DOI:
10.1039/D3SC00132F
(Edge Article)
Chem. Sci., 2023,
14, 3982-3989
Molybdenum chloride double perovskites: dimensionality control of optical and magnetic properties†
Received
9th January 2023
, Accepted 3rd March 2023
First published on 6th March 2023
Abstract
Halide double perovskites are a promising class of semiconducting materials for applications in solar cells and other optoelectronic devices. Recently, there has been a surge of interest in these materials to study phenomena beyond optoelectronics, especially magnetism. Here, we report three new Mo3+ (4d3) based chloride double perovskites: a 3-D rock-salt ordered Cs2NaMoCl6, a 1-D chain (MA)2AgMoCl6 and a Dion–Jacobson type 2-D layered (1,4-BDA)2AgMoCl8 (MA = methylammonium; 1,4-BDA = 1,4-butanediammonium). Their structures and dimensionalities can be tuned by means of the A-cation. The measured bandgaps are relatively narrow (2.0–2.1 eV) which show a blueshift on reducing the dimensionality. At low temperatures, we observe antiferromagnetic coupling between the nearest-neighbour Mo3+ ions in all these systems. Cs2NaMoCl6 shows stronger coupling with a frustration index f of 5 which we attribute to the geometrically frustrating fcc lattice of Mo3+ ions. This work expands the scope of halide double perovskites beyond main group metals and beyond optoelectronics, and we hope that it will lead to future developments in magnetic halide perovskites.
1. Introduction
Halide double perovskites (HDPs) (also known by their alternative name, elpasolite) are typically a class of quaternary compounds of composition A2BIBIIIX6 (A = A-site monocation; BI = B-site monovalent metal; BIII = B-site trivalent metal; X = halide). At the six-coordinated B-site, BI and BIII metal ions show rock-salt ordering, which is often driven by their charge difference.1 In some cases, such as Cs2HgIIMIICl6 (MII = Cu or Pd), coordination geometry can drive the B-site metal ordering even though the charge difference is zero.2 The inorganic lattice of A2BIBIIIX6 could be constructed by transmutation of two divalent metal ions (BII) of a ABIIX3 single perovskite into one BI ion and one BIII ion, i.e. 2BII → BI + BIII.3 The interest in HDPs began to surge in 2016, when three research groups independently reported the synthesis and the optoelectronic properties of Cs2AgBiX6 (X = Cl, Br).1,4,5 Since then, many such materials have been realized, thanks to the rich degree of freedom in selecting BI and BIII metals. Most of the HDPs of current interest are based on post-transition metals with a d10s2 configuration (such as Sb6 and Bi7) or d10s0 configuration (such as In8) at the BIII site and any size-matching monovalent metals (such as Ag, Tl, Na, and K) at the BI site, and have been widely reported for use in photovoltaics,9 X-ray detectors,10 photodetectors,11 and humidity sensors.12 Unfortunately, they have their own limitations mainly due to large band gaps and large effective carrier masses.13,14
During the 1970s to the 1990s, a number of fluoride double perovskites of trivalent 3d metal ions,15–19 and some 4d metal ions such as Mo,20,21 and Rh,22 were reported. Rare-earth metals were also incorporated in fluoride based double perovskites.23,24 At that time, particular interest was focused on synthesis and structures, and in some cases magnetism. In light of the recent surge of interest in semiconducting halide perovskites, there have been reports on incorporating transition metals at the perovskite B-site with the intention to develop novel perovskites with high potential to be used in emerging applications such as spin-electronics, where exchange interaction between unpaired spins is important.25–28 FeIII(3d5),29 and RuIII (4d5)30 have been alloyed into the benchmark double perovskite Cs2AgBiBr6 to decrease its bandgap by introducing an intermediate band inside the pristine bandgaps. Recently, Ning et al. reported interesting magnetic exchange properties in Fe-alloyed Cs2AgBiBr6 and suggested that this material has potential for use in spintronics.31 FeIII based 3D perovskites, Cs2MIFeIIICl6 (MI = Na or Ag) have been reported as narrow bandgap semiconductors (Eg = ∼1.5 eV),32 as well as geometrically frustrating antiferromagnetic systems.33 Ferromagnetic CuI–CuII–InIII based layered halide double perovskites have been recently reported.34 Moving to 4d metals increases the importance of spin–orbit coupling (SOC) in exotic physics of unpaired spins such as magnetic frustration in layered α-RuCl3,35,36 α-MoCl3,37 and other heavy transition metal halides.38 High SOC metal ions such as RuIII, and RhIII have been incorporated in 1-D chain,39,40 and 2-D layered double perovskites.41 The effect of SOC on the magnetic properties has been studied in these low dimensional systems. The RuIII systems display unusual magnetic properties, particularly, deviation from the Kotani model for a low spin d5 metal. By using the Kotani model, single ion magnetic properties of vacancy ordered double perovskites of RuIV,42 and OsIV,43 both low spin d4 ions, have been recently studied. These recent reports indeed suggest that transition metal based HDPs are of potential interest, which researchers have begun to explore.
In this work, we sought to introduce MoIII (4d3) as the magnetic ion into three new chloride based double perovskites: a 3-D rock-salt ordered Cs2NaMoCl6, a 1-D chain (MA)2AgMoCl6 and a 2-D layered (1,4-BDA)2AgMoCl8 (where MA = methylammonium; 1,4-BDA = 1,4-butanediammonium). To our knowledge, Cs2NaMoCl6 is the first example of a chloride based 3-D HDP in which an open-shell 4d metal is incorporated at the B-site. Structurally, it is similar to prototype compound Sr2FeMoO6 which is one of the heavily discussed oxide perovskites in the condensed matter physics community in terms of unusual magnetic and electronic properties arising from the rock salt ordered sub-lattices of FeIII (d5) and MoV (d1) ions.44,45 (MA)2AgMoCl6 and (1,4-BDA)2AgMoCl8 present rare examples of 1-D chain and 2-D layered hybrid double perovskites, respectively, in which both BI and BIII metals are transition metal ions, i.e. Ag and Mo. We find that their structures are tuneable with different A-cations, i.e., Cs, MA, and 1,4-BDA. Their optical and magnetic properties can be controlled with both chemical composition and structural dimensionality. Their X-ray structures, optical absorption, and magnetism are discussed below.
2. Results and discussion
2.1. Synthesis
The synthetic protocol for the compounds reported here is similar to the method reported in the literature by some of us (see the ESI for the method†).39 Typically, to obtain Cs2NaMoCl6 and (MA)2AgMoCl6, a mixture of ACl (A = Cs or MA), BICl (BI = Na or Ag) and MoCl3 was heated hydrothermally in aqueous hydrochloric acid as the solvent. Crystals were obtained on slow cooling of the reaction autoclaves. Similarly, (1,4-BDA)2AgMoCl8 was obtained from the reaction of 1,4-butanediamine, AgCl and MoCl3. Initially in all the reactions, we observed the formation of a green solution likely stemming from the formation of higher valence Mo ions in aqueous HCl. A stoichiometric amount of H3PO2 (reducing agent) was used to control the oxidation. The structures of the as synthesized compounds were determined from single-crystal X-ray diffraction (SCXRD) data. Selected unit-cell parameters are given in Table 1 and the details in Tables S1–S5.† The selected bond parameters are given in Table S6.† The simulated powder X-ray diffraction (PXRD) patterns from the SCXRD data show a good match with the experimental PXRD patterns, confirming that all these compounds can be prepared as phase pure (Fig. S1–S3†).
