
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn(OTf)3-catalyzed reorganization/cycloaddition of two imine units and subsequent modular assembly of acridinium photocatalysts†
Jiang
Nan
 *a,
Guanjie
Huang
a,
Shilei
Liu
a,
Jing
Wang
a,
Yangmin
Ma
a and
Xinjun
Luan
*b
*a,
Guanjie
Huang
a,
Shilei
Liu
a,
Jing
Wang
a,
Yangmin
Ma
a and
Xinjun
Luan
*b
aThe Youth Innovation Team of Shaanxi Universities, Shaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: nanjiang@sust.edu.cn
bKey Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, College of Chemistry & Materials Science, Northwest University, Xi'an 710021, China
First published on 17th April 2023
Abstract
Herein, we disclose a novel reorganization/cycloaddition between two imine units catalyzed by In(OTf)3 Lewis acid that differs from the well-known [4 + 2] cycloaddition version via the Povarov reaction. By means of this unprecedented imine chemistry, a collection of synthetically useful dihydroacridines has been synthesized. Notably, the obtained products give rise to a series of structurally novel and fine-tuneable acridinium photocatalysts, offering a heuristic paradigm for synthesis and efficiently facilitating several encouraging dihydrogen coupling reactions.
Introduction
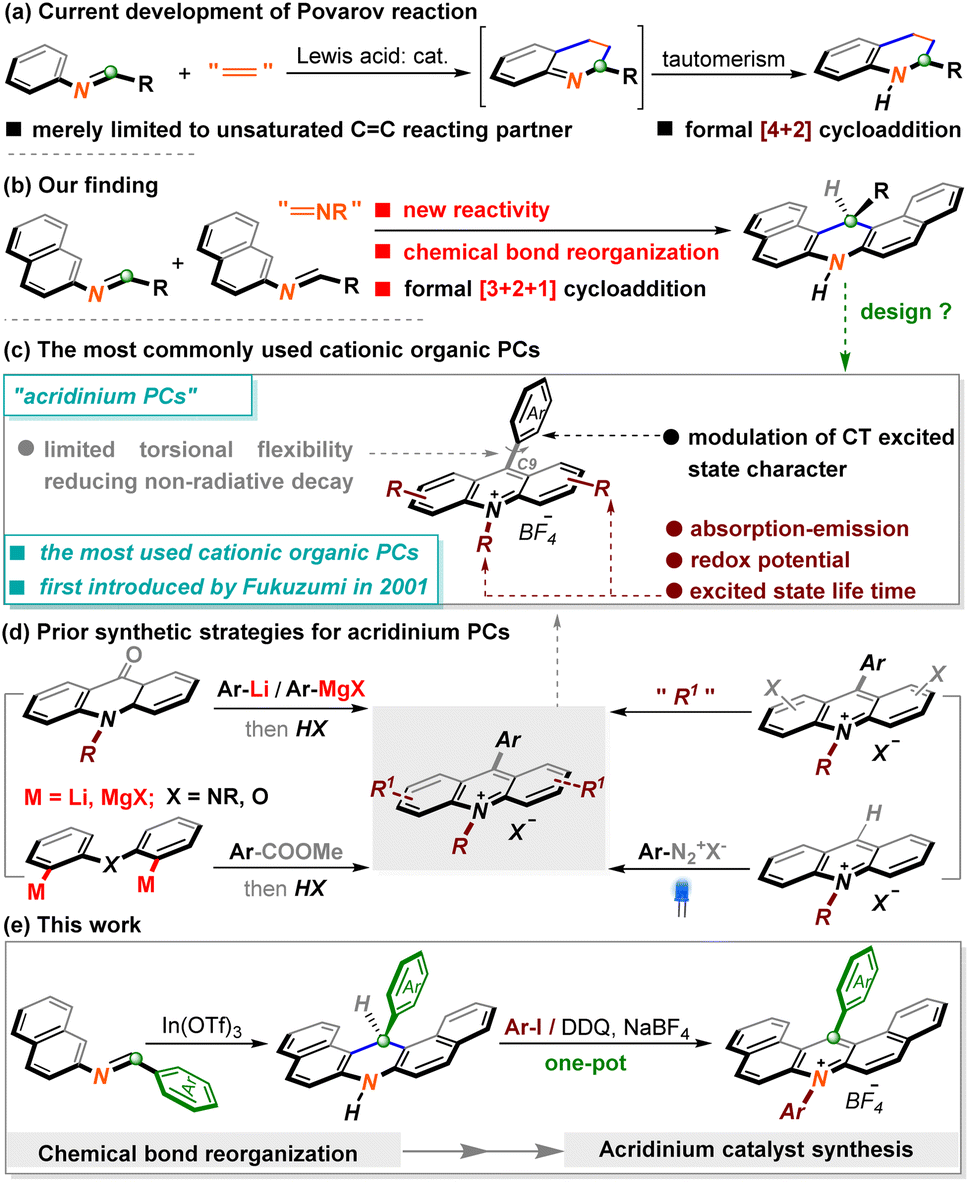
The cycloaddition reaction represents one of the most powerful chemical tools to expeditiously assemble diverse polycarbocycle or polyheterocycle frameworks from readily accessible precursors in a single step.1 A representative example is the Diels–Alder reaction, which serves as a standard strategy to build six-membered rings, especially distinguished by forging all-carbon cyclohexenes.2 Cycloaddition manipulation between N-aryl imines and dienophiles is defined as the Povarov reaction, an extremely significant part of the Diels–Alder reaction, which has benefited academia and industry because of the powerful capability of preparing widely utilized nitrogen-containing cycles.3 Although numerous marvellous reactions have been discovered in this realm over the past decades, this type of cycloaddition appears only feasible in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated systems (Scheme 1a), which somehow restricts the potency to create more contractually intriguing heterocycles.4 In this context, the analogous reactivity of imines with other unsaturated moieties such as CN has not been reported yet, which is likely attributed to the instability of the envisaged dinitrogen six-membered rings. During our research with imine chemistry,5 we discovered an entirely novel reactivity of imine molecules. In sharp contrast to the typical Povarov [4 + 2] cyclization version,6 this observation unusually behaved as a formal [3 + 2 + 1] interaction via the reorganization of multiple chemical bonds. Herein, we will detail this unprecedented chemical reactivity of imines, which leads to the modular assembly of synthetically useful dihydroacridine derivatives under concise reaction conditions (Scheme 1b).
