
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of triarylmethanes by silyl radical-mediated cross-coupling of aryl fluorides and arylmethanes†
Jun
Zhou
 a,
Zhengyu
Zhao
a,
Bingyao
Jiang
b,
Katsuhiro
Yamamoto
b,
Yuji
Sumii
b and
Norio
Shibata
*ab
a,
Zhengyu
Zhao
a,
Bingyao
Jiang
b,
Katsuhiro
Yamamoto
b,
Yuji
Sumii
b and
Norio
Shibata
*ab
aDepartment of Nanopharmaceutical Sciences, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan. E-mail: nozshiba@nitech.ac.jp
bDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan
First published on 21st March 2023
Abstract
Although the cross-couplings of aryl halides with diarylmethanes are mostly achieved by transition-metal catalysis, aryl fluorides are rarely used as coupling partners owing to the high inertness of C–F bonds. Herein, we describe the efficient silylboronate-mediated cross-coupling reaction of aryl fluorides with arylalkanes under transition-metal-free, room-temperature conditions. The combination of silylboronate and KOtBu is critical for driving a radical process via the cleavage of C–F and C–H bonds in two appropriate coupling precursors, resulting in a cross-coupling product. This practical cross-coupling protocol is applicable to a wide variety of aryl fluorides with a C(sp2)–F bond. This method can be extended to other coupling partners with a C(sp3)–H bond, including diarylmethanes, diarylethanes, and monoarylalkanes. Many di- and triarylalkanes with tertiary or quaternary carbon centers can be obtained easily in moderate to high yields. We believe that the developed silylboronate-mediated cross-coupling method is a valuable contribution to C–F and C–H activation chemistry.
Introduction
Benzylic motifs with a C(sp3)–H bond (ArCHR2) are present in many bioactive compounds,1 and ∼25% of the 200 top-selling pharmaceuticals contain these motifs.2 Therefore, the functionalization of such benzylic C–H bonds to new C–C,3 C–N,4 and C–O5 bonds is the logical next step for the further modification of drug candidates. In particular, triarylmethanes (ArCHAr2) and diarylalkanes (Ar2CHR) are some of the most attractive frameworks targeted for benzylic C–H functionalization, as they widely exist in pharmaceuticals,6 functional materials,7 and sensing systems.8 Several representative triarylmethanes and diarylalkane compounds have been utilized as pharmacological agents for treating viral infection, bacterial infection, breast cancer, and diabetes (Fig. 1A). Friedel–Crafts arylations of diarylmethanols are traditionally used for producing these frameworks, but this chemistry limits nucleophilic and electron-rich arenes and occasionally forms regioisomers.9 Walsh et al. reported the first Pd-catalyzed cross-couplings of aryl halides (Ar–Br and Ar–Cl) with diarylmethanes, providing triarylmethanes at room temperature.10 The conditions of Pd(OAc)2, NiXantphos, and KHMDS were able to effectively circumvent the limitations of traditional cross-coupling methods, which require high reaction temperatures (Fig. 1B(i)). Subsequently, several methods for preparing triarylmethanes or diarylalkanes under mild conditions have been reported, most of which involve transition-metal catalysis.11 These protocols require aryl halides (Ar–X, X = I, Br, Cl), but aryl fluorides (Ar–F) are not appropriate as cross-coupling precursors because the C–F bond is rather inert and possesses the highest bond dissociation energy in the series. The chemical transformation of fluorinated moieties into other functional groups is a considerable challenge,12 In 2018, Walsh et al. extended their cross-coupling protocol to aryl fluorides. They found suitable conditions [Ni(COD)2 (10 mol%) and 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene (IMes, 20 mol%) in the presence of NaHMDS (3.0 equiv.) in cyclopentyl methyl ether (CPME)] for producing the desired products, but high temperatures and long reaction times (16 h) were required (Fig. 1B(ii)).13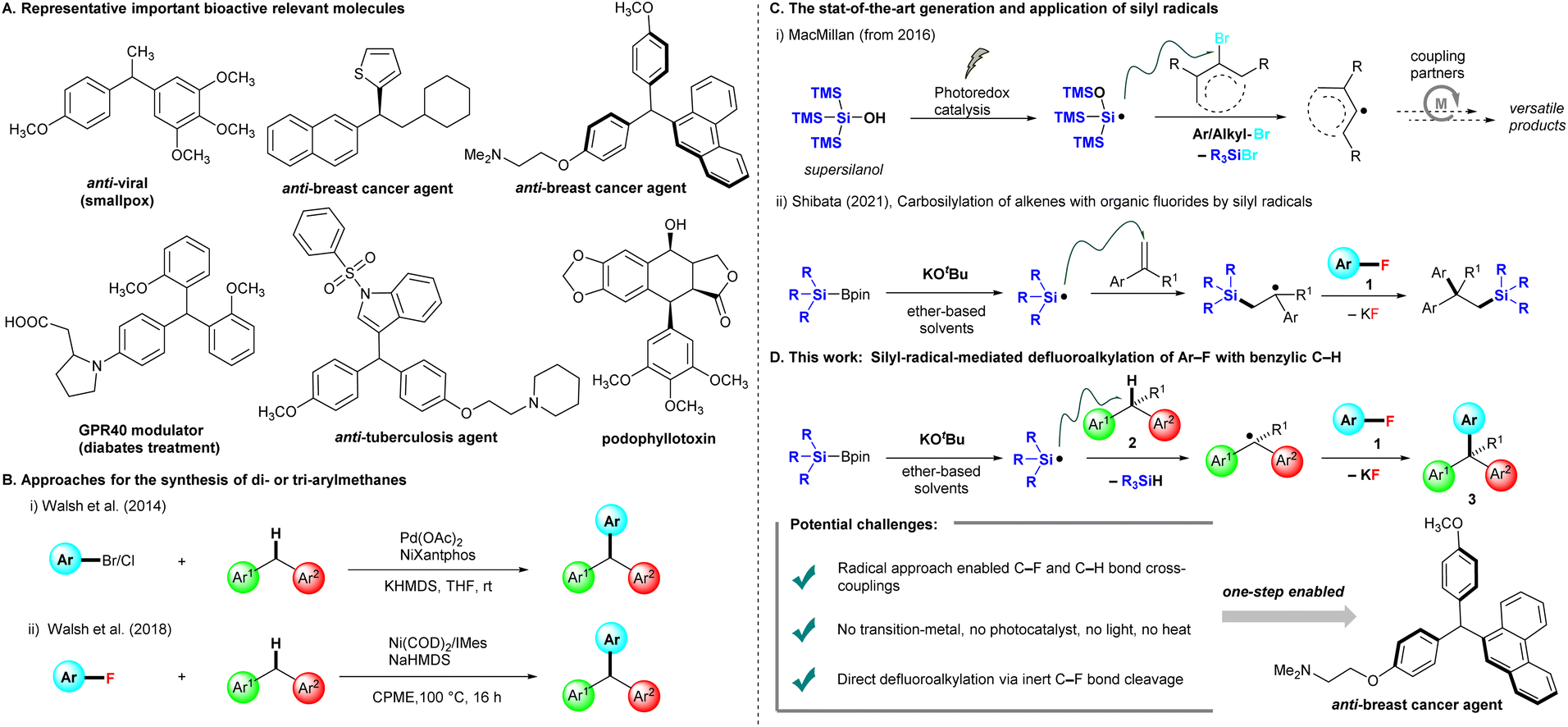 | ||
| Fig. 1 Representative bioactive relevant molecules and approaches for the synthesis of triarylmethanes and variants. (A) Bioactive relevant molecules. (B) Cross-couplings and related reactions of aryl or alkyl halides with diarylalkanes. (C) Generation and applications of silyl radicals. (D) This work: silyl-radical-mediated cross-coupling of benzylic C–H with Ar–F. | ||
