
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomically dispersed Ir catalysts exhibit support-dependent water oxidation kinetics during photocatalysis†
Hongna
Zhang
 a,
Tianying
Liu
a,
Nicholas
Dulock
a,
Benjamin P.
Williams
a,
Yuanxing
Wang
a,
Boqiang
Chen
a,
Haden
Wikar
a,
David Z.
Wang
a,
Gary W.
Brudvig
b,
Dunwei
Wang
*a and
Matthias M.
Waegele
*a
a,
Tianying
Liu
a,
Nicholas
Dulock
a,
Benjamin P.
Williams
a,
Yuanxing
Wang
a,
Boqiang
Chen
a,
Haden
Wikar
a,
David Z.
Wang
a,
Gary W.
Brudvig
b,
Dunwei
Wang
*a and
Matthias M.
Waegele
*a
aDepartment of Chemistry, Boston College, Merkert Chemistry Center, Chestnut Hill, Massachusetts 02467, USA. E-mail: dunwei.wang@bc.edu; waegele@bc.edu
bDepartment of Chemistry and Yale Energy Sciences Institute, Yale University, New Haven, Connecticut 06520-8107, USA
First published on 26th May 2023
Abstract
Heterogeneous water oxidation catalysis is central to the development of renewable energy technologies. Recent research has suggested that the reaction mechanisms are sensitive to the hole density at the active sites. However, these previous results were obtained on catalysts of different materials featuring distinct active sites, making it difficult to discriminate between competing explanations. Here, a comparison study based on heterogenized dinuclear Ir catalysts (Ir-DHC), which feature the same type of active site on different supports, is reported. The prototypical reaction was water oxidation triggered by pulsed irradiation of suspensions containing a light sensitizer, Ru(bpy)32+, and a sacrificial electron scavenger, S2O82−. It was found that at relatively low temperatures (288–298 K), the water oxidation activities of Ir-DHC on indium tin oxide (ITO) and CeO2 supports were comparable within the studied range of fluences (62–151 mW cm−2). By contrast, at higher temperatures (310–323 K), Ir-DHC on ITO exhibited a ca. 100% higher water oxidation activity than on CeO2. The divergent activities were attributed to the distinct abilities of the supporting substrates in redistributing holes. The differences were only apparent at relatively high temperatures when hole redistribution to the active site became a limiting factor. These findings highlight the critical role of the supporting substrate in determining the turnover at active sites of heterogeneous catalysts.
Introduction
The oxygen evolution reaction (OER) provides electrons and protons required in fuel-forming reduction reactions such as hydrogen production1,2 and CO2 fixation3 and is central to the development of artificial photosynthesis.4,5 However, the OER also represents a major challenge as this reaction involves four electrons and four protons and is often sluggish.6 Despite decades of research, a heterogeneous catalyst that is inexpensive, durable, and fast in oxidizing water remains elusive. At the heart of the problem is an insufficient understanding of the reaction mechanisms.The dependence of the mechanism and rate on the surface hole concentration is one key example. Recent studies have shown that the chemical and energetic properties of surface holes on various catalysts are central to understanding the OER mechanisms.7–12 For instance, high concentrations of O-centered holes, created by leaching Ni from IrNiOx, were found to enhance the activity, with IrNiOx being 1.7 times more active than IrOx at an overpotential of 0.3 V.13 Other studies found that the reaction order with respect to holes changes from first-to third-order with increasing hole density at various metal-oxide electrocatalysts, thereby providing a more facile reaction mechanism at higher hole density.10,14,15 Similarly, we have reported a potential-induced mechanistic switch of the OER on Co-oxide based electrocatalysts.16 These results underscore the fundamental importance of hole distribution dynamics among surface sites and surface hole density in OER catalysis.
However, an understanding of the material properties that favor or disfavor hole accumulation on active sites is largely missing. This knowledge is difficult to obtain because these prior studies were carried out on catalysts composed of different materials, making it challenging to discern whether the observed phenomenon was due to the difference in the chemical catalytic properties or distinct charge redistribution behaviors of the materials.
To answer this question, here we employed a novel catalytic system in which the hole distribution properties could be tuned while preserving the same type of active site, which was a molecularly-derived, Ir-based dinuclear heterogeneous catalyst (Ir-DHC). When supported on CeO2 and indium tin oxide (ITO), a drastically different dependence of the reaction kinetics on temperature was observed. At low temperatures (288–298 K), the catalytic activities of Ir-DHC on the two different supports were comparable. By contrast, at elevated temperatures (310–323 K), the OER activities of Ir-DHC on ITO were ca. 100% higher than that of Ir-DHC on CeO2. We rationalized these results in terms of the disparate hole redistribution dynamics of the supports. At low temperatures, the rate of the OER is determined by a thermal barrier associated with the water oxidation chemistry on the active site, rendering the rate insensitive to the hole redistribution dynamics of the support. At elevated temperatures, the thermal barrier is readily overcome, whereas the sluggish hole redistribution in CeO2 limits the rate of OER for Ir-DHC on CeO2. Our results demonstrate that the support plays a key role in channeling holes to the active site and in determining the overall OER behaviors.