Table 1 Unit-cell parameters for Cs2NaMoCl6, (MA)2AgMoCl6, (1,4-BDA)2AgMoCl8 and (MA)4MoCl6·Cl
|
|
Cs2NaMoCl6 |
(MA)2AgMoCl6 |
(1,4-BDA)2AgMoCl8 |
(1,4-BDA)2AgMoCl8_460 K |
(MA)4MoCl6·Cl |
| Space group |
Cubic, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
Trigonal, Pm1 |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
Monoclinic, P2/n |
|
a (Å) |
10.41290(10) |
7.3359(2) |
7.6668(15) |
7.636(2) |
16.0647(9) |
|
b (Å) |
10.41290(10) |
7.3359(2) |
7.7072(14) |
7.671(2) |
7.3477(4) |
|
c (Å) |
10.41290(10) |
6.8017(2) |
9.4490(14) |
9.789(3) |
16.1264(8) |
|
α (°) |
90 |
90 |
102.229(8) |
80.191(12) |
90 |
|
β (°) |
90 |
90 |
91.183(6) |
88.082(12) |
103.599(2) |
|
γ (°) |
90 |
120 |
90.127(6) |
89.402(12) |
90 |
| Volume (Å3) |
1129.05(3) |
317.00(2) |
545.54(17) |
564.7(3) |
1850.17(17) |
| CCDC number |
2234517 |
2234518 |
2234519 |
2234520 |
2234521 |
2.2. Structure of Cs2NaMoCl6
As shown in Fig. 1, Cs2NaMoCl6 crystallizes in a cubic Fmm space group, the 3D inorganic framework of which is composed of corner-shared [NaCl6]5− and [MoCl6]3− units at the B-site with rock-salt ordering. Locally, the Na+ and Mo3+ ions form ideal octahedral units which are interconnected infinitely through Cl bridges along all three axes. The Cs+ cation occupies the A-site interstitials. The Na–Cl (2.756(3) Å) and the Mo–Cl (2.450(3) Å) bond lengths are slightly shorter than those calculated (Na–Cl = 2.813 Å; Mo–Cl = 2.476 Å) from Shannon radii,46 which is due to the increased covalency in the metal–halide bonds. Furthermore, a small displacement parameter (Beq) of 2.16 in the case of a Cs cation, Cs+ size to A-cavity size ratio of 0.99 and tolerance factor of 0.97 collectively suggest that the Cs+ ion fits well in the A-site cavity. Perhaps, due to this reason, the compound shows high thermal and ambient stabilities (Fig. S4 and S5†).
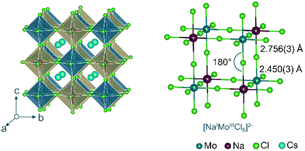 |
| | Fig. 1 SCXRD structure of Cs2NaMoCl6, and the ball and stick model of the corresponding perovskite frameworks is also provided to show corner-sharing connectivity of [NaCl6]5− and [MoCl6]3− units. | |
2.3. Structure of (MA)2AgMoCl6
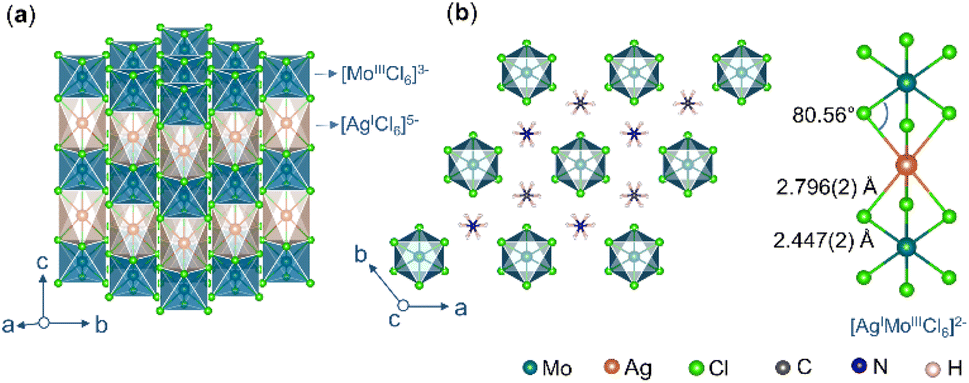
We, recently reported that a double perovskite most likely forms a corner-shared 3-D framework when the tolerance factor 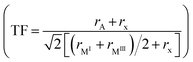 (where rA, rx, rMI, and rMIII are the radii of the A-cation, halide, monovalent metal and trivalent metal, respectively) is in the 0.8–1.0 range, and a face-shared 1-D chain when the TF is slightly more than 1.0.47 Here, we replaced Cs+ with MA and Na+ with Ag+ to obtain (MA)2AgMoCl6 (Fig. 2a and b) which has a TF of 1.03. It crystallizes in a hexagonal phase, similar to previously reported AgI–RuIII halide perovskites.39 The [AgCl6]5− and [MoCl6]3− octahedra are alternatively connected by face-sharing of two opposite faces in 1-D chain running along the c-axis. The MA cation occupies the interchain space and forms N⋯H⋯Cl hydrogen bonds with chlorides (the shortest N⋯Cl distance of 3.37 Å). It is worthwhile to note that both [MoCl6]3− and [AgCl6]5− octahedra are trigonally distorted, with the latter being heavily distorted as suggested by the bond angle variance (σ2) (Table 2).
(where rA, rx, rMI, and rMIII are the radii of the A-cation, halide, monovalent metal and trivalent metal, respectively) is in the 0.8–1.0 range, and a face-shared 1-D chain when the TF is slightly more than 1.0.47 Here, we replaced Cs+ with MA and Na+ with Ag+ to obtain (MA)2AgMoCl6 (Fig. 2a and b) which has a TF of 1.03. It crystallizes in a hexagonal phase, similar to previously reported AgI–RuIII halide perovskites.39 The [AgCl6]5− and [MoCl6]3− octahedra are alternatively connected by face-sharing of two opposite faces in 1-D chain running along the c-axis. The MA cation occupies the interchain space and forms N⋯H⋯Cl hydrogen bonds with chlorides (the shortest N⋯Cl distance of 3.37 Å). It is worthwhile to note that both [MoCl6]3− and [AgCl6]5− octahedra are trigonally distorted, with the latter being heavily distorted as suggested by the bond angle variance (σ2) (Table 2).
