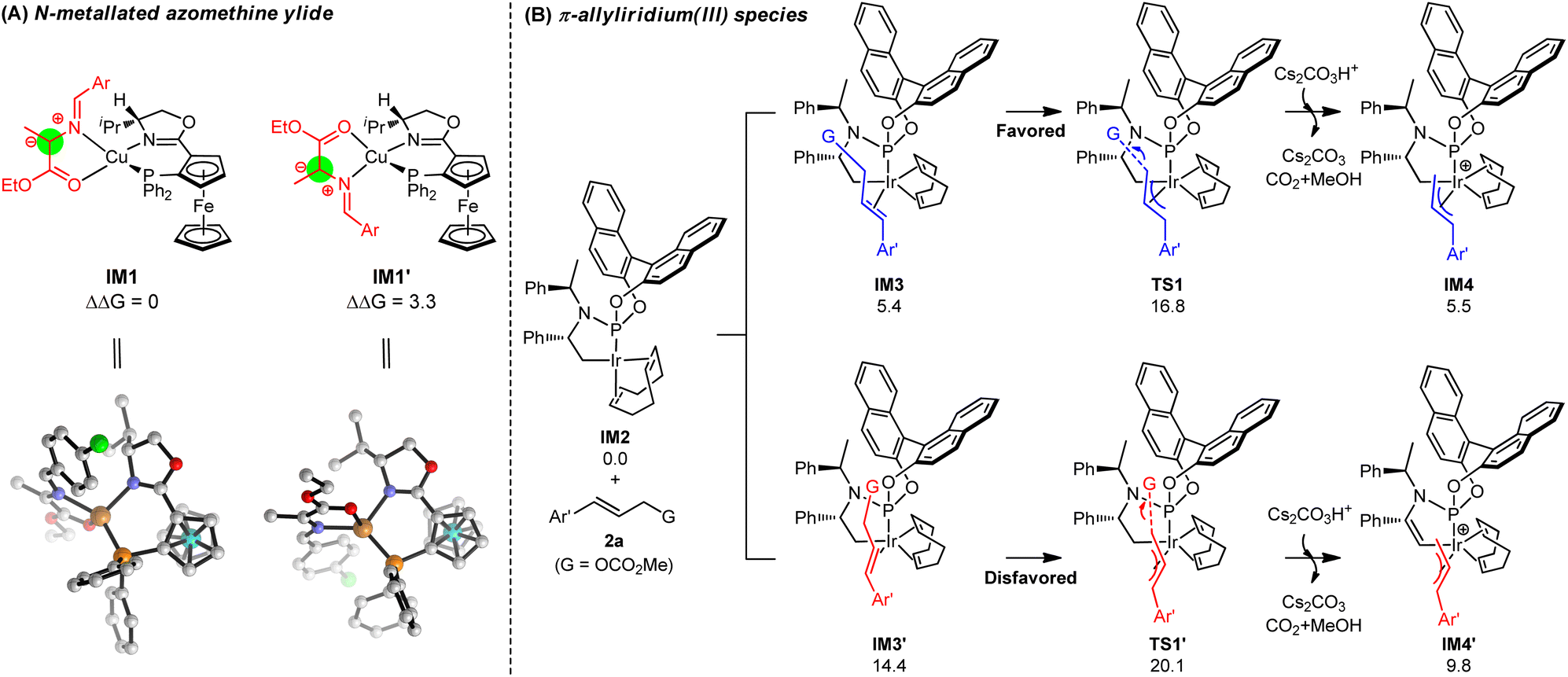
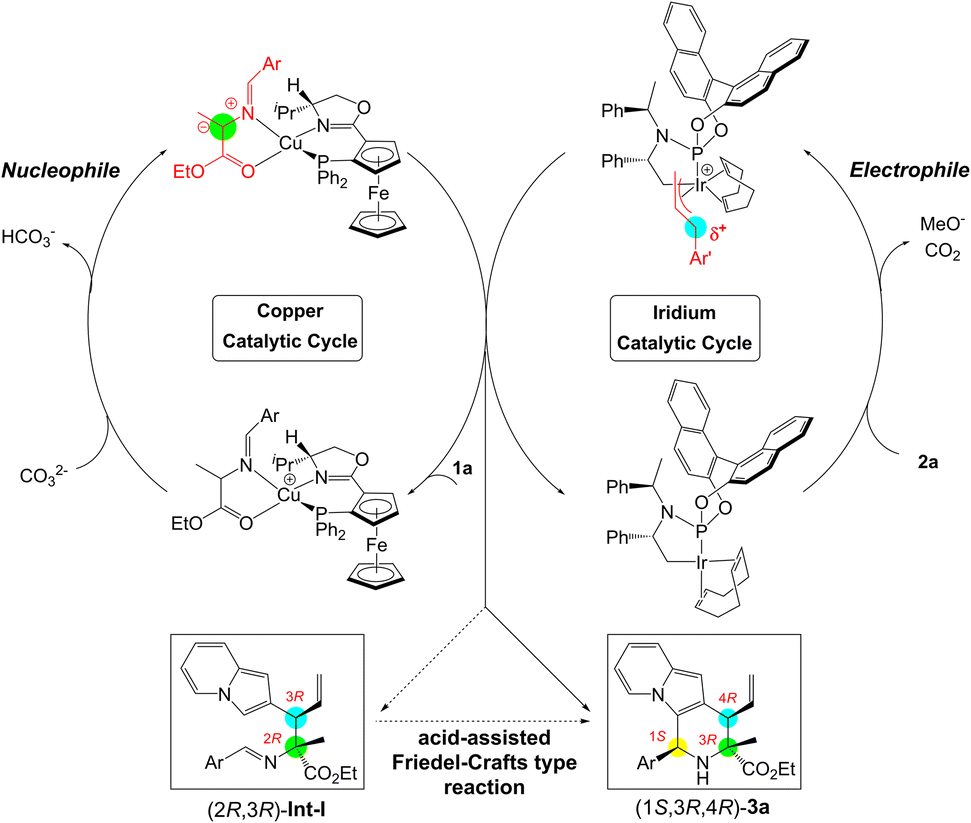
To gain insights into the molecular mechanism and understand the origins of stereoselectivity for this synergistic Cu/Ir catalysis, the catalytic reaction in entry 2, Table 1 (Cu/(R,Rp)-L1 and Ir/(R,R,Ra)-L6) was chosen as a representative for DFT mechanistic study (see the ESI† for details). Fig. 2A exhibits the in situ generation of copper azomethine ylide (AY) nucleophiles IM1 and IM1′. The metallated prochiral nucleophile has two diastereomers that show opposite relative orientations of the azomethine ylide unit with the ligand. The arrangement in which the large aryl group of the azomethine ylide unit is placed on the less encumbered left-hand side of the ligand is energetically favorable, which can be attributed to steric effects directed by the Cp-based group.18 The diastereomeric IM1′ is 3.3 kcal mol−1 less stable than IM1 caused by steric hindrance. As a result, only the more stable diastereomer of the prochiral nucleophile IM1 is considered in the following C–C coupling procedures.18 Note that the formation of Cu(I)-azomethine ylide IM1via deprotonation by inorganic base Cs2CO3 is exergonic by −20.2 kcal mol−1, indicating a strong thermodynamic driving force (see Fig. S3† for details).
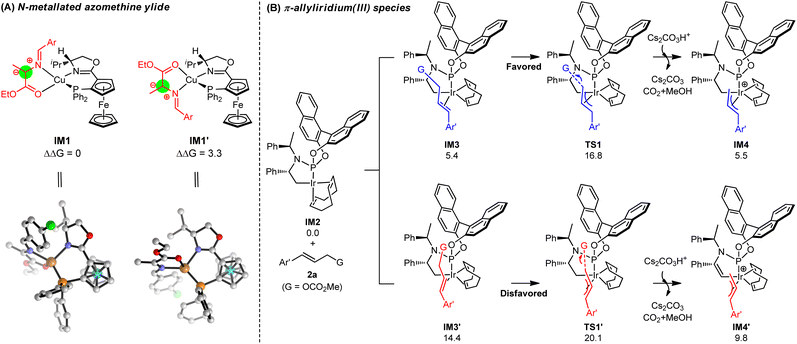 |