In recent years, MacMillan et al. reported that photocatalytically generated silicon-centered radicals from “supersilanol” could effectively abstract a bromine atom from suitable aryl/alkyl bromides to afford the corresponding aryl/alkyl radicals, which are subsequently captured by transition metal catalysis, especially nickel or copper catalysis, to undergo a series of catalytic circles with coupling partners to afford a library of coupling products (Fig. 1C(i)). The state-of-the-art combination of the “supersilyl” group, photoredox catalysis and the transition metal catalysis system has emerged as a powerful strategy in organic chemistry.14 In 2021, our group discovered that silicon-centered radicals are effectively generated by mixed silylboronates (R3SiBpin) and potassium tert-butoxide (KOtBu) without either a photoredox system or high temperature, and then enabled a catalyst-free carbosilylation of alkenes with aryl fluorides 1via the activation of an inert C–F bond at room temperature (Fig. 1C(ii)).15
Inspired by the studies mentioned above14 and our studies on C–F bond activation,15,16 we herein propose a silylboronate-mediated radical cross-coupling reaction of aryl fluorides 1 with arylalkanes 2via the cleavage of both C–F and C–H bonds, which produces triarylmethanes 3 (Fig. 1D). Notably, our approach allows cross-coupling between an aryl C(sp2)–F bond in 1 and a benzylic C(sp3)–H bond in 2. This coupling reaction proceeds smoothly at room temperature without transition-metal catalysis. Supersilyl, photoredox catalysis, or irradiation with LEDs is not required. The substrate scope of arylalkanes 2 is broad, including diarylmethanes, diarylethanes, and monoarylalkanes. Dihydroanthracene and 9H-xanthene are also good coupling partners with aryl fluorides 1, furnishing the desired triarylmethane products 3 in high yields. As many aryl fluorides and arylalkanes are readily available, including complex pharmaceuticals, we expect the radical coupling of aryl fluorides with arylalkanes to be a valuable method for the straightforward preparation of various materials, such as drug candidates and specialty materials.
Results and discussion
Silylboronate-mediated cross-coupling reaction of organic fluorides and arylalkanes
The reaction of 4-fluorobiphenyl (1a) with diphenylmethane (2a) was initially investigated as a model reaction (Table 1). We attempted the reaction in the presence of Et3SiBpin (2.0 equiv.), Ni(COD)2 (10 mol%), and KOtBu (3.0 equiv.) in THF at room temperature; these conditions were also used in our previous defluorosilylation of aryl fluorides.16 The reaction proceeded as expected, producing 4-benzhydrylbiphenyl (3aa) in 37% yield accompanied by the defluorosilylated product, biphenyl-4-yltriethylsilane (4a) (entry 1). Without Ni(COD)2, the yield of 3aa improved to 47% (entry 2). This was followed by base screening (entries 3–6). Weaker bases were not suitable (entries 3 and 4), but strong bases (NaOtBu and KHMDS) did not improve the yield (entries 5 and 6). The yield was slightly increased to 49% with the use of 4.0 equiv. of KOtBu (entry 7). Significantly, the solvent was found to be crucial for obtaining a high yield of 3aa (entries 8–16), and the use of diglyme resulted in a high yield (95%, entry 16). Notably, diglyme effectively suppressed the formation of byproduct 4a (entry 16). The effects of diglyme can be explained by the fact that K+ would be encapsulated by diglyme, giving a more “naked” and stronger base.17 Control experiments revealed that no reaction occurred in the absence of KOtBu or Et3SiBpin (entries 17 and 18). Finally, the reactions were repeated using 0.2 mmol and 4.0 mmol of 1a, respectively, under the same conditions as those for entry 16 to evaluate the scale-up of the coupling process, and product 3aa was successfully obtained in 96% (93% isolated yield, entry 19) and 85% isolated yield (entry 20). Further details of optimization of the conditions are shown in the ESI.†|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | 3aa (%) | 4a (±) |