 |
| | Fig. 2 SCXRD structure of (MA)2AgMoCl6: (a) side view and (b) top view. Ball and stick model of the corresponding perovskite framework is also provided to show face-sharing connectivity of [AgCl6]5− and [MoCl6]3− units. | |
Table 2 Bond distortion level (Δd) and bond angle variance (σ2)
| Compounds |
Δd |
σ
2
|
| Cs2NaMoCl6 |
[MoCl6] = 0 |
[MoCl6] = 0 |
| [NaCl6] = 0 |
[NaCl6] = 0 |
| (MA)2AgMoCl6 |
[MoCl6] = 0 |
[MoCl6] = 0.6136 |
| [AgCl6] = 0 |
[AgCl6] = 217.49 |
| (1,4-BDA)2AgMoCl8 |
[MoCl6] = 1.03 × 10−6 |
[MoCl6] = 0.5047 |
| [AgCl6] = 9.85 × 10−3 |
[AgCl6] = 6.448 |
| (MA)4MoCl6·Cl |
[MoCl6] = 1.59 × 10−6 |
[MoCl6] = 0.1609 |
| [MoCl6] = 1.48 × 10−7 |
[MoCl6] = 0.0979 |
2.4. Structure of (1,4-BDA)2AgMoCl8
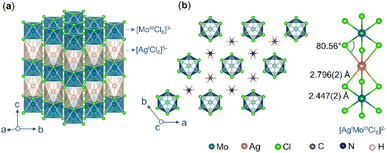
Layers of 2-D perovskite can be thought of as derived from slicing (conceptually) the 3-D analogue along a specific crystallographic plane and stacking the slices over each other when large organic ammonium is used as the A-cation.48 We used 1,4-BDA to obtain (1,4-BDA)2AgMoCl8 which crystallizes in a triclinic P space group as a (100)-oriented Dion–Jacobson (DJ) type double perovskite (Fig. 3a). The perovskite layer is composed of orderly arranged [AgCl6]5− and [MoCl6]3− in the ab-plane which are inter-connected through four equatorial chlorides, while two axial chlorides remain dangling in the interlayer space (Fig. 3b). The [0,0] type of layer stacking pattern is clearly distinguishable without stacking-fault or without mixed stacking of AgI/AgI and AgI/MoIII phases.49 The [AgCl6]5− polyhedra are highly distorted which is commonly ascribed to preferential formation of linear coordination geometry due to the mixing of filled Ag 4d orbitals with empty Ag 5s orbitals.50 The 1,4-BDA spacer adopts a gauche conformation (Fig. 3c). Our previous studies have revealed that the gauche conformation of this spacer is stable in layered Ag–Ru–Br perovskite, (1,4-BDA)2AgRuBr8 and the anti conformation, in layered Ag–Ru–Cl perovskite, (1,4-BDA)2AgRuCl8 at room temperature.41 Here, we show that this spacer can exhibit both gauche and anti conformations in the same compound, but at different temperatures. The calorimetric measurements suggest a reversible phase transition (Fig. 3d) and the SCXRD structure confirms that the phase transition is driven by a gauche ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) anti conformational change. In a 460 K structure (Fig. 3e), half the number of 1,4-BDA cations have changed from the gauche (Fig. 3f) form to the anti (Fig. 3g) form without changing the overall lattice symmetry and the compound formula can therefore be written as [(gauche-1,4-BDA)(anti-1,4-BDA)]AgMoCl8. There is a slight volume expansion due to the increase in the interlayer distance. The octahedral tilting in the perovskite layer is slightly reduced (Fig. 3h). As shown in Fig. S6 and S7,† the perovskite layer has cavities of two different sizes and two crystallographically different 1,4-BDA cations. The BDA-2 which forms N–H⋯Cl hydrogen bonds with the chlorides of the larger cavity is more disordered with larger thermal parameters in the room temperature as well as in the high temperature structures. Furthermore, BDA-2 is found to be longer with more penetration depth below the plane of axial chlorides. We believe that BDA-2 changes the conformation from gauche to anti during the phase transition, whilst BDA-1 remains quiet. We did not observe significant changes in the N⋯Cl distances as a consequence of the change in the 1,4-BDA conformation (Table S7†).
anti conformational change. In a 460 K structure (Fig. 3e), half the number of 1,4-BDA cations have changed from the gauche (Fig. 3f) form to the anti (Fig. 3g) form without changing the overall lattice symmetry and the compound formula can therefore be written as [(gauche-1,4-BDA)(anti-1,4-BDA)]AgMoCl8. There is a slight volume expansion due to the increase in the interlayer distance. The octahedral tilting in the perovskite layer is slightly reduced (Fig. 3h). As shown in Fig. S6 and S7,† the perovskite layer has cavities of two different sizes and two crystallographically different 1,4-BDA cations. The BDA-2 which forms N–H⋯Cl hydrogen bonds with the chlorides of the larger cavity is more disordered with larger thermal parameters in the room temperature as well as in the high temperature structures. Furthermore, BDA-2 is found to be longer with more penetration depth below the plane of axial chlorides. We believe that BDA-2 changes the conformation from gauche to anti during the phase transition, whilst BDA-1 remains quiet. We did not observe significant changes in the N⋯Cl distances as a consequence of the change in the 1,4-BDA conformation (Table S7†).
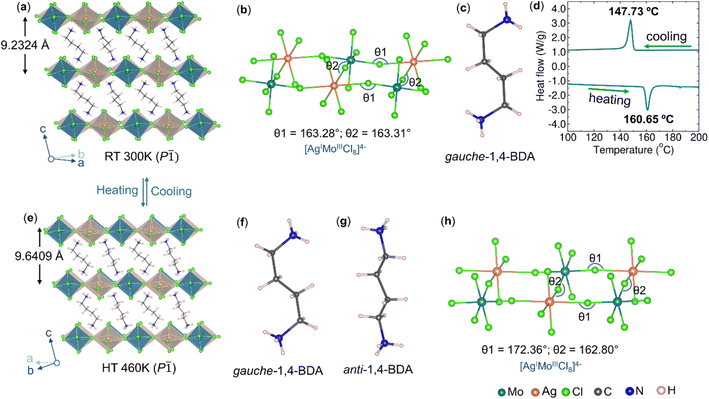 |
| | Fig. 3 (a) Room temperature SCXRD structure of (1,4-BDA)2AgMoCl8. (b) Corresponding ball and stick models for the perovskite layer showing octahedral connectivity between [AgCl6]5− and [MoCl6]3− units. (c) 1,4-BDA spacer in a gauche conformation. (d) DSC curve of (1,4-BDA)2AgMoCl8. (e) SCXRD structure of (1,4-BDA)2AgMoCl8 at 460 K. (f and g) 1,4-BDA spacer in gauche and anti conformations. (h) Ball and stick models for the perovskite layer in the 460 K structure, showing reduced octahedral tilting than that of the room temperature structure. | |
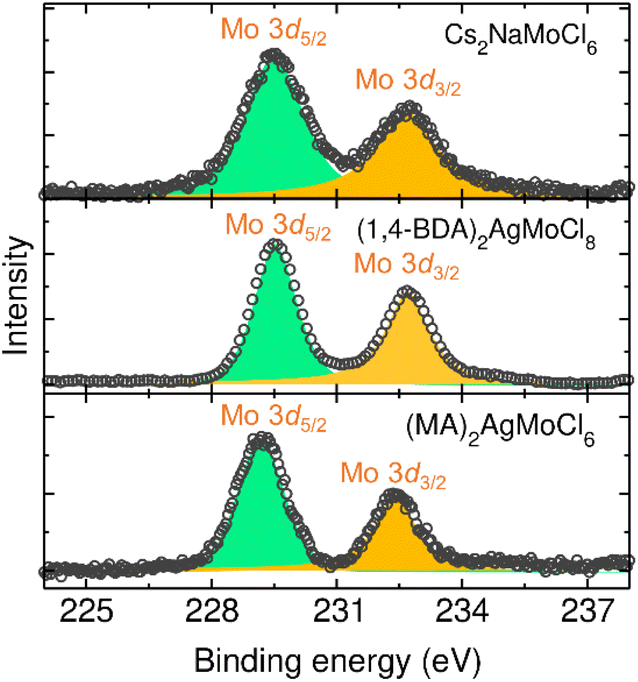
2.5. XPS
It is important to note that Mo is susceptible to oxidation and can therefore exist in multiple oxidation states under ambient conditions. We measured X-ray photoelectron spectroscopy (XPS) spectra to examine the chemical composition as well as the valence state of Mo ions (Fig. S9–S14 and Table S8†). The spectra show the presence of the desired elements. The core level Mo 3d spectra exhibit characteristic doublet peaks at around 229 eV and 232 eV with a peak area ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, corresponding to the Mo 3d5/2 and Mo 3d3/2 states (Fig. 4). These binding energies and the peak area ratios clearly suggest that the Mo is in the trivalent state in all these compounds.51 Furthermore, we did not observe any Mo 3d peaks at higher binding energies, confirming the absence of higher valence states.
:2, corresponding to the Mo 3d5/2 and Mo 3d3/2 states (Fig. 4). These binding energies and the peak area ratios clearly suggest that the Mo is in the trivalent state in all these compounds.51 Furthermore, we did not observe any Mo 3d peaks at higher binding energies, confirming the absence of higher valence states.
