
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelical fluxionality: numerical frustration drives concerted low-barrier screw motions of a tricopper cluster†
Heechan
Kim
 ,
Juhwan
Shin
,
Seyong
Kim
and
Dongwhan
Lee
*
,
Juhwan
Shin
,
Seyong
Kim
and
Dongwhan
Lee
*
Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea. E-mail: dongwhan@snu.ac.kr
First published on 1st March 2023
Abstract
Uneven allocation of resources creates frustration, tension, and conflicts. Confronted with an apparent mismatch between the number of donor atoms and the number of metal atoms to be supported, helically twisted ligands cleverly come up with a sustainable symbiotic solution. As an example, we present a tricopper metallohelicate exhibiting screw motions for intramolecular site exchange. A combination of X-ray crystallographic and solution NMR spectroscopic studies revealed thermo-neutral site exchange of three metal centres hopping back and forth inside the helical cavity lined by a spiral staircase-like arrangement of ligand donor atoms. This hitherto unknown helical fluxionality is a superimposition of translational and rotational movements of molecular actuation, taking the shortest path with an extraordinarily low energy barrier without compromising the overall structural integrity of the metal–ligand assembly.
Introduction
The visually appealing shape of helices continues to inspire the creation of architectural objects and art forms featuring spiral structures.1,2 In addition to macroscopic objects, helical structures abound in biological systems. Examples include double-stranded DNA3,4 or protein α-helices,5,6 in which shape complementarity serves as the underpinning mechanisms of information storage and spontaneous folding. To build structural mimics of such constructs, helical metal–ligand assemblies have been investigated extensively.7–17 Studies on metallohelicates have focused primarily on topological design,10,18–21 stereoselective synthesis,22–26 chiroptical application,26–29 and DNA binding properties.30–34 Within this context, only a handful of works explored the dynamic aspects of helical metal complexes.15,35–37As schematically shown in Fig. 1, a screw-like motion is a superimposition of translation and rotation, both of which have been realized with molecular machines and devices.38–42 While conceptually appealing, spiral motion is difficult to implement with multinuclear metal complexes. Movement along the helical path (Fig. 1) requires concerted breaking and remaking of multiple metal–ligand bonds without compromising the overall structural integrity of the intertwined topology. To draw an analogy, a railroad track must be constantly removed and reinstalled as the train moves. This challenging synchronization is realized for the first time with a tricopper(I) cluster 1 built on a conformationally restricted π-conjugated ligand L (Scheme 1). The design, synthesis, and molecular mechanism of low-barrier spiral motions of the metallohelicate 1 constitute the main topics of this paper.