| a Reactions were attempted under indicated reagents and conditions: 1a (17.2 mg, 0.1 mmol), 2a, KOtBu and solvent (1.0 mL) reacted at room temperature for 8 h. b Determined by 19F NMR and 1H NMR spectroscopy using 3-fluoropyridine as an internal standard. The number in parentheses refers to the isolated yield. c 10 mol% Ni(COD)2 was used. d Performed without Et3SiBpin. e 0.2 mmol scale was performed. f 4.0 mmol scale was performed. | ||||
| 1c | KOtBu (3.0) | THF | 37 | + |
| 2 | KOtBu (3.0) | THF | 47 | + |
| 3 | K2CO3 (3.0) | THF | — | — |
| 4 | Cs2CO3 (3.0) | THF | — | — |
| 5 | NaOtBu (3.0) | THF | 28 | + |
| 6 | KHMDS (3.0) | THF | 30 | + |
| 7 | KOtBu (4.0) | THF | 49 | + |
| 8 | KOtBu (4.0) | c-hexane/THF (8/1, v/v) | 34 | + |
| 9 | KOtBu (4.0) | c-hexane | 9 | + |
| 10 | KOtBu (4.0) | Toluene | 11 | + |
| 11 | KOtBu (4.0) | Dioxane | Trace | + |
| 12 | KOtBu (4.0) | DME | 36 | + |
| 13 | KOtBu (4.0) | CPME | 18 | + |
| 14 | KOtBu (4.0) | MTBE | 12 | + |
| 15 | KOtBu (4.0) | DTBE | Trace | + |
| 16 | KOtBu (4.0) | Diglyme | 95 | — |
| 17 | — | Diglyme | 0 | — |
| 18d | KOtBu (4.0) | Diglyme | 0 | — |
| 19e | KOtBu (4.0) | Diglyme | 96 (93) | — |
| 20f | KOtBu (4.0) | Diglyme | (85) | — |
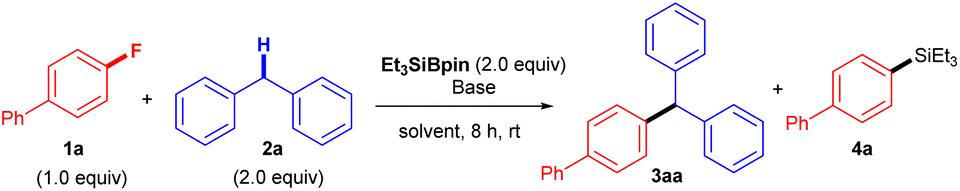
Silylboronate-mediated cross-coupling reaction of organic fluorides and arylalkanes
With the optimal reaction conditions determined (entry 19, Table 1), the substrate scope of this silylboronate-mediated defluorinative cross-coupling reaction was further investigated (Fig. 2). A range of substituted aryl fluorides 1 were investigated with 2a to assess their generality (Fig. 2(I)). As shown, a wide range of fluoroarenes, including π-extended systems 1a–d, fluorobenzene 1e, and methyl- and methoxy-substituted fluorobenzenes 1f–h, were efficiently coupled with 2a to afford corresponding triarylmethanes 3aa–ha in yields of up to 93%. The yields of 3 were slightly lowered in the coupling reactions of aryl fluorides 1 that were affected by steric hindrance (3ca: 46%) and electron-rich substituents (3ga: 42%; 3ha: 46%). A series of p-substituted 4′-fluorobiphenyls 1i–m and dioxole 1n also produced the corresponding triarylmethanes in moderate to good yields under standard conditions: 3ia: 57%, 3ja: 73%, 3ka: 68%, 3la: 40%, 3ma: 45%, and 3na: 47%. These results indicate that the ether (OMe), the benzylic position (OBn), and the C(sp3)–F bond of CF3 are tolerated in this transformation. In addition, aryl fluorides 1o–q with attached heterocycles were evaluated, as C–H activation might be competitively induced by the heterocyclic moiety. Pyrrole- or indole-containing aryl fluorides 1o and 1p successfully reacted with 2a to furnish 1H-pyrrole derivative 3oa (86%) and N-methyl-1H-indole derivative 3pa (73%), respectively, without any C–H activation product detected. In addition, benzofuran-bearing fluoroarene 1q participated in this cross-coupling reaction, although the yield of coupling product 3qa was only 39%. | ||
| Fig. 2 Substrate scope of 1 and 2. (I) Scope of aryl fluorides 1; (II) scope of diarylmethanes 2; (III) scope regards to the combination of 1 and 2; (IV) scope within 1 and DHA or 9H-xanthene. Unless otherwise noted, reactions were conducted using 1 (0.2 mmol), 2a (2.0 equiv.), Et3SiBpin (2.0 equiv.), KOtBu (4.0 equiv.), and diglyme (2.0 mL) at room temperature for 8 h, with isolated yields shown. a Reaction performed using 0.4 mmol of 1. b Reaction performed using Et3SiBpin (3.0 equiv.) and KOtBu (6.0 equiv.). | ||
We next examined the substrate scope of diarylmethanes 2 in the cross-couplings with 4-fluorobiphenyl (1a) (Fig. 2(II)). Electron-rich phenyl-(4-methoxyphenyl)methane (2b), bis(4-methoxyphenyl)methane (2c), and bis(tolyl)methane (2d) smoothly reacted with 1a under the standard conditions to furnish desired triarylmethanes 3ab (77%), 3ac (52%), and 3ad (71%), respectively. Even sterically hindered ortho-methyl-diphenylmethane (2e) produced coupling product 3ae in 57% yield. Notably, Cl-substituted diphenylmethane 3f was tolerated under the same conditions, selectively providing defluorinative coupling product 3af (61%). Furthermore, 2-benzylthiophene (2g) was successfully coupled with 1a under identical reaction conditions to yield 3ag (84%).