C unsaturated systems (Scheme 1a), which somehow restricts the potency to create more contractually intriguing heterocycles.4 In this context, the analogous reactivity of imines with other unsaturated moieties such as CN has not been reported yet, which is likely attributed to the instability of the envisaged dinitrogen six-membered rings. During our research with imine chemistry,5 we discovered an entirely novel reactivity of imine molecules. In sharp contrast to the typical Povarov [4 + 2] cyclization version,6 this observation unusually behaved as a formal [3 + 2 + 1] interaction via the reorganization of multiple chemical bonds. Herein, we will detail this unprecedented chemical reactivity of imines, which leads to the modular assembly of synthetically useful dihydroacridine derivatives under concise reaction conditions (Scheme 1b).
 | ||
| Scheme 1 Cycloaddition of imines and acridinium catalysts. | ||
On the other hand, organic photoredox catalysts (OPCs) have attracted considerable attention due to their more sustainable superiority and the ability to fine-tune photophysical and electronical performance,7 despite the fact that the well-known polypyridyl ruthenium or iridium complexes have long stood at the frontier of PCs in organic synthesis.8 Acridinium salts, seminally identified as OPCs by Fukuzumi9 in 2001 due to their high excited-state reduction potentials and further developed by Nicewicz10 and others,11 have evolved as the most robust and promising cationic OPCs in photoinduced reaction systems (Scheme 1c). Without a doubt, exquisite design is required to afford these architecturally diverse acridinium salt counterparts. Recently, major developments have been made by Sparr12 and other groups,13 which are mainly categorized into two aspects. The first type refers to the construction of an acridinium core through de novo synthesis and is the most mainstream tactic currently (Scheme 1d, left). Despite their great efficacy, the introduction of moisture-sensitive metal reagents consequentially raised the challenge of modularly preparing a broad spectrum of functionalized acridinium PCs. Alternatively, several groups successively disclosed very elegant methodologies through direct late-stage operation with acridinium PCs (Scheme 1d, right).13 While it is efficacious to forge a variety of well-functionalized ones, these issues necessitate the tedious pre-synthesis of the acridinium core. Overall, the existing avenues to access acridinium PCs remain fairly few and, to some extent, problematic.
From the viewpoint of retrosynthetic analysis, the hydroacridine scaffold as a raw material appears to be one of the most versatile choices to access acridinium PCs due to the superiority of the inherent high-functionalized aza-heterocycle units. Accordingly, apart from communicating this novel chemical reactivity with imines, in this paper, the subsequent procedure for preparing acridinium PCs by decorating generated products is also disclosed. Delightedly, modifying products could conveniently assemble an array of architecturally novel acridinium PCs (24 examples) that exhibit more outstanding photophysical properties (Scheme 1e).
Results and discussion
At the outset, the optimization of reaction conditions was executed by choosing N-(naphthalen-2-yl)-1-phenylmethanimine 1a (0.2 mmol) as a representative substrate. After systematically screening several underlying parameters, including commonly utilized Lewis acid catalysts, solvents, and reaction temperatures, the desired reaction conditions were ultimately identified: 20 mmol% loading of In(OTf)3 as the catalyst with environmentally friendly ethylene glycol as the solvent (0.3 M) at 120 °C for 12 h under argon atmosphere, wherein the structurally fascinating 14-phenyl-7,14-dihydrodibenzo[a,j]acridine 2a was obtained in an 87% isolated yield. As expected, the control experiment revealed that the Lewis acid played a vital role in facilitating this target transformation (details in ESI†).Having ascertained the optimized reaction conditions, we attempted to investigate the substrate scope of this method by treating a suite of substituted imine starting materials (1a–z, 1a′–i′). Overall, an assortment of intriguing structure-symmetrical C14-arylated dihydroacridine molecules (2a–z, 2a′–i′) were prepared smoothly under standard or slightly tuned reaction conditions. Gratifyingly, two subunits with imine reagents could be favorably decorated, as displayed in Scheme 2. With regard to the phenyl segment, the para-installed methyl (2b), tertiary butyl (2c), methoxy (2d), thiomethyl (2e), phenyl (2f), chloro (2g), fluoro (2h), trifluoromethyl (2i, i′), trifluorothiomethyl (2j), nitrile (2k), nitro (2l), and triazole (2m) groups, as well as the meta- or ortho-substituted bromo (2n, p) and iodine (2o) moieties, appeared to be quite suitable, delivering the corresponding adducts in 56–81% yields. Strikingly, the more daunting substrate with the potentially reactive 2-naphthyl fragment likewise proceeded to yield 2q in 56% yield. Heterocycles such as the thienyl (2r) and benzofuryl (2s) groups participated well in the target conversion, as expected. Remarkably, the serial successes of electronically discriminating bi-substituted systems (2t–z) indicated that the electron effects with aryl fragments were nearly unbiased. On the other hand, the napththyl parent was also viable and able to accommodate methoxy (2a′), bromo (2b′e′), methyl (2c′), cyclopropyl (2d′), phenyl (2f′), and thienyl (2g′) functionalities. The architecturally more fantastic anthryl unit (2h′) was amenable to the title protocol appropriately. As expected, simultaneously adjusting the aryl and naphthyl units also delivered target molecule 2i′ with satisfactory yield. Meanwhile, products 2a and 2e were characterized by X-ray crystallography. Unsatisfactorily, synthesis of the simple product 2a′′ with the phenyl unit was blocked because of its more stable aromaticity than that of naphthyl imines.