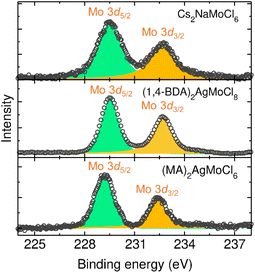 |
| | Fig. 4 Core level XPS spectra of Mo 3d states. | |
2.6. UV-visible absorption spectra
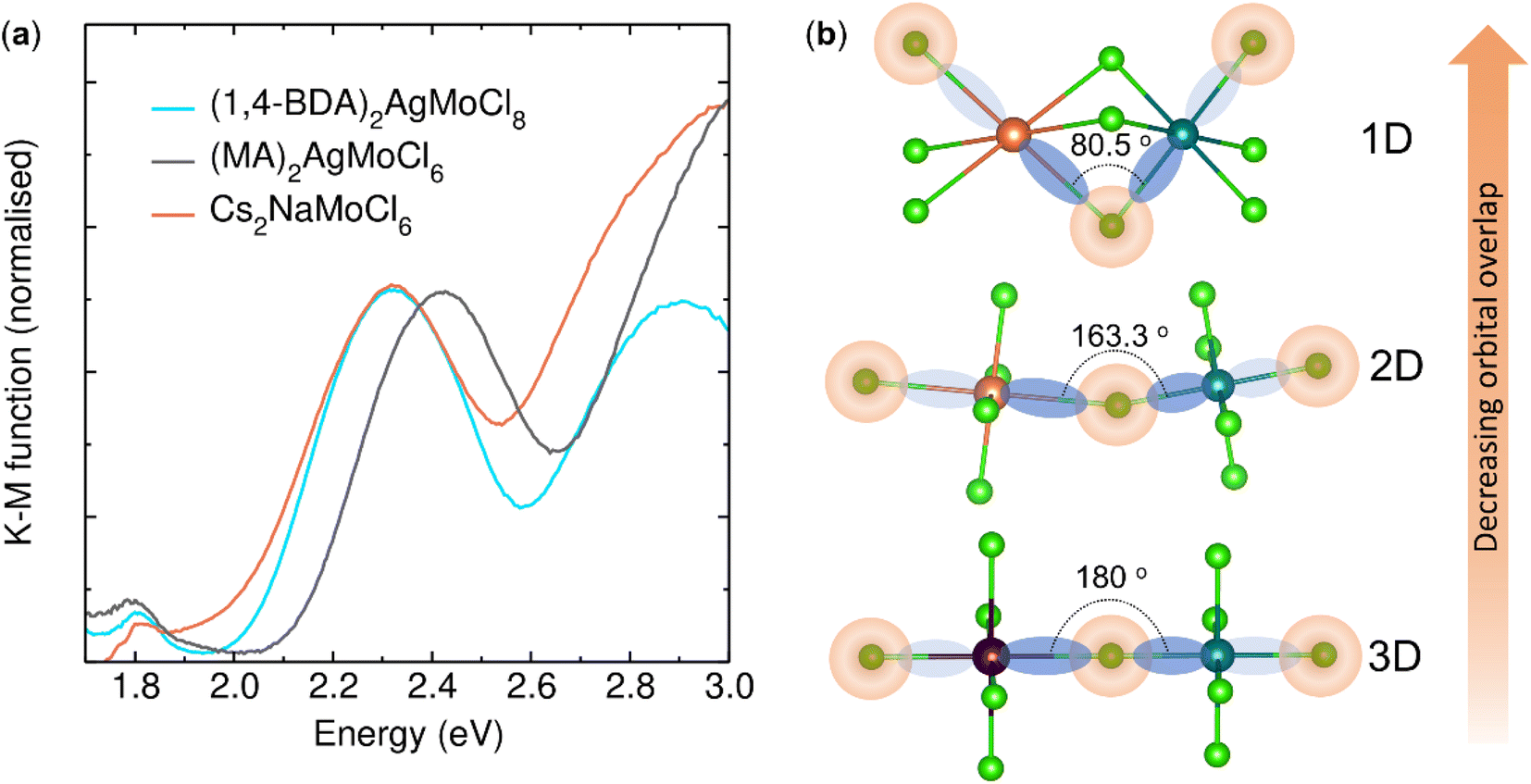
Fig. 5a shows the UV-visible absorption spectra of these double perovskites which were recorded in the reflectance mode and converted to absorbance by using the Kubelka–Munk formula [α/S = (1 − R)2/(2R)] (where α, S, and R represent the absorption coefficient, scattering coefficient, and absolute reflectance, respectively). The spectra suggest that the optical bandgap increases in the order of Cs2NaMoCl6 (Eg = 2.0 eV), (1,4-BDA)2AgMoCl8 (Eg = 2.04 eV) and (MA)2AgMoCl6 (Eg = 2.12 eV). We attribute this bandgap blueshift to the reduced orbital overlap caused by both reduced dimensionality and increased structural distortion (Fig. 5b).14 However, the bandgap shift is minute between the 3-D and the 2-D compounds, which is likely due to the increased bandgap of the former compound as a consequence of poor contribution of Na to the electronic band near the extrema when compared to that of Ag.52 As listed in Table 2, (MA)2AgMoCl6 has the highest bond angle variance (σ2),53 indicating a high octahedral distortion which correlates well with its higher value of bandgap. Furthermore, the dimensionality control of the bandgap shift is undoubtedly clear in the case of the 2-D and the 1-D phases, wherein the chemical constituents of the perovskite frameworks are identical, e.g., Ag and Mo at the B-site. However, the magnitude of the bandgap shift is smaller than those observed in post-transition-metal analogues. Furthermore, the bandgaps are smaller than those of heavily explored halide double perovskites Cs2AgBiBr6 (Eg = 2.17 eV),4 Cs2AgBiCl6 (Eg = 2.77 eV),4 and Cs2AgInCl6 (Eg = 3.2 eV).54 We also observed a weak band at low energy in each spectrum which is consistent with the spin forbidden 4A2g → 4T1g transitions in MoIII chloride.55
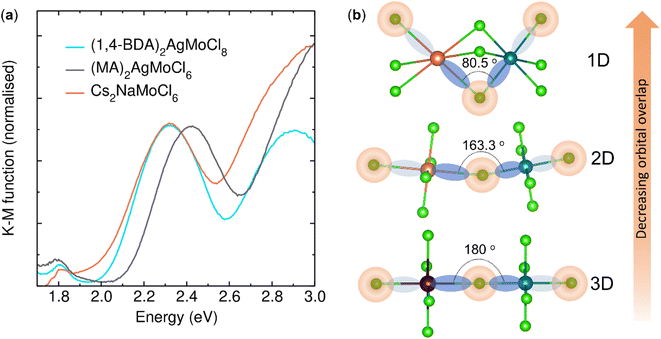 |
| | Fig. 5 (a) Optical absorption spectra. (b) Schematic illustration of reduced orbital overlapping between [MICl6]5− and [MoIIICl6]3− octahedra due to lattice distortion. | |
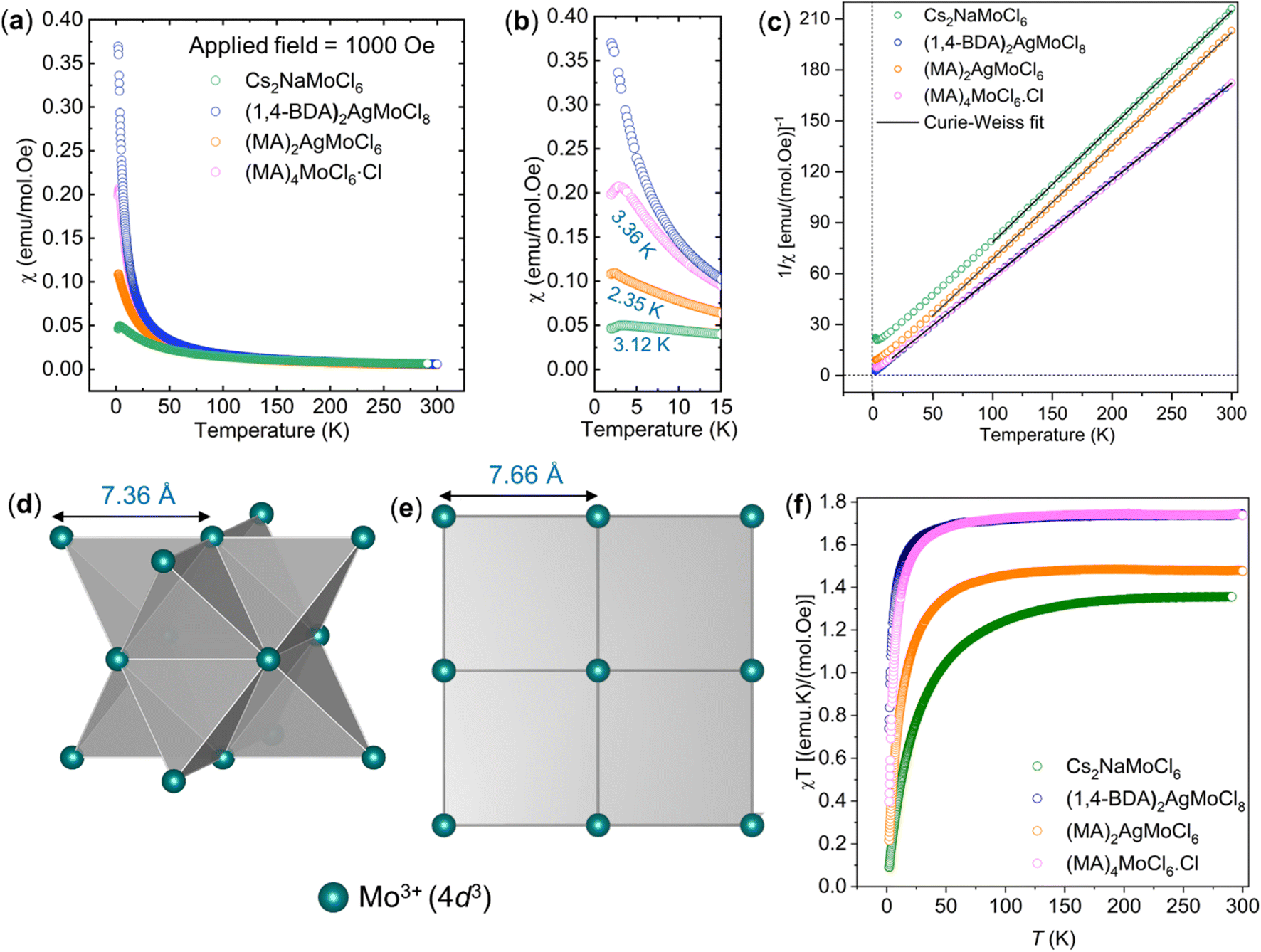
2.7. Magnetism
We measured magnetic susceptibility (χ) for all the three perovskites in the temperature range of 2 K to 300 K by using a superconducting quantum interference device-vibrating sample magnetometer (SQUID-VSM). The susceptibility data were fitted to the modified Curie–Weiss law equation [χ = C/(T − θCW) + χ0], where C is the Curie constant which is proportional to the square of the effective magnetic moment 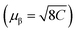 , T is the temperature, θCW is the Curie–Weiss temperature which is a measure of the strength and the nature of magnetic exchange interaction between the nearest-neighbors, and χ0 is the temperature-independent diamagnetic contribution. Fig. 6a and b show the molar susceptibility curves. We shall first discuss the magnetic properties of Cs2NaMoCl6 which exhibits long range antiferromagnetic ordering at a Néel temperature (TN) of 3.12 K, and there is no divergence in zero field cooled (ZFC) and field cooled (FC) curves (Fig. S16†). Above TN, the paramagnetic susceptibility shows an excellent fit to the Curie–Weiss equation with a negative Curie–Weiss temperature (θCW) of −15.8 K, and Curie constant (C) of 1.472 emu Oe−1 K mol−1 (Fig. 6c). The effective magnetic moment µβ is calculated to be 3.43 µB f.u.