We further attempted the coupling reaction for a range of substituted fluoroarenes (1j–p) and substituted diarylmethanes (2b–g) to widen the generality (Fig. 2(III)). Fluoro-biphenyls with Me (1j) or OMe (1k) substituents reacted with diarylmethanes 2b–f under the optimal conditions to furnish the desired triarylmethanes in moderate to good yields (3jb: 55%; 3jc: 65%; 3jg: 74%; 3kb: 66%; 3kc: 86%; 3kd: 78%; 3ke: 33%; 3kf: 57%). The low yield of 3ke can be explained by the steric hindrance of the o-Me group in 2e, and the result was improved to 52% with the use of excess Et3SiBpin (3.0 equiv.) and KOtBu (6.0 equiv.). In addition, dioxole-bearing fluoroarene 1n reacted well with 4-methoxydiphenylmethane (2b) and dianisylmethane (2c) to give triarylmethanes 3nb (68%) and 3nc (56%), respectively. Furthermore, despite possessing several reactive C(sp2)–H bonds in their heterocyclic skeletons, the cross-coupling reactions of pyrrole aryl fluoride 1o and indole aryl fluoride 1p proceeded well to furnish heteroaryl-containing products 3ob (62%), 3og (80%), 3pb (45%), and 3pg (41%) via C–F bond cleavage, without any of the anticipated C–H cross-coupling reactions in the heteroaromatic moiety.
The interesting aspect of this protocol is the usage of dihydroanthracene (DHA, 2h) as a cross-coupling partner with fluoroarenes (Fig. 2(IV)). DHA is known to act as a radical inhibitor.18 Although this transformation should involve a radical process (see the discussion below), the cross-coupling of aryl fluorides 1 with 2h proceeded very well under the standard conditions, giving the desired cross-coupling products in good yields (3ah: 76%; 3jh: 71%; 3kh: 75%; 3lh: 45%; 3nh: 71%). Similarly, 9H-xanthene (2i) also produced good results with aryl fluorides 1 under the same conditions (3ai: 77%; 3ji: 85%; 3ki: 80%; 3ni: 74%; 3pi: 74%).
We next examined another potential limitation of this methodology, focusing on arylalkanes 2 (Fig. 3, top). The cross-coupling of 1a with 1,1-diphenylalkanes 2j–l under the optimal conditions produced products 3aj (79%), 3ak (64%), and 3al (51%), which possessed a quaternary carbon center, in good yields. In comparison, cumene (2m) produced product 3am in only 23% yield. Other arylalkanes with a single aromatic group (2n–p) afforded corresponding cross-coupling products 3an–ap in 20–25% yields (3an: 25%; 3ao: 22%; 3ap: 20%). The low yields of 3 were slightly improved by the use of excess reagents (3am: 37%; 3an: 41%; 3ao: 35%; 3ap: 33%). These results indicated that successful conversion is highly dependent on the stability of the reactive benzylic species. When we attempted the reaction of allylbenzene (2q), we obtained 3aq (Z/E = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3) in 34% yield instead of the expected coupling product 3aq′. While there are several possibilities for its formation, one is the isomerization of 3aq′ to 3aq under the optimal conditions. Indeed, we obtained 3aq (Z/E = 1:1.3) by treating 3aq′ (independently prepared) under the same KOtBu/Et3SiBpin conditions. Interestingly, the isomerization of 3aq′ under the base (KOtBu) gave 3aq with a different ratio (Z/E = 1:1) (see details in the ESI†). Next, the reaction of 1a with toluene was attempted to confirm this hypothesis. As expected, no desired coupling product was observed. However, when p-phenyl-substituted toluene (2r) was used, the desired coupling product 3ar was obtained in 15% yield, supporting the formation of a benzylic radical species (see the Discussion section). Although various substrates 1, 2, and 3 with functional groups and heterocycles have been used successfully (Fig. 2 and 3), some functional limitations were experienced, such as with carbonyls, amines, and free H (OH, NH2, etc.). These limitations are listed in the ESI.†
:1.3) in 34% yield instead of the expected coupling product 3aq′. While there are several possibilities for its formation, one is the isomerization of 3aq′ to 3aq under the optimal conditions. Indeed, we obtained 3aq (Z/E = 1:1.3) by treating 3aq′ (independently prepared) under the same KOtBu/Et3SiBpin conditions. Interestingly, the isomerization of 3aq′ under the base (KOtBu) gave 3aq with a different ratio (Z/E = 1:1) (see details in the ESI†). Next, the reaction of 1a with toluene was attempted to confirm this hypothesis. As expected, no desired coupling product was observed. However, when p-phenyl-substituted toluene (2r) was used, the desired coupling product 3ar was obtained in 15% yield, supporting the formation of a benzylic radical species (see the Discussion section). Although various substrates 1, 2, and 3 with functional groups and heterocycles have been used successfully (Fig. 2 and 3), some functional limitations were experienced, such as with carbonyls, amines, and free H (OH, NH2, etc.). These limitations are listed in the ESI.†
 | ||
| Fig. 3 Further scope and limitations of arylalkanes 2. Unless otherwise noted, reactions were conducted with 1 (0.2 mmol), 2 (2.0 equiv.), Et3SiBpin (2.0 equiv.), KOtBu (4.0 equiv.), and diglyme (2.0 mL) at room temperature for 8 h, with isolated yields shown. a Reaction performed using Et3SiBpin (3.0 equiv.) and KOtBu (6.0 equiv.). | ||
Application of the silylboronate-mediated defluorinative coupling reaction
To highlight the synthetic applications of this silylboronate-mediated defluorinative coupling reaction, we examined the functionalization of several drug derivatives with a fluoroarene moiety or benzylic C–H moiety (Fig. 3, bottom). Estrone-derived fluoroarene 1r successfully underwent a coupling reaction with diphenylmethane 2a to afford desired estrone derivative 3ra in 60% yield. Moreover, the three benzylic C–H bonds containing motif 2s, derived from estrone, were also successfully functionalized at the secondary C–H site using this transformation with 1a to give 3as in 27% yield. Another noteworthy application is that this modular approach also enables the rapid synthesis of anti-breast-cancer agent 3st in one single step rather than several steps.19 By simply employing 9-fluorophenanthrene 1s and 2-(4-(4-methoxybenzyl)phenoxy)-N,N-dimethylethan-1-amine 2t under standard reaction conditions, desired product 3st can be easily fashioned in 68% yield.It should be mentioned that the chemoselectivity of our coupling reaction is slightly poor, whereas it was difficult to efficiently transform 4-chloro-4′-fluoro-1,1′-biphenyl (1t) into desired product 3ta (23%). As a result, borylated product 5 (57%) was obtained preferably via C–Cl bond cleavage (Fig. 4A).