| | Fig. 2 (A) In situ generation and the structures of diastereomeric prochiral nucleophiles IM1 and IM1′. (B) The generation of π-allyliridium(III) species IM4 and IM4′via oxidative addition. Free energies are given in kcal mol−1. | |
In the meantime, the generation of the active π-allyliridium(III) species via decarboxylative oxidative addition was considered and the DFT-calculated results are presented in Fig. 2B. We explored an activated form of the Ir(I) catalyst IM2, which was produced from the activation of the C–H bond by Cs2CO3.18d In the presence of (R,R,Ra)-L6, the pathway of the formation of π-allyl-Ir(III) IM4 (IM2 → IM3 → TS1 → IM4) has overwhelming advantages in kinetics and thermodynamics than that leading to allyl-Ir(III) IM4′ (IM2 → IM3′ → TS1′ → IM4′). Specifically, the competition between TS1 and TS1′ shows a big energy barrier difference of 3.3 kcal mol−1 in support of π-allyl-iridium IM4, and IM4 is more stable than its diastereomeric IM1′ by 4.3 kcal mol−1. The main reason for the energy differences lies in the binding mode of allyl carbonate 2a, which gives rise to more steric hindrances between 2a and the COD ligand in IM3', TS1' and IM4' (see Fig. S4† for details). The computations coupled with the fact that the subsequent C–C coupling is relatively easy (see below) demonstrate that it is reasonable to consider IM4 as the main active allyl-Ir(III) species derived from the oxidation process.