−1 which is slightly less than the spin only moment of 3.87 µB f.u.−1 for a d3 ion [
, T is the temperature, θCW is the Curie–Weiss temperature which is a measure of the strength and the nature of magnetic exchange interaction between the nearest-neighbors, and χ0 is the temperature-independent diamagnetic contribution. Fig. 6a and b show the molar susceptibility curves. We shall first discuss the magnetic properties of Cs2NaMoCl6 which exhibits long range antiferromagnetic ordering at a Néel temperature (TN) of 3.12 K, and there is no divergence in zero field cooled (ZFC) and field cooled (FC) curves (Fig. S16†). Above TN, the paramagnetic susceptibility shows an excellent fit to the Curie–Weiss equation with a negative Curie–Weiss temperature (θCW) of −15.8 K, and Curie constant (C) of 1.472 emu Oe−1 K mol−1 (Fig. 6c). The effective magnetic moment µβ is calculated to be 3.43 µB f.u.−1 which is slightly less than the spin only moment of 3.87 µB f.u.−1 for a d3 ion [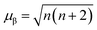 , where n = number of unpaired electrons, n = 3 in this case]. Fig. 6d shows the fcc sub-lattice composed of magnetic Mo ions in Cs2NaMoCl6 which clarifies the edge-shared tetrahedral arrangement of Mo3+ metal ions in a geometrically frustrating system. The tetrahedron is dimensionally symmetric with a nearest-neighbor Mo⋯Mo separation of 7.36 Å. The calculated magnetic frustration index f
, where n = number of unpaired electrons, n = 3 in this case]. Fig. 6d shows the fcc sub-lattice composed of magnetic Mo ions in Cs2NaMoCl6 which clarifies the edge-shared tetrahedral arrangement of Mo3+ metal ions in a geometrically frustrating system. The tetrahedron is dimensionally symmetric with a nearest-neighbor Mo⋯Mo separation of 7.36 Å. The calculated magnetic frustration index f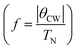 of 5.0 indeed suggests that the system has weak magnetic frustration, and it warrants further studies.56,57 The 1-D compound (MA)2AgMoCl6 features an antiferromagnetic transition temperature TN of 2.35 K and a negative θCW of −2.9 K with an effective magnetic moment of 3.48 µB f.u.−1 The calculated frustration index (f) of 1.2 indicates that this 1-D system is magnetically non-frustrating.
of 5.0 indeed suggests that the system has weak magnetic frustration, and it warrants further studies.56,57 The 1-D compound (MA)2AgMoCl6 features an antiferromagnetic transition temperature TN of 2.35 K and a negative θCW of −2.9 K with an effective magnetic moment of 3.48 µB f.u.−1 The calculated frustration index (f) of 1.2 indicates that this 1-D system is magnetically non-frustrating.