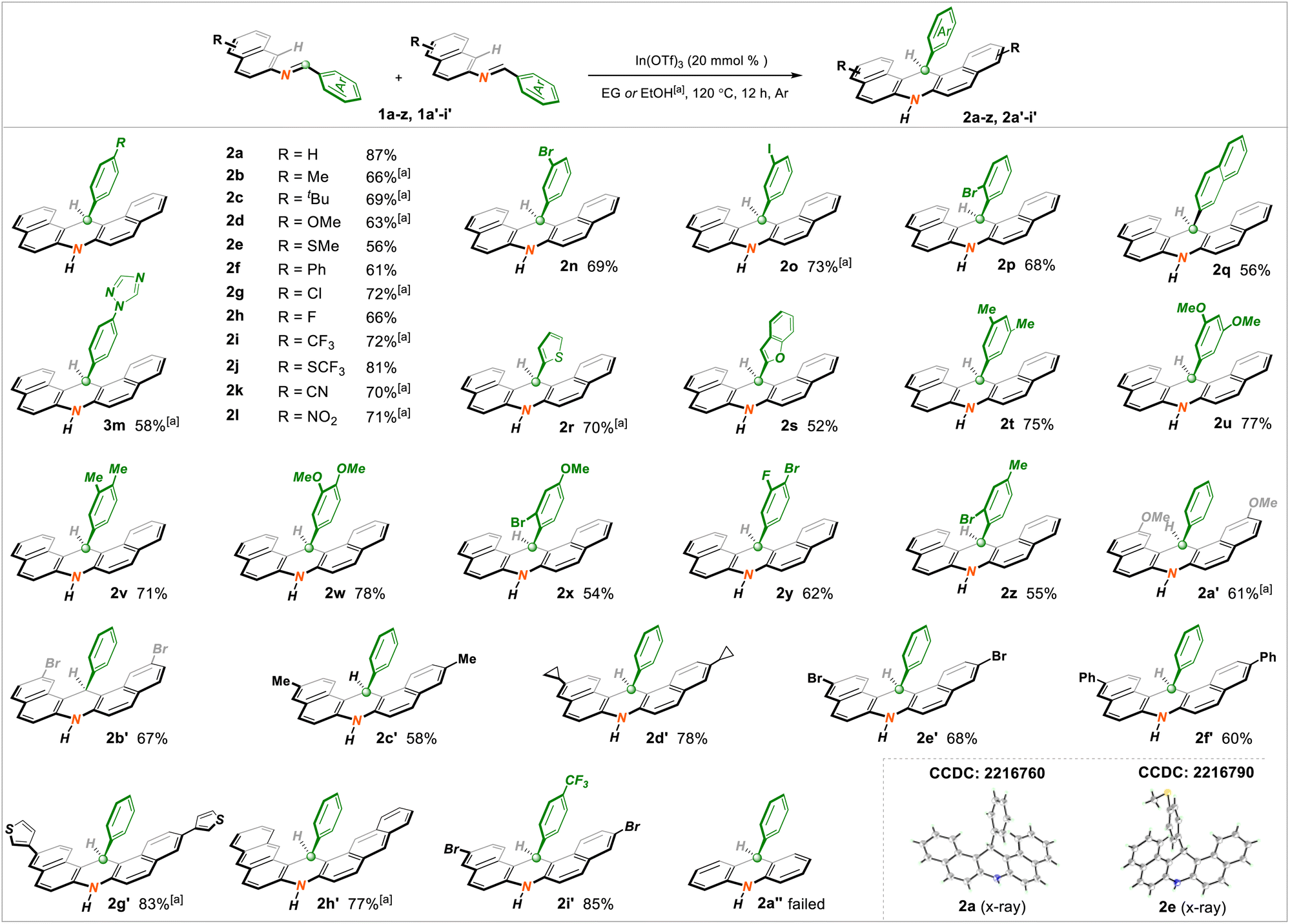 | ||
| Scheme 2 Substrate scope of dihydroacridines.a Ethyl alcohol was used as the solvent. | ||
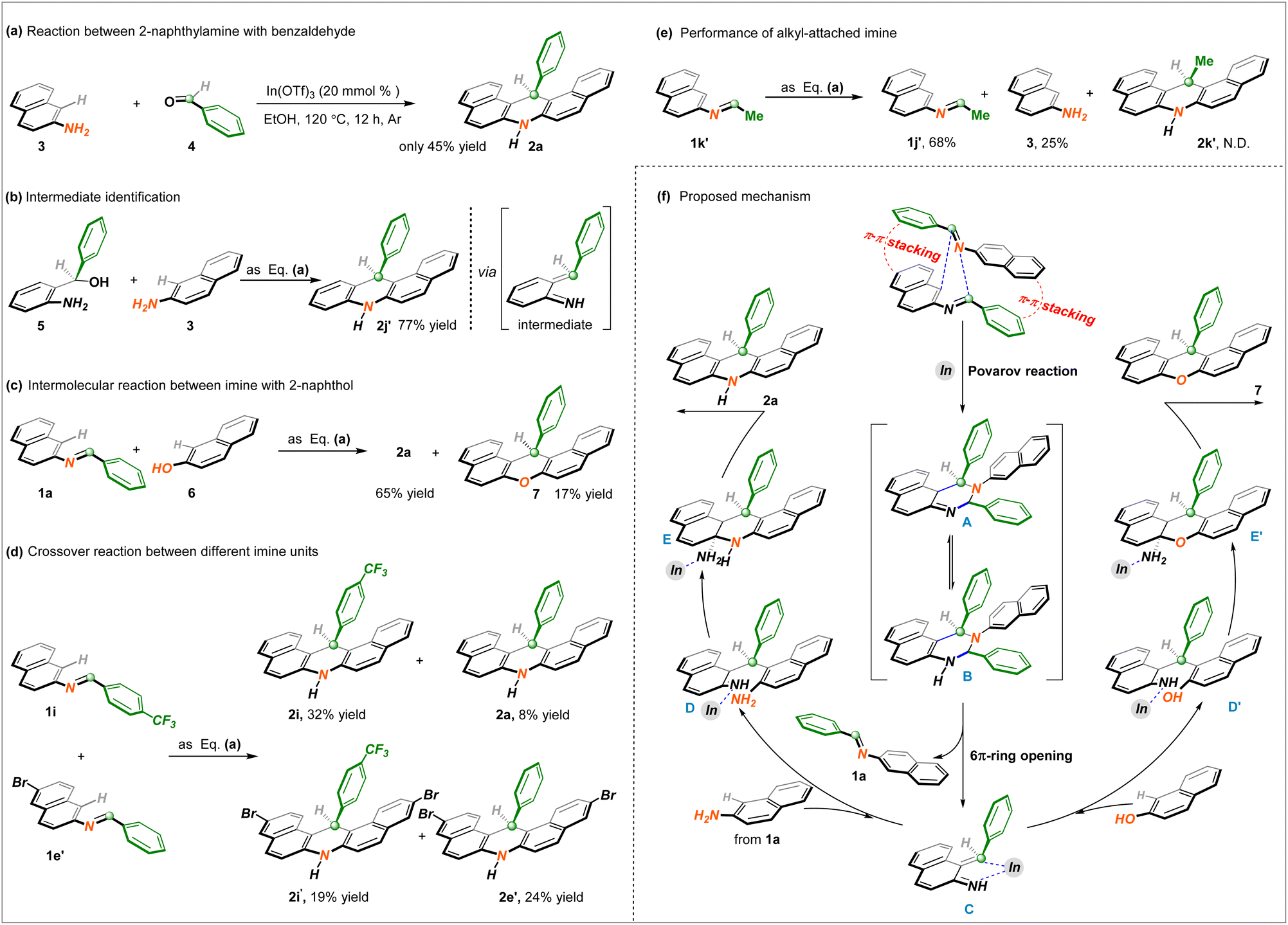
Considering this unprecedented reactivity of imine bond recombination, additional exploration experiments were implemented to illustrate the reaction mechanism of this finding. As described in Scheme 3a, under the identified reaction conditions, the intermolecular reaction between 2-naphthylamine and benzaldehyde remained feasible to generate the anticipated product 2a but significantly diminished, which indicated that the intramolecular interaction of the imine was favored. To probe the intermediacy of unsaturated imines, precursor 5 was employed in the corresponding conversion, and unsymmetrical dihydroacridine 2j′ was obtained in a 77% yield (Scheme 3b). More noteworthy, the involvement of 2-naphthol, representing another nucleophile, detected the formation of analogous 14-phenyl-14H-dibenzo[a,j]xanthene 7 in a yield of 17% (Scheme 3c). These above-outlined results unambiguously demonstrated that the high-activity α, β-unsaturated imine generated in situ likely played a vital role in the desired protocol. A crossover experiment with two distinguished imine units gave rise to four cross-counterparts (2a of 8%, 2i of 32%, 2i′ of 19%, and 2e′ of 24%) (Scheme 3d), which proved