
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn abiotic, tetrameric, eight-helix bundle†
Friedericke S.
Menke
a,
Barbara
Wicher
 b,
Lars
Allmendinger
a,
Victor
Maurizot
c and
Ivan
Huc
*a
b,
Lars
Allmendinger
a,
Victor
Maurizot
c and
Ivan
Huc
*a
aDepartment of Pharmacy, Ludwig-Maximilians-University, Butenandstraße 5-13, 81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
bDepartment of Chemical Technology of Drugs, Poznan University of Medical Sciences, Grunwaldzka 6, 60-780 Poznan, Poland
cCBMN (UMR 5248), Univ. Bordeaux, CNRS, Bordeaux INP, 2 Rue Robert Escarpit, 33600 Pessac, France
First published on 28th February 2023
Abstract
Four helically folded aromatic oligoamide sequences containing either a chiral monomer based on 2-(2-aminophenoxy)-propionic acid, an N-terminal (1H)-camphanyl group, or both, were synthesized. Spectroscopic solution investigations using 1H NMR and circular dichroism (CD) demonstrated that the 2-(2-aminophenoxy)-propionic acid unit biases helix handedness quantitatively in chloroform and dichloromethane. It even quantitatively overcomes an opposing effect of the camphanyl group and thus ensures reliable helix handedness control. A series of nine sequences composed of two helically folded aromatic oligoamide segments separated by a flexible linker based on a di-, tri- or tetraethylene glycol unit were then synthesized. In these sequences, helix handedness was controlled by means of an N-terminal (1H)-camphanyl group or a 2-(2-aminophenoxy)-propionic acid units in either both helical segments, or only in the N-terminal segment, or in none of the segments. The helical segments all displayed hydroxy and carbonyl groups at their surfaces as hydrogen bond donors and acceptors so as to promote helix-to-helix hydrogen bonding. NMR and CD spectroscopic studies showed that, in some cases, well-defined, stable, discrete abiotic helix-turn-helix tertiary folds form in organic solvents. Molecular modelling suggests that these correspond to structures in which the two helix axes are at an angle. In one case, the absence of handedness control resulted in a complex and large aggregate. A solid state structure obtained by single crystal X-ray diffraction analysis revealed a tetrameric assembly composed of eight helices with both right and left handedness arranged in three subdomains consisting of two hydrogen-bonded three-helix bundles and one two-helix-bundle. Several helix-to-helix hydrogen bonds were mediated by bridging water molecules. This structure constitutes an important milestone in the construction of abiotic protein-like architectures.
Introduction
We have engaged in a program aimed at eliciting protein-like folding in organic solvents using abiotic structures, that is, at the exclusion of α-amino acids or analogous aliphatic building blocks. This program is motivated by the prediction that sophisticated functions may emerge in abiotic tertiary or quaternary structures, just like this level of complexity is required for most protein functions. It is also expected that protein-like behaviour in aprotic organic solvents may differ from that of proteins in water because solvation and desolvation phenomena would be completely different. The field is essentially uncharted and caught our curiosity.Foldamer research has produced numerous synthetic backbones that have the ability to fold into helices.1–7 Furthermore, helix–helix interactions are among the best-understood protein structural patterns. Coiled–coiled peptide helix bundles are amenable to design8–17 and have been extended to peptidomimetic structures that do not exist in nature, including 310 α-peptide helices,18 α/β-peptide helices,19 β-peptide helices,20,21 and oligourea helices.22,23 It was thus a logical step to consider abiotic tertiary folds using helices as a starting point. Oligoamides of 8-amino-2-quinoline carboxylic acid (Q in Fig. 1a) constitute an attractive class of abiotic foldamers that adopt stable 2.5 helical conformations.24–26 Structural stability is illustrated, for example, by the persistence of an octaamide helix at 120 °C in DMSO.24 Extreme stability is not necessarily desirable when designing a protein-like object. In the case of Qn oligomers, it can be mitigated by replacing some Q units with more flexible P units that possess additional rotatable bonds and have a smaller surface for aromatic stacking while coding for the same helix