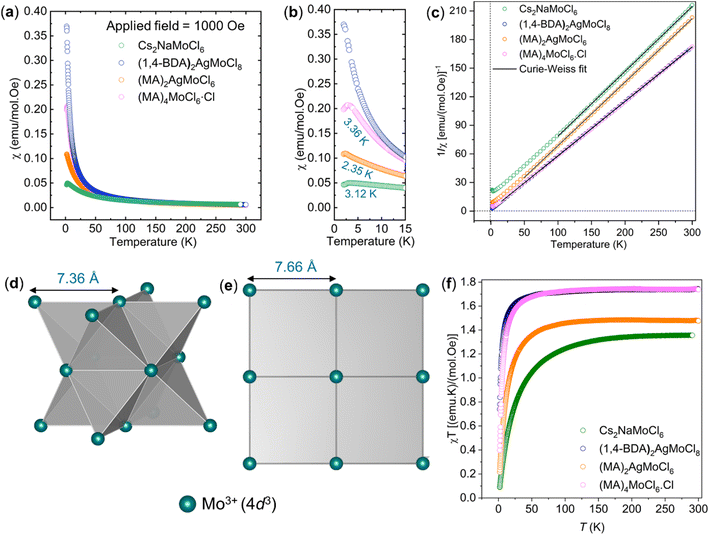 |
| | Fig. 6 (a and b) Temperature dependent magnetic susceptibility curves. (c) Curie–Weiss fitting of the susceptibility data shown in panel (a). (d) Geometrically frustrating tetrahedron based on Mo3+ ions in Cs2NaMoCl6. (e) Square lattice based on Mo3+ ions in (1,4-BDA)2AgMoCl8. (f) Thermal dependence of the χMT products. | |
In the case of (1,4-BDA)2AgMoCl8, we did not observe magnetic ordering down to the lowest measurement temperature of 2 K, but the system obeys the Curie–Weiss law. Only a weak antiferromagnetic exchange was observed with a θCW of −1.7 K and an effective moment of 3.74 µB f.u.−1 We attribute the weakening of AFM exchange to the increased distance between the nearest-neighbor spins (Mo⋯Mo = 7.667 Å) compared to that in Cs2NaMoCl6 (7.363 Å) and (MA)2AgMoCl6 (6.802 Å). The geometric frustration has also significantly relaxed in the 2D system due to the formation of a square lattice (Fig. 6e). The isothermal field dependent M–H curve (Fig. S20c†) of (1,4-BDA)2AgMoCl8 at 2 K adopts a non-linear shape, but with no hysteresis (neither remanence nor coercivity), suggesting paramagnetic behaviour.58 The M–H loops (Fig. S20a and b†) of Cs2NaMoCl6 and (MA)2AgMoCl6 are linear which is characteristic of an antiferromagnetic nature. Additionally, we synthesized (MA)4MoCl6·Cl (Fig. S21†), a molecular compound, with a nearest-neighbor Mo⋯Mo distance of 7.35 Å which is close to that found in Cs2NaMoCl6 (7.363 Å). Also, it shows a TN of 3.36 K, close to that of Cs2NaMoCl6, but the strength of AFM coupling is much weaker as indicated by a θCW of −1.2 K. This suggests that the magnetic coupling in these systems primarily takes place through the –MoIII–Cl–MI–Cl–MoIII– pathway, the strength of which depends not only on the nearest-neighbor Mo⋯Mo distance, but also on the geometry of the magnetic sub-lattice. Fig. 6f shows that on lowering the temperature, χMT values decrease which indicates antiferromagnetic interactions in all these systems.
3. Conclusions
In summary, we have synthesized and structurally characterized three new chloride double perovskites with Na+/Ag+ and Mo3+ metal ions ordering at the perovskite B-site. The X-ray structures suggest that octahedral connectivity and dimensionality of the inorganic sub-lattice can readily be modulated with the choice of the A-site cation. The measured bandgaps are relatively narrow (2.0–2.1 eV) which are even lower than that of the benchmark double perovskite Cs2AgBiBr6 (2.2 eV), and are tuneable with structural dimensionality. Magnetic measurements suggest nearest-neighbor antiferromagnetic coupling at low temperatures which occurs due to super-exchange through the –MoIII–Cl–MI–Cl–MoIII– (MI = Na or Ag) pathway. The magnetic ordering parameters, the Curie–Weiss temperature (θCW) and the Néel temperature (TN) can be tuned with the dimensionality of the magnetic sub-lattice. In the case of 3-D perovskite, |θCW| ≫ TN suggests magnetic frustration as a consequence of the fcc arrangement of Mo3+ (4d3) ion, whilst the 1-D and 2-D analogues do not exhibit such an arrangement. These materials expand the scope of halide double perovskites beyond optoelectronics, and we hope that these results will trigger future developments in magnetic optoelectronic halide perovskites by making use of other A-site cations, B-site metals and halides.
Data availability
Crystallographic information files have been deposited in CCDC (2234517–2234521). All necessary supporting data are provided in the ESI.†
Author contributions
DCB: carried out the experiments, data analysis (except magnetic data), wrote the initial draft of manuscript. PPM and DPP: magnetic data collection and analysis. PV: project design, conceptualization, methodology, data curation, manuscript writing and revision, project administration, and funding acquisition.
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
The authors thank Prof. C. N. R. Rao (FRS) for his kind support and guidance. This work was financially supported by the Science & Engineering Research Board (SERB) of the Govt. of India through a Ramanujan Fellowship (Grant No. RJF/2020/000106), and a Core Research Grant (Grant No. CRG/2022/009230) to PV. We thank the SERB for these grants and the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore, for the research infrastructure. PV also thanks the Sheikh Saqr Laboratory (SSL) for financial support. DCB thanks JNCASR and SSL for fellowships. DPP thanks CSIR for fellowship.
Notes and references
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef PubMed.
- G. Ji and Z. Xiao, Chem. Mater., 2022, 34, 8207–8212 CrossRef CAS.
- X.-G. Zhao, J.-H. Yang, Y. Fu, D. Yang, Q. Xu, L. Yu, S.-H. Wei and L. Zhang, J. Am. Chem. Soc., 2017, 139, 2630–2638 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- G. Volonakis, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 1254–1259 CrossRef CAS PubMed.
- J. Zhou, X. Rong, M. S. Molokeev, X. Zhang and Z. Xia, J. Mater. Chem. A, 2018, 6, 2346–2352 RSC.
- H. Lei, D. Hardy and F. Gao, Adv. Funct. Mater., 2021, 31, 2105898 CrossRef CAS.
- A. Noculak, V. Morad, K. M. McCall, S. Yakunin, Y. Shynkarenko, M. Wörle and M. V. Kovalenko, Chem. Mater., 2020, 32, 5118–5124 CrossRef CAS PubMed.
- F. Igbari, R. Wang, Z.-K. Wang, X.-J. Ma, Q. Wang, K.-L. Wang, Y. Zhang, L.-S. Liao and Y. Yang, Nano Lett., 2019, 19, 2066–2073 CrossRef CAS PubMed.
- J. A. Steele, W. Pan, C. Martin, M. Keshavarz, E. Debroye, H. Yuan, S. Banerjee, E. Fron, D. Jonckheere, C. W. Kim, W. Baekelant, G. Niu, J. Tang, J. Vanacken, M. Van der Auweraer, J. Hofkens and M. B. J. Roeffaers, Adv. Mater., 2018, 30, 1804450 CrossRef PubMed.
- F. Fang, H. Li, S. Fang, B. Zhou, F. Huang, C. Ma, Y. Wan, S. Jiang, Y. Wang, B. Tian and Y. Shi, Adv. Opt. Mater., 2021, 9, 2001930 CrossRef CAS.
- Z. Weng, J. Qin, A. A. Umar, J. Wang, X. Zhang, H. Wang, X. Cui, X. Li, L. Zheng and Y. Zhan, Adv. Funct. Mater., 2019, 29, 1902234 CrossRef.