 | ||
| Fig. 4 Chemoselectivity and parallel experiments. (A) Chemoselectivity of Ar–F over Ar–Cl. (B) Coupling reactions of 2a with 1a (X = F), 6 (X = Cl), 7 (X = Br), and 8 (X = I). | ||
However, the advantage of using aryl fluorides as coupling partners over other aryl halides in the present transformation is evident, as shown in the parallel experiments (Fig. 4B). When we attempted the cross-coupling reactions of diphenylmethane (2a) with biphenyl chloride (6), biphenyl bromide (7), and biphenyl iodide (8), mixtures of borylated product 9 and silylated product 4a were detected, with only traces of desired cross-coupling product 3aa observed under the standard conditions.
Mechanistic study
Several observations in the present study led us to believe that this transformation proceeds via a single-electron transfer (SET) radical process, but not via nucleophilic substitution pathways, such as traditional nucleophilic aromatic substitution (SNAr) and SN2 where arylfluorides can act as electrophiles or benzyne precursors,20–23 because SNAr and SN2 protocols require electron-deficient aryl fluorides under strongly basic conditions. We, therefore, conducted several further experiments to gain insight into the reaction mechanism (Fig. 5). We first examined the coupling reaction of aryl fluoride 1a and diphenylmethane 2a in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) (Fig. 5A(i)). Although coupling product 3aa was obtained in 93% yield under the standard conditions, the yields decreased considerably as the amount of TEMPO increased: 52% (1.0 equiv. of TEMPO), 20% (2.0 equiv. of TEMPO), and trace (4.0 equiv. of TEMPO). Moreover, increasing the quantity of TEMPO led to an increase in the yield of 1-(benzhydryloxy)-2,2,6,6-tetramethylpiperidine (Int-TEMPO) from 11% (1.0 equiv. of TEMPO) to 68% (2.0 equiv. of TEMPO) and 80% (4.0 equiv. of TEMPO). These results suggest that the cross-coupling reaction involves a radical species. Ohmiya and co-workers reported the cross-coupling of aryl fluorides by tertiary benzylic organoboronates with KOtBu at a high temperature of 120 °C via SNAr.24a We therefore attempted the reaction of 1a with pinBCHPh210 in the presence of KOtBu in diglyme at room temperature (Fig. 5A(ii)). We detected only 9% of 3aa, leaving most of 1a. These results indicate the formation of carbanion from pinBCHPh2 and that the SNAr process would not be included.24b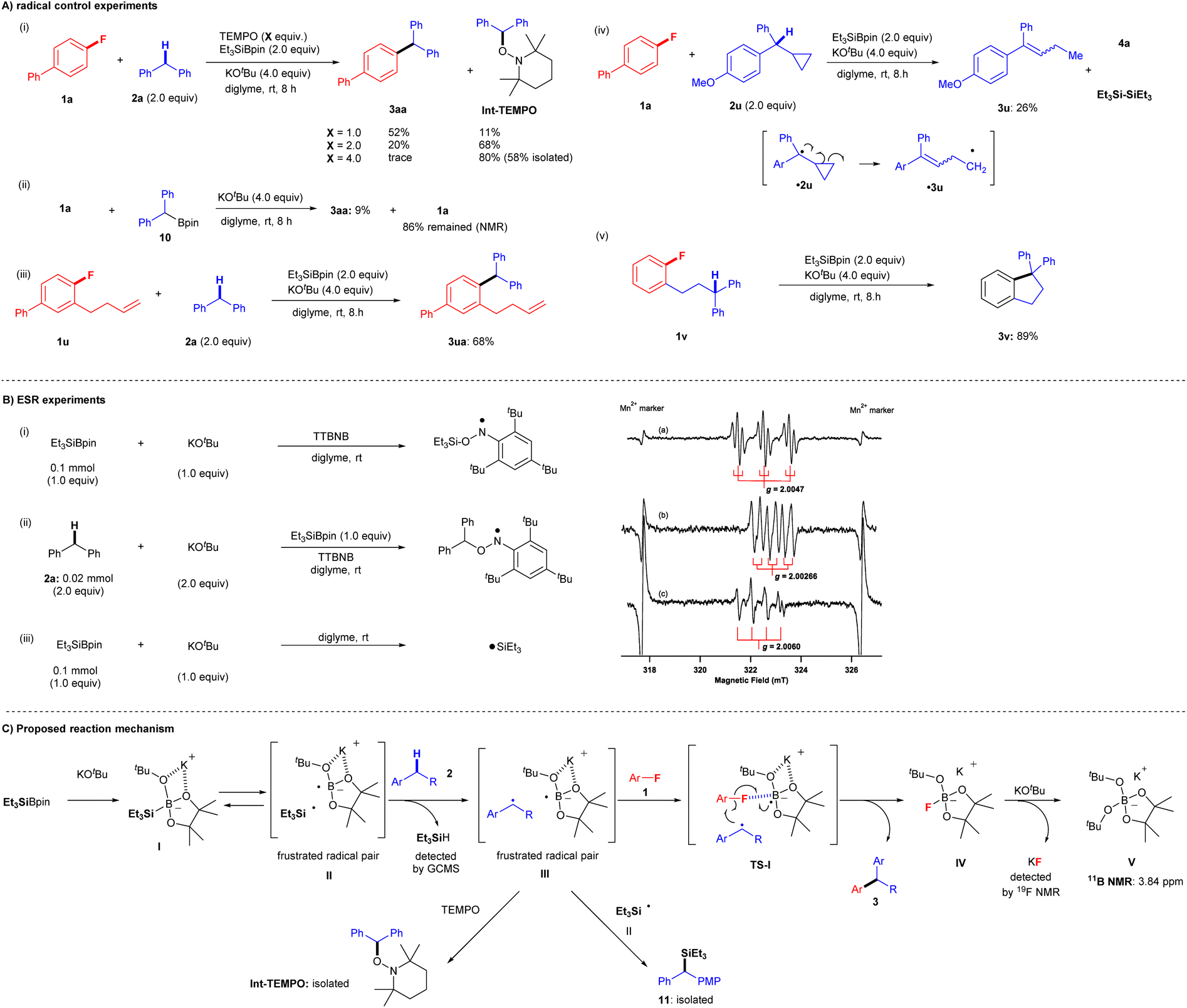 | ||
| Fig. 5 Control experiment study and the proposed reaction mechanism. (A) Radical control experiments. (i) Effect of TEMPO in the silylboronate-mediated coupling reaction. (ii) SNAr conditions using pinBCHPh210 in the presence of KOtBu. (iii) Radical cyclization experiments. (iv) Radical ring-opening experiment. (v) Radical cyclization experiment. (B) ESR experiments and chemical structure. (i) and (a) spin-adduct of TTBNB with triethyl silyl radical (anilino-type) and (ii) and (b) spin-adduct of TTBNB with diphenyl methyl radical (anilino-type). (iii) and (c) triethyl silyl radical. (C) Proposed reaction mechanism. | ||
