
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotophysics of the red-form Kaede chromophore†
Kiri
Addison
,
Palas
Roy‡
,
Giovanni
Bressan
,
Karolina
Skudaite
,
Josh
Robb
,
Philip C.
Bulman Page
,
Eleanor K.
Ashworth
 ,
James N.
Bull
and
Stephen R.
Meech
*
,
James N.
Bull
and
Stephen R.
Meech
*
School of Chemistry, University of East Anglia, Norwich, Norfolk NR4 7TJ, UK. E-mail: s.meech@uea.ac.uk
First published on 9th March 2023
Abstract
The green fluorescent protein (GFP) drove revolutionary progress in bioimaging. Photoconvertible fluorescent proteins (PCFPs) are an important branch of the FP family, of which Kaede is the prototype. Uniquely, PCFPs can be permanently switched from green to red emitting forms on UV irradiation, facilitating applications in site-specific photolabelling and protein tracking. Optimisation and exploitation of FPs requires understanding of the photophysical and photochemical behaviour of the chromophore. Accordingly, the principal GFP chromophore has been the subject of intense experimental and theoretical investigation. In contrast, the photophysics of the red emitting PCFP chromophore are largely unstudied. Here we present a detailed investigation of the excited-state properties of the Kaede chromophore in solution, utilising steady state measurements, ultrafast time-resolved electronic and vibrational spectroscopies, and electronic structure theory. Its excited state dynamics are very different to those of the parent GFP. Most remarkably, the PCFP chromophore has highly complex wavelength-dependent fluorescence decays and a mean lifetime an order of magnitude longer than the GFP chromophore. Transient electronic and vibrational spectroscopies suggest that these dynamics arise from a range of excited-state conformers that are spectrally and kinetically distinct but chemically similar. These conformers are populated directly by excitation of a broad thermal distribution of ground state structures about a single conformer, suggesting an excited-state potential surface with several minima. Temperature-dependence confirms the existence of barriers on the excited-state surface and reveals the radiationless decay mechanism to be internal conversion. These experimental observations are consistent with a model assuming a simple ground state potential energy surface accessing a complex excited state possessing multiple minima.
Introduction
The discovery of the green fluorescent protein (GFP) drove a revolution in bioimaging, affording greater sensitivity and specificity to studies of functioning cells.1–4 A wide variety of GFP mutants were developed, yielding faster maturation, superior brightness, and a wide range of colours. An important step forward was the discovery and optimisation of photoactivatable FPs. Some of these – the reversibly switchable (rs) FPs based on the Dronpa family – can be switched between bright and dark forms by irradiation with UV and visible light, respectively.5,6 While the detailed mechanism remains a topic of debate,7–11 the key structural change in the chromophore is a cis to trans isomerisation coupled with a proton-transfer reaction.12–14 The rsFPs were critical in developing super-resolution fluorescence microscopy.15–17 A second important FP family – the photoconvertible (PC) FPs – was developed following discovery of the Kaede protein.18 Initially Kaede has the same principal chromophore as GFP and the rsFPs, but on irradiation with UV light undergoes an irreversible photochemical reaction to yield a new chromophore with extended conjugation (Fig. 1).19–22 This photoreaction results in an FP with enhanced emission and red-shifted absorption and emission spectra. Such a permanent colour change permits ‘optical highlighting’ of spatially-selected populations of labelled proteins and tracking of their evolution over time.23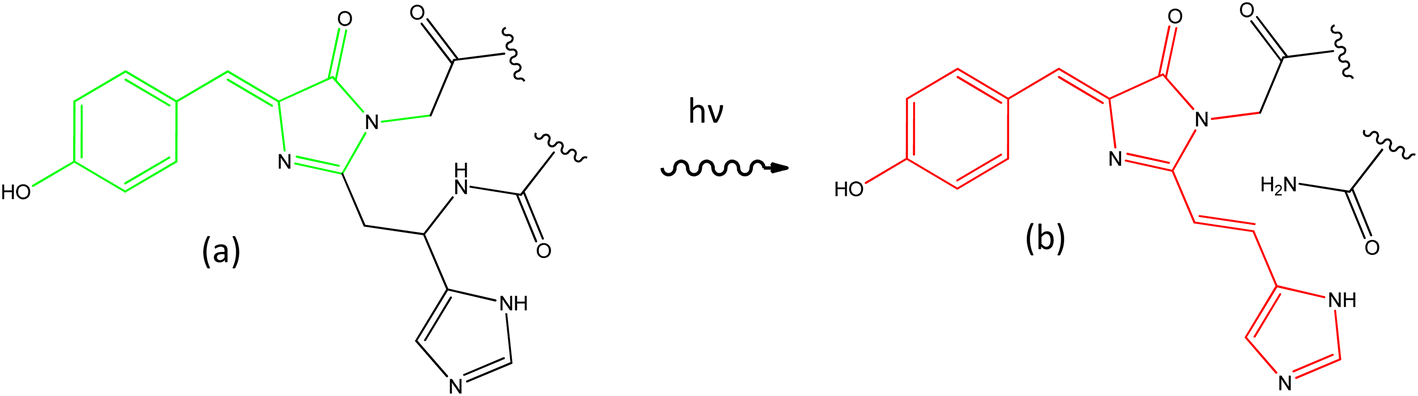 | ||
| Fig. 1 Photoconversion of the chromophore in Kaede. The principal GFP chromophore (a, HBDI) is present in the green form of Kaede, which photoconverts under UV irradiation to the red form (b, rK1H). | ||
Considerable experimental24–34 and theoretical35–43 effort has been invested in resolving the excited state dynamics associated with the principal GFP chromophore (p-hydroxybenzylidene-2,3-dimethylimidazolinone, HBDI, Fig. 1a). Its excited state properties are dominated by ultrafast internal conversion, weakly dependent on solvent polarity and viscosity, sensitive to substituent, and markedly different in free solution compared with when expressed in its protein environment.25,26,44 In contrast, there have been very few studies of the photophysics of the red form of the Kaede chromophore (here called rK1H to indicate it is the red protonated form of the Kaede chromophore, Fig. 1b). The chromophore has been synthesised by a number of groups,45–48 and Baranov, Lukyanov and their co-workers reported several derivatives.49–51 Lukyanov and co-workers also characterised the steady state electronic spectra of rK1H and some of its derivatives.46 Absorption and fluorescence spectra were reported in a range of solvents and as a function of pH. We and others investigated the fundamental spectroscopy of the anion rK1− in the gas phase.48,52,53 Here, we present a detailed study of the condensed phase photophysics of both rK1H and rK1−, including ultrafast time-resolved fluorescence (TRF), transient absorption (TA), and femtosecond stimulated Raman spectroscopy (FSRS). These experiments are applied to investigate effects of solvent polarity and viscosity on excited state spectroscopy and dynamics. The measurements reveal complex, multicomponent picosecond timescale excited state relaxation, which depends significantly on whether the chromophore is in its neutral or anionic form. Steady state measurements are extended to low temperature, where spectra exhibit well-resolved vibronic structure, which is modelled by Franck–Condon–Hertzberg–Teller (FCHT) simulations. The results are discussed in terms of a multiple conformer excited state potential energy surface. Similarities and differences with the GFP chromophore are described.
Results and analysis
Steady state absorption and emission
Fig. 2 shows the peak normalised solvent dependent absorption spectra of rK1H and rK1−, where the latter was generated by adding a drop of aqueous NaOH to the rK1H solutions. The NMR spectrum of rK1H was measured in D2O, revealing a single ground state conformer (ESI,† Fig. S1). The pKa of rK1H in H2O was 7.5, in good agreement with Yampolsky et al.46 The similarity of the rK1H pKa with that for HBDI suggests that deprotonation is at the phenolic ring, a conclusion supported by our earlier electronic structure calculations with explicit modelling of water molecules.53 The rK1H absorption spectra show a moderate solvent dependence, with the aprotic solvents acetonitrile (MeCN) and tetrahydrofuran (THF) being most blue shifted. Attempts to measure spectra in nonpolar solvents (cyclohexane, toluene) were frustrated by a lack of solubility and aggregation. Among the polar protic solvents, higher polarities led to a small blue shift in the absorption spectra (Fig. 2). For rK1−, solvent polarity and H-bonding effects on the absorption spectra are smaller, with most spectra being essentially superimposable.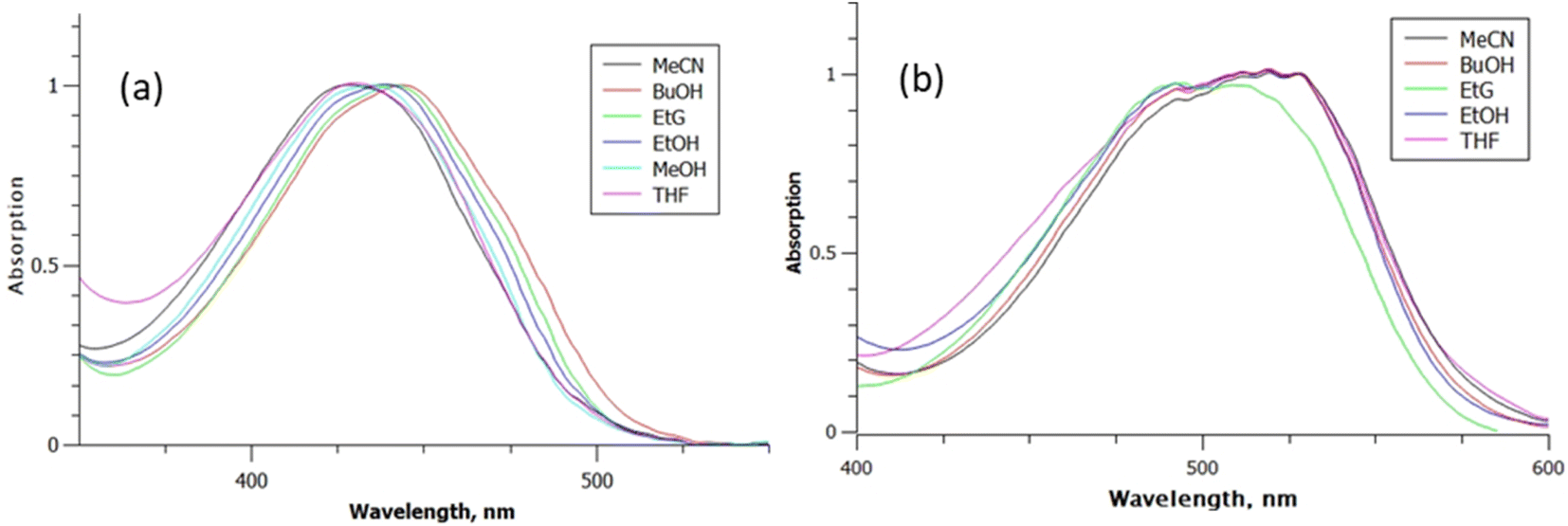 | ||
| Fig. 2 Absorption spectra for: (a) rK1H, and (b) rK1− as a function of solvent: MeCN; butan-1-ol (BuOH); ethylene glycol (EtG); ethanol (EtOH); THF. | ||
The corresponding fluorescence emission spectra shown in Fig. 3 were normalised to have the same absorbance at the common excitation wavelength, such that the observed variation in intensity in each series reflects the relative fluorescence quantum yield, ΦrF. The fluorescence quantum yield of rK1− in ethanol was measured to be ca. six-fold lower than that for rK1H. The emission maximum for rK1H again exhibits a weak dependence on solvent polarity (as better highlighted by peak normalised spectra shown in the ESI,† Fig. S2). The emission spectrum is almost featureless, although weak vibronic structure is observed, which is evident in aprotic THF but unresolved in the polar protic solvents; the vibronic structure becomes much more prominent in temperature-dependent data discussed below. The weak solvatochromism, i.e., the small changes in Stokes shift with solvent polarity (compare Fig. 2 and 3),54,55 points to the absence of large changes in dipole moment between ground and excited electronic state. In turn, this suggests the absence of large charge-transfer contributions to the fluorescent state of rK1H. The ΦrF is, however, sensitive to solvent, with protic solvents leading to excited-state quenching. Solvent polarity evidently plays a secondary role to H-bonding in determining ΦrF.