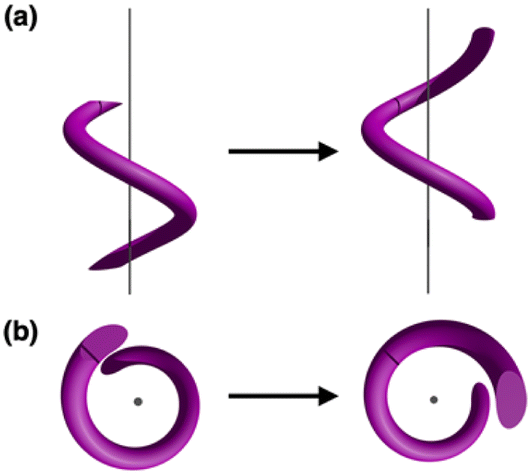 | ||
| Fig. 1 Helical motion as a superimposition of translational and rotational motions viewed (a) parallel to, and (b) perpendicular to, the spiral axis. | ||
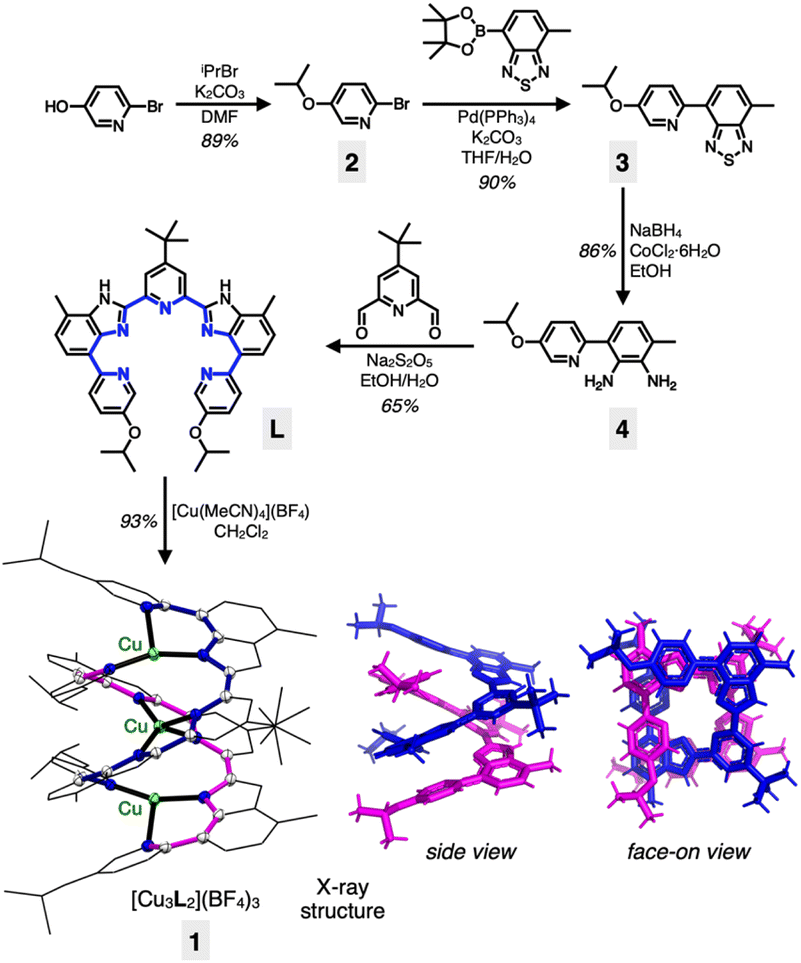 | ||
| Scheme 1 Synthetic routes to L and [Cu3L2](BF4)3 (1), and the X-ray structure of the cation. In the capped-stick representations (bottom right), the two intertwined ligand strands are color-coded (in blue and magenta) for better visualization of the double-helical topology; the metal atoms are omitted for clarity. | ||
Results and discussion
Ligand design and cluster assembly
Our entry into the chemistry of the tricopper(I) complex 1 was aided by the ligand L (Scheme 1). Multiple aryl–aryl junctions of L not only restrict the conformational freedom of bond rotation, but also promote tight π–π stacking upon metallation-induced helical assembly. The solubility-enhancing isopropoxy and tert-butyl groups also served as convenient spectroscopic handles to probe the intriguing dynamic behaviour in solution (vide infra).A pyridine-appended benzothiadiazole 3 was prepared by a Suzuki coupling reaction between bromopyridine 2 and the corresponding boronic ester (Scheme 1). The reduction of 3 to diamine 4, followed by an oxidative condensation reaction with the pyridine dialdehyde afforded L in a straightforward manner. The reaction between L and [Cu(MeCN)4](BF4) in CH2Cl2 produced 1 in high yield (93%). The [3 + 2]-type metal-to-ligand binding stoichiometry of this assembly was initially suggested by a Job plot analysis (Fig. S1†), and the resulting double-stranded helical structure of 1 was unambiguously established by single-crystal X-ray diffraction analysis on dark brown crystals grown by vapor diffusion of Et2O to a saturated CH3CN solution of the material.
As shown in Scheme 1 and Fig. S2†, the tricopper(I) cluster 1 features a C2-symmetric double-helical core with the tightly positioned ten N-donor atoms supporting an essentially linear alignment of the trimetallic array with ∠Cu–Cu–Cu = 170.41(2)°. The Cu⋯Cu intermetallic distances of 2.8000(5) Å are close to the sum of their van der Waals (vdW) radii (2.80 Å).43 The metal–ligand bond distances range from 1.945(2) Å to 2.174(2) Å, typical for CuI–Nsp2 bonds.10 The helical ligand assembly is also aided by the tightly stacked pyridine⋯benzimidazole aromatic ring pairs with a centroid-to-centroid distance of 3.3894(14) Å.
With two pentadentate ligands supporting three metal centres, a numerical mismatch is inevitable. As shown in Scheme 1, ten N-donor atoms within 1 are allocated to three copper(I) centres to produce a combination of {3, 4, 3} as the coordination numbers. Such inequality in the coordination environments, in conjunction with the kinetic lability of the d10 electronic configuration of copper(I), promotes rapid site-exchanging motions, which is the topic of the following section.