that the CN bond was subject to chemical reassociation. In addition, the incompatibility and non-interaction of the alkyl-attached imine 1k′ illustrated a crucial function with the aryl fragment to realize this envisioned process (Scheme 3e).
 | ||
| Scheme 3 Insight into the reaction mechanism. | ||
Based on these mechanistic observations and relevant literature about imine chemistry,14 a tentative reaction pathway for CN bond reorganization/cyclization is proposed. As illustrated in Scheme 3f, depending on the double π–π stacking interaction of two imine substrates supported by the outcome of Scheme 2e, an intermolecular [4 + 2] cycloaddition takes place to generate the C–H/N–H cyclic adduct A in the version of the Povarov reaction. Its imine–enamine tautomer B performs 6π-ring opening to deliver the key α, β-unsaturated imine C, which further undergoes a Michael addition with 2-naphthamine from imine hydrolysis with the assistance of In(OTf)3 Lewis acid and gives rise to the formation of complex D. Then, intramolecular condensation of species D provides polycycle-fused intermediate E, followed by an aromatization process to access the final dihydroacridine product 2a through C–N dissociation. At the stage of high-activity intermediate C, likewise, the assembly of 14-phenyl-14H-dibenzo[a,j]xanthene 7 is also clarified via a similar subsequent process.