Experimental
Materials
For catalyst synthesis, the following chemicals were obtained from commercial sources and used without further purification unless otherwise specified: 2-(pyridine-2-yl)propan-2-ol (95%, Strem Chemicals), dichloro-(pentamethylcyclopentadienyl)-iridium(III) dimer ([Cp*IrCl2]2, 98%, Strem Chemicals), sodium bicarbonate (99.7%, Sigma), acetone (99.9%, Sigma), magnesium sulfate anhydrous (95%, Fisher), NaIO4 (99.8%, Sigma), ITO (99.5%, light green, 17–28 nm particle size, 30–50 m2 g−1, Alfa Aesar) and CeO2 (light yellow, <25 nm particle size, 55 m2 g−1, Sigma).For the photocatalytic system, the following chemicals were used: sodium sulfate decahydrate (>99%, Acros Organics), sodium bicarbonate (>99.7%, powder, Sigma-Aldrich), tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate (>98%, Strem Chemicals), sodium hexafluorosilicate (powder, Aldrich Chemistry), sodium persulfate (>99.7%, Honeywell).
Synthesis of Ir-DHC
The precursor, Cp*Ir[pyalc(Cl)] (Cp*: pentamethylcyclopentadienyl, pyalc:2-(2′-pyridyl)-2-propanoate), was synthesized as reported.17 Briefly, [Cp*IrCl2]2 (0.10265 g), sodium bicarbonate (0.08625 g), and 2-(pyridine-2-yl)propan-2-ol (0.0355 g) were combined in a 100 mL flask with an attached reflux condenser, and 12.5 mL of degassed acetone was added. The air in the flask was removed by connecting the flask to vacuum and refilling it with N2; this process was repeated 3 times. The reaction mixture was then heated to reflux for 2 h under a slow flow of nitrogen gas through the reaction vessel's headspace. The solution lightened in color to a pale orange during the reaction. Then, the solution was cooled to room temperature and treated with a small quantity of anhydrous magnesium sulfate to remove excess water. The resulting solution was concentrated on a rotary evaporator to a total volume of ca. 2 mL, then stored in the freezer overnight, yielding orange crystals that were separated from the remaining supernatant and were then dried in a vacuum oven at 313 K.A desired amount of Cp*Ir[pyalc(Cl)] (0.024 g for deposition on CeO2; 0.0088 g for deposition on ITO) was first dissolved in 50.0 mL water, forming a red solution. The loading of Ir-DHC depends on the adsorption of the dimeric precursor on the support. The amount of Cp*Ir[pyalc(Cl)] was chosen to achieve a desired loading of 0.1% Ir (by weight). Subsequently, dimerization of Cp*Ir[pyalc(Cl)] in solution was induced by adding 1.07 g of NaIO4. The reaction mixture was stirred for 2 h at room temperature, yielding a blue solution, indicative of Cp*Ir[pyalc(Cl)] dimerization. The desired oxide powder (0.780 g for CeO2 or ITO) was soaked in the blue Ir dimer solution for 24 h and then thoroughly rinsed with deionized water, producing the heterogenized Ir catalyst. The organic ligands of the heterogenized Ir catalyst were removed by treating the sample with ultraviolet (UV) light (240 W) in air and at room temperature for 20 min in a UV ozone cleaner system (Jelight Company, Inc.).
Catalyst characterization
In a typical experiment, the DRIFTS reactor was loaded with ca. 60 mg of catalyst powder. Under Ar flow (50 standard cubic feet per min (sccm), 99.99%; Airgas), the reaction chamber was heated to 423 K for 2 h to remove volatile species from the catalyst's surface and was then cooled to 298 K. Under constant Ar flow and at 298 K, single-beam reference spectra were collected. Then, the gas stream was switched to CO (10 sccm, 99.9%; Airgas) for 30 min. The gas stream was switched back to pure Ar for 30 min to remove gas-phase CO and reversibly bound CO. Afterwards, single-beam sample spectra of irreversibly bound CO on the catalyst were collected.
1.5 mL of a freshly prepared catalyst suspension (10 mg mL−1) was added to a cylindrical quartz vessel (diameter of 1 cm), whose temperature was controlled by a water-jacket. Under stirring (150 rotations per min), the suspension was allowed to equilibrate to the desired temperature for 10 min. Then, Ar was bubbled through the suspension for 3 min to remove dissolved O2. After a steady baseline in O2 concentration was achieved as monitored with the Clark electrode, the cell was sealed with a rubber stopper. Water oxidation was initiated with a 1 s light pulse (beam diameter = 7 mm (1/e2 value); wavelength = 450 nm) from a laser diode (Thorlabs, model number: L450P1600MM). The ensuing evolution of O2 was monitored over 1.5 min. The initial rate of O2 evolution was obtained by fitting a linear function to the initial rise of the O2 concentration.