Helix is dynamic, not static: solvent-controlled and temperature-dependent fluxional motions
To probe whether the helical structure of 1 is also maintained in the solution phase, we proceeded to carry out NMR spectroscopic studies. In CDCl3 at T = 298 K, the 1H NMR spectrum of 1 featured 12 different proton resonances in the aromatic region, which is consistent with the solid-state structure (Fig. 2a). In support of this notion, the 2D-ROESY NMR spectrum of 1 revealed “inter”ligand correlations between H2a/H2b and HMe protons (Fig. S4†) expected for the double-helical structure comprising two strands of L. The hydrodynamic radius of 1 cation determined by DOSY at different temperatures (Table S2; Fig. S5 and S6†) are also in good agreement with the crystallographically determined molecular dimensions (Fig. S7†).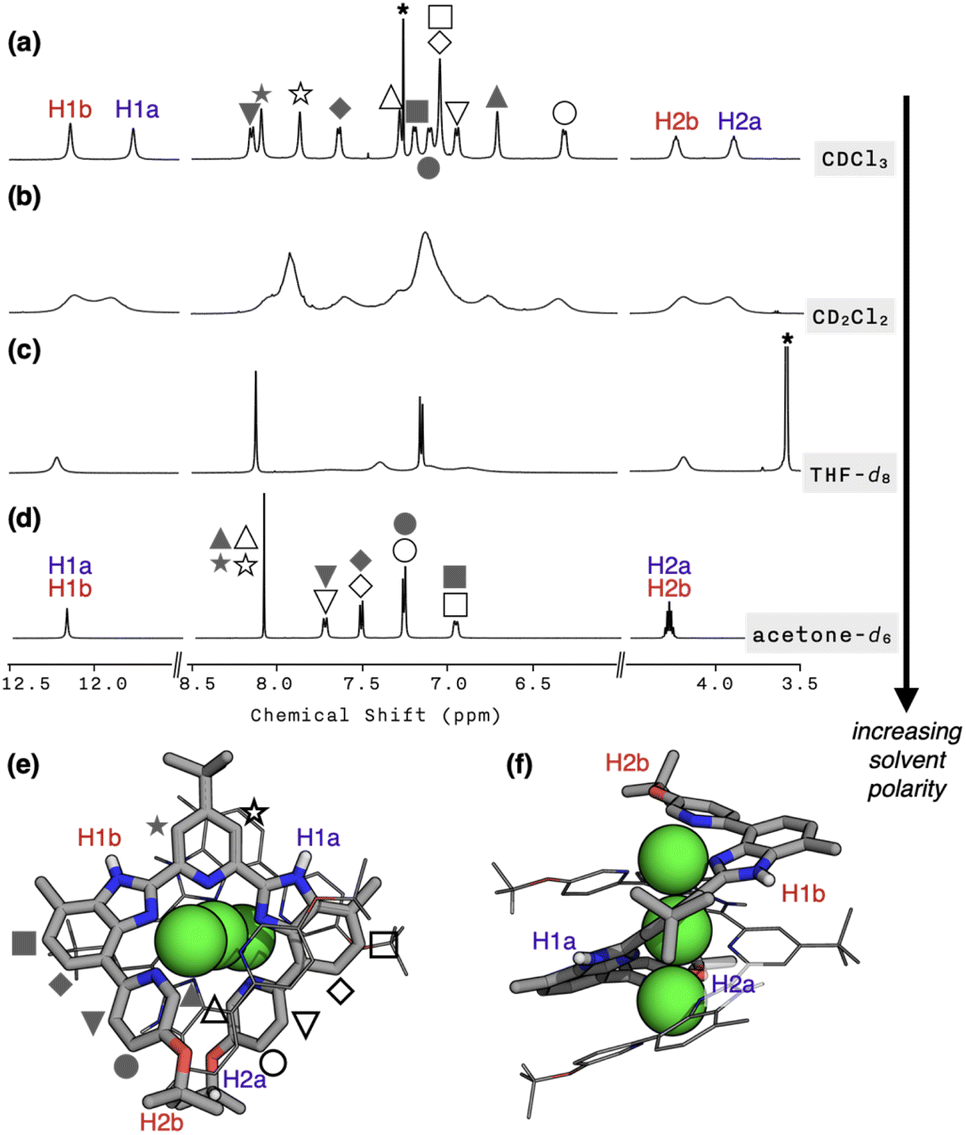 | ||
| Fig. 2 1H NMR spectra of 1 in (a) CDCl3, (b) CD2Cl2, (c) THF-d8, and (d) acetone-d6 at T = 298 K. The residual solvent peaks are indicated by asterisks. (e) Top view, and (f) side view of the capped-stick (for one strand) and wireframe (for the other strand) representation of the X-ray structure of 1 cation, with the representative protons labelled. Green spheres are copper atoms. | ||
Upon metallation to become part of the helicate, the symmetry-related proton pairs in the 1D 1H NMR spectrum of the free ligand L (Fig. S8†) become no longer equivalent. For example, the two isopropyl C–H protons of 1 are now in different environments: one in the “inner groove” (H2a in Fig. 2e and f) and the other at the “outer end” (H2b in Fig. 2e and f) of the helix. In stark contrast, a highly symmetric spectral pattern emerged in a polar solvent such as acetone-d6; only six resonances were observed in the aromatic region (Fig. 2d). Solvents of