curvature (Fig. 1a).27–29 Thus, tertiary fold design was initially considered using P- and Q-containing oligomers as predictable construction modules. Preliminary work essentially involved connecting helices with short linkers.30–32 This first generation did not qualify as true tertiary folds in that no defined helix–helix interactions occurred. Next, we considered functionalizing Q and P monomers with 4-hydroxy substituents as hydrogen bond donors and produced analogous monomers X and Y (Fig. 1). Acidic phenols are excellent hydrogen bond donors33 and amide carbonyl groups diverge from Qn helices, providing good hydrogen bond acceptors. We thus planned to mediate helix-to-helix hydrogen bonding in organic solvents using these donors and acceptors. During synthesis, hydroxy groups are protected. Protected monomers are referred to as ![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) and
and ![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) .
.
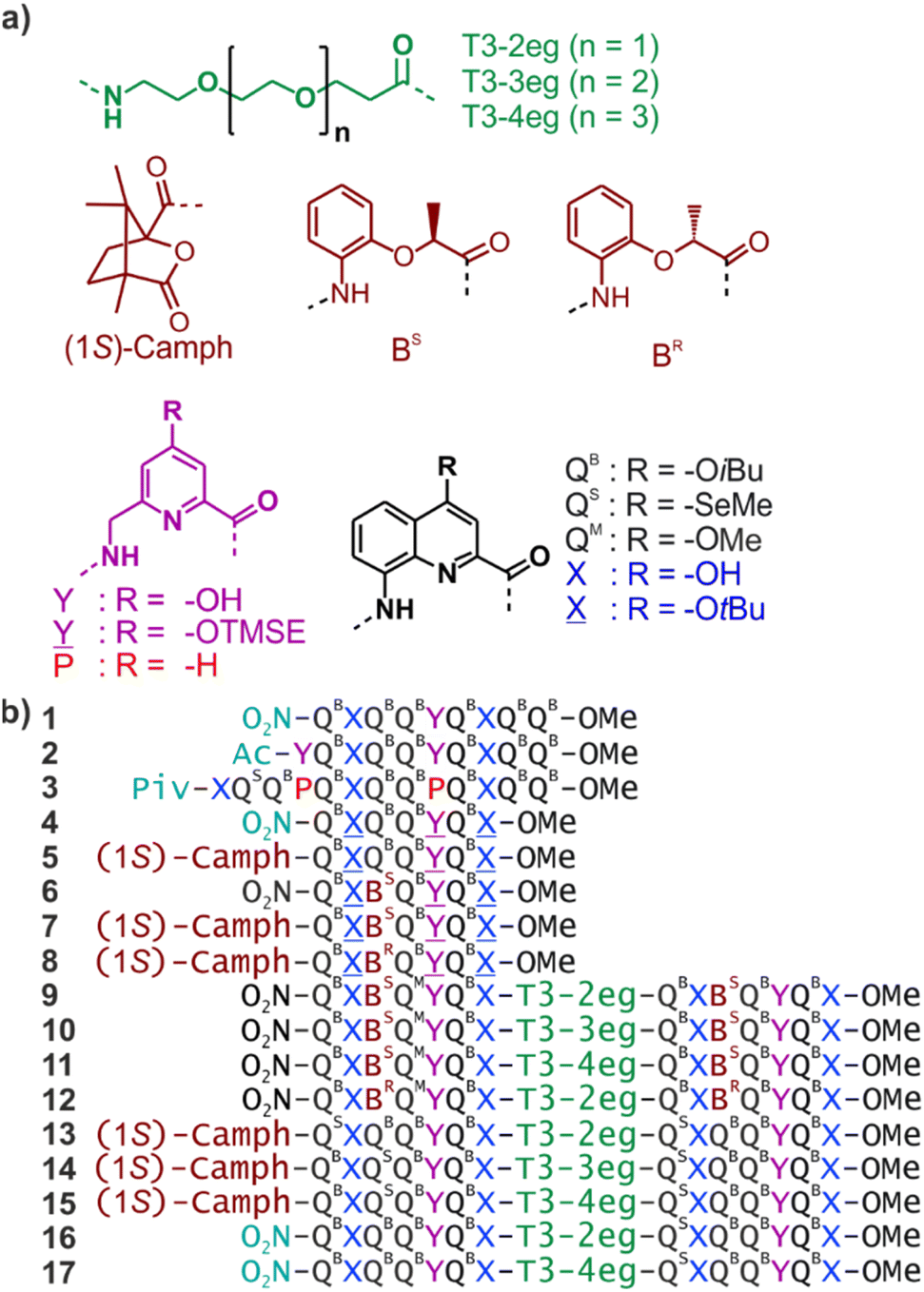 | ||
| Fig. 1 (a) Structures of di-, tri- and tetraethylene glycol T3 spacers, chiral (1S)-camph, BS and BR units, and Q, X, P and Y amino acid monomers. QB carries organic solubilizing side chains. QS was introduced in some sequences to assist crystallographic structure elucidation using the anomalous scattering of Se, though it turned out to be unneeded. QM is isosteric to QS. and are the protected precursors of hydrogen-bonding monomers X and Y, respectively. TMSE = 2-trimethylsilylethyl. (b) Oligoamide foldamer sequences. Piv = pivaloyl. In sequences ending with an 8-nitro group, this group replaces the terminal amine. | ||
Progress towards abiotic tertiary folds has been made by a mix of successful designs and serendipitous discoveries. For example, we rationally designed the first abiotic helix-turn-helix motifs based on careful modelling of the structures.34 After choosing relatively rigid turn units of appropriate length as linkers, hydroxy groups were introduced at precise locations so as to mediate helix-to-helix hydrogen bonding between X units, and between Y units (Fig. 2a, b and S1†). Depending on the type of turn unit, a parallel‡ head-to-head homochiral arrangement (i.e. with both helices having the same handedness) or a parallel‡ head-to-tail heterochiral arrangement (i.e. with one P helix and one M helix) could be rationally produced (Fig. 3a, b and S1†).34–37 In contrast, the self-assembly of the same or similar sequences not linked by a turn unit was full of surprises. Thus, sequence 2 (Fig. 1) forms not a dimeric but a trimeric parallel‡ head-to-head homochiral bundle involving similar hydrogen bonding to that of the helix-turn-helix structures, as well as other species (Fig. 2a).34 Sequence 1 forms (again, among other species) a homochiral tilted dimer where the two helix axes are not parallel but instead tilted at an angle of 120° (Fig. 2a, c, 3 and S1†).34 This is made possible by the fact that a stack of two X units and one Y at the surface of a helix creates an array of three hydrogen bond donors and three acceptors arranged in a sort of hexagon: donors and acceptors may meet in three different configurations by rotating the array by ± 120°, potentially generating parallel dimers, one clockwise and one counterclockwise tilted dimer (Fig. 3 and S1†). Furthermore, removing some of the hydroxy groups through Y → P mutations, as in 3, resulted in the formation of unexpected, shifted parallel‡ dimers (Fig. 2a).38 The term shifted was used because, in these structures, X units did not face X units of the other helices but were instead shifted by one helix turn and hydrogen-bonded to a P unit. Both PM heterochiral or PP/MM homochiral shifted dimers have been observed. These aggregates are similar and are stabilized by one type of helix-to-helix hydrogen bond (Fig. 2d and S1†). Eventually, we found that the arrangement of rationally designed helix-turn-helix structures never forms in the absence of a rigid turn unit, indicating that the rigid turn mediates some sort of strain while simultaneously preventing other helix–helix interaction patterns.37
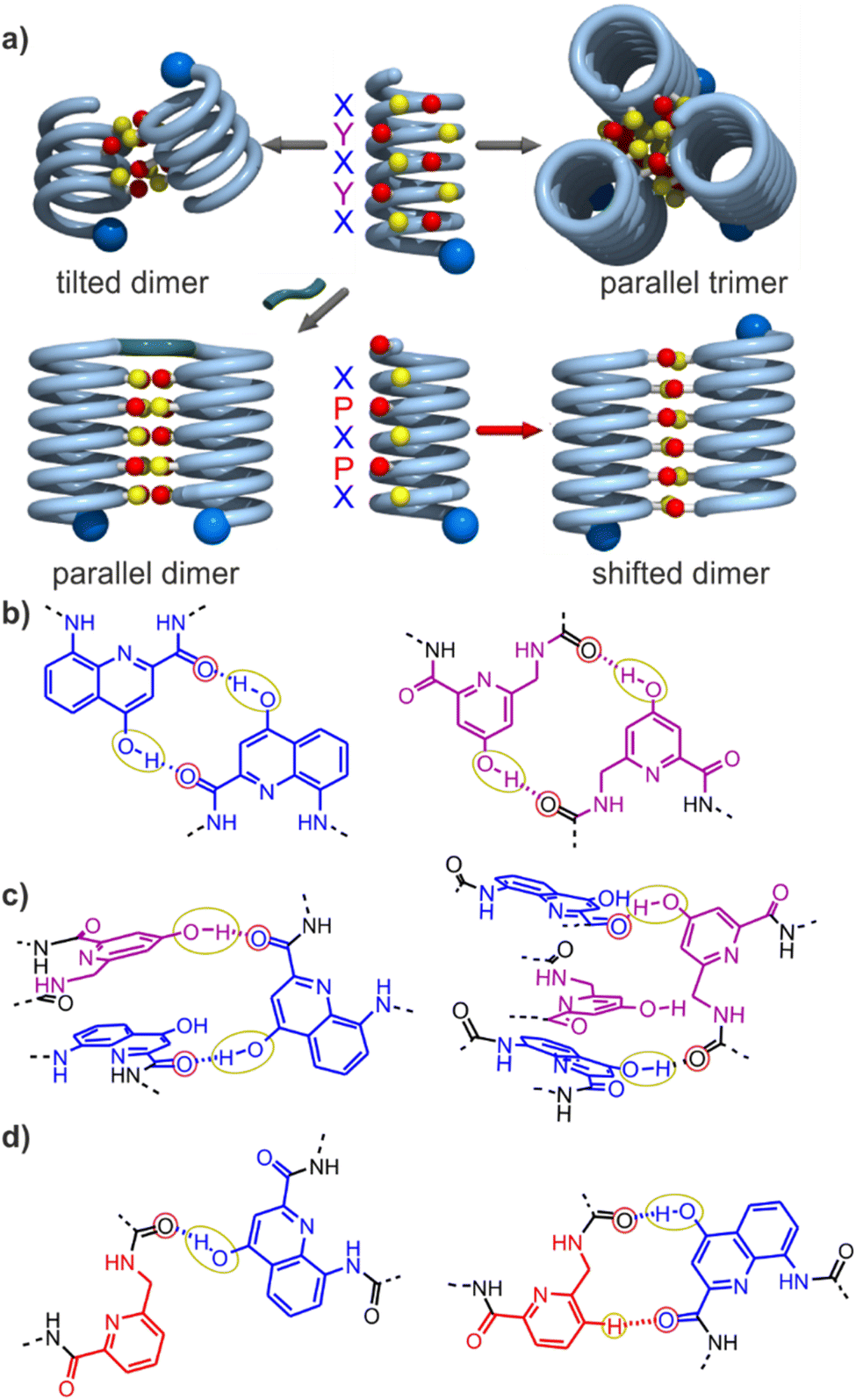 | ||