As a logical but challenging extension, employing this new tactic as a key step to expeditiously construct the invaluable acridinium salt PCs widely used in radical reactions was attempted. After much design and endeavor, we were delighted that the sequential Buckwald coupling of dihydronacidine 2a with iodobenzene followed by one-pot DDQ oxidation and anion exchange procedures was found practicable, which provided a powerful synthetic platform to obtain acridinium salt 8a in 79% yield (Scheme 4). Foreseeably, the diversity of the PCs was able to significantly expand the chemical toolkit of photo–catalytic reactions; thus, we began examining the substrate scope and exploiting numerous acridinium PCs using this novel procedure. To our delight, almost all the compounds mentioned above were amenable to this run. This upgraded protocol tolerates a broad spectrum of electronically and sterically unbiased functional groups at both the phenyl ring and the acridine skeleton, giving rise to the formation of highly functionalized acridinium salt counterparts (8b–o) with decent yields. Furthermore, as a key fragment, it is recognized that the N-substituent of the acridinium salt significantly influences the redox potential and excited-state lifetime. In this context, we aimed to expand the library of acridinium catalysts by introducing a plethora of aromatic units at the nitrogen atom. Collectively, the N-substituent could be diversely modified by arenes bearing methyl (8p), chloro (8q), ester (8r), and nitro (8s) groups. Remarkably, upon introducing several more electron-rich (8t–u) or electron-poor (8v) bi-substituted units, and even larger conjugated systems (8w–x), this target conversion remained compatible. The reaction is currently incompatible with the corresponding N-alkyl photocatalysts.
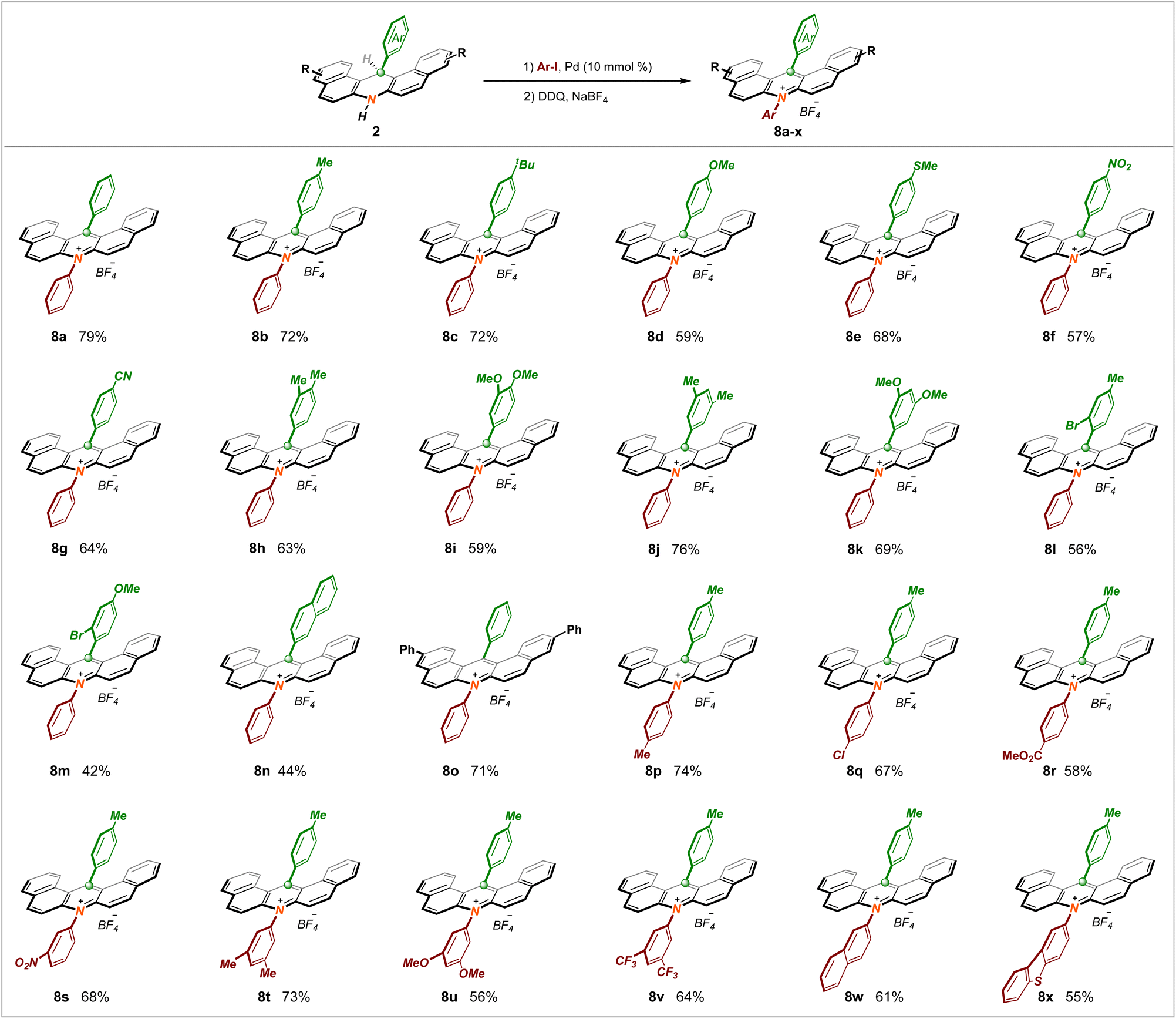 | ||
| Scheme 4 Substrate scope of acridinium PCs. | ||
By virtue of the pre-existing great application potency of acridinium PCs in modern organic reactions, we next examined the relevant photophysical and electrochemical properties of these structurally new ones (8a–x). As summarized in Table 1, a comparative wide range of excited state redox potentials were observed, from an E1/2(PC*/P−) of +1.28 V (8k) to an E1/2(PC*/PC−) of +2.07 V (8j), which lay in between the commercial transition-metal-based photocatalysts [Ir[dF-(CF3)ppy]2(dtbbpy)]PF6 [E1/2(PC*/PC−) = +1.21 V vs. SCE]15 and the Fukuzumi catalyst [MesMeAcr+·BF4−] [E1/2(PC*/PC−) = +2.06 V vs. SCE].16 In addition, in sharp contrast to the Fukuzumi acridinium catalyst, these PCs exhibited longer fluorescence lifetimes that were likely sufficient for photoredox catalysis. Collectively, these data implied that our synthesized serial acridinium salt PCs could theoretically facilitate more classes of photoinduced organic conversions via tuning the substituents of the acridinium core to match related reaction systems.