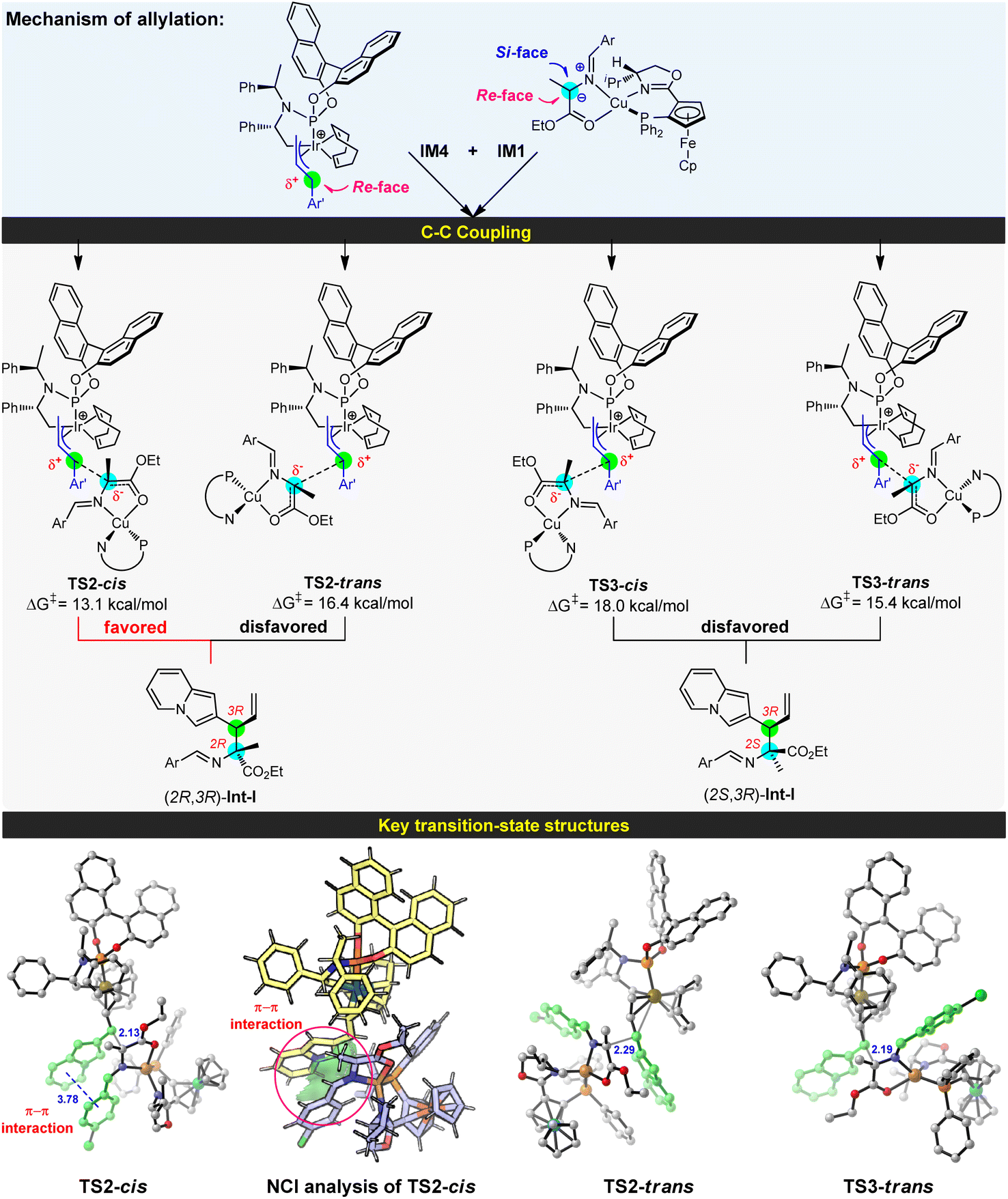
With an in-depth investigation of the generation of the active Cu/Ir catalysis, we examined the C–C coupling of the nucleophilic Cu(I)-azomethine ylide IM1 with the electrophilic allyl-iridium(III) IM4 to disclose the stereoselectivity of two vicinal stereocenters (see Fig. 3). In the allyl-iridium(III) species IM4, the Si-face was hindered by the cyclometallated moiety and the COD ligand, and lost its reactivity towards the Cu(I)-ylide IM1. Correspondingly, the Re-face region was exposed and easily attacked by the nucleophile. As a result, the R-configuration of C4 in product 3a can be preliminarily determined. On the other hand, in the Cu(I)-ylide IM1, the Si-face was congested by the oxazoline ring and its bulky isopropyl substituent; comparatively, the Re-face space was somewhat unimpeded and allowed the reaction with the electrophile. Hence, this indicated that the C3 site is more favored to be the R-configuration. On the basis of these analyses, we have located four types of C–C coupling transition states to form the alkylation compounds (see Fig. 3). TS2-cis originating from the reaction between the Re-face of IM1 and the Re-face of IM4 was the most favorable pathway and provided the allylation complex (2R,3R)-Int-I, which would undergo Friedel–Crafts type cyclization to give the experimental product (1S,3R,4R)-3a (see below). TS2-cis is 2.3 kcal mol−1 lower than TS3-trans, the favored transition state to give the allylation complex (2S,3R)-Int-I, and the energy differences evidently support the formation of (2R,3R)-Int-I (>20:1 dr). As a result, the computationally predicted stereoselectivity is consistent with the experimental results (entry 2, Table 1). Herein, TS2-cis builds on the favorable Re-face–Re-face stereoscopic configuration with a cis Ar⋯Ar′ arrangement, which could avoid strong ligand/substrate steric repulsions and gain attractive π–π stacking interactions to stabilize the C–C coupling process. This type of π–π dispersion force was further verified by a visual analysis of noncovalent interactions (NCIs).19 In the absence of this type of π–π interaction, TS2-trans has higher barrier than TS2-cis owing to the mismatched Ar⋯Ar′ structure. Additional Re-face–Si-face reaction pathways (TS3) enclosed strong steric hindrances between the allyl moiety and oxazoline fragment, so they are unfavorable (see Fig. S5† for details).