| Fig. 2 (a) Cartoons representing identified assembly modes of aromatic helices displaying hydrogen bond acceptors and donors shown as red and yellow balls, respectively. The bottom left structure exists only when the helices are covalently linked. (b) Hydrogen-bonding patterns involving X and Y units as observed in helix-turn-helix tertiary structures in which sequences 1 or 2 are connected by a rigid linker.34–36 (c) Hydrogen-bonding patterns involving X and Y units as observed in a tilted dimer of 1.34 (d) Hydrogen-bonding patterns involving X and P units as observed in a PM (left) and in PP or MM (right) shifted dimers of 3.38 In (a–d), yellow and red circles around the hydrogen bond donors and acceptors correspond to the yellow cups and red knobs in (a) and Fig. 3, and to the yellow and red balls in Fig. 7. | ||
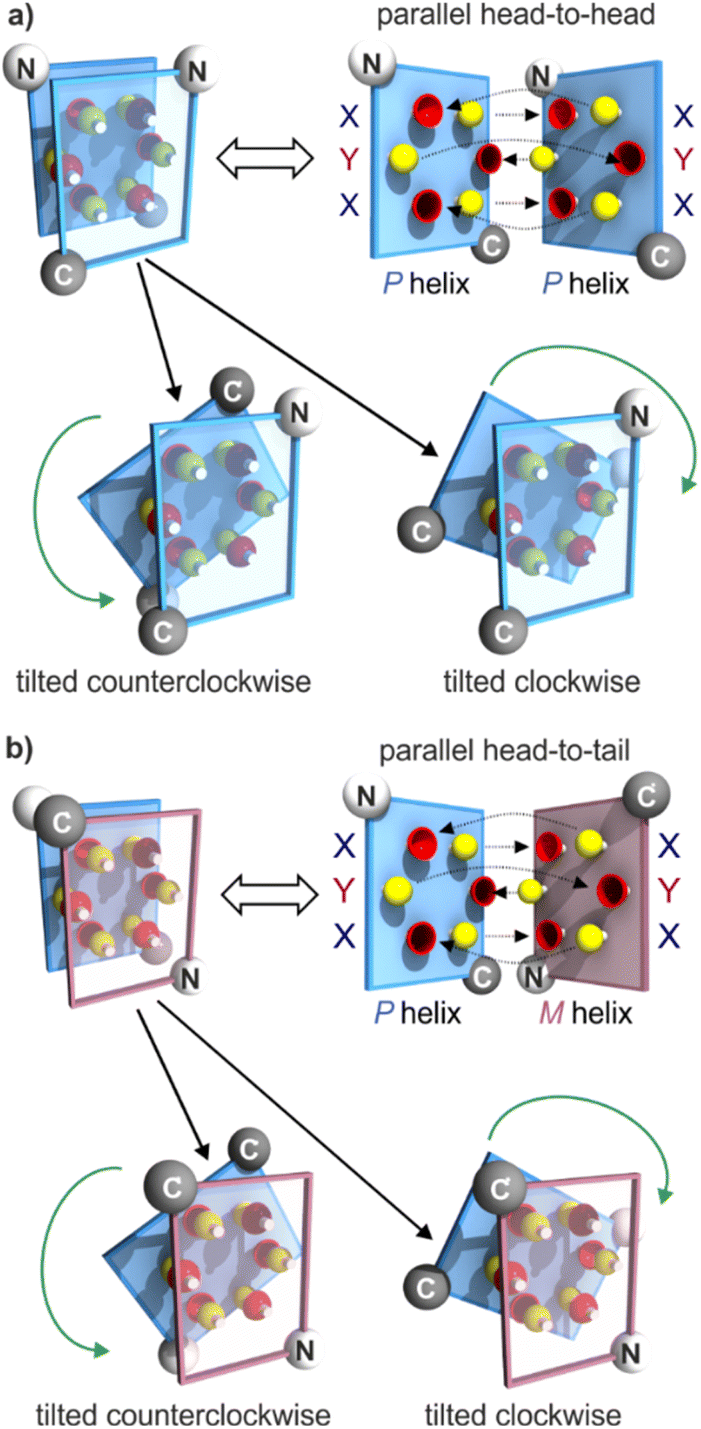 | ||
| Fig. 3 Schematic representations of helix–helix hydrogen bonding when three hydrogen bond donors (yellow knobs) and three acceptors (red cups) are arranged in a sort of hexagon at the surface of a helix, as in 1.34 The hexagonal arrangement stems from two X units flanking a Y unit at the surface of a helix, with the hydrogen bond donor and acceptor being closer in X than in Y units (Fig. 2b). The helix face is represented as a plane, with the N- and C-termini shown as white and grey balls, respectively. Planes with a blue and red frame correspond to P and M helices, respectively. (a) “Open-book view” and stacked view of a hydrogen-bonded parallel‡ head-to-head arrangement of two P helices and of the related clockwise and counterclockwise dimers. Hydrogen bonds are shown as dashed lines in the open-book view. In the stacked view, the top plane is transparent so that one can see the six hydrogen bonds (knobs into cups) and the plane behind. The parallel head-to-head arrangement has been observed only when the two helices are linked by a rigid turn unit (Fig. 2a and S1†).34,35 The PP clockwise tilted dimer of 1 has been characterized in the solid state.34 (b) Similar parallel arrangement, but this time head-to-tail, between a P and an M helix, as well as the clockwise and counterclockwise tilted dimers. The parallel arrangement has also been seen only when the helices are connected by a rigid linker.36 There is no evidence for the existence of PM tilted dimers, but models show plausible structures (see the ESI†). | ||
We now report the folding and assembly of helix-turn-helix structures in which the turn unit possesses considerable flexibility. The initial intention was to identify links that would stabilize the tilted dimers in an intramolecular tertiary fold. While this objective was met with good confidence (albeit without crystallographic evidence), we also discovered one case where four helix-linker-helix modules assemble into a 12.9 kDa quaternary-like object composed of eight helices. The resulting eight-helix bundle involves some already identified helix–helix arrays of hydrogen bonds, such as a sort of parallel trimer and a shifted dimer. It also revealed new patterns involving multiple water molecule bridges. Altogether, these results represent an important milestone on the way to increasingly large and complex abiotic tertiary and quaternary folds.
Results and discussion
Absolute helix handedness control
New heptaamide 4 was derived from sequence 1 and used as the elementary helical building block in this study. It contains protected and units at positions suitable to form, after deprotection, the hydrogen-bonded parallel or tilted dimers mentioned in the introduction. With respect to 1, sequence 4 has been shortened by two QB units at the C-terminus and should thus span a little less than three helix turns. It does not contain any stereogenic center and must, therefore, fold as a racemic mixture of right-handed (P) or left-handed (M) helices. In order to explore the possible occurrence of well-defined intramolecular helix-to-helix hydrogen bonding patterns (as opposed to dimeric assemblies), we considered sequences that would comprise two segments analogous to the sequence of 4 linked by a flexible T3 linker containing two, three or four ethylene glycol units (see next section for design considerations). In such sequences, each helical segment can, in principle, be P or M. Therefore, PP/MM and PM/MP diastereomeric conformers may coexist.31,39 After deprotection of and , we anticipated that different types of helix-to-helix hydrogen bonding may occur within each of the PP/MM and PM/MP conformers (not taking into account possible intermolecular hydrogen bonding) and that the overall behaviour may be difficult to analyse.