Results and discussion
This work was enabled by a unique catalyst design as illustrated in Fig. 1A. The active center of the catalyst consists of two Ir cations bridged by oxygen and is stably bound to a metal-oxide support. The synthesis protocol of Ir-DHC supported on various oxides was reported previously17,21–23 and is described in the Experimental section.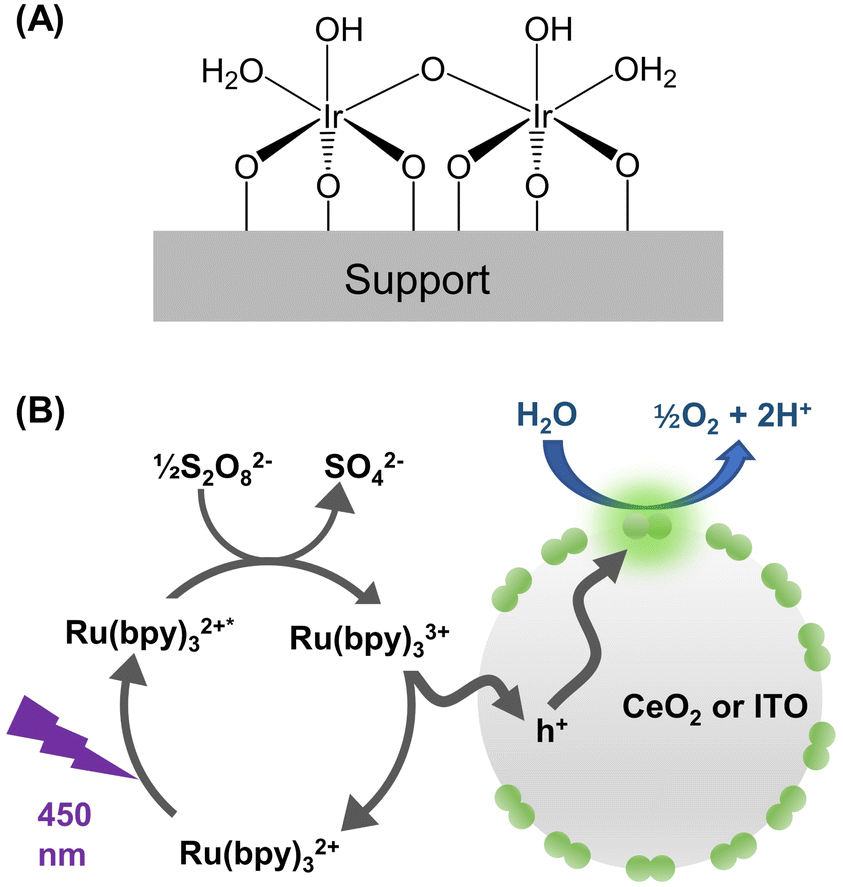 | ||
| Fig. 1 Design of the catalyst and photochemical water oxidation. (A) Catalyst structure as established in ref. 23. (B) Scheme showing the photocatalytic cycle involving excitation of the light absorber ([Ru(bpy)3]2+), oxidation of the triplet state of the light absorber ([Ru(bpy)3]2+*), and subsequent hole transfer to the metal oxide nanoparticle (grey circle) decorated with Ir-DHC (green pairs of circles). | ||
For this work, we focused on two types of supports, CeO2 and ITO, to take advantage of their distinctly different hole distribution properties (vide infra). The dispersed nature of Ir-DHC was confirmed by DRIFTS of Ir-bound CO (Fig. S1 of the ESI†). A detailed discussion of the spectra and their interpretation is provided in Note S1 of the ESI.† The infrared spectra and XPS (Fig. S2 and Note S2 of ESI†) of the catalyst before and after catalysis suggested that the catalyst structure was stable under photocatalytic conditions.