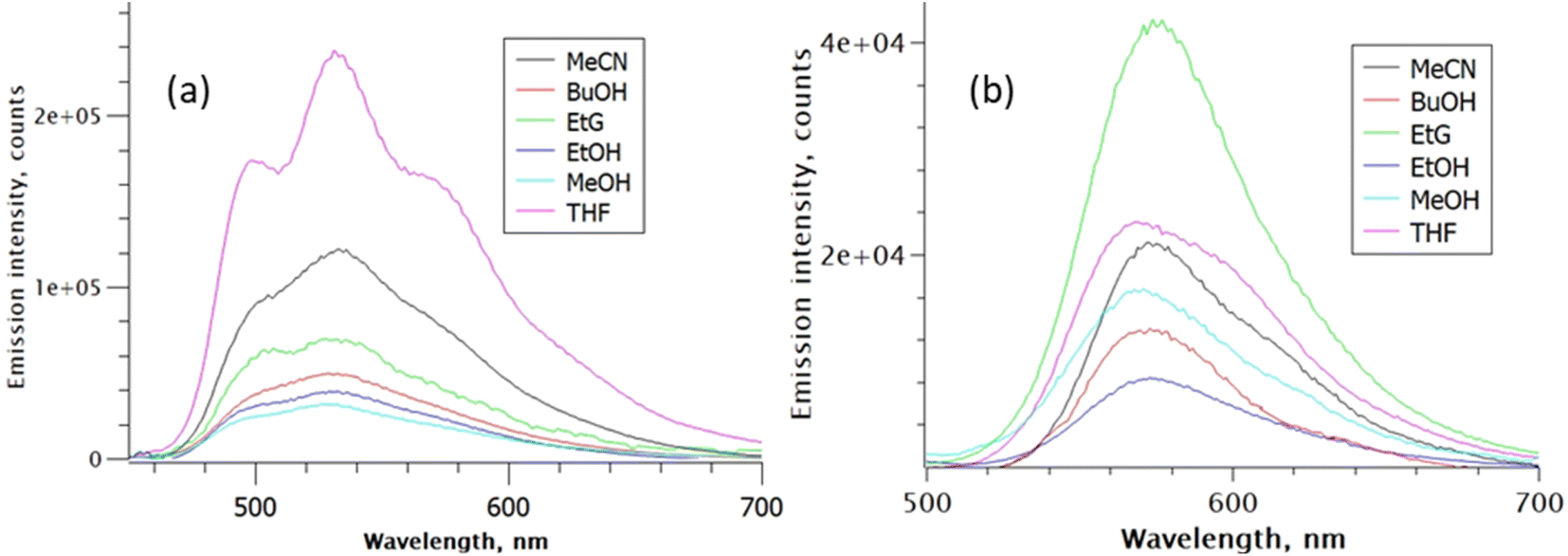 | ||
| Fig. 3 Solvent-dependent emission spectra: (a) rK1H, and (b) rK1−. Spectra are normalised for constant absorbance to reveal the relative quantum yield of fluorescence. Excitation was at 420 nm. | ||
The fluorescence spectrum of rK1− is red shifted from rK1H, as expected from the absorption spectrum, and the peak wavelength again shows little polarity dependence (Fig. 3b, and S2 in the ESI†). Thus, similar to the situation for rK1H, charge-transfer configurations leading to a dipole moment change do not play a major role in the rK1− emissive state. The solvent dependent ΦrF for rK1− is, however, different to that for rK1H, with viscosity becoming a significant factor; for example, the most viscous alcohol studied, ethylene glycol (EtG), has the largest ΦrF. However, aprotic solvents give rise to a larger quantum yield than protic solvents of similar polarity and viscosity, suggesting the H-bonding effect on ΦrF, found for rK1H, still operates.
The viscosity effect was investigated further through measurements of ΦrF in a series of ethylene glycol:ethanol mixtures, where polarity and H-bonding ability are similar but the viscosity varies by a factor of 10. The results are shown in Fig. 4, where the quantum yields for both rK1H and rK1− increase linearly with viscosity, with the slope for rK1− being larger, consistent with the viscosity effect seen in Fig. 3b. Such a viscosity-dependent ΦrF suggests that the radiationless decay coordinate involves a structural reorganisation in the excited state that displaces a significant volume of solvent.56–58 In that case, solvent friction opposes motion along the reaction coordinate, enhancing fluorescence. The weaker viscosity dependence in rK1H may suggest that the coordinate leading to radiationless decay displaces a smaller volume of solvent. A common viscosity-dependent excited state reaction coordinate in stilbenes,59 and polymethine dyes60 is isomerisation about a double bond that has reduced bond order in the excited electronic state. This can lead to a crossing with the electronic ground state (in which double bond rotation is energetically unfavourable) promoting internal conversion. rK1H has structural similarities with these chromophores, suggesting double bond isomerisation as a likely radiationless decay pathway (as was also found for HBDI).35,38
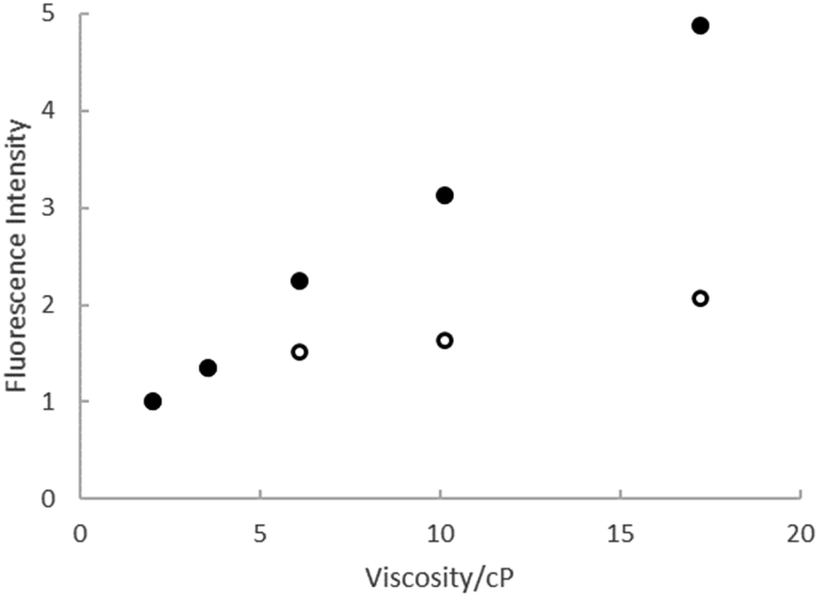 | ||
| Fig. 4 Relative fluorescence intensity from rk1H (open symbols) and rK1− (solid symbols) as a function of the viscosity of ethanol:ethylene glycol mixtures. | ||
In summary, steady state electronic spectra suggest that there is a negligible change in dipole moment between ground and excited states of rK1H and rK1−. Fluorescence is quenched by H-bonding interactions (especially in rK1H) but enhanced by increasing solvent viscosity (especially in rK1−), consistent with a radiationless decay coordinate involving excited-state structure change.