To reduce, at least at the start, the number of conformational degrees of freedom in these molecules, we decided to introduce an absolute handedness control of the helical segments. We have shown before that a (1S)-camphanyl group (Fig. 1a) at the N-terminus biases helix handedness quantitatively (as far as NMR can detect) in favour of P helicity.40 This bias is effective in any solvent that we tested from water to chlorinated solvents. We have also shown that a BS unit inserted within a Qn oligomer biases handedness quantitatively (as far as NMR can detect) in favour of P helicity.41 Conversely, BR favours M helicity. However, the effect of BR or BS units had only been demonstrated in water, methanol and DMSO using Qn oligomers bearing polar side chains. It was unknown whether handedness bias would also be effective in chloroform. We thus prepared sequences 5–8 that contain a (1S)-camphanyl group, a chiral B unit, or both, while all having and units in the same positions as 1 (see the ESI† for details). Note that the more flexible B unit was introduced between two Q units and not next to the also flexible Y because consecutive flexible units may result in a loss of helix stability.35
The 1H NMR spectra of all these compounds in CDCl3 and CD2Cl2 show one single set of signals (Fig. S2 and S3†). For such long sequences, helix handedness reversal is slow on the NMR timescale, as shown, for example, by anisochronous CH2 protons of iBu side chains.29 The P and M diastereomeric helices of chiral compounds 5–8 would thus appear as distinct sets of signals, should they both exist in significant amounts. The presence of a single set of signals is, therefore, indicative of the prevalence of a single species. This was expected for 5, with its terminal camphanyl group. The behaviour of 6 shows that a single chiral BS unit is also effective in biasing handedness quantitatively in chlorinated solvents. In 7, the (1S)-camphanyl and BS groups favour the same handedness. That one species prevails is, therefore, unsurprising. In contrast, the prevalence of a single diastereomeric helix in 8, in which the (1S)-camphanyl and BR groups compete to favour one handedness or the other, indicates that the effect of one of the two groups completely overtakes the effect of the other. CD spectra of 5–8 show bands of equal intensities consistent with quantitative handedness bias (Fig. S4†).§ The sign of the bands allows for the absolute assignment of helix handedness40 and indicates that 5–7 are P helical whereas 8 is M helical. The latter result demonstrates that, in 8, the effect of the BR unit overtakes the effect of the (1S)-camphanyl group, thus highlighting a particularly strong handedness bias effect.
Linker design
The initial purpose of this work was to explore which linker could eventually promote an intramolecular helix-turn-helix structure with the two helices hydrogen-bonded in a tilted arrangement. The art of installing a linker between two entities without perturbing the interactions in which each of them is involved is a delicate one, whether it is between helices42–45 or between smaller molecules bound to proteins, as in PROTACs.46 Actually, when designing the first abiotic helix-turn-helix structures,34–37 we did not adjust the linker according to the interactions but instead implemented interactions, that is, introduced X and Y units, after having chosen the linker. Within tilted dimers, the helix N- and C-termini are not located at positions obvious to connect by a linker (Fig. 3). We thus decided to test a set of rather flexible loops possessing two, three or four ethylene glycol units, namely T3-2eg, T3-3eg and T3-4eg (Fig. 1), that have the advantage of being commercially available as Fmoc-protected amino acids ready for solid phase synthesis.We first systematically evaluated molecular models of sequences composed of two heptameric segments analogous to that of 4 (in its deprotected form with free OH groups). For each linker, we considered parallel,‡ clockwise tilted, counterclockwise tilted, and shifted dimers, both when the helices have the same handedness (e.g. PP) or when they have opposite handedness. As explained below, few of these configurations were predicted to be realistic. In a first step, an energy-minimized model of two hydrogen-bonded helices in the desired configuration was produced using Maestro (see the ESI† for details).47 Starting from one helix, the linker was then built in a plausible conformation, a step which ended with the creation of a single bond between the end of the linker and the other helix. In many cases, an energy minimization at this stage ended in the disruption of a helix structure or of the array of hydrogen bonds. In the latter case, energy minimization was repeated upon imposing hydrogen bonding through distance constraints. Typically, the consequence was a perturbation of the helical structure indicating the inadequacy of the linker for that particular arrangement. Upon repeating energy minimization after removing the constraints, the perturbation persisted. Only in a few cases, the array of hydrogen bonds appeared to be compatible with unaltered helix structures and a reasonable turn conformation.
The results can be summarized as follows: (i) all shifted arrangements can be ruled out because they leave four out of six hydroxy groups not involved in hydrogen bonds (Fig. S5 and S6†); (ii) parallel PP arrangements were not considered since they must be head-to-head34,36 placing the C-terminus of one helix too far from the N-terminus of the other helix (Fig. 3a); (iii) parallel PM arrangements are head-to-tail36 and in principle compatible with the T3 linker lengths with no or minute helix distortions (Fig. 3b and S7†). However, the fact that they do not occur in the absence of a linker makes them unlikely to happen with a flexible linker that does not impose a particular geometry.34,38 Parallel PM arrangements were thus also dismissed; (iv) all linkers are too short for PM clockwise tilted arrangements (Fig. S8†); (iv) the two shorter linkers but not the longer T3-4eg perturb the arrangement of PP counterclockwise tilted arrangements (Fig. S9†); (v) all linkers seem compatible with PM counterclockwise tilted arrangements albeit in extended conformations unusual for oligoethylene glycols which normally prefer gauche conformations (Fig. S10†);48 (vi) all linkers seem compatible with the PP clockwise tilted arrangements (Fig. S11†). Therefore, at best, two hydrogen-bonded helix–helix arrangements are compatible with the two shorter linkers (PP clockwise and PM counterclockwise tilted arrangements) and three with the longest linker (PP counterclockwise tilted arrangement on top of the former two). As a representative example, a model of the PP clockwise tilted arrangement with a T3-2eg linker is shown in Fig. 4.
 | ||
| Fig. 4 Energy minimized model of a PP clockwise tilted helix-T3-2eg-helix arrangement. The sequence is analogous to that of 16 using Q units without side chains. (a) Space-filling representation with T3-2eg shown in green and the helices shown in blue. Amide carbonyl oxygen and hydroxy oxygen atoms involved in hydrogen bonding are shown in red. Hydroxy protons are shown in white. (b) Same view as in (a) showing only the outer rim of the backbone in tube representation. Hydrogen bond acceptors (carbonyl oxygen atoms) and donors (hydroxy protons) are shown as balls. (c) Same representation as in (b) but a different view showing the pseudo-symmetry of the structure (with the exclusion of the T3-2eg linker) and approximately hexagonal arrangement of the hydrogen bond donors and acceptors. | ||
Sequence design, synthesis and solution behaviour
Since the homochiral (PP or MM) tilted clockwise arrangements seem the most likely candidates, we first considered sequences 9–11, which only differ in their linker length and in which all helix segments contain a BS unit that should promote P helix handedness. Compounds 9–11 were assembled first with a solid phase using QB, QM, BS,, and T3 monomers having a main chain Fmoc-amine protection, a free carboxylic acid, and TMSE and tBu-ether protections of the hydroxy group in and , respectively (see the ESI†).49 Coupling was mediated by acid chloride activation, and Fmoc was removed after each coupling using 20% piperidine in DMF. The last monomer introduced at the N-terminus was QB, with a nitro group at position 8. Synthesis was performed on an acid labile Sasrin® resin so that a mild acid (hexafluoroisopropanol) allowed for resin cleavage while preserving TMSE and tBu-ether protections, yielding protected precursors of 9–11 having a C-terminal acid function. These precursors were purified by crystallization in ethyl acetate/n-hexane before being converted to methyl esters in solution using TMSCHN2 in CHCl3/MeOH. If purity was insufficient, another crystallization using ethyl acetate/n-hexane was performed at this stage. Finally, treatment with TFA in CH2Cl2 removed protecting groups from and units. The deprotected sequences were used without further purification. All other sequences were prepared in a similar manner, with small modifications, e.g. to introduce a N-terminal (1S)-camphanic group (see the ESI†).