Water oxidation activity
We next illuminated the samples to induce photocatalysis (Fig. 1B). Our goal was to quantify the initial rates of O2 formation as a measure of the OER kinetics. Briefly, the Ir-DHC-loaded catalysts (in powder form) were suspended in a pH-buffered aqueous solution (pH 5.7) containing a light absorber, tris(bipyridine)-ruthenium ([Ru(bpy)3]2+), and a sacrificial reagent, persulfate (S2O82−). Following excitation of the suspension with a monochromatic light pulse at 450 nm (duration: 1 s), the rate of O2 formation was monitored by a Clark electrode. Representative traces of O2 evolution are shown in Fig. S3 and S4 of the ESI.† The initial rate was determined by linear regression of the initial rise of O2 formation. Further details of the experiment and analysis are described in the Experimental section and Note S3 of the ESI.† All rates presented in this work represented the initial OER rates obtained on fresh catalysts. Prior research has shown that anions, such as SO42−, could affect the water oxidation kinetics of Ir catalysts.24 Therefore, we studied fresh catalysts to avoid confounding influences from exposure to different ligands.It is apparent from Fig. 2A and B that the OER kinetics of Ir-DHC on CeO2 and ITO (labeled as Ir-DHC/CeO2 and Ir-DHC/ITO, respectively) were comparable at low temperatures (≤298 K). For instance, at 298 K with a fluence of 151 mW cm−2, a turnover frequency (TOF) per Ir atom of ca. 0.040 s−1 was measured on Ir-DHC/CeO2, compared to ca. 0.037 s−1 on Ir-DHC/ITO. These values are also comparable to other reported values of Ir-based catalysts measured under similar conditions.20,25–27 Although higher TOFs have been reported on Ir catalysts, they were measured under electrocatalytic conditions, which are fundamentally different from the testing conditions employed here. It is noted that the absolute measured rates may also depend on the details of the experimental setup. We focus here on the relative rates rather than the absolute values. Within this context, when the temperature was increased to 310 K, a clear difference was observed. With a fluence of 151 mW cm−2, a TOF of ca. 0.126 s−1 was measured on Ir-DHC/ITO, compared with ca. 0.060 s−1 on Ir-DHC/CeO2. A similar difference was observed at 323 K, as well. To better illustrate the stark contrast, we plot the ratios of the OER rates between Ir-DHC/ITO and Ir-DHC/CeO2 in Fig. 2C. It can be seen that Ir-DHC/ITO consistently exhibited a faster OER than Ir-DHC/CeO2 by a factor of ca. 2 at temperatures ≥310 K.
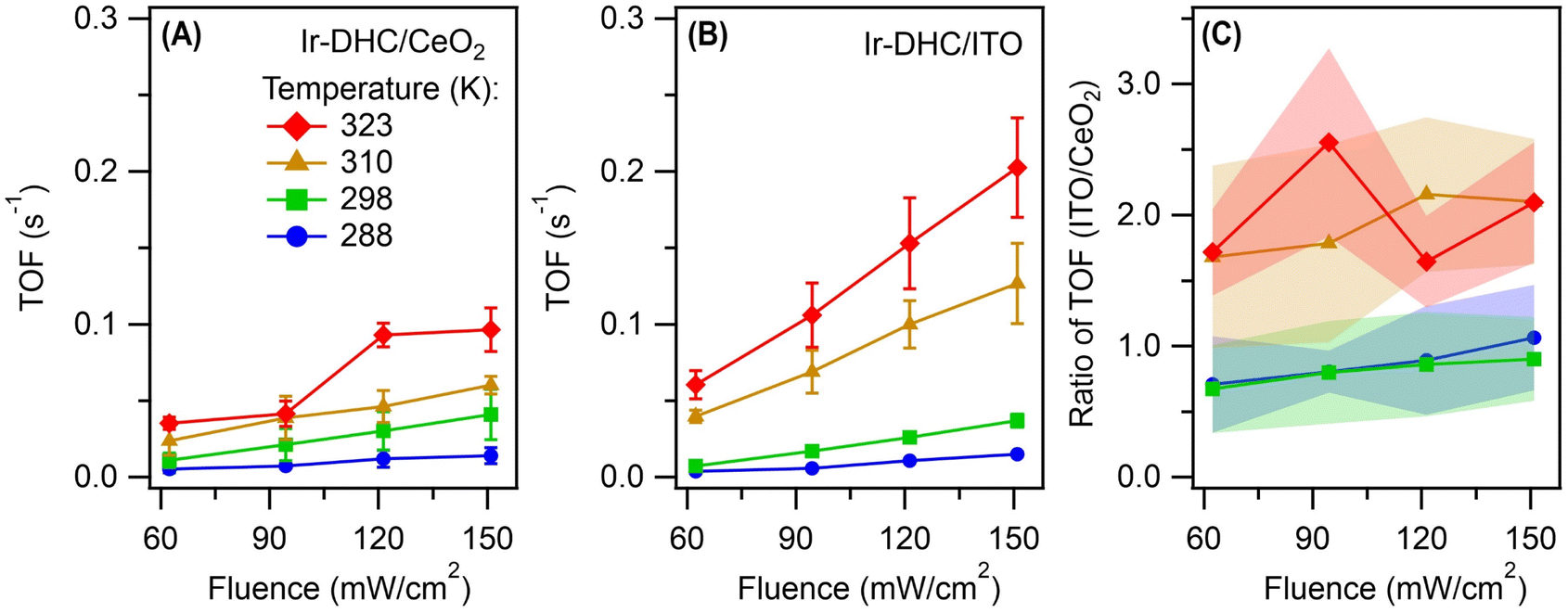 | ||
| Fig. 2 Comparison of catalyst performance. Turnover frequencies (TOFs) per Ir atom derived from the initial rates of water oxidation on (A) Ir-DHC/CeO2 and (B) Ir-DHC/ITO as a function of fluence and temperature. (C) A direct comparison of OER kinetics between Ir-DHC on CeO2 and ITO at different temperatures. Error bars and bands represent standard errors and were calculated on the basis of at least three independent experiments using fresh catalysts in fresh solutions. The legend for all panels is given in panel (A). The methods of data analysis are described in Note S3 of the ESI.† | ||
Hole transfer