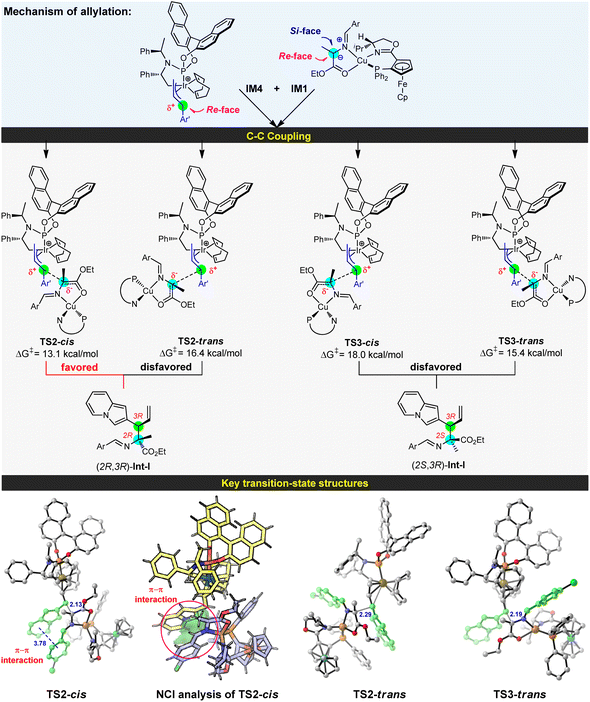 |
| | Fig. 3 Mechanism for the allylation of Cu(I)-azomethine ylide IM1 by allyliridium(III) IM4, and the key structures of bond-forming transition states. Free energies are given in kcal mol−1 (energy zero: IM1 and IM4), and selected distances are shown in Å. | |
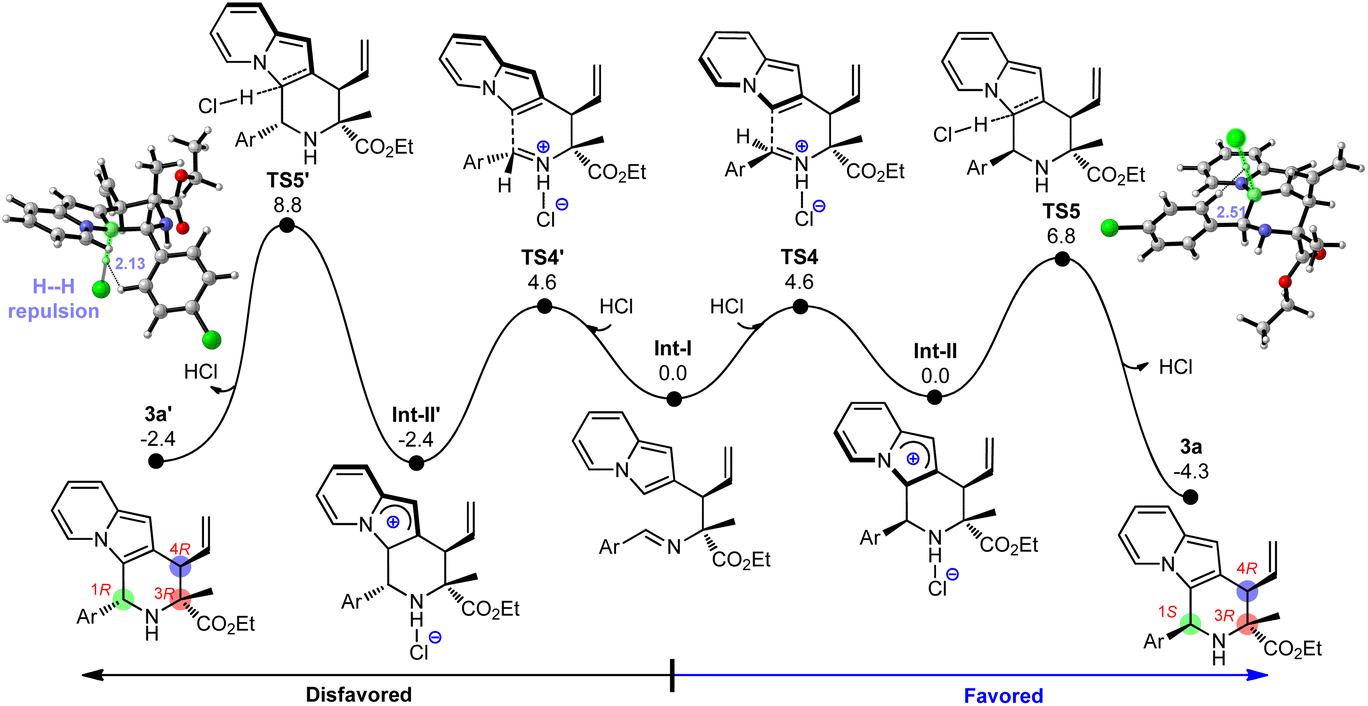
After revealing the formation of C3 and C4 stereocenters in product 3a by Cu/Ir synergistic catalysis, we tried to understand the source of chirality at the C1 center. In view of the fact that catalytic amounts of protonic acids (e.g., HCl) were generated during the catalyst activation (e.g., IM2),11q,18c the protonated imine on the allylated intermediate Int-I can activate the electrophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and thus stimulate the ring-closure to yield the desired 2,3-fused indolizine product. Fig. 4 displays the DFT-computed free energy profile for the Friedel–Crafts type reaction, which consists of acid-enabled C–C bond formation (TS4) and base (Cl−)-facilitated deprotonation (TS5). Because of the low kinetic barriers (4.6 kcal mol−1 and 6.8 kcal mol−1) and favorable thermodynamic driving force (−4.3 kcal mol−1), this Friedel–Crafts type cyclization is facile to carry out, which elucidates its high yield (93%, Table 1, entry 2). Although the energy barriers of C–C formation are similar (TS4vs.TS4′), TS5 is 2.0 kcal mol−1 lower than TS5′ during the deprotonation process, which illustrates the stereoselectivity favoring the (1S,3R,4R)-3a over (1R,3R,4R)-3a. The main factor that makes TS5′ unstable is that it contains severe H⋯H steric repulsions labeled at 2.13 Å between the leaving H atom and the Ar group, while there is no H⋯H repulsion at the corresponding region in TS5. What's more, (1R,3R,4R)-3a has greater thermodynamic driving force than that of (1S,3R,4R)-3a (−4.3 kcal mol−1vs. −2.4 kcal mol−1), which is further conducive to the formation of product (1S,3R,4R)-3a. These analyses reveal the origins of stereochemistry. In addition, we explored the cascade allylation/Friedel–Crafts type reaction with the set of catalyst combination of (Cu(I)/(S,Sp)-L1+ Ir(I)/(R,R,Ra)-L6), and the computed results are analogous to those discussed above, and are in good agreement with the experimentally observed results in Table 3 (see Fig. S6–S7† for details).