A standard radical clock experiment using 1-(but-3-en-1-yl)-2-fluorobenzene (1u) was performed with 2a under identical cross-coupling conditions (Fig. 5A(iii)). Significantly, corresponding cross-coupling products 3ua (no cyclization; 68% yield) were obtained. The result is suggestive of C–F bond cleavage proceeding via a cascade, concerted process in the final part of the reaction mechanism, without generating a free aryl radical. Moreover, two more radical clock experiments were conducted to give further insight into the reaction process. First, the reaction of 1a and cyclopropyl benzyl derivative 2u was attempted, but the reaction did not yield the corresponding cross-coupling product. Instead, the ring-opening product 3u was isolated (26% yield) presumably via a cyclopropyl benzyl radical ˙2u, and we also detected the associated by-products such as 4a and hexaethyldisilane ((Et3Si)2) (Fig. 5A(iv)). Additionally, an intramolecular cross-coupling reaction was achieved by treatment of diphenylpropyl-substituted fluorobenzene 1v under identical conditions, and cyclization product 3v was furnished in 89% yield (Fig. 5A(v)).
Finally, ESR experiments were carried out to confirm the generation of radicals under the optimized conditions (Fig. 5B). First, an ESR experiment was performed with the spin trap tri-tert-butylnitrosobenzene (TTBNB). The ESR spectrum (triple–triplet) for the reaction of Et3SiBpin and KOtBu in diglyme at room temperature (Fig. 5B(i)(a)) corresponded to that of the spin adduct of the triethylsilyl radical (˙SiEt3) trapped by TTBNB. The hyperfine splitting (hfs) constant due to nitrogen (AN; spin quantum number I = 1) was 1.03 mT, and the small splitting constant due to the two hydrogens at the meta position of the TTBNB benzene ring (AHm; I = 1/2) was 0.175 mT. The g-value of 2.0047 was assigned to an anilino-type radical (Fig. 5B(i)).25 The observed spectrum is the similar triple-triplet spectrum reported for SiEt3-TTBNB previously.26,27 We next investigated the reaction of diphenylmethane (2a), Et3SiBpin, and KOtBu in diglyme at room temperature (Fig. 5B(ii)(b)). The ESR spectrum (double-triplet; sextet line) was assigned to a benzyl-type radical (˙CHPh2) trapped by TTBNB. The hfs constants AN and AHα (due to an α-proton) were 0.62 and 0.34 mT, respectively. The splitting due to the meta-hydrogens was too small to be resolved (<0.06 mT). The g-value of 2.00266 was assigned to an anilino-type radical (Fig. 5B(ii))25 In addition, we investigated the reaction of Et3SiBpin and KOtBu in diglyme at room temperature (Fig. 5B(iii)(c)). The quartet line with a g-value of 2.0060 should be assigned to a silyl radical ˙SiEt3. This allowed us to directly detect the generated silyl radical ˙SiEt3 (see the ESI† for more details on the discussion of the ESR experiments).