To rationalize the observations summarized in Fig. 2, we considered the water oxidation process as discribed in Fig. 1B.28–30 The transfer of holes from [Ru(bpy)3]3+ to the catalyst was first examined. Direct hole transfer from SO4˙− radicals, which intermediately formed in the photocatalytic cycle, was previously shown to be negligible under similar experimental conditions.20,31,32 The amount of photogenerated [Ru(bpy)3]3+ was determined by the amplitude of the 1496 cm−1 band of the dye (Fig. S5 in the ESI†).32,33 Within a 200 ms photolysis pulse, similar amounts of [Ru(bpy)3]3+ were generated in the presence of the bare CeO2 or ITO supports, as well as in their absence. By contrast, in the presence of Ir-DHC/CeO2 or Ir-DHC/ITO, the amplitude of the 1496 cm−1 was significantly reduced, suggesting fast transfer of holes from [Ru(bpy)3]3+ to the Ir-DHC decorated supports. These results are consistent with prior reports that hole transfer from [Ru(bpy)3]3+ to IrOx catalysts occurs within <200 μs for electrostatically adsorbed dye molecules and in a few milliseconds for freely diffusing dyes.20 Taken together, these results suggest that the disparate water oxidation kinetics arise from steps that occur after the transfer of holes to the catalysts.The holes could transfer to the support and/or directly to Ir-DHC; hole transfer from [Ru(bpy)3]3+ to the bare supports may not manifest itself in our measurements because of possible fast back transfer to the dye in the absence of Ir-DHC. Irrespective of the site of hole transfer, once on the catalyst surface, the holes are expected to undergo dynamic redistribution among surface sites,15 a process that has also been termed as “surface hole hopping.”34 The accumulation of holes at active sites has been suggested as a key factor determining water oxidation rates.13,15,16,35–37 Efficient redistribution of surface holes is thus essential for a sufficient number of holes to accumulate on a single active site to trigger water oxidation. Therefore, we hypothesized that different hole redistribution behavior in CeO2 and ITO may give rise to the distinct water oxidation kinetics.
A critical difference between CeO2 and ITO is that the former is reducible because both Ce4+ and Ce3+ are stable cations.38,39 We therefore hypothesized that the presence of Ce3+ may hinder hole redistribution. To test this hypothesis, we studied the water oxidation activity of another Ir-DHC/CeO2 sample, the Ce3+ concentration of which was increased by annealing of commercial CeO2 in H2.38,39 Annealing was carried out prior to loading Ir-DHC. In the following, the annealed sample is referred to as Ir-DHC/CeO2,red. The properties of Ir-DHC/CeO2 and Ir-DHC/CeO2,red were characterized with XPS and XRD (Fig. S6 of the ESI†). As discussed in Note S2 of the ESI,† the data indicated that annealing increased the content of Ce3+ from ca. 9 to ca. 17% under preservation of the crystal structure. Consistent with our hypothesis, the activity of Ir- DHC/CeO2,red at 323 K is lower relative to that of Ir-DHC/CeO2 (Fig. S6D of the ESI†). These results indicate that the presence of Ce3+ sites hinders hole redistribution, thereby giving rise to the lower activity of Ir-DHC/CeO2 compared with that of Ir-DHC/ITO.
In summary, at relatively low temperatures (e.g., ≤298 K), when the rate of water oxidation by Ir is slow, the difference between hole redistribution by CeO2 and ITO is concealed. That is why the apparent OER rates for Ir-DHC/CeO2 and Ir-DHC/ITO are comparable at 288 K and 298 K for all fluences tested (Fig. 2). The slight depression of the TOF for Ir-DHC/ITO relative to that of Ir-DHC/CeO2 at low temperatures and fluences is likely due to a low background activity of CeO2 for water oxidation. At relatively high temperatures (e.g., ≥310 K), when the rate of water oxidation is fast, the hole redistribution between the support and Ir active center becomes a limiting factor. This understanding explains the observation made in Fig. 2 and is summarized in Fig. 3. Subtle differences in the local environment and coordination of Ir-DHC sites may also contribute to the observed differences.