Time-resolved studies
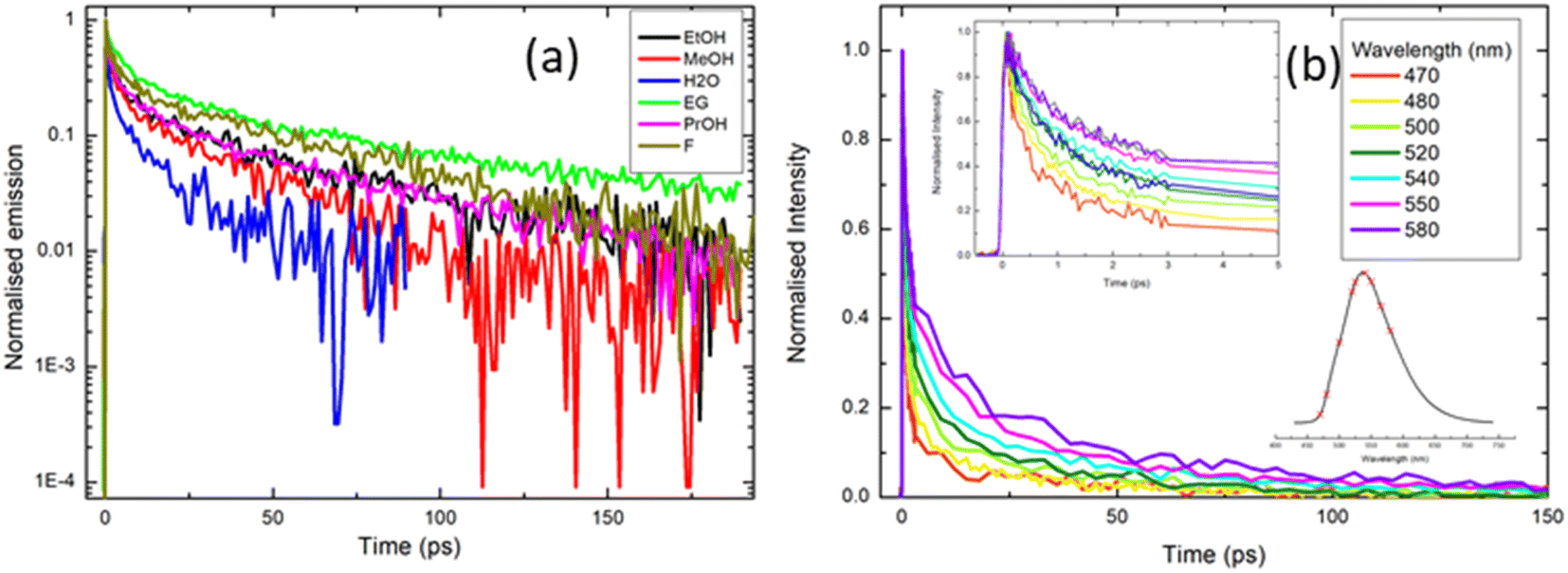 | ||
| Fig. 5 Ultrafast time-resolved fluorescence of rK1H excited at ≈400 nm: (a) fluorescence decay profiles recorded in a range of solvents (EG = ethylene glycol; F = formamide). Note that data are presented on a logarithmic intensity scale to highlight the non-single exponential decay. (b) Emission wavelength dependence of the decay profile in methanol. The insets in (b) show the early time data and the wavelengths spanned by the measurement (470 nm–580 nm) respectively. Fitting parameters from the analysis of these data are given in the ESI.† | ||
To further explore the complex fluorescence decay kinetics, we recorded the wavelength dependence of the time-resolved fluorescence of rK1H in methanol (Fig. 5b). As the detection wavelength is tuned from blue to red across the emission spectrum, 〈τF〉 changes from <4 ps on the blue side to >10 ps on the red side, but the decay remains multi-exponential at all wavelengths. Three of the four fitted time constants increase with increasing observation wavelength, while the weights for the two shorter components decrease and those for the two longer components increase (full fitting details are given in the ESI,† Table S2). Significantly, there is no evidence of a risetime on the red edge of the emission; all components decay monotonically from t = 0. Solvation dynamics are a common source of multi-exponential wavelength dependent fluorescence decays, but such processes are, invariably, characterised by a risetime on the red side of the emission on the timescale of the solvation time correlation function (a few ps for methanol).54 The lack of a clear risetime feature and, consequently, the absence of solvation dynamics, is consistent with the observed weak solvatochromism (Fig. 2 and 3). Thus, the wavelength dependence (Fig. 5b) is more consistent with direct excitation of a broad range of emissive states (e.g. excited-state conformations) with different emission spectra which do not interconvert over the tens of picoseconds timescale. The states emitting at shorter wavelengths are associated with shorter lifetimes, so that the overall effect is a collapse of the initial broad spectrum to leave a longer-lived ensemble with a red-shifted fluorescence spectrum. The time-resolved emission spectra were reconstructed from the data in Fig. 5b and reveal an ≈35 nm red shift that is complete in <10 ps (ESI,† Fig. S3).
The complexity of the fluorescence decay is surprising given the simplicity of the electronic spectra and the presence of only a single ground state conformer (as judged from NMR, see the ESI,† Fig. S1). A possible origin is the presence of solute aggregates formed at the high concentrations used for fluorescence up-conversion. This was ruled-out by measuring the time-resolved emission at 525 nm with solution concentrations between 0.5 mM and 50 μM. The emission curves were all superimposable (ESI,† Fig. S4). Another possible artifact is photoconversion in the 400 nm excitation beam to yield a transient population of multiple ground states, for example, through an excited-state isomerisation reaction, which are then re-excited and decay with a different lifetime. The presence of multiple photo-induced isomers was tested by performing the up-conversion measurement with the sample in either a static or a flow cell and recording the lifetimes as a function of the power of the 400 nm excitation light, which was varied between 2 and 5 mW. Again, all measured decay curves were essentially superimposable (ESI,† Fig. S5). Thus, the complex decay kinetics must arise from discrete rK1H molecules. This conclusion, coupled with the absence of a clear risetime at any wavelength, suggests that the Franck–Condon excited state distribution relaxes rapidly (<100 fs) to populate a distribution of emitting conformers with distinct decay times. We return to this assignment in the discussion of temperature-dependent fluorescence.
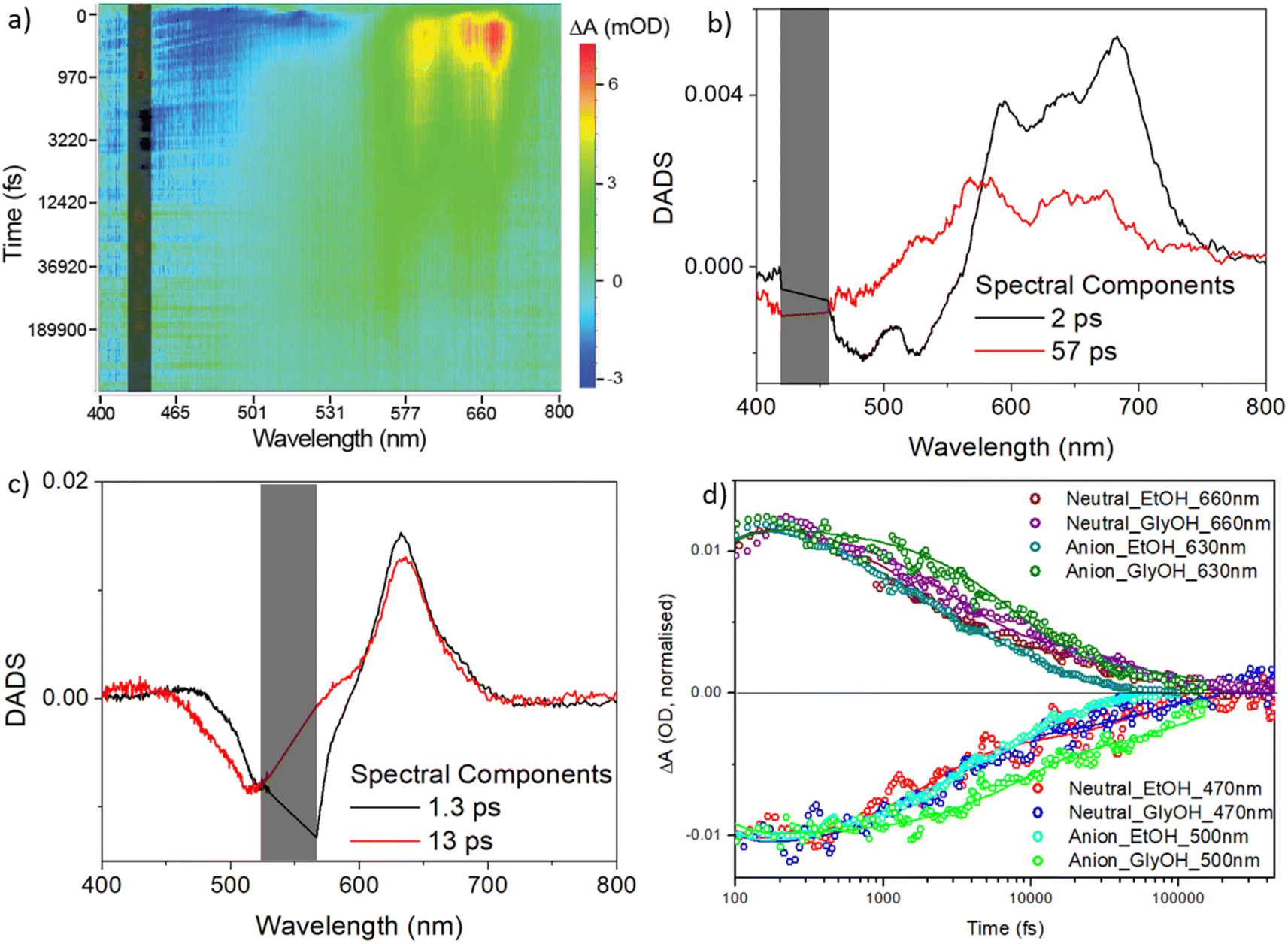 | ||
| Fig. 6 Transient absorption spectroscopy for rK1H and rK1−. (a) Heat map for rK1H in ethanol excited at 440 nm. Blue indicates ground state bleach plus stimulated emission, and red/yellow indicates the excited-state absorption. (b) DADS from a global analysis of the data in (a) using the sum of two decay components. (c) DADS for rK1− (see heat map in the ESI,† Fig. S6). In (a), (b), and (c) the scattered light contribution is shaded out. (d) The recovery kinetics at specific wavelengths, reflecting the excited state decay and ground state recovery in ethanol and ethylene glycol (solid lines are the global fits). | ||
The decay associated difference spectra (DADS) recovered from the two-state global analysis show a bleach (negative ΔOD) at the absorption/stimulated emission wavelength, overlapped with a red-shifted excited state absorption (between 550 and 750 nm for rK1H, and at ca. 630 nm for rK1−, Fig. 6b and c, respectively). In both cases the fast (ca. 2 ps) and slow (tens of ps) DADS in ethanol are similar to one another, although the spectral profile of the excited-state absorption of rK1H shows minor evolution, shifting to the blue in the slower DADS. These data are consistent both with the distribution of lifetimes being associated with the decay of spectrally similar states, and with the small red-shift observed in the time resolved emission (Fig. S3†).