Based on both our experimental results and previous reports,15,16 we proposed a reaction mechanism involving a radical-mediated defluorinative cross-coupling reaction (Fig. 5C).28Fig. 5C shows a schematic of the mechanism for the representative reaction of aryl fluorides 1 and diarylmethanes 2. First, Et3SiBpin reacts with a molecule of KOtBu to form intermediate I; the formation of this intermediate was previously confirmed by the Avasare group based on density functional theory calculations.29 We also confirmed the existence of intermediate I by 11B NMR and 29Si NMR spectroscopy.15,16 In this step, due to the inherent steric repulsion of this ate complex I, the intermediate I splits into a sterically demanding and frustrated radical pair II30 consisting of a triethylsilyl radical (˙SiEt3) and boron-radical species via the homolytic scission of the Si–B bond. Hydrogen abstraction from diarylmethane 2 by ˙SiEt3 affords a frustrated radical pair III accompanied by the formation of HSiEt3 (detected by GC-MS). Then, aryl fluorides 1 participate in the cascade process via transition state TS-I, where the C–F bond of aryl fluorides 1 is activated by the boron atom in Bpin. Subsequently, the boron-radical side of a frustrated radical pair III in TS-I promotes a radical reaction; aryl fluorides 1 are transformed into aryl radicals via C–F bond cleavage by SET.30–32 In the meantime, the benzyl radical approaches the aryl radicals. Then, C–C bond formation is completed by the release of IV ([Bpin(OtBu)F]K) to furnish desired cross-coupling product 3.15,16 Finally, the IV ([Bpin(OtBu)F]K) further reacts with the second mole of KOtBu to provide a stable V ([Bpin(OtBu)2]K) (detected by 11B NMR) and KF (detected by 19F NMR). Some benzyl radical species in the frustrated radical pair III are competitively captured by ˙SiEt3 from II, providing 11, which was occasionally detected as a by-product in the experiments. The benzyl radical in III is also evidenced by the formation of Int-TEMPO. The lower yields of the coupling reactions using monoarylalkanes can be understood based on the lower stabilities of the corresponding radical species.
Conclusions
In summary, we developed the first silylboronate-mediated radical cross-coupling reaction of aryl fluorides with arylalkanes, in which C–F bond cleavage is concomitant with the initial cleavage of a C–H bond to form a new C–C bond. A variety of triaryl- and diarylalkanes were efficiently and smoothly synthesized in moderate to excellent yields under very mild conditions at room temperature. Another important feature of the present coupling system is that it relies on C–F and C–H bond activation occurring at room temperature. This method obviates the use of transition metals and specialized ligands with high reaction temperatures. A radical reaction mechanism was suggested by the experimental results and confirmed by ESR analysis. The library of arylalkanes obtained by this method can be used as valuable scaffolds for pharmaceuticals and functional materials. As many organic fluorides are readily available, including complex pharmaceuticals,33 and agrochemicals34 we expect the radical coupling of organic fluorides with arylalkanes to be a valuable method for the straightforward preparation of various materials, such as drug candidates and specialty materials.Data availability
The data that support the findings of this study are available within the article and the ESI.† Details about materials and methods, experimental procedures, characterization data, mechanistic studies, ESR studies and NMR spectral are included. All relevant data are also available from the authors.Author contributions
JZ, ZZ, and BJ performed the experiments and analyzed the data. ZZ and KY performed ESR experiments. JZ, ZZ, BJ, KY, YS, and NS discussed the results. JZ and NS wrote the manuscript. NS supervised the project. All authors contributed to the manuscript and have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by the Japan Science and Technology Agency's (JST) CREST program entitled “Precise Material Science for Degradation and Stability” (JPMJCR21L1) Japan.Notes and references
- (a) J. Zhang, C. Stanciu, B. Wang, M. M. Hussain, C.-S. Da, P. J. Carroll, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2011, 133, 20552–20560 CrossRef CAS PubMed; (b) L. J. Scott, Drugs, 2012, 72, 249–272 CrossRef CAS PubMed.
- (a) N. A. Mcgrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS; (b) W. Xu, W. Wang, T. Liu, J. Xie and C. Zhu, Nat. Commun., 2019, 10, 4867–4874 CrossRef PubMed.
- (a) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed; (b) S. Guo, D. I. Abusalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS PubMed; (c) Q. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed; (d) E. Le Saux, M. Zanini and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 1113–1118 CrossRef CAS PubMed.
- (a) J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583–591 CrossRef CAS PubMed; (b) S. E. Suh, S. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS PubMed; (c) T. Kato and K. Maruoka, Chem. Commun., 2022, 58, 1021–1024 RSC.
- (a) L. Tanwar, J. Börgel and T. Ritter, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS PubMed; (b) H. Hu, S. Chen, M. Mandal, S. M. Pratik, J. A. Buss, S. W. Krska, C. J. Cramer and S. S. Stahl, Nat. Catal., 2020, 3, 358–367 CrossRef CAS PubMed; (c) B. J. Lee, K. S. Deglopper and T. P. Yoon, Angew. Chem., Int. Ed., 2020, 59, 197–202 ( Angew. Chem. , 2020 , 132 , 203–208 ) CrossRef CAS PubMed.
- (a) K. N. Babu, F. Massarwe, I. Shioukhi and A. Masarwa, Angew. Chem., Int. Ed., 2021, 60, 26199–26209 CrossRef CAS PubMed; (b) S. Singh, R. Mahato, P. Sharma, N. Yadav, N. Vodnala and C. Kumar Hazra, Chem. Eur. J., 2022, 28, e202104545 CrossRef CAS PubMed; (c) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed.
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381–433 CrossRef CAS; (b) M. S. Shchepinov and V. A. Korshun, Chem. Soc. Rev., 2003, 32, 170–180 RSC.
- (a) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC; (b) T. T. Divya, K. Ramshad, V. C. Saheer and L. Chakkumkumarath, New J. Chem., 2018, 42, 20227–20238 RSC.
- R. Kshatriya, V. P. Jejurkar and S. Saha, Eur. J. Org. Chem., 2019, 2019, 3818–3841 CrossRef CAS.
- J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276–6287 CrossRef CAS PubMed.
- M. Nambo and C. M. Crudden, ACS Catal., 2015, 5, 4734–4742 CrossRef CAS.