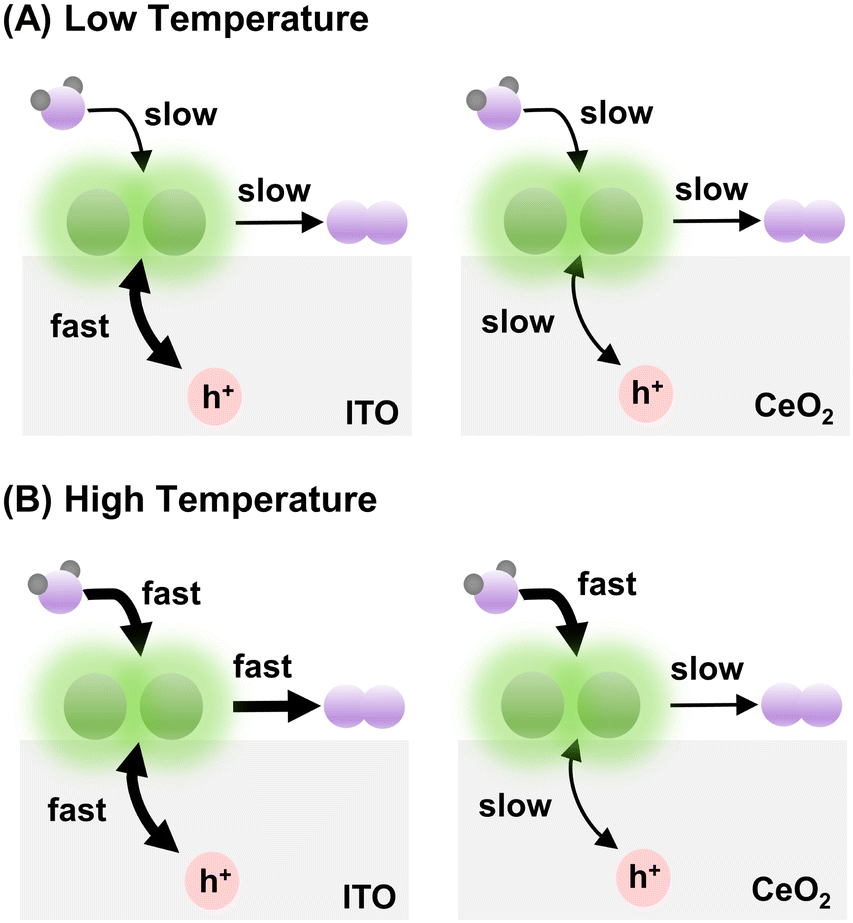 | ||
| Fig. 3 Illustration of support-dependent OER activity. (A) At low temperatures, the overall OER activity is limited by the surface chemical step, concealing the differences of hole redistribution between CeO2 and ITO and yielding comparable OER activities for Ir-DHC/ITO and Ir-DHC/CeO2. (B) At high temperatures, the surface chemistry is accelerated, revealing the differences of hole redistribution between CeO2 and ITO and yielding faster overall OER activities by Ir-DHC/ITO than Ir-DHC/CeO2 (Green ball pairs represent Ir-DHC; purple and grey balls represent O atoms and H atoms, respectively.). | ||
Temperature dependence
A careful examination of Fig. 2C reveals a step in the rate of water oxidation for Ir-DHC/ITO between 298 K and 310 K. To further study this observation, we carried out water oxidation kinetics measurements between 278 K and 333 K with a 5 K increment at a fixed fluence of 151 mW cm−2. As shown in Fig. 4A, it was observed that below 308 K, the TOFs of Ir-DHC/ITO were indeed comparable to those of Ir-DHC/CeO2. However, at 308 K a step in the rate of water oxidation catalyzed by Ir-DHC/ITO was observed, corroborating the results shown in Fig. 2C. Arrhenius analysis of these data yielded apparent activation barriers (Eapp). As shown in Fig. 4B, an inflection point was observed at ca. 308 K for both Ir-DHC/ITO and Ir-DHC/CeO2. At temperatures lower than 303 K, the Eapp of 76 kJ mol−1 and 59 kJ mol−1 was obtained for Ir-DHC/ITO and Ir-DHC/CeO2, respectively; at temperatures above 308 K, the Eapp changed to 21 kJ mol−1 and 23 kJ mol−1 for Ir-DHC/ITO and Ir-DHC/CeO2, respectively. We caution that apparent activation barriers obtained by Arrhenius analysis for multi-step reactions can be complicated functions of various thermodynamic quantities of multiple steps. Therefore, a more detailed interpretation of these quantities requires in-depth knowledge of the underlying mechanisms that is currently missing. These complexities notwithstanding, the clear inflection at ca. 308 K is consistent with our hypothesis that a change of the rate determining step (RDS) takes place with the rise of temperature.40 The understanding as proposed here highlights that hole accumulation plays a central role in water oxidation kinetics, which has been a focal point of recent studies.13,15,16,36,37,41 It has been reported that when the surface hole concentration increases, the rate of water oxidation accelerates. While the mechanistic interpretation of the acceleration of the rates varies, a commonality is that the accumulation of surface holes on an active site facilitates water oxidation, thereby accelerating the overall rate.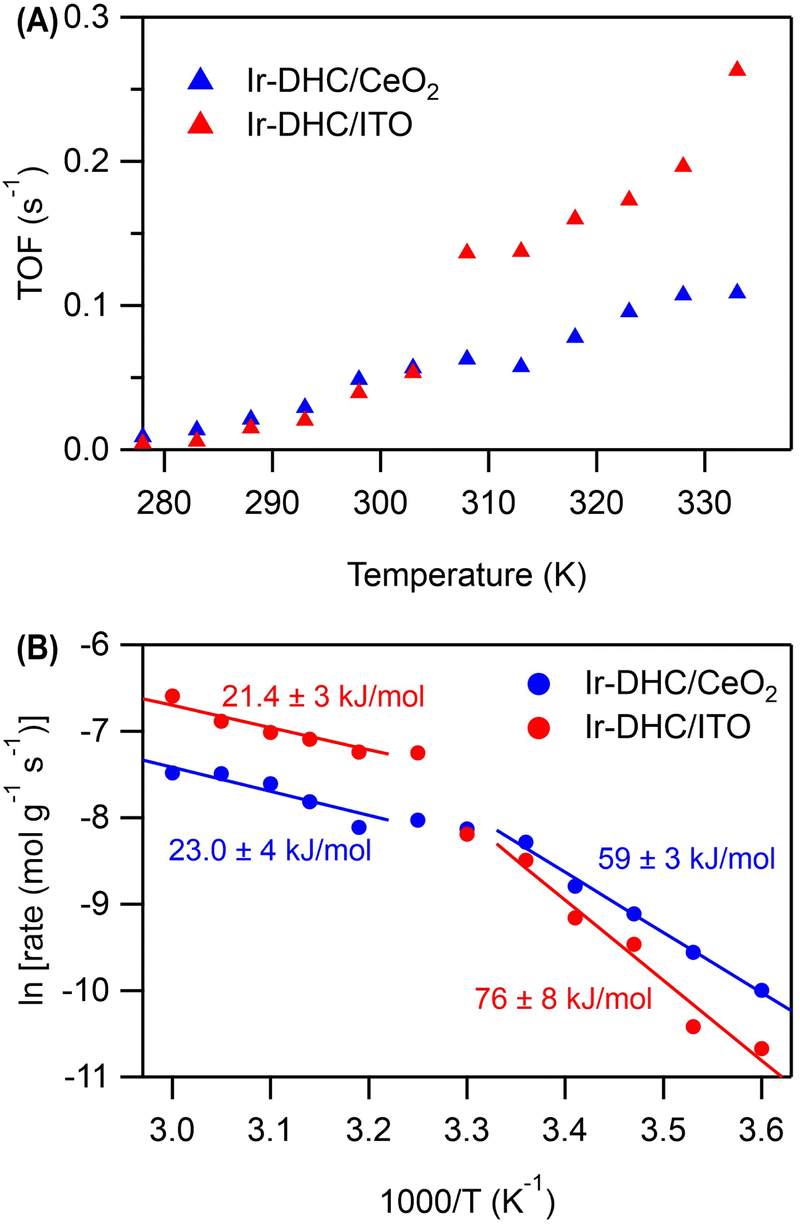 | ||