intermediate polarities, such as CD2Cl2 or THF-d8, produced unusually broadened spectra (Fig. 2b and c). Regardless of the choice of solvent, however, molecular ion peaks of m/z = 804 and 1694, corresponding to [Cu3L2](BF4)2+ and [Cu3L2](BF4)2+, respectively, were consistently observed by CSI-MS (Fig. S9†).
To understand this peculiar solvent-dependent behaviour of 1, temperature-dependent 1H NMR spectra were obtained in various solvents (Fig. 3a and S10−S13†). At sufficiently low temperatures, regardless of the choice of solvent, the spectral patterns of twelve well-resolved aromatic resonances are consistent with the solid-state structure of 1. Upon increasing the temperature, however, the sharp peaks began to collapse into broad signals (Fig. 3a). In polar solvents such as THF-d8 or acetone-d6, the resonances eventually coalesce into simple and symmetric patterns. Such spectral transitions could not be observed in CDCl3 or CD2Cl2 (Fig. S10 and S11†), presumably because the coalescence temperatures (Tc) lie above the solvent's boiling point. Nonetheless, the overall trend of this fluxional behaviour unambiguously points toward the involvement of dynamic intramolecular ligand exchange in solution.
Further evidence for the fluxionality came from 2D 1H–1H NMR exchange spectroscopy (EXSY) on 1 in CD2Cl2 and CDCl3 (Fig. 3b, S14, and S15†). We found that every proton of the ligand has a one-to-one correspondence to the equivalent proton of the other ligand constituting the double-helical 1. This observation implies that the protons at the inner groove rapidly exchange positions with the corresponding protons at the outer end of the double helix, which is clearly seen for the imidazolyl N–H (i.e., H1a and H1b) and isopropyl C–H (i.e., H2a and H2b) protons denoted in Fig. 2e, f, and 3b.