- (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (c) T. Stahl, H. F. Klare and M. Ostreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (d) T. Ahrens, T. J. Kohlmann and M. Ahrens, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (e) J. D. Hamel and J. F. Paquin, Chem. Commun., 2018, 54, 10224–10239 RSC.
- J. Li, C. Wu, B. Zhou and P. J. Walsh, J. Org. Chem., 2018, 83, 2993–2999 CrossRef CAS PubMed.
- (a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (b) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed; (c) G. H. Lovett, S. Chen, X.-S. Xue, K. N. Houk and D. W. C. Macmillan, J. Am. Chem. Soc., 2019, 141, 20031–20036 CrossRef CAS PubMed; (d) R. W. Pipal, K. T. Stout, P. Z. Musacchio, S. Ren, T. J. A. Graham, S. Verhoog, L. Gantert, T. G. Lohith, A. Schmitz, H. S. Lee, D. Hesk, E. D. Hostetler, I. W. Davies and D. W. C. MacMillan, Nature, 2021, 589, 542–547 CrossRef CAS PubMed; (e) X. Zhao and D. W. C. Macmillan, J. Am. Chem. Soc., 2020, 142, 19480–19486 CrossRef CAS PubMed; (f) N. W. Dow, A. Cabré and D. W. C. Macmillan, Chem, 2021, 7, 1827–1842 CrossRef CAS PubMed; (g) T. M. Faraggi, C. Rouget-Virbel, J. A. Rincón, M. Barberis, C. Mateos, S. García-Cerrada, J. Agejas, O. De Frutos and D. W. C. Macmillan, Org. Process Res. Dev., 2021, 25, 1966–1973 CrossRef CAS; (h) W. Liu, M. N. Lavagnino, C. A. Gould, J. Alcázar and D. W. C. MacMillan, Science, 2021, 374, 1258–1263 CrossRef CAS.
- J. Zhou, B. Jiang, Y. Fujihira, Z. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749–3757 CrossRef CAS PubMed.
- (a) B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393–4400 CrossRef PubMed; (b) J. Zhou, Z. Zhao and N. Shibata, Front. Chem., 2021, 9, 771473 CrossRef CAS PubMed; (c) J. Zhou, B. Jiang, Z. Zhao and N. Shibata, Org. Lett., 2022, 24, 5084–5089 CrossRef CAS PubMed.
- T. Saito, J. Wang, E. Tokunaga, S. Tsuzuki and N. Shibata, Sci. Rep., 2018, 8, 11501 CrossRef PubMed.
- (a) E. Yamamoto, K. Izumi, Y. Horita, S. Ukigai and H. Ito, Top. Catal., 2014, 57, 940–945 CrossRef CAS; (b) B. Górski, A. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef; (c) S. Das, S. Roy, A. Bhowmik, W. Sarkar, I. Mondal, A. Mishra, S. J. Saha, S. Karmakar and I. Deb, Chem. Commun., 2022, 58, 2902–2905 RSC.
- (a) M. Nambo and C. M. Crudden, Chem. Rec., 2021, 21, 3978–3989 CrossRef CAS PubMed; (b) M. Miao, W. Yin, L. Wang, Z. Chen, J. Xu and H. Ren, J. Org. Chem., 2018, 83, 10602–10612 CrossRef CAS PubMed.
- S. Caron, E. Vazquez and J. M. Wojcik, J. Am. Chem. Soc., 2000, 122, 712–713 CrossRef CAS.
- M. Ueno, M. Yonemoto, M. Hashimoto, A. E. H. Wheatley, H. Naka and Y. Kondo, Chem. Commun., 2007, 2264–2266 RSC.
- N. P. Bizier, J. W. Wackerly, E. D. Braunstein, M. Zhang, S. T. Nodder, S. M. Carlin and J. L. Katz, J. Org. Chem., 2013, 78, 5987–5998 CrossRef CAS PubMed.
- X. Ji, T. Huang, W. Wu, F. Liang and S. Cao, Org. Lett., 2015, 17, 5096–5099 CrossRef CAS PubMed.
- (a) M. Takeda, K. Nagao and H. Ohmiya, Angew. Chem., Int. Ed., 2020, 59, 22460–22464 CrossRef CAS PubMed; (b) S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16368–16388 CrossRef CAS PubMed.
- B. J. Qu, Y. H. Xu, W. F. Shi and B. Rånby, Macromolecules, 1992, 25, 5215–5219 CrossRef CAS.
- H. G. Gasanov and S. Kh. Dotdaev, Russ. Chem. Bull., 1986, 35, 1801–1805 CrossRef.
- J. C. Evans, P. Hupfiled, C. C. Rowlands and S. E. Cray, J. Chem. Soc., Faraday Trans., 1990, 68, 3221–3227 RSC.
- As the research progressed (ref. 15 and 16), we gradually modified the reaction mechanisms proposed in each paper based on new results and discussions. We believe that the mechanism presented here is the most appropriate for understanding the reaction process so far.
- P. Jain, S. Pal and V. Avasare, Organometallics, 2018, 37, 1141–1149 CrossRef CAS.
- (a) L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC; (b) D. Schilter, Nat. Rev. Chem., 2018, 2, 255 CrossRef CAS; (c) A. Dasgupta, E. Richards and R. L. Melen, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed; (d) F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. Leest, B. Bruin and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS PubMed; (e) F. Holtrop, A. R. Jupp and J. C. Slootweg, in Frustrated Lewis Pairs, Molecular Catalysis, ed. J. C. Slootweg and A. R. Jupp, Springer, Cham, 2021, vol. 2, pp. 361–385 Search PubMed; (f) T. H. Warren and G. Erker, Top. Curr. Chem., 2013, 334, 219–238 CrossRef PubMed.
- G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751–9757 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00154g |
| This journal is © The Royal Society of Chemistry 2023 |