The TA kinetics probed at wavelengths monitoring the excited-state decay and the ground-state recovery of rK1H and rK1− are shown in Fig. 6d along with the global fits, for both ethanol and ethylene glycol solvents (the fit data are tabulated in ESI,† Table S3). The key features are: (i) ground-state recovery and excited-state decay show the same kinetics, and (ii) ground-state recovery is complete on the sub-nanosecond timescale. For both species, these features are consistent with the radiationless decay mechanism being internal conversion back to the electronic ground state. The viscosity effect identified in the fluorescence up-conversion measurements is also evident in the solvent dependent TA data (Fig. 6d, and Table S3 in the ESI†); both fitted decay components increase between ethanol and ethylene glycol solvents and the effect of viscosity is larger for the anionic form.
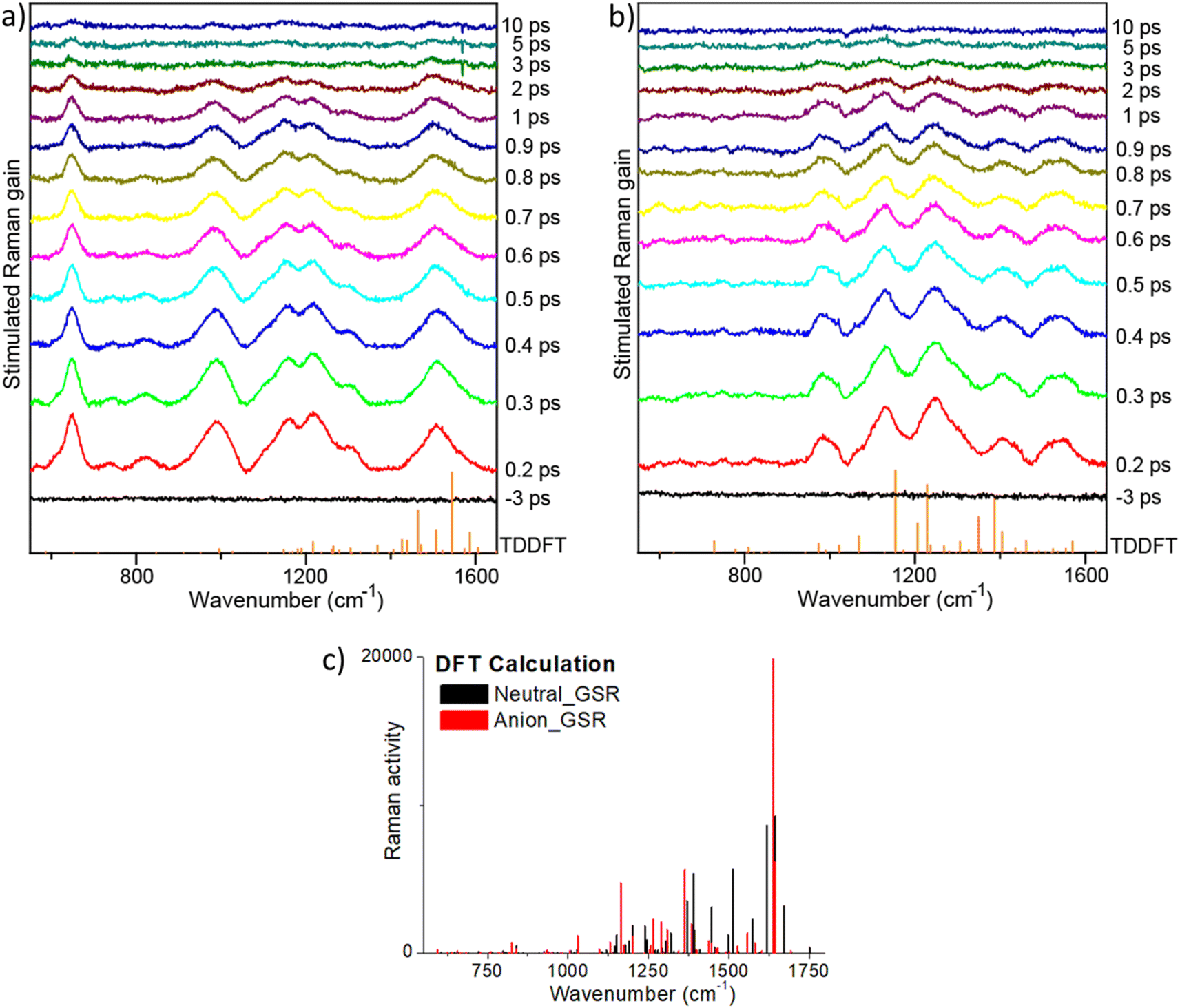 | ||
| Fig. 7 FSRS spectra of (a) rK1H, and (b) rK1− in methanol. Spectra were measured as a function of time after an actinic pulse (440 nm for rK1H and 535 nm for rK1−) with Raman pulses at different wavelengths (700 nm for rK1H and 650 nm for rK1−). The vertical stick spectra (orange) at the bottom of panels (a) and (b) show the TDDFT calculated excited state Raman spectra. (c) The DFT calculated ground state Raman spectra for the two forms are shown for reference. | ||
The highest frequency modes calculated for ground state rK1H and rK1− involve C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, although these are weak in the simulated Raman spectra for both ground (Fig. 7c) and excited states, and are absent in the resonant FSRS spectra (Fig. 7a and b), suggesting a small resonance Raman cross section. Precise assignments of the Raman transitions observed in a ‘large’ molecule such as rK1H is difficult because the modes have delocalised character, with each involving motion of many nuclei. However, in general, the strongly Raman active modes in the 1380–1600 cm−1 region can be assigned to CC stretching coupled with ring-breathing modes. Raman active modes calculated to lie in the 900–1350 cm−1 range are mostly associated with in-plane CH bends coupled to CC stretches. This assignment is true for both ground and excited electronic states, although the excited state modes are usually at lower frequency, consistent with weaker CC bonds following ππ* excitation.
O stretching, although these are weak in the simulated Raman spectra for both ground (Fig. 7c) and excited states, and are absent in the resonant FSRS spectra (Fig. 7a and b), suggesting a small resonance Raman cross section. Precise assignments of the Raman transitions observed in a ‘large’ molecule such as rK1H is difficult because the modes have delocalised character, with each involving motion of many nuclei. However, in general, the strongly Raman active modes in the 1380–1600 cm−1 region can be assigned to CC stretching coupled with ring-breathing modes. Raman active modes calculated to lie in the 900–1350 cm−1 range are mostly associated with in-plane CH bends coupled to CC stretches. This assignment is true for both ground and excited electronic states, although the excited state modes are usually at lower frequency, consistent with weaker CC bonds following ππ* excitation.
Comparing the observed FSRS with the calculations, we note that agreement for the anion is good, with both calculation and measurement showing activity in the CH bend and CC stretch regions. For rK1H, agreement of the FSRS with calculation is not as good, with the measured spectra having more intensity in the CH bend/CC stretch region. Thus, the experimental spectra both show similar activity in this region, suggesting that these modes are enhanced when the Raman pump is tuned into resonance with the S1–Sn transient absorption. The most striking difference between the measured FSRS of rK1H and rK1− is the strong Raman activity around 650 cm−1 in the neutral form, which is entirely absent for the anion. Inspection of the (weakly) Raman active modes in the calculation shows that this wavenumber is likely associated with an in-plane ring deformation mode. The enhancement of this mode specifically for rK1H probably reflects a difference in the resonant Sn states, with the ring deformation being significantly displaced between S1 and Sn only in the neutral form, leading to the larger enhancement observed. A more detailed assignment requires calculation of mode displacements in the upper state,66,69,70 which are difficult for a ‘flexible’ molecule of this size. The result does, however, suggest that FSRS could be a powerful tool for studying excited-state proton transfer reactions in Kaede, such as have been observed in GFP.71,72 Turning to the time dependence of the FSRS spectra (Fig. 7b and c), no significant evolution between 100 fs and 10 ps was observed for either species. This result is consistent with our interpretation of the TA and TRF measurements, in that the distribution of lifetimes is associated with chemically and structurally similar, but kinetically distinct, excited-state conformations.