The CD spectra of 9–11 all showed a positive band near 375 nm, confirming that P helicity was quantitatively preserved in all helical segments (Fig. S12†). The 1H NMR spectra of 9 and 10 showed one set of signals (Fig. 5), indicating the prevalence of one species in solution.¶ The prevalence of one species remained true at both higher (>5 mM) and lower (<1 mM) concentrations (Fig. S13–S15†). The signals of the six OH protons could be identified as being exchangeable with deuterium and not correlated to nitrogen in 1H,15N-HSQC spectra (Fig. S16–S18†). The chemical shift values above 8.5 ppm of the OH protons indicated their involvement in hydrogen bonding.34 A DOSY NMR spectrum of 9 mixed with its fully protected precursor showed that they have the same diffusion coefficient, i.e. that both species have the same size and thus that 9 is monomeric in solution (Fig. S19 and S20†). A DOSY NMR spectrum of 10 measured under similar conditions indicated a similar diffusion coefficient (Fig. S21†). This was confirmed using a DOSY NMR spectrum of 10 mixed with its fully protected precursor (Fig. S22†). Based on the molecular models discussed above, the single species observed in NMR spectra can be tentatively assigned to monomeric helix-turn-helix structures with a PP tilted clockwise arrangement (Fig. 4). In contrast, the 1H NMR spectrum of 11 showed two sets of signals (Fig. 5). DOSY NMR suggested that the two species have the same size as 9 and 10 (Fig. S23 and S24†). As mentioned above, molecular models suggested that, with the longer T3-3eg linker, both clockwise and counterclockwise PP tilted arrangements may form, possibly corresponding to the observed sets of signals.
 | ||
| Fig. 5 Part of the 500 MHz 1H NMR spectra of sequences 9–17 at 2.3 mM in CDCl3 at 25 °C showing the amide and hydrogen-bonded OH proton resonances. Signals assigned to OH protons are marked with a blue diamond. A few OH protons could not be unambiguously assigned due to signal overlap. For 11, 15 and 17, signals assigned to different aggregates are shown in different colors. | ||
Attempts to produce single crystals of 9–11 suitable for X-ray diffraction analysis were unsuccessful. Racemic crystallographic approaches have frequently been successful with helical structures,50–54 and sequence 12, the enantiomer of 9, was synthesized for this purpose. The 1H NMR spectrum of 12 was identical to that of 9 and to that of the 9 + 12 racemic mixture (Fig. S25–S27†), indicating that 9 and 12 fold without interfering with one another. Unfortunately, no suitable crystals were produced from the racemic mixture either.
We next considered sequences 13–15. These are analogues of 9–11, respectively, in which handedness is controlled only at the N-terminal helix segment by a (1S)-camphanyl group. Sequences 13–15 can thus exist as PP or PM conformers but a priori not as MM or MP conformers. In 13–15, the two chiral B monomers of 9–11 have been replaced by Q monomers. Although Q and B contribute similarly to helix curvature, small differences in helix shape and properties might also result from this change. The NMR spectrum of 14 showed the coexistence of numerous species (Fig. 5). For this compound with a T3-3eg linker, releasing handedness control at the C-terminal helix allowed access to new unidentified conformers or aggregates. In the absence of a prevalent species, investigations were not pursued in this series. In contrast, the 1H NMR spectra of 13 and 15 showed one dominant set of signals (Fig. 5). Band intensity in CD spectra confirmed that these species must be PP and not PM (Fig. S28†). Here, the handedness preference of the C-terminal helix is determined by its interaction with the N-terminal helix. DOSY NMR spectra indicated that they have the same size as 9–11 (Fig. S29–S32†). The assignment of OH resonances again suggested their involvement in hydrogen bonding (Fig. S33 and S34†). The conformations of 13 and 15 in solution may thus be assigned to the same structures as the prevailing conformations of 9 and 11, respectively (Fig. 4). The fact that 15 seems better behaved than 11 (one clearly prevalent species vs. two coexisting species) may result from the differences imparted by Q monomers in the former vs. B monomers in the latter.55
In another attempt to obtain single crystals, sequences 16 and 17 were produced as achiral, nitro-terminated analogues of 13 and 15, respectively. Nitro-terminated aromatic helices have frequently shown good crystallization abilities.38 Sequences 16 and 17 can be PP, MM, PM, or MP. However, because of the preference of 13 and 15 for PP conformations, we expected 16 and 17 to exist as a racemic PP/MM mixture. The 1H NMR spectrum of 17 is similar to that of 15 (Fig. 5 and S35–S37†), supporting a similar behaviour in the absence of handedness control. Yet no suitable crystals were obtained to elucidate its structure unambiguously. In contrast, the NMR spectrum of 16 showed twice as many resonances as that of 13, as if two monomeric species coexisted in equal proportions, or as if an aggregate containing four inequivalent helices had formed. A DOSY NMR spectrum of 16 mixed with its fully protected precursor showed that 16 has a smaller diffusion coefficient despite having lost the protecting groups (Fig. S38†). Furthermore, DOSY NMR showed that all protons of 16 belonged to entities of similar size. The only possibility for 16 to be larger than its precursor is to form some kind of discrete aggregate. The absence of aggregation for 13, in which the handedness of the N-terminal helix is biased to P, suggests that the aggregate formed by 16 may not be produced from PP and PM conformers alone. It must involve PP and MM conformers, or PM and MP conformers, or all four, that is, combinations not accessible to 13. The aggregate shows no sign of dissociation upon diluting down to 0.46 mM (Fig. S39†) and no significant change upon cooling to −40 °C or heating to 40 °C (Fig. S40†). Multiple hydrogen-bonded OH signals could be identified (Fig. 5 and S41†) but the full assignment is complicated by overlap due to a large number of resonances.