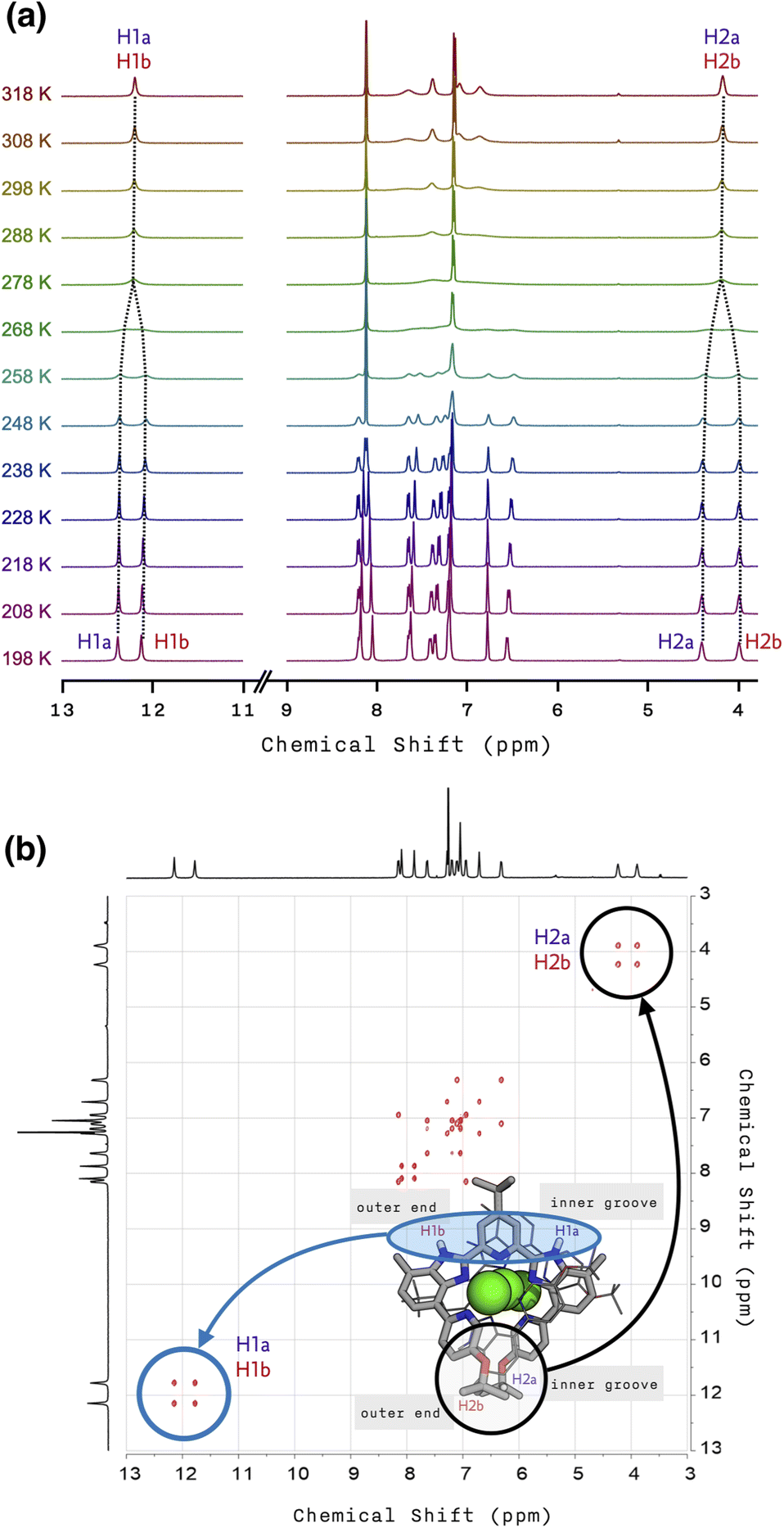 | ||
| Fig. 3 (a) Temperature-dependent (from T = 198 K to 318 K) 1H NMR spectra of 1 in THF-d8. (b) 2D 1H–1H EXSY NMR spectrum of 1 with tm = 100 ms in CDCl3 at T = 298 K. | ||
Mechanism and energetics of helical fluxionality
Variable-temperature (VT) and 2D NMR spectroscopic studies (Fig. 3) suggest that the thermoneutral site exchange of 1 occurs by sliding motions of one ligand along the slope of a spiral path defined by the other ligand (Fig. 4a and b). To take the path of least resistance, this intramolecular screw motion is coupled to a concerted hopping of the three copper(I) centres up and down inside the helical cavity. The traveling distance of the ligand along the screw-axis is estimated to be 2.8 Å, which corresponds to the Cu⋯Cu intermetallic separation (Scheme 1; Fig. S1†).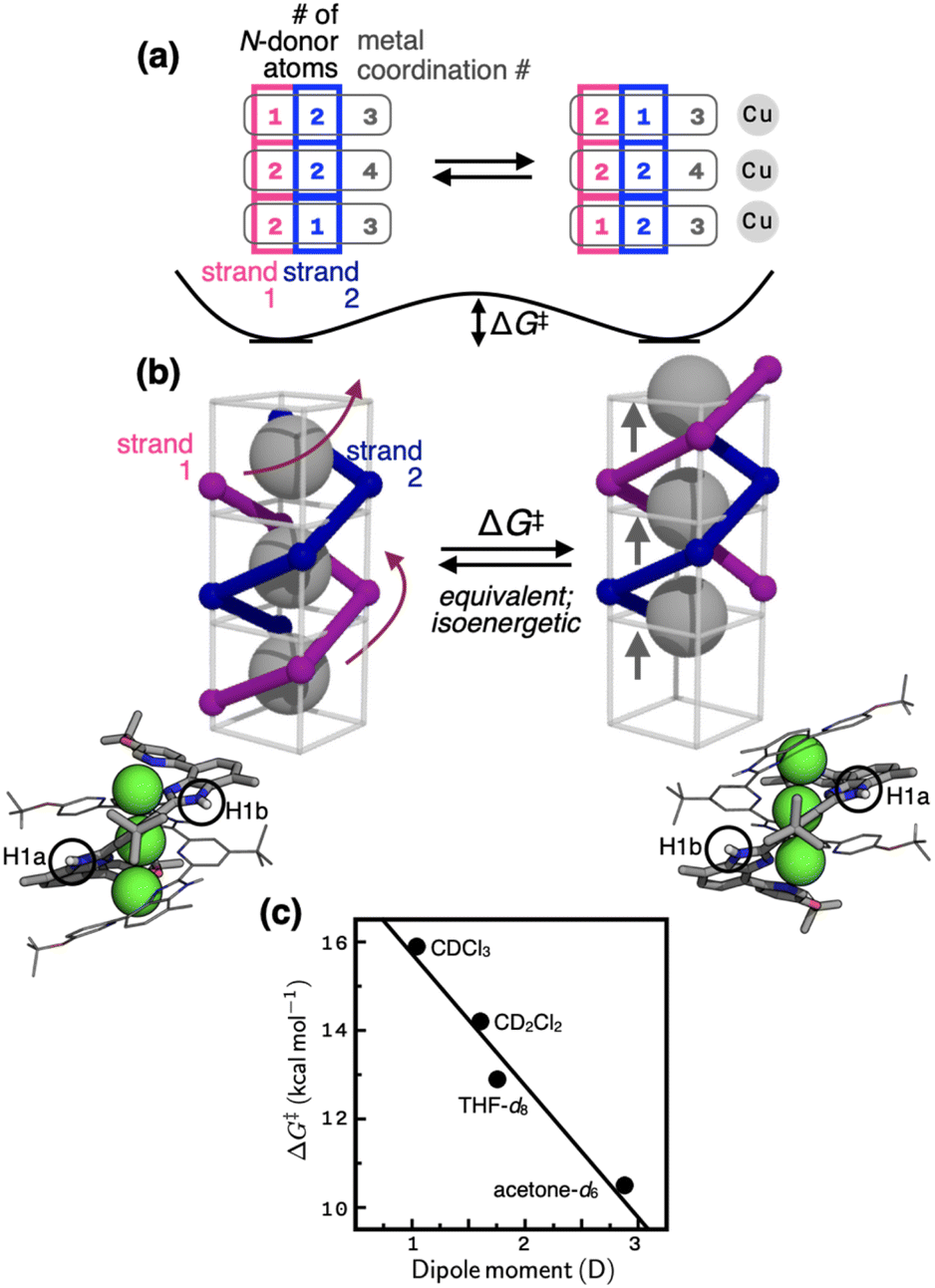 | ||
| Fig. 4 (a) Changes in the number of N-donor atoms contributed by each ligand strand (blue and magenta) to the individual copper centres (gray) during the fluxional process schematically shown in (b). On each side of the schematic model in (b) is shown the corresponding view of the X-ray structure; the N–H protons are labelled as H1a (inner groove) and H1b (outer end), which exchange positions (see the EXSY spectrum in Fig. 3b) by the screw motion. (c) The relationship between the solvent dipole moments46 and the activation barrier (ΔG‡) defined in (b). | ||
The energetics of this site-exchange was investigated by NMR spectroscopy. Using the Δν value obtained at the slow-exchange limit in VT 1D NMR and the coalescence temperature (Tc), the activation barrier ΔG‡ of the screw motion was estimated in THF-d8 and acetone-d6 (Table S3†).44 For low boiling point solvents CDCl3 and CD2Cl2, the ΔG‡ values were determined by VT 2D EXSY NMR (see ESI for details†). With the ΔG‡ value of 14.2 kcal mol−1 obtained by VT 2D EXSY NMR (Fig. S18†) and the Δν value of 100 Hz determined by VT 1D NMR (Fig. S11†), the coalescence temperature of 1 in CD2Cl2 is estimated to be 298 K. This prediction is consistent with the data shown in Fig. S11†, with Tc lying just above this temperature point.