| PC | E 0.0 (eV) | E 1/2(PC/PC−)b (V vs. SCE) | E 1/2(PC*/PC−)c (V vs. SCE) | τ(1PC)d (ns) |
|---|---|---|---|---|
| a Determined at the intersection between normalized absorption and emission spectra, with E = 1240/λ. b Ground-state reduction potentials determined by cyclic voltammetry. c Excited-state reduction potential, estimated with ground-state reduction potentials and excited-state energies. d Time-correlated single-photon counting technique, λex = 340 nm. | ||||
| 8a | 2.68 | −0.79 | 1.89 | 3.98 |
| 8b | 2.68 | −0.78 | 1.90 | 3.98 |
| 8c | 2.70 | −0.79 | 1.91 | 4.10 |
| 8d | 2.53 | −0.85 | 1.68 | 8.09 |
| 8e | 2.34 | −0.81 | 1.53 | 4.84 |
| 8f | 2.64 | −0.86 | 1.78 | 4.25 |
| 8g | 2.65 | −0.79 | 1.86 | 4.36 |
| 8h | 2.63 | −0.77 | 1.86 | 3.84 |
| 8i | 2.44 | −0.81 | 1.63 | 5.40 |
| 8j | 2.85 | −0.78 | 2.07 | 4.45 |
| 8k | 2.06 | −0.78 | 1.28 | 4.39 |
| 8l | 2.66 | −0.80 | 1.86 | 4.05 |
| 8m | 2.52 | −0.84 | 1.68 | 6.73 |
| 8n | 2.38 | −0.84 | 1.54 | 4.42 |
| 8o | 2.58 | −0.80 | 1.78 | 7.14 |
| 8p | 2.72 | −0.81 | 1.91 | 4.20 |
| 8q | 2.66 | −0.77 | 1.89 | 3.88 |
| 8r | 2.40 | −0.81 | 1.59 | 3.93 |
| 8s | 2.36 | −0.80 | 1.56 | 3.86 |
| 8t | 2.66 | −0.79 | 1.87 | 4.22 |
| 8u | 2.70 | −0.82 | 1.88 | 4.15 |
| 8v | 2.36 | −0.82 | 1.54 | 4.17 |
| 8w | 2.69 | −0.79 | 1.90 | 4.06 |
| 8x | 2.70 | −0.78 | 1.92 | 4.00 |
Indeed, to our delight, these new-scaffold acridinium catalysts were capable of efficiently inducing several quite fantastic and environmentally benign organic conversions under a blue LED-irradiated system. For example, capitalizing on the acridinium catalyst 8c, the intramolecular dehydrogenation operation of 2-phenyl carboxylic acid 9 ran smoothly to deliver 6H-benzo[c]chromen-6-one 10 in a 64% yield, in which our synthesized catalyst gave tremendous catalytic potency compared to the commercially available acridinium catalyst (Scheme 5a). Additionally, catalyst 8c equally stimulated the more elusive intermolecular dehydrogenation coupling reaction between anisole and pyrazole, yielding biaryl 13 through C–N bond creation (Scheme 5b). By using this catalyst, the oxidation of methyl-benzyl alcohol 14 to acetophenone 15 was realized successfully (Scheme 5c). In a desire to value the practicality of our identified new methodology, a scale-up conversion with imine 1a was first carried out. The experimental outcomes showcased that the productivities of dihydronacridine 2a and subsequent acridine salt PC 8a all behaved without significant erosion (Scheme 5d). Finally, reacting imine 1b′ with naphthylamine could yield target molecule 1l′, followed by proceeding smoothly through the identified cascade reaction system to give unsymmetric acridinium photocatalyst 8y (Scheme 5e).