Temperature-dependent measurements
To elucidate the origin of complexity in the excited-state decay of rK1H and rK1−, we recorded their temperature dependent fluorescence spectra in ethanol (Fig. 8). Both show two prominent features: (i) a strong temperature dependence of ΦrF, with the relative yield increasing by at least two orders of magnitude between room temperature and T = 80 K, and (ii) the emission spectra at low temperature show clear vibronic structure.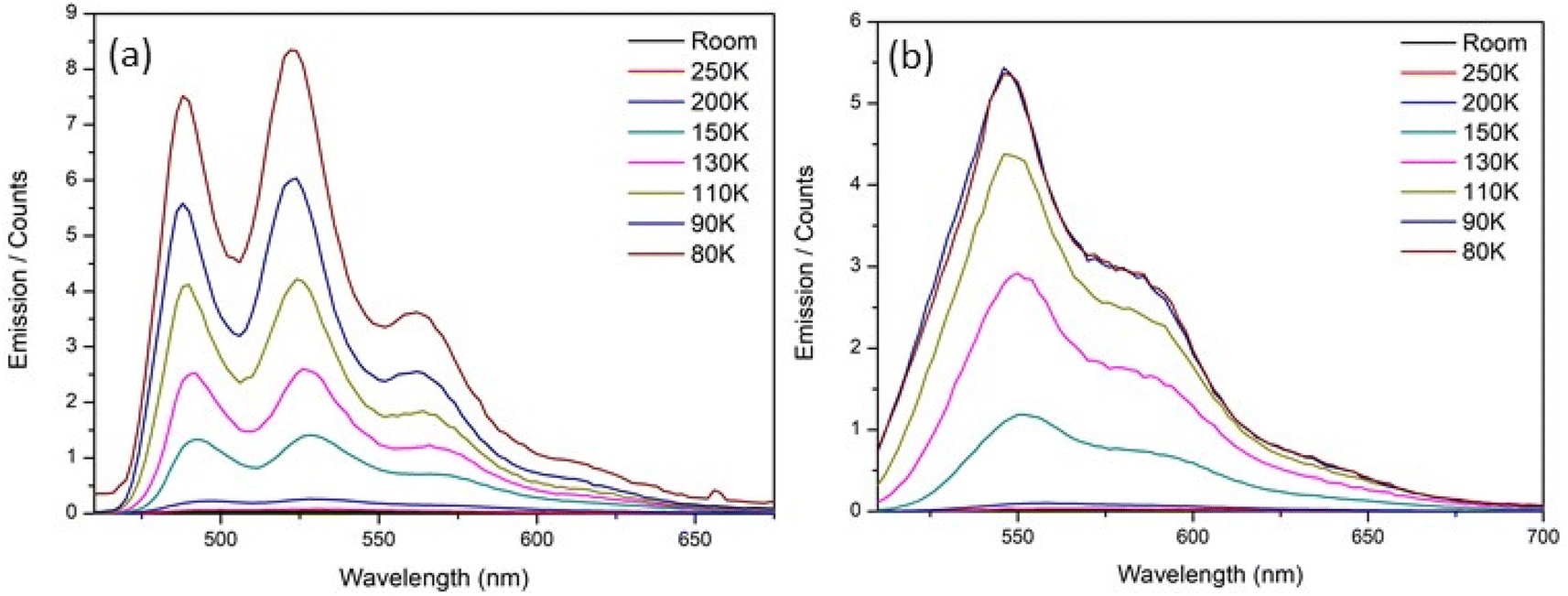 | ||
| Fig. 8 Temperature dependence of the fluorescence emission spectra for (a) rK1H and (b) rK1− in ethanol. | ||
Given a reported room temperature fluorescence quantum yield for rK1H of ∼0.5%,46 the near 100-fold enhancement with decreasing temperature suggests convergence to near unit quantum efficiency. Evidently, the protein structure surrounding rK1H chromophore in Kaede causes an enhancement of the emission yield, to an extent similar to freezing to the glass transition temperature (for Kaede ΦF = 0.33 at 298 K).18 A similar effect was noted for HBDI− when compared to GFP.24 It seems unlikely that internal motion of the chromophore in the protein is hindered to the same extent as in an ethanol glass at T = 77 K, in-turn suggesting that protein-matrix-specific enhancement mechanism operates. The origin of the similarly dramatic enhancement in GFP has been widely discussed,25,44 with recent experimental and theoretical data pointing to an important role for protein-chromophore electrostatic interactions.73–75 It is significant that electrostatic effects were earlier implicated in theoretical investigations of the radiationless decay of HBDI. In that case the excited state lifetime was calculated to be longer in vacuum than in a polar (aqueous) medium.37,76 The effect arose from a polar environment suppressing the barrier to the conical intersection (CI) with the ground state. Similar polarity effect have been seen in other photobiological chromophores.76,77 A similar electrostatic enhancement mechanism as in GFP may operate in Kaede, but confirmation requires calculations of the electric field environment of the chromophore in the protein. The near unit quantum efficiency of rK1H at low temperature is consistent with the observed full recovery of the ground electronic state in the TA data (Fig. 6a and d), and confirms that the radiationless mechanism is internal conversion with a negligible role for intersystem crossing.
The strong temperature dependence of fluorescence emission in ethanol parallels the similarly strong temperature dependence for HBDI and its anion, in which internal conversion is known to be the dominant decay mechanism (albeit with a larger rate constant).24 For rK1H, an Arrhenius analysis (as described previously for HBDI24) yields an activation energy for the radiationless (internal conversion) rate coefficient of 10 ± 1 kJ mol−1 (9 ± 1 kJ mol−1 for rK1−, see Fig. S7 in the ESI†). These activation energies are significant and contrast with the near barrierless excited-state decay of HBDI.24 It was shown above that the excited-state decay is sensitive to viscosity, so the measured activation energy should not be interpreted as arising solely from a potential energy barrier to internal conversion, but rather comprises the influence of both thermal motion over the barrier and the temperature dependent viscosity; the latter can often be treated as an Arrhenius-like activated process (sufficiently far above the glass transition). The rigorous separation of these two contributions is difficult, requiring an isoviscosity analysis.57,78 However, we can conclude that the measured activation energy is a sum of these two contributions, and that the actual potential energy barriers to internal conversion will be lower than the measured activation energies. The coexistence of two mechanisms controlling the radiationless decay is highlighted by temperature-dependent studies in glass-forming 2-methyl-THF solvent, which gave rise to a non-linear Arrhenius plot (ESI,† Fig. S8), presumably due to the interplay of these two contributions.
We now consider the vibronic structure apparent in the temperature-dependent fluorescence emission spectra (Fig. 8). As the temperature decreases and the fluorescence quantum yield increases, vibronic structure, which is absent in the room-temperature absorption and barely discernible in the emission spectra (Fig. 2 and 3), becomes increasingly clear. A similar but less well resolved change was seen for HBDI in 2-methyl-THF.79 Fluorescence excitation spectra measured as a function of temperature also show the appearance of vibronic structure in the S1 ← S0 transition at low temperature (ESI,† Fig. S9). The mirror image correspondence between the fluorescence excitation and emission spectra is consistent with the involvement of a single absorbing and emitting electronic state in both rK1H and its anion. Although the NMR shows a single conformer (ESI,† Fig. S1), we suggest that the broad room temperature spectra with weak or absent vibronic structure implies a thermally populated distribution of ground state structures about the equilibrium structure, with that structural distribution being coupled to the transition energy. In principle, this distribution may reflect both intramolecular (structural) and intermolecular (i.e., solvent) disorder. However, the weak solvent dependence of the electronic spectra (Fig. 2 and 3) infers an intramolecular origin. We return to this point below.
The separation of vibronic peaks in the low temperature emission shows a ground state spacing of 1460 ± 30 cm−1, which is in the region expected for a CC stretch. A more detailed analysis is possible by a Franck–Condon–Hertzberg–Teller simulation, which is shown in Fig. 9. Good agreement with the low temperature spectra is obtained. In particular, the spectra for rK1− are dominated by the 0–0 transition, while the rK1H spectra has more marked vibronic structure, consistent with the data in Fig. 8. Analysis shows that the vibronic activity is indeed dominated by CC stretching modes, notably those associated with the bridging bonds. Spectra were also simulated for the equilibrium geometry at T = 300 K, which showed the expected enhanced activity in low frequency modes (hot bands) that broaden the vibronic transitions (ESI,† Fig. S10). However, the extent of the broadening in the experimental spectra (Fig. 2 and 3) was not reproduced. Thus, the temperature-dependent spectral broadening is more consistent with contributions from a range of structures (both intramolecular, e.g. about the CC single bonds, and intermolecular, e.g. solvent – solute orientations, H-bonding interactions, etc.).
 | ||
| Fig. 9 Franck–Condon–Herzberg–Teller simulations of the absorption (black) and emission (red) spectra, at T = 0 K, for (a) rK1H and (b) rK1−. The 0–0 transition has been shifted to match the experimental spectra. | ||
Finally, the fluorescence lifetimes of both rK1H and rK1− were measured at T = 77 K using the time correlated single photon counting method. As expected from the high fluorescence quantum yield, the lifetime is in the nanosecond range, at 3.2 ns for rK1H and 3.3 ns for rK1−. Significantly, and in sharp contrast to the room temperature result, the fluorescence decay at T = 77 K is a single exponential (ESI,† Fig. S11).
Discussion
The apparently straightforward weakly solvent-dependent electronic spectra of rK1H and rK1− hide surprisingly complex excited-state relaxation dynamics. The relaxation pathway was shown to be internal conversion but, at room temperature, the excited-state decay is wavelength dependent and is clearly multi-exponential, with lifetime components ranging from subpicosecond to tens of picoseconds. These dynamics were shown to be a property of the isolated molecule in solution rather than originating from ground or excited state intermolecular interactions. Further, the NMR data show that, on average, there is only a single conformer in the ground state, although the broad featureless electronic spectra at room temperature are indicative of a broad thermal distribution of structures about that equilibrium conformation.A model which reproduces all the experimental observations is illustrated in Fig. 10. At room temperature in the electronic ground state, there is a single equilibrium conformer that samples a broad distribution of intermolecular structures and solvent-chromophore orientations. We propose that, in the excited state, the barriers to accessing other conformations are lower, so that multiple minima are accessible. The Franck–Condon excited state is initially populated at t = 0 with the broad room temperature distribution from the ground state. This initial distribution can relax in <100 fs to directly populate the different minima on the excited state surface, consistent with the absence of a risetime in fluorescence at any wavelength. The complex multi-exponential decay kinetics observed in both TRF and TA experiments then arise from each minimum being associated with a different spectrum and lifetime. The absence of a risetime suggests these states do not interconvert on the picosecond timescale, which in-turn suggests interconformer barriers that are >kT. The wavelength dependence of the fluorescence (Fig. 5b) showed that blue emitting states have a shorter lifetime than red emitting states. This difference in lifetime can arise from the distinct minima having different energies and pathways to the conical intersection(s) leading to internal conversion. The population of multiple excited state minima results in multiple activation energies, with each minimum having a different potential barrier to a conical intersection with S0 and its own dependence on the medium viscosity (depending on, amongst other factors, the solvent volume displaced along the pathway); this is illustrated schematically in Fig. 10.