N bond and thus stimulate the ring-closure to yield the desired 2,3-fused indolizine product. Fig. 4 displays the DFT-computed free energy profile for the Friedel–Crafts type reaction, which consists of acid-enabled C–C bond formation (TS4) and base (Cl−)-facilitated deprotonation (TS5). Because of the low kinetic barriers (4.6 kcal mol−1 and 6.8 kcal mol−1) and favorable thermodynamic driving force (−4.3 kcal mol−1), this Friedel–Crafts type cyclization is facile to carry out, which elucidates its high yield (93%, Table 1, entry 2). Although the energy barriers of C–C formation are similar (TS4vs.TS4′), TS5 is 2.0 kcal mol−1 lower than TS5′ during the deprotonation process, which illustrates the stereoselectivity favoring the (1S,3R,4R)-3a over (1R,3R,4R)-3a. The main factor that makes TS5′ unstable is that it contains severe H⋯H steric repulsions labeled at 2.13 Å between the leaving H atom and the Ar group, while there is no H⋯H repulsion at the corresponding region in TS5. What's more, (1R,3R,4R)-3a has greater thermodynamic driving force than that of (1S,3R,4R)-3a (−4.3 kcal mol−1vs. −2.4 kcal mol−1), which is further conducive to the formation of product (1S,3R,4R)-3a. These analyses reveal the origins of stereochemistry. In addition, we explored the cascade allylation/Friedel–Crafts type reaction with the set of catalyst combination of (Cu(I)/(S,Sp)-L1+ Ir(I)/(R,R,Ra)-L6), and the computed results are analogous to those discussed above, and are in good agreement with the experimentally observed results in Table 3 (see Fig. S6–S7† for details).
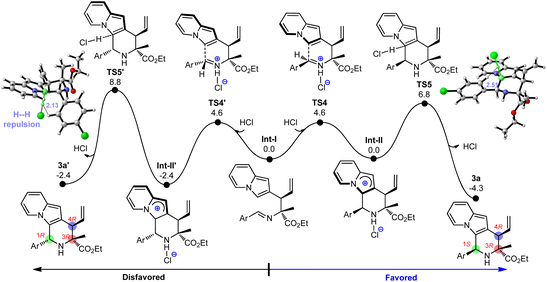 |
| | Fig. 4 Free energy profile for Friedel–Crafts type cyclization leading to the indolizine product 3a. | |
As shown in Scheme 3, a reasonable cooperative catalytic cycle for this asymmetric cascade allylation/Friedel–Crafts type reaction to access enantioenriched 2,3-fused indolizine derivatives enabled by a synergistic Cu/Ir catalysis was established. This protocol could proceed through the asymmetric allylation between π-allyl-Ir(III) species derived from 2-indolizine allyl carbonates and Cu(I)-azomethine ylide, followed by a proton-assisted stereoselective Friedel–Crafts type cyclization to complete the annulation reaction and deliver the final 2,3-fused indolizine products.