- X.-G. Zhao, D. Yang, J.-C. Ren, Y. Sun, Z. Xiao and L. Zhang, Joule, 2018, 2, 1662–1673 CrossRef CAS.
- Z. Xiao, W. Meng, J. Wang, D. B. Mitzi and Y. Yan, Mater. Horiz., 2017, 4, 206–216 RSC.
- E. Alter and R. Hoppe, Z. Anorg. Allg. Chem., 1975, 412, 110–120 CrossRef CAS.
- G. Siebert and R. Hoppe, Z. Anorg. Allg. Chem., 1972, 391, 117–125 CrossRef CAS.
- E. Alter and R. Hoppe, Z. Anorg. Allg. Chem., 1974, 403, 127–136 CrossRef CAS.
- E. Alter and R. Hoppe, Z. Anorg. Allg. Chem., 1974, 407, 305–312 CrossRef CAS.
- E. Alter and R. Hoppe, Z. Anorg. Allg. Chem., 1974, 405, 167–175 CrossRef CAS.
- R. Hoppe and K. Lehr, Z. Anorg. Allg. Chem., 1975, 416, 240–250 CrossRef CAS.
- L. M. Toth, G. D. Brunton and G. P. Smith, Inorg. Chem., 1969, 8, 2694–2697 CrossRef CAS.
- V. Wilhelm and R. Hoppe, Z. Anorg. Allg. Chem., 1975, 414, 91–96 CrossRef CAS.
- R. Acevedo and V. Poblete, Powder Diffr., 1995, 10, 241–242 CrossRef CAS.
- M. E. Villafuerte-Castrejón, M. R. Estrada, J. Gómez-Lara, J. Duque and R. Pomés, J. Solid State Chem., 1997, 132, 1–5 CrossRef.
- L. Mao, J. Chen, P. Vishnoi and A. K. Cheetham, Acc. Mater. Res., 2022, 3, 439–448 CrossRef CAS.
- J. Wang, C. Zhang, H. Liu, R. McLaughlin, Y. Zhai, S. R. Vardeny, X. Liu, S. McGill, D. Semenov, H. Guo, R. Tsuchikawa, V. V. Deshpande, D. Sun and Z. V. Vardeny, Nat. Commun., 2019, 10, 129 CrossRef PubMed.
- L. T. Nguyen and R. J. Cava, Chem. Rev., 2021, 121, 2935–2965 CrossRef CAS PubMed.
- L. A. Muscarella and E. M. Hutter, ACS Energy Lett., 2022, 7, 2128–2135 CrossRef CAS PubMed.
- S. Thawarkar, S. R. Rondiya, N. Y. Dzade, N. Khupse and S. Jadkar, Chem.–Eur. J., 2021, 27, 7408–7417 CrossRef CAS PubMed.
- Z. Zhang, G. Liu, W. Guo, X. Li, Y. Zhang, C. Wu, B. Qu, J. Shi, Z. Chen and L. Xiao, Mater. Adv., 2022, 3, 4932–4937 RSC.
- W. Ning, J. Bao, Y. Puttisong, F. Moro, L. Kobera, S. Shimono, L. Wang, F. Ji, M. Cuartero, S. Kawaguchi, S. Abbrent, H. Ishibashi, R. De Marco, I. A. Bouianova, G. A. Crespo, Y. Kubota, J. Brus, D. Y. Chung, L. Sun, W. M. Chen, M. G. Kanatzidis and F. Gao, Sci. Adv., 2020, 6, eabb5381 CrossRef CAS PubMed.
- H. Yin, Y. Xian, Y. Zhang, W. Chen, X. Wen, N. U. Rahman, Y. Long, B. Jia, J. Fan and W. Li, Adv. Funct. Mater., 2020, 30, 2002225 CrossRef CAS.
- J. Xue, Z. Wang, A. Comstock, Z. Wang, H. H. Y. Sung, I. D. Williams, D. Sun, J. Liu and H. Lu, Chem. Mater., 2022, 34, 2813–2823 CrossRef CAS.
- B. A. Connor, R. W. Smaha, J. Li, A. Gold-Parker, A. J. Heyer, M. F. Toney, Y. S. Lee and H. I. Karunadasa, Chem. Sci., 2021, 12, 8689–8697 RSC.
- S. Mashhadi, Y. Kim, J. Kim, D. Weber, T. Taniguchi, K. Watanabe, N. Park, B. Lotsch, J. H. Smet, M. Burghard and K. Kern, Nano Lett., 2019, 19, 4659–4665 CrossRef CAS PubMed.
- A. Banerjee, C. A. Bridges, J.-Q. Yan, A. A. Aczel, L. Li, M. B. Stone, G. E. Granroth, M. D. Lumsden, Y. Yiu, J. Knolle, S. Bhattacharjee, D. L. Kovrizhin, R. Moessner, D. A. Tennant, D. G. Mandrus and S. E. Nagler, Nat. Mater., 2016, 15, 733–740 CrossRef CAS PubMed.
- M. A. McGuire, J. Yan, P. Lampen-Kelley, A. F. May, V. R. Cooper, L. Lindsay, A. Puretzky, L. Liang, S. KC, E. Cakmak, S. Calder and B. C. Sales, Phys. Rev. Mater., 2017, 1, 64001 CrossRef.
- H. Lu, J. R. Chamorro, C. Wan and T. M. McQueen, Inorg. Chem., 2018, 57, 14443–14449 CrossRef CAS PubMed.
- P. Vishnoi, J. L. Zuo, T. A. Strom, G. Wu, S. D. Wilson, R. Seshadri and A. K. Cheetham, Angew. Chem., Int. Ed., 2020, 59, 8974–8981 CrossRef CAS PubMed.
- N. P. Holzapfel, A. Milder and P. M. Woodward, Chem. Mater., 2022, 34, 7705–7711 CrossRef CAS.
- P. Vishnoi, J. L. Zuo, X. Li, D. C. Binwal, K. E. Wyckoff, L. Mao, L. Kautzsch, G. Wu, S. D. Wilson, M. G. Kanatzidis, R. Seshadri and A. K. Cheetham, J. Am. Chem. Soc., 2022, 144, 6661–6666 CrossRef CAS PubMed.
- P. Vishnoi, J. L. Zuo, J. A. Cooley, L. Kautzsch, A. Gómez-Torres, J. Murillo, S. Fortier, S. D. Wilson, R. Seshadri and A. K. Cheetham, Angew. Chem., Int. Ed., 2021, 60, 5184–5188 CrossRef CAS PubMed.
- M. Saura-Múzquiz, M. Avdeev, H. E. A. Brand and B. J. Kennedy, Inorg. Chem., 2022, 61, 15961–15972 CrossRef PubMed.
- D. D. Sarma, E. V. Sampathkumaran, S. Ray, R. Nagarajan, S. Majumdar, A. Kumar, G. Nalini and T. N. Guru Row, Solid State Commun., 2000, 114, 465–468 CrossRef CAS.
- A. Arulraj, K. Ramesha, J. Gopalakrishnan and C. N. R. Rao, J. Solid State Chem., 2000, 155, 233–237 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Vishnoi, R. Seshadri and A. K. Cheetham, J. Phys. Chem. C, 2021, 125, 11756–11764 CrossRef CAS.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- B. Vargas, G. Rodríguez-López and D. Solis-Ibarra, ACS Energy Lett., 2020, 5, 3591–3608 CrossRef CAS.
- R. Hooijer, A. Weis, A. Biewald, M. T. Sirtl, J. Malburg, R. Holfeuer, S. Thamm, A. A. Y. Amin, M. Righetto, A. Hartschuh, L. M. Herz and T. Bein, Adv. Opt. Mater., 2022, 10, 2200354 CrossRef CAS.