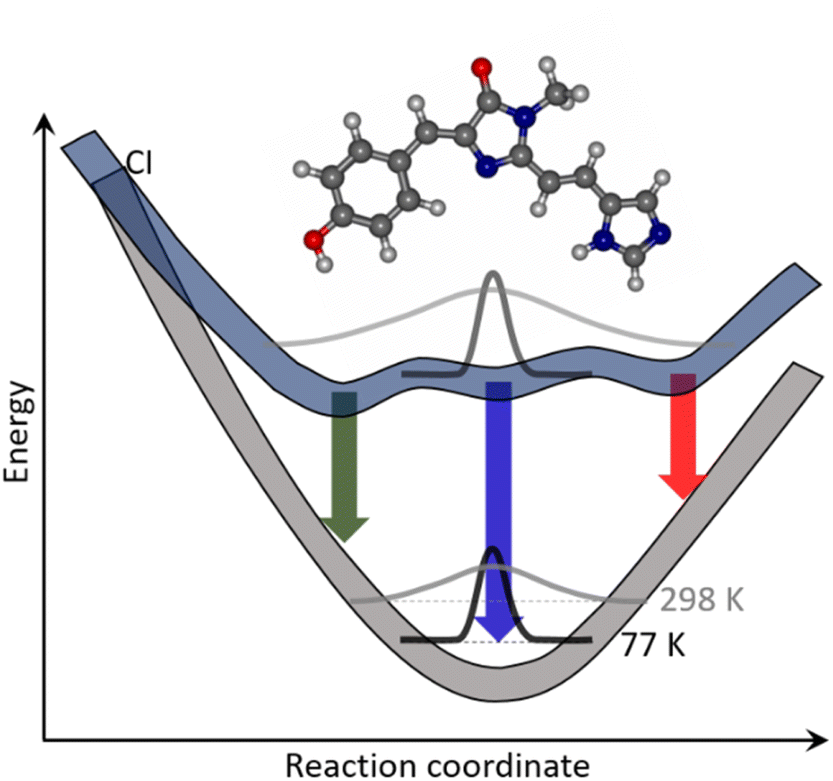 | ||
| Fig. 10 Schematic potential energy surfaces (blue lines) illustrating the origins of observed complex photophysics of rK1H and rK1−. A single ground state conformer exists, but with a distribution of geometries (including solvent molecules) around the average conformation due to a shallow potential energy surface. This distribution is broader at higher temperature (grey Gaussian lines). Electronic excitation transfers this distribution to the Franck–Condon excited state. The excited state surface is more complex, with multiple minima separated by low barriers. The broad distribution relaxes in <100 fs to directly populate different minima, where each decays with its distinct fluorescence spectrum (down arrows) and has a lifetime determined by its pathway and energy barrier to a conical intersection (CI) at which internal conversion occurs. At low temperature, the ground state distribution is narrowed and only a single excited state minimum is populated from the Franck–Condon state, which then decays with a long but single exponential decay time. | ||
As the temperature is reduced, the ground state distribution about the equilibrium position narrows. Consequently, on excitation to the Franck–Condon state, only a single excited state minimum is populated, which then emits back to the original ground state, showing vibronic structure. Both the low temperature and the very high viscosity near to the glass transition prevent access from the Franck–Condon state to any other minima on the excited state surface, and also to the conical intersection(s) that lead to internal conversion. As a result, the excited state decays with a nanosecond lifetime single exponential decay (Fig. S11†).
Finally, we note some similarities and differences between rK1H from Kaede and the more widely studied HBDI from GFP. The electronic spectra of rK1H are substantially red-shifted from those HBDI, as expected from the extended conjugation. Both have electronic spectra that display a weak dependence on solvent polarity,79 which indicates that intramolecular charge transfer does not make a major contribution to the excited state electronic structure (or at least does not lead to a large change in permanent dipole moment). However, the fluorescence quantum yield (and mean lifetime) of rK1H is in the few to tens of picosecond range, much longer than the sub-picosecond decay of HBDI.80,81 This can be traced to a greater activation energy for internal conversion and a greater sensitivity to solvent viscosity in rK1H. This, in turn, suggests differences in the coordinates leading to radiationless decay in the two chromophores. On formation of rK1−, the fluorescence lifetime and yield are decreased, which is opposite to HBDI.62 Most surprisingly, the fluorescence decay kinetics of rK1H (Fig. 5) are much more complex than those for HBDI. The decay of HBDI is also non-single exponential but the distribution of decay times is much narrower than in rK1H. In addition, unlike for rK1H, the HBDI fluorescence decay is wavelength independent.81 We assigned the complex behaviour of rK1H and rK1− to a broad distribution of ground state structures, which, on transfer to the excited state, directly populate distinct minima on the upper electronic state potential surface. Each of these minima has a different lifetime and spectrum. In contrast, HBDI is thought to have a single excited state structure that relaxes along an essentially barrierless pathway to one of two conical intersections.36,38 Understanding the apparently complex excited state potential of rK1H will require detailed quantum chemical calculations similar to those applied to HBDI. Significantly, there is one feature that both chromophores share: their fluorescence quantum yields and excited state dynamics are dramatically different in the protein compared to any solvent studied. This points to the exquisite control that the protein environments exercise over the photophysical properties of their respective chromophores.
Conclusion
A detailed investigation of the dynamics of the Kaede chromophore rK1H (and rK1−) reveal striking differences from the parent chromophore, HBDI. Although the electronic spectra are broad, featureless, and only a weak function of solvent polarity, ultrafast fluorescence reveals that the excited state decay dynamics are remarkably complex. This is confirmed by femtosecond transient absorption and stimulated Raman spectroscopy, which further show that the decay pathway is internal conversion, and that the distribution of lifetimes is associated with chemically similar species. Temperature-dependent fluorescence reveals activation energies to internal conversion and inhomogeneous broadening at higher temperatures. The data are adequately described by a model which invokes a single minimum ground state and a complex multi-minima excited state. In the electronic ground state at room temperature a broad distribution of structures exists, which, on transfer to the Franck–Condon excited state directly relaxes into a range of minima that are isolated on the tens of picoseconds timescale. Each minimum decays with a characteristic spectrum and lifetime, leading to the observed complex kinetics and time-dependent emission spectrum.Methods
Spectroscopy
The ultrafast methods used here for fluorescence up-conversion,61 transient absorption82 and femtosecond stimulated Raman spectroscopy83 have all been described elsewhere and are further detailed in the ESI.† The sub nanosecond photo counting measurements at 77 K are also detailed in ESI.†Computational
Geometry optimisations and vibrational frequency calculations for rK1H and rK1− were performed at the ωB97X-D/cc-pVDZ and ωB97X-D/aug-cc-pVDZ level of theory, respectively, in Gaussian 16B.01.84 Franck–Condon–Herzberg–Teller simulations of absorption and emission spectra for the anionic and neutral species were performed at T = 0 K and T = 300 K.Data availability
All data are available on request to the authors.Author contributions
SRM conceived the project and wrote the draft. KA collected and analysed steady-state and time-resolved fluorescence data. PR collected and analysed time-resolved absorption and excited state Raman data and performed DFT/TDDFT calculations for geometry optimisation and calculating Raman spectra. PCBP synthesized the chromophore. GB and JR collected and analysed temperature dependent steady-state fluorescence data. KS collected and analysed viscosity, pH and solvent dependent steady-state fluorescence data. EKA and JNB simulated the absorption and emission spectra and carried out the 77 K photon counting experiment. All authors contributed to the discussion of the data and revising and editing the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to EPSRC for funding (EP/J009148/1, EP/R042357/1, EP/H025715/1).References
- D. M. Chudakov, S. Lukyanov and K. A. Lukyanov, Fluorescent proteins as a toolkit for in vivo imaging, Trends Biotechnol., 2005, 23, 605–613 CrossRef CAS PubMed
.