Solid state structure of 16 as a tetrameric eight-helix bundle
Single crystals of 16 suitable for X-ray crystallographic analysis grew from a mixture of chloroform/toluene/acetonitrile and diffracted at 1.05 Å resolution, allowing for the elucidation of its solid state structure (Fig. 6, 7a and S42–S44†). The structure revealed a tetramer composed of two PP conformers of 16 (which we named A and B) and two MM conformers of 16 (which we named A′ and B′). With eight helices involved, the structure is larger and far more complex than any previously characterized abiotic foldamer. It admits a crystallographic centre of symmetry (Fig. 6b). A′ and B′ are thus the inverted structures of A and B, respectively. The structure is thus consistent with the number of signals observed by 1H NMR spectroscopy (Fig. 5). It is composed of three domains stacked on top of one another. Two peripheral domains consist of a parallel bundle of three helices composed of the two helices of A (respectively A′) and the N-terminal helix of B (respectively the N-terminal helix of B′). The central domain, across the centre of inversion, is a head-to-tail PM parallel shifted helix bundle of two C-terminal helical segments of B and B′. The domains are held together by helix-to-helix hydrogen bonds involving mostly (but not only) hydroxy donors of X and Y residues and amide carbonyl acceptors. Remarkably, no fewer than six of these contacts are mediated by a bridging water molecule.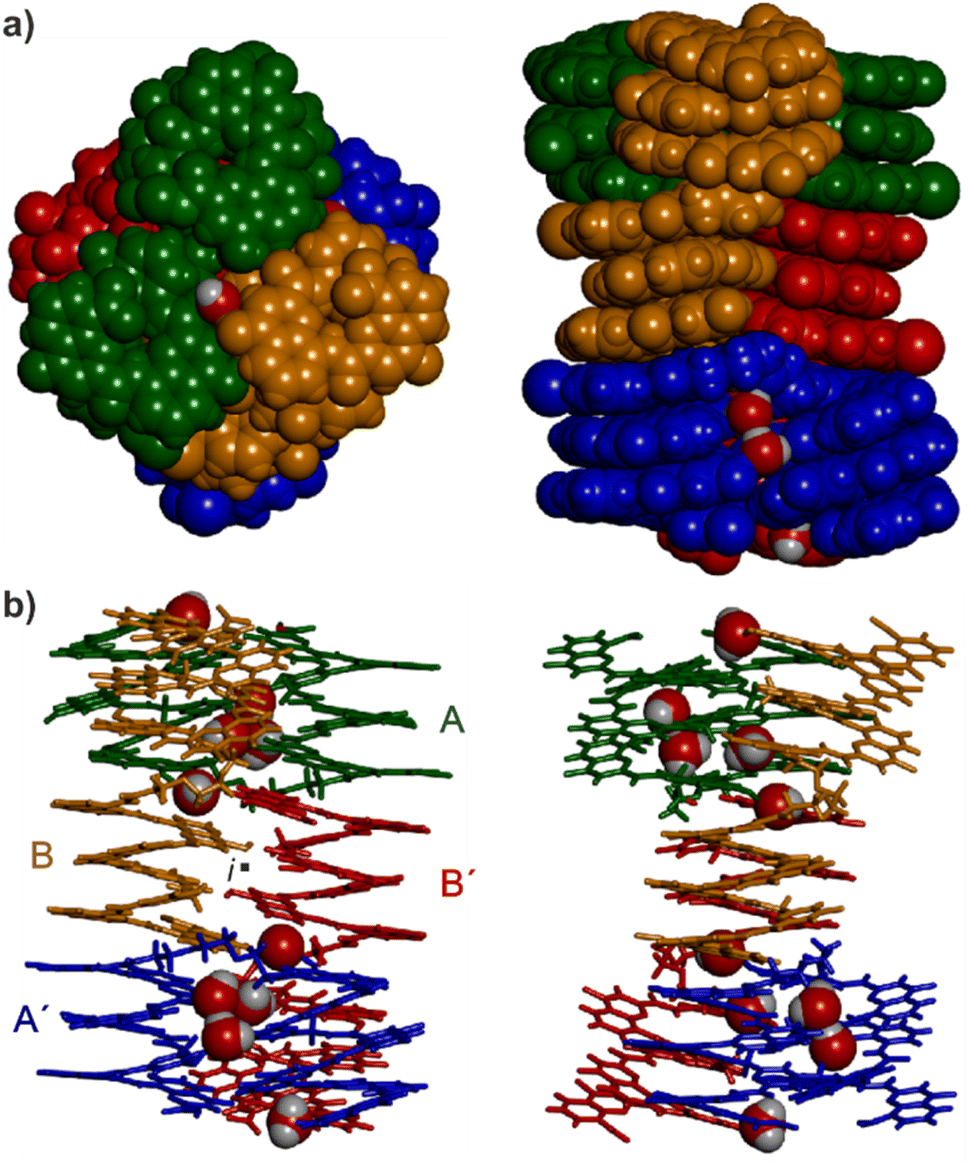 | ||
| Fig. 6 Structure of 164 in the solid state. (a) Top view and side view of the tetramer shown in a space-filling representation. The four molecules are shown in blue, red, gold and green. (b) Side view and front view of the tetramer in tube representation. The same colors are used as in (a). The inversion center is indicated by a black dot (i). Water molecules hydrogen bonded at helix–helix interfaces are shown in space-filling representation. Side chains and other included solvent molecules are omitted for clarity. | ||
The three-helix bundle domains can only be remotely compared to the trimeric helix bundle that was previously characterized (Fig. 7b and S42†).34 The latter was homochiral (PPP or MMM), with all three helices parallel and oriented head-to-head. We have previously shown that inverting both the handedness and the orientation of a helix within a parallel aggregate produces a similar arrangement of hydrogen bond donors and acceptors. One could thus imagine a parallel trimeric bundle in which two P helices would be oriented head-to-head and a third helix would be M helical and have the opposite orientation. However, the three-helix bundles of the structure of 16 show yet another pattern: two helices are P and one M in one bundle (two M and one P in the other bundle), but the two P helices (resp. M) are arranged head-to-tail. Besides, the three helices are not strictly parallel. One helix axis forms an angle with the other two. This helix is also shifted along the bundle axis, i.e. protruding a bit outwards (Fig. 7a). This arrangement results from the presence of molecules A within the bundle (respectively A′ in the other bundle), which have two segments of the same handedness but are arranged head-to-tail since connected by the T3-2eg spacer. The outcome of this complex arrangement is that each three-helix bundle involves only three direct helix-to-helix hydrogen bonds, including the hydroxy group of a Y unit that serves both as a donor and acceptor to bind to the two other helices. Other contacts are mediated by five bridging water molecules per bundle. Some of the water bridges occur within one molecule of A (or A′), others between A and B (respectively A′ and B′). Two water molecules bridge simultaneously three helices (one is shown in Fig. 7d). Bridging water molecules are common at the interface between biopolymers in water but much less so in organic solvents. We have before observed only one instance of a water molecule insertion in a helix–helix hydrogen-bonding interface in an organic solvent.35 Bridging water molecules have also been reported in self-assembled calix[4]resorcinarene-based capsules.56 Their occurrence within 164 shows how they can give access to complex, hard to predict interfaces in organic solvents.