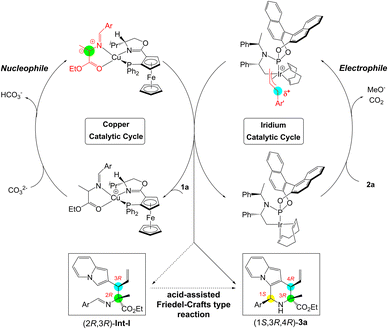 |
| | Scheme 3 Catalytic cycle for the cascade allylation/Friedel–Crafts type reaction via synergistic Cu/Ir catalysis. | |
Conclusions
In conclusion, we present herein a highly diastereoselective and enantioselective assembly of 2,3-fused indolizine derivatives empowered by a synergistic Cu/Ir catalysis with two readily available chiral ligands. The designed cascade allylation/Friedel–Crafts type reaction of in situ-formed nucleophilic Cu-azomethine ylides with electrophilic π-allyl-Ir species provides an unprecedented route to enantiomerically enriched indolizines bearing three stereogenic centers. The scope of this protocol, especially toward the aldimine esters, is remarkably broad. More importantly, through the pairwise combination of copper and iridium catalysts, this method is capable of rapidly constructing four stereoisomers of the corresponding products in a predictable manner. DFT mechanistic studies established a reasonable cooperative catalytic cycle for the synergistic Cu/Ir catalysis to stereoselectively construct 2,3-fused indolizine derivatives bearing three stereogenic centers in moderate to high yields with excellent stereoselective control.
Data availability
All experimental procedures, characterisation data, mechanistic investigations, NMR spectra and HPLC spectra can be found in the ESI.†
Author contributions
C. J. W. conceptualized the project. C. J. W. and X. Q. D. supervised the investigation. B. K. Z., L. X., X. C. and L. W. performed the research. Y. D. directed the DFT calculation, and H. X. performed the DFT calculation research. C. J. W., X. Q. D. and Y. D. co-wrote the paper. All authors analyzed the data, discussed the results, and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by NSFC (22071186, 22071187, 22073067, 22101216, and 22271226), National Youth Talent Support Program, Hubei Province NSF (2020CFA036 and 2021CFA069), and Fundamental Research Funds for the Central Universities (2042022kf1180).
Notes and references
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
-
(a) V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 CrossRef CAS;
(b) L.-L. Gundersen, A. H. Negussie, F. Rise and O. B. Østby, Arch. Pharm., 2003, 336, 191–195 CrossRef CAS PubMed;
(c) K. Bedjeguelal, H. Bienaymé, A. Dumoulin, S. Poigny, P. Schmitt and E. Tam, Bioorg. Med. Chem. Lett., 2006, 16, 3998–4001 CrossRef CAS PubMed;
(d) D. A. James, K. Koya, H. Li, S. Chen, Z. Xia, W. Ying, Y. Wu and L. Sun, Bioorg. Med. Chem. Lett., 2006, 16, 5164–5168 CrossRef CAS PubMed;
(e) Y. Xue, J. Tang, X. Ma, Q. Li, B. Xie, Y. Hao, H. Jin, K. Wang, G. Zhang, L. Zhang and L. Zhang, Eur. J. Med. Chem., 2016, 115, 94–108 CrossRef CAS PubMed;
(f) S. Park, E. H. Kim, J. Kim, S. H. Kim and I. Kim, Eur. J. Med. Chem., 2018, 144, 435–443 CrossRef CAS PubMed.
-
(a) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed;
(b) E. Kim, M. Koh, J. Ryu and S. B. Park, J. Am. Chem. Soc., 2008, 130, 12206–12207 CrossRef CAS PubMed;
(c) J. Li, S. Zhang and H. Zou, Org. Chem. Front., 2020, 7, 1218–1223 RSC;
(d) W. E. Meador, S. A. Autry, R. N. Bessetti, J. N. Gayton, A. S. Flynt, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2020, 85, 4089–4095 CrossRef CAS PubMed.
- N. K. Ratmanova, I. A. Andreev, A. V. Leontiev, D. Momotova, A. M. Novoselov, O. A. Ivanova and I. V. Trushkov, Tetrahedron, 2020, 76, 131031 CrossRef CAS.
-
(a) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237–5257 CrossRef CAS PubMed;
(b) S. Dong, X. Fu and X. Xu, Asian J. Org. Chem., 2020, 9, 1133–1143 CrossRef CAS;
(c) I. Khan, A. Ibrar and S. Zaib, Top. Curr. Chem., 2021, 379, 3 CrossRef CAS PubMed;
(d) J. S. S. Neto and G. Zeni, Asian J. Org. Chem., 2021, 10, 1282–1318 CrossRef CAS.
-
(a) J. T. M. Correia, B. List and F. Coelho, Angew. Chem., Int. Ed., 2017, 56, 7967–7970 CrossRef CAS PubMed;
(b) H. Chen, L. Zhu, K. Zhong, X. Yue, L.-B. Qu, R. Bai and Y. Lan, Chin. Chem. Lett., 2018, 29, 1237–1241 CrossRef CAS;
(c) P.-J. Yang, L. Qi, Z. Liu, G. Yang and Z. Chai, J. Am. Chem. Soc., 2018, 140, 17211–17217 CrossRef CAS PubMed;
(d) L. Yang, X. Pu, D. Niu, Z. Fu and X. Zhang, Org. Lett., 2019, 21, 8553–8557 CrossRef CAS PubMed;
(e) K. Li and C. Li, Org. Lett., 2020, 22, 9456–9461 CrossRef CAS PubMed;
(f) P. Yang, C.-X. Liu, W.-W. Zhang and S.-L. You, Acta Chim. Sin., 2021, 79, 742–746 CrossRef CAS;
(g) J. Lu, M. Wang, R. Xu, H. Sun, X. Zheng, G. Zhong and X. Zeng, Asian J. Org. Chem., 2021, 10, 1500–1507 CrossRef CAS;
(h) Q. Ni, Z. Zhu, Y. Fan, X. Chen and X. Song, Org. Lett., 2021, 23, 9548–9553 CrossRef CAS PubMed;
(i) X. Song, Y. Fan, Z. Zhu and Q. Ni, Org. Lett., 2022, 24, 2315–2320 CrossRef CAS PubMed.