| Fig. 4 Temperature-dependent kinetics of water oxidation. (A) The turn-over-frequencies of Ir-DHC/ITO and Ir-DHC/CeO2 at a fixed fluence of 151 mW cm−2. (B) Arrhenius plots of the water oxidation rate as a function of temperature. Generated rate of oxygen was normalized by mass of Ir. The values in the graph represent Eapp. | ||
Conclusions
In summary, we have studied the role of the catalyst support on photocatalytic water oxidation by using a novel catalytic system in which the hole redistribution properties can be tuned while maintaining the same type of active site, which is a molecularly-derived, Ir-based dinuclear heterogeneous catalyst (Ir-DHC). We observed that the rate of water oxidation exhibits a different dependence on temperature as a function of the properties of the support. Specifically, the catalytic activities of Ir-DHC on CeO2 and indium tin oxide (ITO) are comparable at low temperatures (288–298 K). By contrast, at elevated temperatures (310–323 K), the OER activity of Ir-DHC on ITO is ca. 100% higher than that of Ir-DHC on CeO2. We rationalized these results in terms of the disparate hole redistribution dynamics of the supports. At low temperatures, the rate of the OER is determined by a thermal barrier associated with the water oxidation chemistry on the active site, rendering the rate insensitive to the hole redistribution dynamics of the support. At elevated temperatures, the thermal barrier is readily overcome, whereas the sluggish hole redistribution in CeO2 limits the rate of OER for Ir-DHC on CeO2.Our results demonstrate that the support plays a key role in channeling holes to the active site and in determining the overall OER behaviors. The unique phenomenon can be readily explained by the relative ease or difficulty of hole redistribution by the support. The results corroborate well with the literature that hole accumulation plays a critical role in defining water oxidation reaction kinetics. It also represents a new strategy for studying the reaction by systematically altering the hole transport properties of the catalyst support without altering the chemical properties of the active centers.
Data availability
The data supporting the findings of this work are available within the main article and the ESI.† Additional data are available from the corresponding authors upon request.Author contributions
D. Wang and M. M. Waegele conceived and supervised the research. D. Wang, M. M. Waegele and H. Zhang designed the experiments. H. Zhang and N. Dulock performed the measurements. T. Liu performed the catalyst synthesis. H. Zhang, D. Wang and M. M. Waegele performed most of the data analysis. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under contract DE-SC0020261 (Boston College) and DE-FG02-07ER15909 (Yale University). We thank James Wilkes for his help with XPS measurements.Notes and references
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger III, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS.