- W. B. Frommer, M. W. Davidson and R. E. Campbell, Genetically encoded biosensors based on engineered fluorescent proteins, Chem. Soc. Rev., 2009, 38, 2833–2841 RSC
- A. Miyawaki, A. Sawano and T. Kogure, Lighting up cells: labelling proteins with fluorophores, Nat. Rev. Mol. Cell Biol., 2003, S1–S7 CAS
- R. Y. Tsien, The green fluorescent protein, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed
- P. Dedecker, J. Hotta, R. Ando, A. Miyawaki, Y. Engelborghs and J. Hofkens, Fast and reversible photoswitching of the fluorescent protein Dronpa as evidenced by fluorescence correlation spectroscopy, Biophys. J., 2006, 91, L45–L47 CrossRef CAS PubMed
- S. Habuchi, R. Ando, P. Dedecker, W. Verheijen, H. Mizuno, A. Miyawaki and J. Hofkens, Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9511–9516 CrossRef CAS PubMed
- M. Kaucikas, M. Tros and J. J. van Thor, Photoisomerization and Proton Transfer in the Forward and Reverse Photoswitching of the Fast-Switching M159T Mutant of the Dronpa Fluorescent Protein, J. Phys. Chem. B, 2015, 119, 2350–2362 CrossRef CAS PubMed
- M. M. Warren, M. Kaucikas, A. Fitzpatrick, P. Champion, J. Timothy Sage and J. J. van Thor, Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa, Nat. Commun., 2013, 4, 1461 CrossRef PubMed
- A. Lukacs, A. Haigney, R. Brust, K. Addison, M. Towrie, G. M. Greetham, G. A. Jones, A. Miyawaki, P. J. Tonge and S. R. Meech, Protein Photochromism Observed by Ultrafast Vibrational Spectroscopy, J. Phys. Chem. B, 2013, 117, 11954–11959 CrossRef CAS PubMed
- S. P. Laptenok, A. A. Gil, C. R. Hall, A. Lukacs, J. N. Iuliano, G. A. Jones, G. M. Greetham, P. Donaldson, A. Miyawaki, P. J. Tonge and S. R. Meech, Infrared spectroscopy reveals multi-step multi-timescale photoactivation in the photoconvertible protein archetype dronpa, Nat. Chem., 2018, 10, 845–852 CrossRef CAS PubMed
- N. Coquelle, M. Sliwa, J. Woodhouse, G. Schiro, V. Adam, A. Aquila, T. R. M. Barends, S. Boutet, M. Byrdin, S. Carbajo, E. De la Mora, R. B. Doak, M. Feliks, F. Fieschi, L. Foucar, V. Guillon, M. Hilpert, M. S. Hunter, S. Jakobs, J. E. Koglin, G. Kovacsova, T. J. Lane, B. Levy, M. N. Liang, K. Nass, J. Ridard, J. S. Robinson, C. M. Roome, C. Ruckebusch, M. Seaberg, M. Thepaut, M. Cammarata, I. Demachy, M. Field, R. L. Shoeman, D. Bourgeois, J. P. Colletier, I. Schlichting and M. Weik, Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography, Nat. Chem., 2018, 10, 31–37 CrossRef CAS PubMed
- H. Mizuno, T. K. Mal, M. Walchli, A. Kikuchi, T. Fukano, R. Ando, J. Jeyakanthan, J. Taka, Y. Shiro, M. Ikura and A. Miyawaki, Light-dependent regulation of structural flexibility in a photochromic fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9227–9232 CrossRef CAS PubMed
- M. Andresen, A. C. Stiel, S. Trowitzsch, G. Weber, C. Eggeling, M. C. Wahl, S. W. Hell and S. Jakobs, Structural basis for reversible photoswitching in Dronpa, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13005–13009 CrossRef CAS PubMed
- M. Andresen, M. C. Wahl, A. C. Stiel, F. Grater, L. V. Schafer, S. Trowitzsch, G. Weber, C. Eggeling, H. Grubmuller, S. W. Hell and S. Jakobs, Structure and mechanism of the reversible photoswitch of a fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13070–13074 CrossRef CAS PubMed
- D. M. Shcherbakova, P. Sengupta, J. Lippincott-Schwartz and V. V. Verkhusha, Photocontrollable Fluorescent Proteins for Superresolution Imaging, Annu. Rev. Biophys., 2014, 43, 303–329 CrossRef CAS PubMed
- X. X. Zhou and M. Z. Lin, Photoswitchable Fluorescent Proteins: Ten Years of Colorful Chemistry and Exciting Applications, Curr. Opin. Chem. Biol., 2013, 17, 682–690 CrossRef CAS PubMed
- V. Adam, B. Moeyaert, C. C. David, H. Mizuno, M. Lelimousin, P. Dedecker, R. Ando, A. Miyawaki, J. Michiels, Y. Engelborghs and J. Hofkens, Rational Design of Photoconvertible and Biphotochromic Fluorescent Proteins for Advanced Microscopy Applications, Chem. Biol., 2011, 18, 1241–1251 CrossRef CAS PubMed
- R. Ando, H. Hama, M. Yamamoto-Hino, H. Mizuno and A. Miyawaki, An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12651–12656 CrossRef CAS PubMed
- H. Mizuno, T. K. Mal, K. I. Tong, R. Ando, T. Furuta, M. Ikura and A. Miyawakil, Photo-induced peptide cleavage in the green-to-red conversion of a fluorescent protein, Mol. Cell, 2003, 12, 1051–1058 CrossRef CAS PubMed
- I. Hayashi, H. Mizuno, K. I. Tong, T. Furuta, F. Tanaka, M. Yoshimura, A. Miyawaki and M. Ikura, Crystallographic evidence for water-assisted photo-induced peptide cleavage in the stony coral fluorescent protein kaede, J. Mol. Biol., 2007, 372, 918–926 CrossRef CAS PubMed
- H. Tsutsui, H. Shimizu, H. Mizuno, N. Nukina, T. Furuta and A. Miyawaki, The E1 Mechanism in Photo-Induced beta-Elimination Reactions for Green-to-Red Conversion of Fluorescent Proteins, Chem. Biol., 2009, 16, 1140–1147 CrossRef CAS PubMed
- M. Lelimousin, V. Adam, G. U. Nienhaus, D. Bourgeois and M. J. Field, Photoconversion of the Fluorescent Protein EosFP: A Hybrid Potential Simulation Study Reveals Intersystem Crossings, J. Am. Chem. Soc., 2009, 131, 16814–16823 CrossRef CAS PubMed
- N. Griffiths, E.-A. Jaipargas, M. R. Wozny, K. A. Barton, N. Mathur, K. Delfosse and J. Mathur, Photo-convertible fluorescent proteins as tools for fresh insights on subcellular interactions in plants, J. Microsc., 2016, 263, 148–157 CrossRef CAS PubMed
- K. L. Litvinenko, N. M. Webber and S. R. Meech, Internal conversion in the chromophore of the green fluorescent protein: Temperature dependence and isoviscosity analysis, J. Phys. Chem. A, 2003, 107, 2616–2623 CrossRef CAS
- S. R. Meech, Excited state reactions in fluorescent proteins, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC
- P. J. Tonge and S. R. Meech, Excited state dynamics in the green fluorescent protein, J. Photochem. Photobiol., A, 2009, 205, 1–11 CrossRef CAS
- N. M. Webber, K. L. Litvinenko and S. R. Meech, Radiationless relaxation in a synthetic analogue of the green fluorescent protein chromophore, J. Phys. Chem. B, 2001, 105, 8036–8039 CrossRef CAS
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, Conformationally Locked Chromophores as Models of Excited-State Proton Transfer in Fluorescent Proteins, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed
- J. Conyard, M. Kondo, I. A. Heisler, G. Jones, A. Baldridge, L. M. Tolbert, K. M. Solntsev and S. R. Meech, Chemically Modulating the Photophysics of the GFP Chromophore, J. Phys. Chem. B, 2011, 115, 1571–1576 CrossRef CAS PubMed
- J. Dong, K. M. Solntsev and L. M. Tolbert, Solvatochromism of the green fluorescence protein chromophore and its derivatives, J. Am. Chem. Soc., 2006, 128, 12038–12039 CrossRef CAS PubMed
- K. M. Solntsev, O. Poizat, J. Dong, J. Rehault, Y. B. Lou, C. Burda and L. M. Tolbert, Meta and para effects in the ultrafast excited-state dynamics of the green fluorescent protein chromophores, J. Phys. Chem. B, 2008, 112, 2700–2711 CrossRef CAS PubMed
- A. D. Kummer, C. Kompa, H. Niwa, T. Hirano, S. Kojima and M. E. Michel-Beyerle, Viscosity-dependent fluorescence decay of the GFP chromophore in solution due to fast internal conversion, J. Phys. Chem. B, 2002, 106, 7554–7559 CrossRef CAS
- S. A. Boulanger, C. Chen, L. T. Tang, L. D. Zhu, N. S. Baleeva, I. N. Myasnyanko, M. S. Baranov and C. Fang, Shedding light on ultrafast ring-twisting pathways of halogenated GFP chromophores from the excited to ground state, Phys. Chem. Chem. Phys., 2021, 23, 14636–14648 RSC
- M. A. Taylor, L. D. Zhu, N. D. Rozanov, K. T. Stout, C. Chen and C. Fang, Delayed vibrational modulation of the solvated GFP chromophore into a conical intersection, Phys. Chem. Chem. Phys., 2019, 21, 9728–9739 RSC
- C. M. Jones, N. H. List and T. J. Martinez, Resolving the ultrafast dynamics of the anionic green fluorescent protein chromophore in water, Chem. Sci., 2021, 12, 11347–11363 RSC
- N. H. List, C. M. Jones and T. J. Martinez, Internal conversion of the anionic GFP chromophore: in and out of the I-twisted S-1/S-0 conical intersection seam, Chem. Sci., 2022, 13, 373–385 RSC
- A. Toniolo, S. Olsen, L. Manohar and T. J. Martinez, Conical intersection dynamics in solution: the chromophore of Green Fluorescent Protein, Faraday Discuss., 2004, 127, 149–163 RSC
- M. E. Martin, F. Negri and M. Olivucci, Origin, nature, and fate of the fluorescent state of the green fluorescent protein chromophore at the CASPT2//CASSCF resolution, J. Am. Chem. Soc., 2004, 126, 5452–5464 CrossRef CAS PubMed
- A. Sinicropi, T. Andruniow, N. Ferre, R. Basosi and M. Olivucci, Properties of the emitting state of the green fluorescent protein resolved at the CASPT2//CASSCF/CHARMM level, J. Am. Chem. Soc., 2005, 127, 11534–11535 CrossRef CAS PubMed
- C. Filippi, M. Ziccheddu and F. Buda, Absorption Spectrum of the Green Fluorescent Protein Chromophore: A Difficult Case for ab Initio Methods?, J. Chem. Theory Comput., 2009, 5, 2074–2087 CrossRef CAS PubMed
- I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Nemukhin, Potential Energy Landscape of the Electronic States of the GFP Chromophore in Different Protonation Forms: Electronic Transition Energies and Conical Intersections, J. Chem. Theory Comput., 2010, 6, 2377–2387 CrossRef CAS PubMed