 | ||
| Scheme 5 Applicability studies and scale-up reaction. | ||
Conclusions
In conclusion, we have disclosed unprecedented imine chemistry associated with CN bond recombination catalyzed by In(OTf)3 Lewis acid, wherein an intriguing class of dihydroacridines with excellent functional group compatibility was synthesized. The obtained products can be exquisitely converted into a collection of exceedingly invaluable acridinium salt photocatalysts, which provides a robust and practical synthetic technique for commercially high-priced acridinium photocatalysts. Remarkably, the structurally new photocatalysts exhibit outstanding catalytic performance and will present many promising applications.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the corresponding ESI.†Author contributions
J. N. and Y. M.M. conceived the concept and designed the experiments. G. J. H., S. L. L. and J. W. conducted the chemical reactions described in the manuscript. J. N. and X. J. L. wrote the manuscript and all authors contributed to the reading and editing of the manuscript. G. J. H. compiled the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (21801159, 22171171), the Innovative Talent Promotion Plan-Young Science and Technology Star Project (2022KJXX-15), and the Young Talent Fund of the University Association for Science and Technology in Shaanxi, China (20200605).Notes and references
- (a) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed; (b) A. Lledó, A. P. Quintana and A. Roglans, Chem. Soc. Rev., 2016, 45, 2010 RSC; (c) Z. Yin, Y. He and P. Chiu, Chem. Soc. Rev., 2018, 47, 8881 RSC; (d) T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121, 6850 CrossRef CAS PubMed; (e) R. S. Doerksen, T. Hodík, G. Hu, N. O. Huynh, W. G. Shuler and M. J. Krische, Chem. Rev., 2021, 121, 4045 CrossRef CAS PubMed; (f) M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, Nature, 2020, 586, 64 CrossRef CAS PubMed; (g) J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown and F. Glorius, Science, 2021, 371, 1338 CrossRef CAS PubMed.
- (a) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359 CrossRef CAS PubMed; (b) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983 RSC; (c) B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055 RSC; (d) A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. D. Perez and M. L. Coote, Nature, 2016, 531, 88 CrossRef PubMed; (e) T. Gatzenmeier, M. V. Gemmeren, Y. Xie, D. höfler, M. Leutzsch and B. List, Science, 2016, 351, 949 CrossRef CAS PubMed.
- For selected examples, see: (a) V. V. Kouznetsov, Tetrahedron Lett., 2009, 65, 2721 CrossRef CAS; (b) I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057 CrossRef CAS PubMed; (c) H. Liu, G. Dagousset, G. Masson, P. Retailleau and J. Zhu, J. Am. Chem. Soc., 2009, 131, 4598 CrossRef CAS PubMed; (d) G. Dagousset, J. Zhu and G. Masson, J. Am. Chem. Soc., 2011, 133, 14804 CrossRef CAS PubMed; (e) H. Xu, H. Zhang and E. N. Jacobsen, Nat. Protoc., 2014, 9, 1860 CrossRef CAS PubMed; (f) J. Yu, H. Jiang, Y. Zhou, S. Luo and L. Gong, Angew. Chem., Int. Ed., 2015, 54, 11209 CrossRef CAS PubMed; (g) C. Min, C. Lin and D. Seidel, Angew. Chem., Int. Ed., 2015, 54, 6608 CrossRef CAS PubMed; (h) S. K. Thompson and T. R. Hoye, J. Am. Chem. Soc., 2019, 141, 19575 CrossRef CAS PubMed; (i) C. Chen and R. Liu, Angew. Chem., Int. Ed., 2019, 58, 9831 CrossRef CAS PubMed; (j) X. Ren, B. Bai, Q. Zhang, Q. Hao, Y. Guo, L. Wan and D. Wang, J. Am. Chem. Soc., 2022, 144, 2488 CrossRef CAS PubMed.
- For selected examples, see: (a) G. C. Fu, Acc. Chem. Res., 2006, 39, 853 CrossRef CAS PubMed; (b) C. A. Carsona and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (c) R. Suter, Z. Benkő, M. Bispinghoff and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 11226 CrossRef CAS PubMed; (d) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 36, 354 RSC.
- (a) Z. Zuo, J. Liu, J. Nan, L. Fan, W. Sun, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 15385 CrossRef CAS PubMed; (b) Yi. Ge, C. Qin, L. Bai, J. Hao, J. Liu and X. Luan, Angew. Chem., Int. Ed., 2020, 59, 18985 CrossRef CAS PubMed; (c) Y. Hu, J. Nan, J. Yin, G. Huang, X. Ren and Y. Ma, Org. Lett., 2021, 23, 8527 CrossRef CAS PubMed; (d) P. Chen, J. Nan, Y. Hu, Y. Kang, B. Wang, Y. Ma and M. Szostak, Chem. Sci., 2021, 12, 803 RSC; (e) L. Bai, X. Luo, Y. Ge, H. Wang, J. Liu, Y. Wang and X. Luan, CCS Chem., 2022, 4, 1054 CrossRef CAS.