- D. Zhang, Z. Su, Q. He, Z. Wu, Y. Zhou, C. Pan, X. Liu and X. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed.
-
(a) J. Caldwell, J. Clin. Pharmacol., 1992, 32, 925–929 CrossRef CAS PubMed;
(b) B. Waldeck, Chirality, 1993, 5, 350–355 CrossRef CAS PubMed;
(c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed;
(d) K. M. Rentsch, J. Biochem. Biophys. Methods, 2002, 54, 1–9 CrossRef CAS PubMed;
(e)
K. Jozwiak, W. J. Lough and I. W. Wainer, Drug Stereochemistry: Analytical Methods and Pharmacology, 3rd edn, Informa, New York, 2012 CrossRef.
- For perspectives and reviews on stereodivergent synergistic catalysis, see:
(a) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed;
(b) L. Wei and C. J. Wang, Chin. J. Chem., 2021, 39, 15–24 CrossRef CAS;
(c) X. Huo, G. Li, X. Wang and W. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210086 CAS;
(d) L. Wei and C.-J. Wang, Chem. Catal., 2023, 3, 100455 CrossRef CAS.
- For reviews on synergistic catalysis, see:
(a) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC;
(b) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC;
(c) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed;
(d) S. M. Inamdar, V. S. Shinde and N. T. Patil, Org. Biomol. Chem., 2015, 13, 8116–8162 RSC;
(e) L. Lin and X. Feng, Chem. - Eur. J., 2017, 23, 6464–6482 CrossRef CAS PubMed;
(f) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed;
(g) Y. Wu, X. Huo and W. Zhang, Chem.–Eur. J., 2020, 26, 4895–4916 CrossRef CAS PubMed;
(h) U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S.-g. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed;
(i) S. Martínez, L. Veth, B. Lainer and P. Dydio, ACS Catal., 2021, 11, 3891–3915 CrossRef;
(j) X. Liu, H. Zheng, Y. Xia, L. Lin and X. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed.
- For selected examples of stereodivergent synergistic catalysis, see:
(a) S. Krautwald, D. Sarlah, A. Schafroth Michael and M. Carreira Erick, Science, 2013, 340, 1065–1068 CrossRef CAS PubMed;
(b) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020–3023 CrossRef CAS PubMed;
(c) T. Sandmeier, S. Krautwald, H. F. Zipfel and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 14363–14367 CrossRef CAS PubMed;
(d) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093–11096 CrossRef CAS PubMed;
(e) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029–1032 CrossRef CAS PubMed;
(f) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819–9822 CrossRef CAS PubMed;
(g) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef CAS PubMed;
(h) R. He, X. Huo, L. Zhao, F. Wang, L. Jiang, J. Liao and W. Zhang, J. Am. Chem. Soc., 2020, 142, 8097–8103 CrossRef CAS PubMed;
(i) Y. Peng, X. Huo, Y. Luo, L. Wu and W. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24941–24949 CrossRef CAS PubMed;
(j) J. Zhang, X. Huo, J. Xiao, L. Zhao, S. Ma and W. Zhang, J. Am. Chem. Soc., 2021, 143, 12622–12632 CrossRef CAS PubMed;
(k) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS PubMed;
(l) X. Jiang, P. Boehm and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1239–1242 CrossRef CAS PubMed;
(m) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508–1513 CrossRef CAS PubMed;
(n) Z.-T. He, X. Jiang and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066–13073 CrossRef CAS PubMed;
(o) M.-M. Zhang, Y.-N. Wang, B.-C. Wang, X.-W. Chen, L.-Q. Lu and W.-J. Xiao, Nat. Commun., 2019, 10, 2716 CrossRef PubMed;
(p) Q. Zhang, H. Yu, L. Shen, T. Tang, D. Dong, W. Chai and W. Zi, J. Am. Chem. Soc., 2019, 141, 14554–14559 CrossRef CAS PubMed;
(q) S.-M. Xu, L. Wei, C. Shen, L. Xiao, H.-Y. Tao and C.-J. Wang, Nat. Commun., 2019, 10, 555 CrossRef PubMed;
(r) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48–54 CrossRef CAS;
(s) H. Wang, R. Zhang, Q. Zhang and W. Zi, J. Am. Chem. Soc., 2021, 143, 10948–10962 CrossRef CAS PubMed;
(t) M. Zhu, Q. Zhang and W. Zi, Angew. Chem., Int. Ed., 2021, 60, 6545–6552 CrossRef CAS PubMed;
(u) B. Kim, Y. Kim and S. Y. Lee, J. Am. Chem. Soc., 2021, 143, 73–79 CrossRef CAS PubMed;
(v) Y.-H. Wen, Z.-J. Zhang, S. Li, J. Song and L.-Z. Gong, Nat. Commun., 2022, 13, 1344 CrossRef CAS PubMed;
(w) S. Q. Yang, Y. F. Wang, W. C. Zhao, G. Q. Lin and Z. T. He, J. Am. Chem. Soc., 2021, 143, 7285–7291 CrossRef CAS PubMed;
(x) D. X. Zhu, J. G. Liu and M. H. Xu, J.