As shown in Fig. 4c, the energy barrier of the fluxional motion correlates inversely with the solvent dipole moment, with polar solvent facilitating the site exchange. In the most polar solvent acetone-d6, the ΔG‡ value is as low as 10.5 kcal mol−1. For comparison, the energy barrier of cyclohexane ring flip is 10.8 kcal mol−1.45 Considering that the fluxional motion of 1 requires concomitant dissociation and reassociation of at least four Cu–N bonds (Fig. 4a and b), this low-barrier ligand substitution cascade is quite remarkable. The spiral staircase-like arrangement of the ligand donor atoms inside the helical cavity plays a critical function in this low-barrier fluxional motion. Buried inside the tightly π–π stacked double helix, they enable rapid dissociation, migration, and reassociation of the Cu–N bond as the metal centres hop through the shortest path along the axis of the helix (Fig. 4). Apparently, polar solvent environments facilitate this process by assisting metal–ligand bond polarization (Fig. 4c).
Aside from the mechanistic aspects of the helical fluxionality, what is the origin of the stereochemical instability of this particular metallohelicate? Within 1, the two intertwined pentadentate ligands provide a total of ten donor atoms that need to be shared by three metal centres. This apparent numerical mismatch creates inequalities in the coordination numbers as {3, 4, 3} (Scheme 1). A constant movement of each ligand tries to resolve this imbalance by distributing its five N-donor atoms as a set of either 2–2–1 or 1–2–2 (Fig. 4a). The palindromic nature of these number combinations helps maintain the overall coordination number set of {3, 4, 3} for the three metal centres, each exchanging ligand partners as the screw oscillates. Equally important for the molecular motion here is an ideal balance between the non-stretchability (of the π-conjugated backbone) and the conformational adaptability (through aryl–aryl rotations) of the ligand L. This structural feature ensures that as the head advances, the tail always follows by the same distance. Such a mechanical coupling is difficult to realize with typical metallohelicates, which are built with segmented bi/tridentate ligands connected by flexible alkyl/ether linkers.7–12,18,37
Conclusions
In sum, a double-stranded trimetallic helicate undergoes low-barrier screw-like motions to alleviate the numerical imbalance of metal–ligand pairing. Through this hitherto unknown helical fluxionality, the metal atoms take the shortest path that traverses a spiral staircase-like arrangement of ligand donor atoms constituting the helical inner cavity.By definition, fluxionality involves multiple conformers with no particular structural preference. An externally triggered directionality is thus needed to turn the current system into a new type of molecular actuator with programmable motions. Efforts are currently underway in our laboratory in such directions.
Data availability
The X-ray crystallographic coordinates for 1 have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2178526. Experimental procedures, NMR spectra, and supplementary figures are available in the ESI.†Author contributions
H. Kim: conceptualization, formal analysis, investigation, methodology, visualization, writing-original draft, writing-review & editing. J. Shin and S. Kim: conceptualization, formal analysis, investigation, methodology. D. Lee: conceptualization, funding acquisition, project administration, supervision, validation, writing-original draft, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (2020R1A2C2006381 and 2021R1A5A1030054). H. K. is a recipient of the NRF Global PhD Fellowship (2018H1A2A1060348). We thank Dr Mi Sook Seo and Prof. Wonwoo Nam for assistance in CSI-MS analysis, and Prof. Jung Ho Lee for helpful discussions.Notes and references
- E. Hamilton, Mythology: Timeless Tales of Gods and Heroes, Little, Brown and Company, New York, 1942 Search PubMed.
- S. N. Kramer, Sumerian Mythology, University of Pennsylvania Press, Philadelphia, 1961 Search PubMed.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- C. R. Calladine, H. R. Drew, B. F. Luisi and A. A. Travers, in Understanding DNA, Academic Press, Oxford, 2004 Search PubMed.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1988, 201, 601–619 CrossRef CAS PubMed.
- N. D. Socci, W. S. Bialek and J. N. Onuchic, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1994, 49, 3440–3443 CrossRef CAS PubMed.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed.
- A. F. Williams, C. Piguet and G. Bernardinelli, Angew. Chem., Int. Ed., 1991, 30, 1490–1492 CrossRef.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS PubMed.