- For selected examples, see: (a) C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 13544 CrossRef CAS PubMed; (b) H. Xu, H. Zhang and E. N. Jacobsen, Nat. Protoc., 2014, 9, 1860 CrossRef CAS PubMed; (c) M. K. Škopić, K. Götte, C. Gramse, M. Dieter, S. Pospich, S. Raunser, R. Weberskirch and A. Brunschweiger, J. Am. Chem. Soc., 2019, 141, 10546 CrossRef PubMed.
- For selected examples, see: (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997 CrossRef CAS PubMed; (c) J. Xuan, X. He and W. Xiao, Chem. Soc. Rev., 2020, 49, 2546 RSC; (d) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526 CrossRef CAS PubMed; (e) A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082 CrossRef PubMed; (f) D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) X. Yu, J. Chen and W. Xiao, Chem. Rev., 2021, 121, 506 CrossRef CAS PubMed; (c) L. Liao, G. Cao, J. Ye, G. Sun, W. Zhou, Y. Gui, S. Yan, G. Shen and D. Yu, J. Am. Chem. Soc., 2018, 140, 17338 CrossRef CAS PubMed; (d) G. Cao, X. Hu, L. Liao, S. Yan, L. Song, J. J. Chruma, L. Gong and D. Yu, Nat. Commun., 2021, 12, 3306 CrossRef CAS PubMed; (e) J. R. Dorsheimer, M. A. Ashley and T. Rovis, J. Am. Chem. Soc., 2021, 143, 19294 CrossRef CAS PubMed; (f) S. K. Kariofillis, S. Jiang, A. M. Żurański, S. S. Gandhi, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 1045 CrossRef CAS PubMed; (g) H. Huang, P. Bellotti, J. E. Erchinger, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2022, 144, 1899 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- (a) N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326 CrossRef CAS PubMed; (b) K. A. Margrey, A. Levens and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 15644 CrossRef CAS PubMed; (c) V. A. Pistritto, M. E. S. Horton and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 17187 CrossRef CAS PubMed; (d) N. J. Venditto, Y. S. Liang, R. K. E. Mokadem and D. A. Nicewicz, J. Am. Chem. Soc., 2022, 144, 11888 CrossRef CAS PubMed.
- (a) J. Xuan, X. Xia, T. Zeng, Z. Feng, J. Chen, L. Lu and W. Xiao, Angew. Chem., Int. Ed., 2014, 53, 5653 CrossRef CAS PubMed; (b) Q. Zhou, Y. Zou, L. Lu and W. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed; (c) F. Tan, X. He, W. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS PubMed; (d) Z. Wu and D. A. Pratt, Angew. Chem., Int. Ed., 2021, 60, 15598 CrossRef CAS PubMed; (e) E. L. Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 5357 CrossRef PubMed; (f) Y. Du, B. Chen and W. Shu, Angew. Chem., Int. Ed., 2021, 60, 9875 CrossRef CAS PubMed; (g) A. Selmani, M. D. Schoetz, A. E. Queen and F. Schoenebeck, ACS Catal., 2022, 12, 4833 CrossRef CAS; (h) Q. Xu, L. Wei and B. Xiao, Angew. Chem., Int. Ed., 2022, 61, e202115592 CAS; (i) X. Wang, W. Tong, B. Huang, S. Cao, Y. Li, J. Jiao, H. Huang, Q. Yi, S. Qu and X. Wang, J. Am. Chem. Soc., 2022, 144, 4952 CrossRef CAS PubMed.
- (a) C. Fischer and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 2436 CrossRef CAS PubMed; (b) C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2020, 10, 210 CrossRef CAS; (c) B. Zilate, C. Fischer and C. Sparr, Chem. Commun., 2020, 56, 1767 RSC; (d) V. Hutskalova and C. Sparr, Org. Lett., 2021, 23, 5143 CrossRef CAS PubMed; (e) X. Wu and C. Sparr, Angew. Chem., Int. Ed., 2022, 61, e202201424 CAS.
- (a) H. Yan, J. Song, S. Zhu and H. Xu, CCS Chem., 2021, 3, 317 CrossRef CAS; (b) H. Yan, S. Zhu and H. Xu, Org. Process Res. Dev., 2021, 25, 2608 CrossRef CAS; (c) Y. Cao, G. Zhu, Y. Li, N. L. BretonNolwenn, C. Gourlaouen, S. Choua, J. Boixel, H. J. D. Rouville and J. Soulé, J. Am. Chem. Soc., 2022, 144, 5902 CrossRef CAS PubMed.
- (a) J. Esquivias, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2007, 129, 1480 CrossRef CAS PubMed; (b) V. A. Osyanin, S. A. Pavlov, D. V. Osipov and Y. N. Klimochkin, Chem. Heterocycl. Compd., 2014, 50, 1199 CrossRef CAS; (c) S. Pang, X. Yang, Z. Cao, Y. Zhang, Y. Zhao and Y. Huang, ACS Catal., 2018, 8, 5193 CrossRef CAS; (d) Y. Q. Zhang, Y. P. Zhang, Y. Zheng, Z. Li and L. Ye, Cell Rep. Phys. Sci., 2021, 2, 100448 CrossRef CAS; (e) A. Ngamnithiporn, P. Chuentragool, P. Ploypradith and S. Ruchirawat, Org. Lett., 2022, 24, 4192 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2016, 138, 1724 CrossRef CAS PubMed.
- A. J. Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. Campeau, D. A. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216760 and 2216790. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00163f |
| This journal is © The Royal Society of Chemistry 2023 |