Am. Chem. Soc., 2021, 143, 8583–8589 CrossRef CAS PubMed;
(y) L. Xiao, L. Wei and C. J. Wang, Angew. Chem., Int. Ed., 2021, 60, 24930–24940 CrossRef CAS PubMed;
(z) Y. Xu, H. Wang, Z. Yang, Y. Zhou, Y. Liu and X. Feng, Chem, 2022, 8, 2011–2022 CrossRef CAS;
(a
a) X. Chang, X. Cheng, X. T. Liu, C. Fu, W. Y. Wang and C. J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202206517 CAS;
(a
b) R. Jiang, L. Ding, C. Zheng and S.-L. You, Science, 2021, 371, 380–386 CrossRef CAS PubMed;
(a
c) Q. Hu, Z. He, L. Peng and C. Guo, Nat. Synth., 2022, 1, 322–331 CrossRef;
(a
d) Z. He, L. Peng and C. Guo, Nat. Synth., 2022, 1, 393–400 CrossRef;
(a
e) L. Xiao, B. Li, F. Xiao, C. Fu, L. Wei, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2022, 13, 4801–4812 RSC;
(a
f) L. Xiao, X. Chang, H. Xu, Q. Xiong, Y. Dang and C. J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212948 CAS;
(a
g) J. Han, R. Liu, Z. Lin and W. Zi, Angew. Chem., Int. Ed., 2022, 61, e202215714 Search PubMed;
(a
h) W. Wang, F. Zhang, Y. Liu and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202208837 CAS;
(a
i) H. Wang, Y. Xu, F. Zhang, Y. Liu and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202115715 CAS;
(a
j) X. Hu, X. Tang, X. Zhang, L. Lin and X. Feng, Nat. Commun., 2021, 12, 3012 CrossRef PubMed;
(a
k) Y. Chen, Y. Liu, Z. Li, S. Dong, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052–8056 CrossRef CAS PubMed;
(a
l) C. Xu, K. Wang, D. Li, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 18438–18442 CrossRef CAS PubMed;
(a
m) H. Zheng, Y. Wang, C. Xu, Q. Xiong, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327–5331 CrossRef CAS PubMed;
(a
n) S. Ge, W. Cao, T. Kang, B. Hu, H. Zhang, Z. Su, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 4017–4021 CrossRef CAS PubMed;
(a
o) Y. Chen, S. Dong, X. Xu, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554–16558 CrossRef CAS PubMed.
-
(a) L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084–1100 CrossRef CAS PubMed;
(b) Z.-Y. Xue, Q.-H. Li, H.-Y. Tao and C.-J. Wang, J. Am. Chem. Soc., 2011, 133, 11757–11765 CrossRef CAS PubMed;
(c) H.-L. Teng, F.-L. Luo, H.-Y. Tao and C.-J. Wang, Org. Lett., 2011, 13, 5600–5603 CrossRef CAS PubMed;
(d) L. Wei, L. Xiao, Y. Hu, Z. Wang, H. Tao and C. Wang, Chin. J. Org. Chem., 2019, 39, 2119–2130 CrossRef CAS.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- W. B. Liu, C. Zheng, C. X. Zhuo, L. X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812–4821 CrossRef CAS PubMed.
-
(a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed;
(b) J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed.
- C. J. Richards and A. W. Mulvaney, Tetrahedron: Asymmetry, 1996, 7, 1419–1430 CrossRef CAS.
- CCDC 2221435 ((1S,3R,4R)-3k) contains the supplementary crystallographic data for this paper..
-
(a) X.-X. Yan, Q. Peng, Y. Zhang, K. Zhang, W. Hong, X.-L. Hou and Y.-D. Wu, Angew. Chem., Int. Ed., 2006, 45, 1979–1983 CrossRef CAS PubMed;
(b) J. Xia, T. Hirai, S. Katayama, H. Nagae, W. Zhang and K. Mashima, ACS Catal., 2021, 11, 6643–6655 CrossRef CAS;
(c) H. Xu, B. Li, Z. Liu and Y. Dang, ACS Catal., 2021, 11, 9008–9021 CrossRef CAS;
(d) L. Xiao, B. Li, F. Xiao, C. Fu, L. Wei, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2022, 13, 4801–4812 RSC;
(e) B. Li, H. Xu, Y. Dang and K. N. Houk, J. Am. Chem. Soc., 2022, 144, 1971–1985 CrossRef CAS PubMed.
-
(a) S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed;
(b) C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC;
(c) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2221435. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00118k |
| ‡ These two authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Huailong
Teng
a,
Huailong
Teng