- Z. Wang, Y. Han, J. Liang, H. Huang, C. Hu, P. Liu, J. Xiang, Z. Qi, Y. Lu, K. Liu, J. Jiang and B. Xiang, ACS Appl. Electron. Mater., 2020, 2, 2678–2684 CrossRef CAS.
- R. S. Lamba, P. Basera, S. Bhattacharya and S. Sapra, J. Phys. Chem. Lett., 2019, 10, 5173–5181 CrossRef CAS PubMed.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- J. Luo, S. Li, H. Wu, Y. Zhou, Y. Li, J. Liu, J. Li, K. Li, F. Yi, G. Niu and J. Tang, ACS Photonics, 2018, 5, 398–405 CrossRef CAS.
- R. V. Kamalov, V. A. Volkovich, I. B. Polovov and B. D. Vasin, Russ. Metall., 2012, 2012, 114–118 CrossRef.
- H. Karunadasa, Q. Huang, B. G. Ueland, P. Schiffer and R. J. Cava, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8097–8102 CrossRef CAS PubMed.
- S. Mugiraneza and A. M. Hallas, Commun. Phys., 2022, 5, 95 CrossRef.
- A. Lodi Rizzini, C. Krull, T. Balashov, A. Mugarza, C. Nistor, F. Yakhou, V. Sessi, S. Klyatskaya, M. Ruben, S. Stepanow and P. Gambardella, Nano Lett., 2012, 12, 5703–5707 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization methods, crystallographic information files (CIFs), PXRD patterns, XPS data, and additional figures of magnetic data. CCDC 2234517–2234521. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00132f |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Prashurya Pritam
Mudoi
a,
Prashurya Pritam
Mudoi

 (where rA, rx, rMI, and rMIII are the radii of the A-cation, halide, monovalent metal and trivalent metal, respectively) is in the 0.8–1.0 range, and a face-shared 1-D chain when the TF is slightly more than 1.0.47 Here, we replaced Cs+ with MA and Na+ with Ag+ to obtain (MA)2AgMoCl6 (Fig. 2a and b) which has a TF of 1.03. It crystallizes in a hexagonal phase, similar to previously reported AgI–RuIII halide perovskites.39 The [AgCl6]5− and [MoCl6]3− octahedra are alternatively connected by face-sharing of two opposite faces in 1-D chain running along the c-axis. The MA cation occupies the interchain space and forms N⋯H⋯Cl hydrogen bonds with chlorides (the shortest N⋯Cl distance of 3.37 Å). It is worthwhile to note that both [MoCl6]3− and [AgCl6]5− octahedra are trigonally distorted, with the latter being heavily distorted as suggested by the bond angle variance (σ2) (Table 2).
(where rA, rx, rMI, and rMIII are the radii of the A-cation, halide, monovalent metal and trivalent metal, respectively) is in the 0.8–1.0 range, and a face-shared 1-D chain when the TF is slightly more than 1.0.47 Here, we replaced Cs+ with MA and Na+ with Ag+ to obtain (MA)2AgMoCl6 (Fig. 2a and b) which has a TF of 1.03. It crystallizes in a hexagonal phase, similar to previously reported AgI–RuIII halide perovskites.39 The [AgCl6]5− and [MoCl6]3− octahedra are alternatively connected by face-sharing of two opposite faces in 1-D chain running along the c-axis. The MA cation occupies the interchain space and forms N⋯H⋯Cl hydrogen bonds with chlorides (the shortest N⋯Cl distance of 3.37 Å). It is worthwhile to note that both [MoCl6]3− and [AgCl6]5− octahedra are trigonally distorted, with the latter being heavily distorted as suggested by the bond angle variance (σ2) (Table 2).

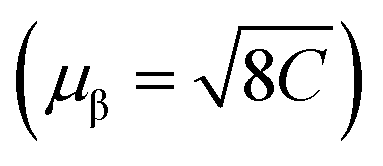 , T is the temperature, θCW is the Curie–Weiss temperature which is a measure of the strength and the nature of magnetic exchange interaction between the nearest-neighbors, and χ0 is the temperature-independent diamagnetic contribution. Fig. 6a and b show the molar susceptibility curves. We shall first discuss the magnetic properties of Cs2NaMoCl6 which exhibits long range antiferromagnetic ordering at a Néel temperature (TN) of 3.12 K, and there is no divergence in zero field cooled (ZFC) and field cooled (FC) curves (Fig. S16†). Above TN, the paramagnetic susceptibility shows an excellent fit to the Curie–Weiss equation with a negative Curie–Weiss temperature (θCW) of −15.8 K, and Curie constant (C) of 1.472 emu Oe−1 K mol−1 (Fig. 6c). The effective magnetic moment µβ is calculated to be 3.43 µB f.u.−1 which is slightly less than the spin only moment of 3.87 µB f.u.−1 for a d3 ion [
, T is the temperature, θCW is the Curie–Weiss temperature which is a measure of the strength and the nature of magnetic exchange interaction between the nearest-neighbors, and χ0 is the temperature-independent diamagnetic contribution. Fig. 6a and b show the molar susceptibility curves. We shall first discuss the magnetic properties of Cs2NaMoCl6 which exhibits long range antiferromagnetic ordering at a Néel temperature (TN) of 3.12 K, and there is no divergence in zero field cooled (ZFC) and field cooled (FC) curves (Fig. S16†). Above TN, the paramagnetic susceptibility shows an excellent fit to the Curie–Weiss equation with a negative Curie–Weiss temperature (θCW) of −15.8 K, and Curie constant (C) of 1.472 emu Oe−1 K mol−1 (Fig. 6c). The effective magnetic moment µβ is calculated to be 3.43 µB f.u.−1 which is slightly less than the spin only moment of 3.87 µB f.u.−1 for a d3 ion [ , where n = number of unpaired electrons, n = 3 in this case]. Fig. 6d shows the fcc sub-lattice composed of magnetic Mo ions in Cs2NaMoCl6 which clarifies the edge-shared tetrahedral arrangement of Mo3+ metal ions in a geometrically frustrating system. The tetrahedron is dimensionally symmetric with a nearest-neighbor Mo⋯Mo separation of 7.36 Å. The calculated magnetic frustration index f
, where n = number of unpaired electrons, n = 3 in this case]. Fig. 6d shows the fcc sub-lattice composed of magnetic Mo ions in Cs2NaMoCl6 which clarifies the edge-shared tetrahedral arrangement of Mo3+ metal ions in a geometrically frustrating system. The tetrahedron is dimensionally symmetric with a nearest-neighbor Mo⋯Mo separation of 7.36 Å. The calculated magnetic frustration index f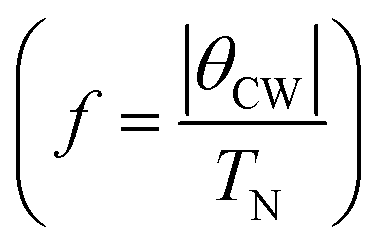 of 5.0 indeed suggests that the system has weak magnetic frustration, and it warrants further studies.56,57 The 1-D compound (MA)2AgMoCl6 features an antiferromagnetic transition temperature TN of 2.35 K and a negative θCW of −2.9 K with an effective magnetic moment of 3.48 µB f.u.−1 The calculated frustration index (f) of 1.2 indicates that this 1-D system is magnetically non-frustrating.
of 5.0 indeed suggests that the system has weak magnetic frustration, and it warrants further studies.56,57 The 1-D compound (MA)2AgMoCl6 features an antiferromagnetic transition temperature TN of 2.35 K and a negative θCW of −2.9 K with an effective magnetic moment of 3.48 µB f.u.−1 The calculated frustration index (f) of 1.2 indicates that this 1-D system is magnetically non-frustrating.