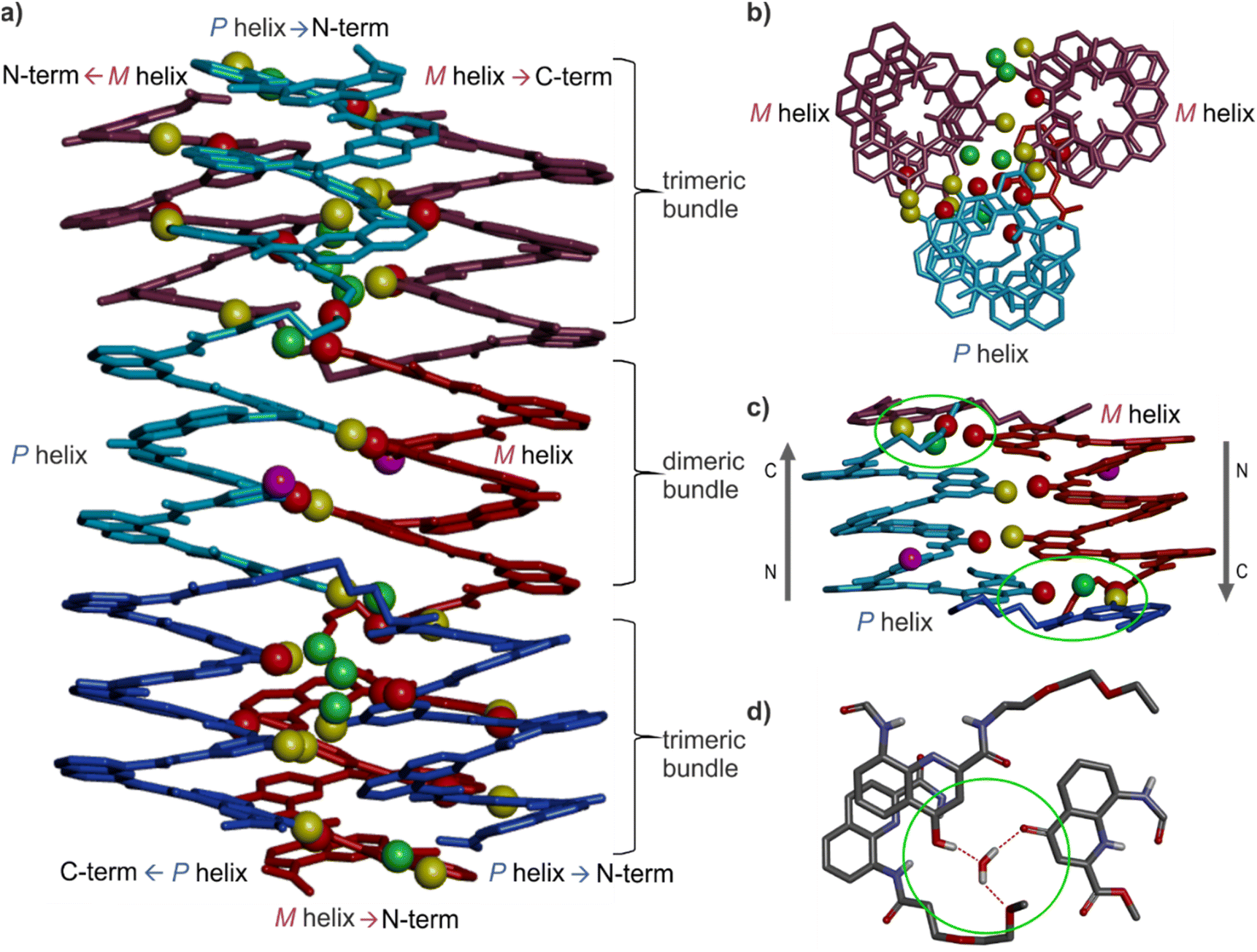 | ||
| Fig. 7 (a) Side-view of the crystal structure of 164 in tube representation indicating the three different domains. The four molecules are shown in brown (A), light blue (B) red (B′) and dark blue (A′). The oxygen atoms at position 4 of X and Y units involved in hydrogen bonding are shown as yellow balls. Those not involved in hydrogen bonding are shown as purple balls. The oxygen atoms of amide carbonyl groups involved in hydrogen bonding are shown as red balls. Bridging water molecules are shown as green balls. Hydroxy protons and carbonyl oxygen atoms of the hydrogen-bonding arrays are shown as yellow and red balls, respectively. Hydrogen atoms and side chains are not shown for clarity. (b) Top view of a three-helix domain with the same representation as in (a). (c) Side view of the central two-helix domain with the same representation as in (a). Arrows indicate the N → C orientation of the sequences. Green circles highlight areas of interest that are shown in (d). (d) Water molecule hydrogen bonded simultaneously to three different helices, including one T3-2eg linker and the carbonyl at position 4 of the C-terminal X unit of molecule B, found as its (1H)-4-quinolinone tautomer. | ||
The central two-helix domain in the middle of the structure is composed of the C-terminal helices of B and B′. B and B′ thus have an extended conformation with one helix involved in the two-helix domain and the other involved in a three-helix domain, in contrast with A and A′, which are back-folded on themselves and are entirely involved in a three-helix domain. The central part of the two helix domain is typical of a head-to-tail heterochiral (PM) parallel shifted dimer,38 giving rise to two intermolecular hydrogen bonds (Fig. 7c and S43†). This arrangement leaves the hydroxy groups of two Y units not involved in hydrogen bonding, the only two in the entire structure (purple balls in Fig. 7a and c). At the periphery of the two-helix domain, the C-terminal X units of B and B′ are found as 4-(1H)-quinolinone tautomers instead of the usual 4-hydroxyquinoline (Fig. 7d and S44†). The quinolinone NH is involved in hydrogen bonds with the adjacent ester and amide carbonyl groups. The latter entails a 180° rotation about the NH–aryl bond, which results in the quinolinone being flipped away from the helix (Fig. S44†). This tautomer should, in principle, give rise to an additional correlation in the 1H,15N-HSQC spectra, but the spectrum shows too much signal overlap to ascertain a clear assignment (Fig. S41†). However, we noted that the number of hydrogen-bonded OH signals identified in the NMR spectrum of 16 – ten (Fig. 5) – matches the number of hydrogen-bonded OH groups in the crystal structure. The carbonyl at position 4 of the flipped 4-(1H)-quinolinone hydrogen bonds to a water molecule that simultaneously bridges monomers from three different helices (Fig. 7d).
Overall the eight-helix bundle structure is held together by hydrogen bonds, notably those involving B and B′ molecules, which span across the trimeric and dimeric domains, by aromatic stacking at domain–domain interfaces, and by the bridging water molecules. While some of its structural features have been observed before, it is novel in several aspects. The structure may be termed a sort of pseudo-quaternary structure. It is indeed composed of multiple subunits but may perhaps not qualify as a true quaternary structure in that it is not composed of independent, self-assembled, tertiary folds.
Conclusion
Using an approach that combined molecular modelling, the screening of three different T3 linkers, and selective handedness control in helix-linker-helix sequences, we have identified sequences that fold in discrete homochiral clockwise tilted arrangements in which helix-to-helix hydrogen bonds maintain the helices at an angle. This arrangement complements earlier work in which other linkers had been shown to promote helix-turn-helix structures where helices were parallel. Combining both parallel and tilted arrangements in future designs will allow for a significant enhancement of the complexity of abiotic tertiary folds that can be produced.In one instance, removing all handedness control, that is, allowing all helix segments to be P or M helical, led to the unexpected formation of a large (12.9 kDa) heterochiral aggregate. A crystal structure revealed an architecture of unprecedented complexity in the field of abiotic folding. The architecture consists of four molecules and a total of eight helices arranged in three distinct domains: two three-helix-bundle domains and one two-helix-bundle domain. The domains contain some previously reported hydrogen-bonded helix–helix arrangements as well as new ones. Several helix-to-helix hydrogen bonds were mediated by bridging water molecules. This object constitutes an important milestone as it gives a glimpse of the vastness of the chemical space available to abiotic foldamers. The field will undoubtedly keep developing through combinations of design by modelling on the one hand and discovery on the other.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
FM synthesized new compounds. FM and LA performed solution studies. VM produced suitable crystals. BW solved and refined the crystal structure. FM and VM performed modelling studies. All authors contributed to experiment design and interpretation. IH supervised the research. FM and IH wrote the manuscript. All authors proofread and improved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DFG (Excellence Cluster 114, CIPSM). D. Gill is gratefully acknowledged for contributing synthetic precursors and monomer synthesis. We also thank S. Wang, L. Wang and D. Bindl for monomer synthesis, C. Glas and C. Ober for assistance with NMR measurements, and B. Kauffmann for crystallographic data collection. This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform (BPCS) at IECB, CNRS UMS3033, Inserm US001, and Bordeaux University.Notes and references
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- R. V. Nair, K. N. Vijayadas, A. Roy and G. J. Sanjayan, Eur. J. Org. Chem., 2014, 2014, 7763–7780 CrossRef CAS.
- I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726 RSC.
- I. Huc, Eur. J. Org. Chem., 2004, 2004, 17–29 CrossRef.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- C. W. Wood and D. N. Woolfson, Protein Sci., 2018, 27, 103–111 CrossRef CAS PubMed.
- F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS PubMed.
- G. G. Rhys, J. A. Cross, W. M. Dawson, H. F. Thompson, S. Shanmugaratnam, N. J. Savery, M. P. Dodding, B. Höcker and D. N. Woolfson, Nat. Chem. Biol., 2022, 18, 999–1004 CrossRef CAS PubMed.