- S. Ardo, D. Fernandez Rivas, M. A. Modestino, V. Schulze Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J.-P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J.-W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- H. S. Shafaat and J. Y. Yang, Nat. Catal., 2021, 4, 928–933 CrossRef.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS PubMed.
- N. S. Lewis, Nat. Nanotechnol., 2016, 11, 1010–1019 CrossRef CAS PubMed.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS.
- M. Zhang and H. Frei, Annu. Rev. Phys. Chem., 2017, 68, 209–231 CrossRef CAS.
- H. Lyle, S. Singh, M. Paolino, I. Vinogradov and T. Cuk, Phys. Chem. Chem. Phys., 2021, 23, 24984–25002 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS.
- J. Li, W. Wan, C. A. Triana, H. Chen, Y. Zhao, C. K. Mavrokefalos and G. R. Patzke, Nat. Commun., 2021, 12, 1–9 CrossRef.
- I. Vinogradov, S. Singh, H. Lyle, M. Paolino, A. Mandal, J. Rossmeisl and T. Cuk, Nat. Mater., 2022, 21, 88–94 CrossRef CAS.
- J. Li and M. M. Waegele, Curr. Opin. Electrochem., 2022, 33, 100932 CrossRef CAS.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Nat. Chem., 2020, 12, 82–89 CrossRef CAS PubMed.
- C. Lang, J. Li, K. R. Yang, Y. Wang, D. He, J. E. Thorne, S. Croslow, Q. Dong, Y. Zhao, G. Prostko, G. W. Brudvig, V. S. Batista, M. M. Waegele and D. Wang, Chem, 2021, 7, 2101–2117 CAS.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS PubMed.
- M. Souada, C. Louage, J.-Y. Doisy, L. Meunier, A. Benderrag, B. Ouddane, S. Bellayer, N. Nuns, M. Traisnel and U. Maschke, Ultrason. Sonochem., 2018, 40, 929–936 CrossRef CAS PubMed.
- N. Um, M. Miyake and T. Hirato, in Zero-Carbon Energy Kyoto 2010, Springer, 2011, pp. 165–170 Search PubMed.
- N. Sivasankar, W. W. Weare and H. Frei, J. Am. Chem. Soc., 2011, 133, 12976–12979 CrossRef CAS.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- Y. Zhao, S. Dai, K. R. Yang, S. Cao, K. L. Materna, H. M. C. Lant, L. C. Kao, X. Feng, J. Guo, G. W. Brudvig, M. Flytzani-Stephanopoulos, V. S. Batista, X. Pan and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2206850120 Search PubMed.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS.
- D. M. Stanbury, Adv. Inorg. Chem., 1989, 33, 69–138 CrossRef CAS.
- H. S. White, W. G. Becker and A. J. Bard, J. Phys. Chem., 1984, 88, 1840–1846 CrossRef CAS.
- P. G. Hoertz, Y.-I. Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111, 6845–6856 CrossRef CAS.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324–7331 Search PubMed.
- M. Zhang, M. De Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS.
- K. M. Omberg, J. R. Schoonover, J. A. Treadway, R. M. Leasure, R. B. Dyer and T. J. Meyer, J. Am. Chem. Soc., 1997, 119, 7013–7018 Search PubMed.
- H. H. Pham, M.-J. Cheng, H. Frei and L.-W. Wang, ACS Catal., 2016, 6, 5610–5617 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- X. Chen, D. J. Aschaffenburg and T. Cuk, Nat. Catal., 2019, 2, 820–827 CrossRef CAS.
- C. Bozal-Ginesta, R. R. Rao, C. A. Mesa, Y. Wang, Y. Zhao, G. Hu, D. Antón-García, I. E. L. Stephens, E. Reisner, G. W. Brudvig, D. Wang and J. R. Durrant, J. Am. Chem. Soc., 2022, 144, 8454–8459 CrossRef CAS PubMed.
- M. J. Manto, P. Xie and C. Wang, ACS Catal., 2017, 7, 1931–1938 CrossRef CAS.
- Q. Zhang, J. Bu, J. Wang, C. Sun, D. Zhao, G. Sheng, X. Xie, M. Sun and L. Yu, ACS Catal., 2020, 10, 10350–10363 CrossRef CAS.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS.
- R. Massad, T. P. Cheshire, C. Fan and F. A. Houle, Chem. Sci., 2023, 14, 1997–2008 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of catalysts by DRIFTS and XPS; analysis of O2 traces; representative O2 traces; spectroscopy of [Ru(bpy)3]3+; study of Ce3+ effect. See DOI: https://doi.org/10.1039/d3sc00603d |
| This journal is © The Royal Society of Chemistry 2023 |