- S. Olsen and S. C. Smith, Bond selection in the photoisomerization reaction of anionic green fluorescent protein and kindling fluorescent protein chromophore models, J. Am. Chem. Soc., 2008, 130, 8677–8689 CrossRef CAS PubMed
- S. Olsen, L. Manohar and T. J. Martinez, Features of interest on the S-0 and S-1 potential energy surfaces of a model green fluorescent protein chromophore, Biophys. J., 2002, 82, 359A Search PubMed
- J. J. van Thor, Photoreactions and dynamics of the green fluorescent protein, Chem. Soc. Rev., 2009, 38, 2935–2950 RSC
- S. Oumouch, M. Bourotte, M. Schmitt and J.-J. Bourguignon, An Expeditious Synthesis of 2,4-Disubstituted 2-Imidazolin-5-ones, Synthesis, 2005, 2005, 25–27 Search PubMed
- I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Synthesis and properties of the red chromophore of the green-to-red photoconvertible fluorescent protein Kaede and its analogs, Bioorg. Chem., 2008, 36, 96–104 CrossRef CAS PubMed
- W.-T. Chuang, B.-S. Chen, K.-Y. Chen, C.-C. Hsieh and P.-T. Chou, Fluorescent protein red Kaede chromophore; one-step, high-yield synthesis and potential application for solar cells, Chem. Commun., 2009, 6982–6984, 10.1039/B908542D
- J. Tay, M. A. Parkes, K. Addison, Y. Chan, L. Zhang, H. C. Hailes, P. C. Bulman Page, S. R. Meech, L. Blancafort and H. H. Fielding, The Effect of Conjugation on the Competition between Internal Conversion and Electron Detachment: A Comparison between Green Fluorescent and Red Kaede Protein Chromophores, J. Phys. Chem. Lett., 2017, 765–771, DOI:10.1021/acs.jpclett.7b00174
- N. S. Baleeva, K. A. Myannik, I. V. Yampolsky and M. S. Baranov, Bioinspired Fluorescent Dyes Based on a Conformationally Locked Chromophore of the Fluorescent Protein Kaede, Eur. J. Org. Chem., 2015, 2015, 5716–5721 CrossRef CAS
- I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Synthesis and properties of the red chromophore of the green-to-red photoconvertible fluorescent protein Kaede and its analogs, Bioorg. Chem., 2008, 36, 96–104 CrossRef CAS PubMed
- S. O. Zaitseva, E. R. Zaitseva, A. Y. Smirnov, N. S. Baleeva and M. S. Baranov, Synthesis and Optical Properties of the New Kaede Chromophore Analog, Russ. J. Bioorg. Chem., 2020, 46, 120–123 CrossRef CAS
- E. K. Ashworth, M. H. Stockett, C. Kjaer, P. C. B. Page, S. R. Meech, S. B. Nielsen and J. N. Bull, Complexation of Green and Red Kaede Fluorescent Protein Chromophores by a Zwitterion to Probe Electrostatic and Induction Field Effects, J. Phys. Chem. A, 2022, 126, 1158–1167 CrossRef CAS PubMed
- N. J. A. Coughlan, M. H. Stockett, C. Kjaer, E. K. Ashworth, P. B. C. Page, S. R. Meech, S. B. Nielsen, L. Blancafort, W. S. Hopkins and J. N. Bull, Action spectroscopy of the isolated red Kaede fluorescent protein chromophore, J. Chem. Phys., 2021, 155, 124304 CrossRef CAS PubMed
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, Subpicosecond Measurements of Polar Solvation Dynamics - Coumarin-153 Revisited, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS
- P. Suppan, Invited review solvatochromic shifts: The influence of the medium on the energy of electronic states, J. Photochem. Photobiol., A, 1990, 50, 293–330 CrossRef CAS
- D. C. Todd and G. R. Fleming, Cis-Stilbene Isomerization - Temperature-Dependence and the Role of Mechanical Friction, J. Chem. Phys., 1993, 98, 269–279 CrossRef CAS
- S. P. Velsko, D. H. Waldeck and G. R. Fleming, Breakdown Of Kramer Theory Description Of Photochemical Isomerization And The Possible Involvement Of Frequency-Dependent Friction, J. Chem. Phys., 1983, 78, 249–258 CrossRef CAS
- M. J. van der Meer, H. Zhang and M. Glasbeek, Femtosecond fluorescence upconversion studies of barrierless bond twisting of auramine in solution, J. Chem. Phys., 2000, 112, 2878–2887 CrossRef CAS
- S. K. Kim and G. R. Fleming, Reorientation and Isomerization of Trans-Stilbene in Alkane Solutions, J. Phys. Chem., 1988, 92, 2168–2172 CrossRef CAS
- A. C. Benniston, P. Matousek, I. E. McCulloch, A. W. Parker and M. Towrie, Detailed picosecond Kerr-gated time-resolved resonance Raman spectroscopy and time-resolved emission studies of merocyanine 540 in various solvents, J. Phys. Chem. A, 2003, 107, 4347–4353 CrossRef CAS
- I. A. Heisler, M. Kondo and S. R. Meech, Reactive Dynamics in Confined Liquids: Ultrafast Torsional Dynamics of Auramine O in Nanoconfined Water in Aerosol OT Reverse Micelles, J. Phys. Chem. B, 2009, 113, 1623–1631 CrossRef CAS PubMed
- K. Addison, J. Conyard, T. Dixon, P. C. B. Page, K. M. Solntsev and S. R. Meech, Ultrafast Studies of the Photophysics of Cis and Trans States of the Green Fluorescent Protein Chromophore, J. Phys. Chem. Lett., 2012, 3, 2298–2302 CrossRef CAS PubMed
- P. Kukura, D. W. McCamant and R. A. Mathies, Annu. Rev. Phys. Chem., 2007, 58, 461–488 CrossRef CAS PubMed
- R. R. Frontiera and R. A. Mathies, Femtosecond stimulated Raman spectroscopy, Laser Photonics Rev., 2011, 5, 102–113 CrossRef CAS
- M. A. Taylor, L. Zhu, N. D. Rozanov, K. T. Stout, C. Chen and C. Fang, Delayed vibrational modulation of the solvated GFP chromophore into a conical intersection, Phys. Chem. Chem. Phys., 2019, 21, 9728–9739 RSC
- T. J. Quincy, M. S. Barclay, M. Caricato and C. G. Elles, Probing Dynamics in Higher-Lying Electronic States with Resonance-Enhanced Femtosecond Stimulated Raman Spectroscopy, J. Phys. Chem. A, 2018, 122, 8308–8319 CrossRef CAS PubMed
- C. R. Hall, A. S. Romanov, M. Bochmann and S. R. Meech, Ultrafast Structure and Dynamics in the Thermally Activated Delayed Fluorescence of a Carbene-Metal-Amide, J. Phys. Chem. Lett., 2018, 9, 5873–5876 CrossRef CAS PubMed
- B. G. Oscar, C. Chen, W. M. Liu, L. D. Zhu and C. Fang, Dynamic Raman Line Shapes on an Evolving Excited-State Landscape: Insights from Tunable Femtosecond Stimulated Raman Spectroscopy, J. Phys. Chem. A, 2017, 121, 5428–5441 CrossRef CAS PubMed
- M. S. Barclay, T. J. Quincy, D. B. Williams-Young, M. Caricato and C. G. Elles, Accurate Assignments of Excited-State Resonance Raman Spectra: A Benchmark Study Combining Experiment and Theory, J. Phys. Chem. A, 2017, 121, 7937–7946 CrossRef CAS PubMed
- D. Green, P. Roy, C. R. Hall, J. N. Iuliano, G. A. Jones, A. Lukacs, P. J. Tonge and S. R. Meech, Excited State Resonance Raman of Flavin Mononucleotide: Comparison of Theory and Experiment, J. Phys. Chem. A, 2021, 125, 6171–6179 CrossRef CAS PubMed
- T. Fujisawa, H. Kuramochi, H. Hosoi, S. Takeuchi and T. Tahara, Role of Coherent Low-Frequency Motion in Excited-State Proton Transfer of Green Fluorescent Protein Studied by Time-Resolved Impulsive Stimulated Raman Spectroscopy, J. Am. Chem. Soc., 2016, 138, 3942–3945 CrossRef CAS PubMed
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy, Nature, 2009, 462, 200–U274 CrossRef CAS PubMed
- J. W. Park and Y. M. Rhee, Electric Field Keeps Chromophore Planar and Produces High Yield Fluorescence in Green Fluorescent Protein, J. Am. Chem. Soc., 2016, 138, 13619–13629 CrossRef CAS PubMed
- C. Y. Lin, M. G. Romei, I. I. Mathews and S. G. Boxer, Energetic Basis and Design of Enzyme Function Demonstrated Using GFP, an Excited-State Enzyme, J. Am. Chem. Soc., 2022, 144, 3968–3978 CrossRef CAS PubMed
- M. G. Romei, C. Y. Lin, I. I. Mathews and S. G. Boxer, Electrostatic control of photoisomerization pathways in proteins, Science, 2020, 367, 76–79 CrossRef CAS PubMed
- T. J. Martínez, Insights for Light-Driven Molecular Devices from Ab Initio Multiple Spawning Excited-State Dynamics of Organic and Biological Chromophores, Acc. Chem. Res., 2006, 39, 119–126 CrossRef PubMed
- C. Ko, A. M. Virshup and T. J. Martínez, Electrostatic control of photoisomerization in the photoactive yellow protein chromophore: Ab initio multiple spawning dynamics, Chem. Phys. Lett., 2008, 460, 272–277 CrossRef CAS
- P. Roy, A. S. Sardjan, W. Danowski, W. R. Browne, B. L. Feringa and S. R. Meech, Photophysics of First-Generation Photomolecular Motors: Resolving Roles of Temperature, Friction, and Medium Polarity, J. Phys. Chem. A, 2021, 125, 1711–1719 CrossRef CAS PubMed
- N. M. Webber and S. R. Meech, Electronic Spectroscopy And Solvatochromism In The Chromophore Of GFP And The Y66F Mutant, Photochem. Photobiol. Sci., 2007, 9, 276–281 Search PubMed
- D. Mandal, T. Tahara and S. R. Meech, Excited-state dynamics in the green fluorescent protein chromophore, J. Phys. Chem. B, 2004, 108, 1102–1108 CrossRef CAS
- D. Mandal, T. Tahara, N. M. Webber and S. R. Meech, Ultrafast fluorescence of the chromophore of the green fluorescent protein in alcohol solutions, Chem. Phys. Lett., 2002, 358, 495–501 CrossRef CAS
- P. Roy, G. Bressan, J. Gretton, A. N. Cammidge and S. R. Meech, Ultrafast Excimer Formation and Solvent Controlled Symmetry Breaking Charge Separation in the Excitonically Coupled Subphthalocyanine Dimer, Angew. Chem., Int. Ed., 2021, 60, 10568–10572 CrossRef CAS PubMed
- C. R. Hall, J. Conyard, I. A. Heisler, G. Jones, J. Frost, W. R. Browne, B. L. Feringa and S. R. Meech, Ultrafast Dynamics in Light-Driven Molecular Rotary Motors Probed by Femtosecond Stimulated Raman Spectroscopy, J. Am. Chem. Soc., 2017, 139, 7408–7414 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00368j |
| ‡ Present address: School of Basic Sciences, Indian Institute of Technology Bhubaneswar, Odisha 752050, India. |
| This journal is © The Royal Society of Chemistry 2023 |