- A. J. Scott, A. Niitsu, H. T. Kratochvil, E. J. M. Lang, J. T. Sengel, W. M. Dawson, K. R. Mahendran, M. Mravic, A. R. Thomson, R. L. Brady, L. Liu, A. J. Mulholland, H. Bayley, W. F. Degrado, M. I. Wallace and D. N. Woolfson, Nat. Chem., 2021, 13, 643–650 CrossRef CAS PubMed.
- M. J. Chalkley, S. I. Mann and W. F. Degrado, Nat. Rev. Chem., 2022, 6, 31–50 CrossRef CAS PubMed.
- M. Mravic, J. L. Thomaston, M. Tucker, P. E. S. Lijun Liu and W. F. Degrado, Science, 2019, 363, 1418–1423 CrossRef CAS PubMed.
- F. Pirro, N. Schmidt, J. Lincoff, Z. X. Widel, N. F. Polizzi, L. Liu, M. J. Therien, M. Grabe, M. Chino, A. Lombardi and W. F. Degrado, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33246–33253 CrossRef CAS PubMed.
- T. Lebar, D. Lainšček, E. Merljak, J. Aupič and R. Jerala, Nat. Chem. Biol., 2020, 16, 513–519 CrossRef CAS PubMed.
- F. Lapenta, J. Aupič, M. Vezzoli, Ž. Strmšek, S. Da Vela, D. I. Svergun, J. M. Carazo, R. Melero and R. Jerala, Nat. Commun., 2021, 12, 939 CrossRef CAS PubMed.
- F. Lapenta, J. Aupič, Ž. Strmšek and R. Jerala, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- P. Kumar, N. G. Paterson, J. Clayden and D. N. Woolfson, Nature, 2022, 607, 387–392 CrossRef CAS PubMed.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 4178–4180 CrossRef CAS PubMed.
- E. J. Petersson, C. J. Craig, D. S. Daniels, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 5344–5345 CrossRef CAS PubMed.
- D. S. Daniels, E. J. Petersson, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532–1533 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–878 CrossRef CAS PubMed.
- G. W. Collie, R. Bailly, K. Pulka-Ziach, C. M. Lombardo, L. Mauran, N. Taib-Maamar, J. Dessolin, C. D. Mackereth and G. Guichard, J. Am. Chem. Soc., 2017, 139, 6128–6137 CrossRef CAS PubMed.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC.
- F. Devaux, X. Li, D. Sluysmans, V. Maurizot, E. Bakalis, F. Zerbetto, I. Huc and A.-S. Duwez, Chem, 2021, 7, 1333–1346 CAS.
- N. Delsuc, F. Godde, B. Kauffmann, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348–11349 CrossRef CAS PubMed.
- D. Sánchez-García, B. Kauffmann, T. Kawanami, H. Ihara, M. Takafuji, M.-H. Delville and I. Huc, J. Am. Chem. Soc., 2009, 131, 8642–8648 CrossRef PubMed.
- M. Vallade, P. Sai Reddy, L. Fischer and I. Huc, Eur. J. Org. Chem., 2018, 2018, 5489–5498 CrossRef CAS.
- V. Maurizot, C. Dolain, Y. Leydet, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2004, 126, 10049–10052 CrossRef CAS PubMed.
- N. Delsuc, S. Massip, J.-M. Léger, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2011, 133, 3165–3172 CrossRef CAS PubMed.
- C. Dolain, J.-M. Léger, N. Delsuc, H. Gornitzka and I. Huc, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16146–16151 CrossRef CAS PubMed.
- J. Graton, F. Besseau, A.-M. Brossard, E. Charpentier, A. Deroche and J.-Y. Le Questel, J. Phys. Chem. A, 2013, 117, 13184–13193 CrossRef CAS PubMed.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51–57 CrossRef CAS PubMed.
- D. Mazzier, S. De, B. Wicher, V. Maurizot and I. Huc, Chem. Sci., 2019, 10, 6984–6991 RSC.
- D. Mazzier, S. De, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 1606–1610 CrossRef CAS PubMed.
- F. S. Menke, D. Mazzier, B. Wicher, L. Allmendinger, B. Kauffmann, V. Maurizot and I. Huc, Org. Biomol. Chem., 2023, 21, 1275–1283 RSC.
- F. S. Menke, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2023, 62, e202217325 CrossRef CAS PubMed.
- C. Dolain, J.-M. Léger, N. Delsuc, H. Gornitzka and I. Huc, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16146–16151 CrossRef CAS PubMed.
- A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2011, 76, 195–200 CrossRef CAS PubMed.
- D. Bindl, E. Heinemann, P. K. Mandal and I. Huc, Chem. Commun., 2021, 57, 5662–5665 RSC.
- S. P. Ho and W. F. Degrado, J. Am. Chem. Soc., 1987, 109, 6751–6758 CrossRef CAS.
- A. D. Nagi and L. Regan, Folding Des., 1997, 2, 67–75 CrossRef CAS PubMed.
- J. P. Schneider and J. W. Kelly, Chem. Rev., 1995, 95, 2169–2187 CrossRef CAS.
- E. J. Petersson and A. Schepartz, J. Am. Chem. Soc., 2008, 130, 821–823 CrossRef CAS PubMed.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Targeted Anti-Tumor Ther., 2020, 1, 273–312 Search PubMed.
- Maestro, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- K.-J. Liu and J. L. Parsons, Macromolecules, 1969, 2, 529–533 CrossRef CAS.
- B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175–7185 CrossRef CAS PubMed.
- P. K. Mandal, G. W. Collie, B. Kauffmann and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 14424–14427 CrossRef CAS PubMed.
- D. E. Mortenson, J. D. Steinkruger, D. F. Kreitler, D. V. Perroni, G. P. Sorenson, L. Huang, R. Mittal, H. G. Yun, B. R. Travis, M. K. Mahanthappa, K. T. Forest and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13144–13149 CrossRef CAS PubMed.
- C. Toniolo, C. Peggion, M. Crisma, F. Formaggio, X. Shui and D. S. Eggleston, Nat. Struct. Biol., 1994, 1, 908–914 CrossRef CAS PubMed.
- G. Lautrette, B. Kauffmann, Y. Ferrand, C. Aube, N. Chandramouli, D. Dubreuil and I. Huc, Angew. Chem., Int. Ed., 2013, 52, 11517–11520 CrossRef CAS PubMed.
- M. Lee, J. Shim, P. Kang, I. A. Guzei and S. H. Choi, Angew. Chem., Int. Ed., 2013, 52, 12564–12567 CrossRef CAS PubMed.
- D. Bindl, P. K. Mandal and I. Huc, Chem.–Eur. J., 2022, 28, e202200538 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures, detailed experimental protocols and characterization of new compounds. CCDC 2216678. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00267e |
| ‡ Throughout the manuscript, the term “parallel” is used in its geometric definition and refers to helical axes having a parallel orientation without prejudice of the head-to-head or head-to-tail relative arrangement of the oligoamide chains. The term “anti-parallel” is avoided. This terminology differs from that classically used in peptide and protein science where parallel and anti-parallel refer to N → C chain orientation, but is more adapted to discuss the geometrical parameters of the structures. Although well-established, the peptide terminology can at times be confusing. For instance, in a so-called “dimeric parallel coiled-coil peptide” the axes of the two α-helices do not define parallel lines. At any given point, they are slightly tilted. |
| § Slight hyper- and hypsochromic shifts are observed for 5 because of the larger number of contiguous pseudo-conjugated quinolinecarboxamide units (Fig. S4†). |
| ¶ Exchange between the various types of dimers is always slow on the 1H NMR time scale. See ref. 34–37. |
| This journal is © The Royal Society of Chemistry 2023 |