- V. Amendola, M. Boiocchi, V. Brega, L. Fabbrizzi and L. Mosca, Inorg. Chem., 2010, 49, 997–1007 CrossRef CAS PubMed.
- M. Boiocchi and L. Fabbrizzi, Chem. Soc. Rev., 2014, 43, 1835–1847 RSC.
- L. R. Holloway, H. H. McGarraugh, M. C. Young, W. Sontising, G. J. O. Beran and R. J. Hooley, Chem. Sci., 2016, 7, 4423–4427 RSC.
- S. P. Argent, F. C. Jackson, H. M. Chan, S. Meyrick, C. G. P. Taylor, T. K. Ronson, J. P. Rourke and M. D. Ward, Chem. Sci., 2020, 11, 10167–10174 RSC.
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 664–670 CrossRef CAS PubMed.
- J. L. Greenfield and J. R. Nitschke, Acc. Chem. Res., 2022, 55, 391–401 CrossRef CAS PubMed.
- S. Okuda, N. Ousaka, T. Iwata, R. Ishida, A. Urushima, N. Suzuki, S. Nagano, T. Ikai and E. Yashima, J. Am. Chem. Soc., 2022, 144, 2775–2792 CrossRef CAS PubMed.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem.–Eur. J., 2006, 12, 4069–4076 CrossRef CAS PubMed.
- J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Angew. Chem., Int. Ed., 2014, 53, 7823–7827 CrossRef CAS PubMed.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed.
- C. S. Wood, T. K. Ronson, A. M. Belenguer, J. J. Holstein and J. R. Nitschke, Nat. Chem., 2015, 7, 354–358 CrossRef CAS PubMed.
- O. Mamula, A. von Zelewsky and G. Bernardinelli, Angew. Chem., Int. Ed., 1998, 37, 289–293 CrossRef PubMed.
- H.-L. Kwong, H.-L. Yeung, W.-S. Lee and W.-T. Wong, Chem. Commun., 2006, 4841–4843 RSC.
- J. Crassous, Chem. Commun., 2012, 48, 9687–9695 RSC.
- T. Y. Bing, T. Kawai and J. Yuasa, J. Am. Chem. Soc., 2018, 140, 3683–3689 CrossRef CAS PubMed.
- Z. Yao, Y. Zhou, T. Gao, P. Yan and H. Li, RSC Adv., 2021, 11, 10524–10531 RSC.
- Y. Imai and J. Yuasa, Chem. Commun., 2019, 55, 4095–4098 RSC.
- Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653–17661 CrossRef CAS PubMed.
- T. Ono, K. Ishihama, A. Taema, T. Harada, K. Furusho, M. Hasegawa, Y. Nojima, M. Abe and Y. Hisaeda, Angew. Chem., Int. Ed., 2021, 60, 2614–2618 CrossRef CAS PubMed.
- I. Gamba, G. Rama, E. Ortega-Carrasco, J.-D. Maréchal, J. Martínez-Costas, M. Eugenio Vázquez and M. V. López, Chem. Commun., 2014, 50, 11097–11100 RSC.
- S. J. Allison, D. Cooke, F. S. Davidson, P. I. P. Elliott, R. A. Faulkner, H. B. S. Griffiths, O. J. Harper, O. Hussain, P. J. Owen-Lynch, R. M. Phillips, C. R. Rice, S. L. Shepherd and R. T. Wheelhouse, Angew. Chem., Int. Ed., 2018, 57, 9799–9804 CrossRef CAS PubMed.
- C. A. J. Hooper, L. Cardo, J. S. Craig, L. Melidis, A. Garai, R. T. Egan, V. Sadovnikova, F. Burkert, L. Male, N. J. Hodges, D. F. Browning, R. Rosas, F. Liu, F. V. Rocha, M. A. Lima, S. Liu, D. Bardelang and M. J. Hannon, J. Am. Chem. Soc., 2020, 142, 20651–20660 CrossRef CAS PubMed.
- X. Li, J. Wu, L. Wang, C. He, L. Chen, Y. Jiao and C. Duan, Angew. Chem., Int. Ed., 2020, 59, 6420–6427 CrossRef CAS PubMed.
- M. Hutin, R. Frantz and J. R. Nitschke, Chem.–Eur. J., 2006, 12, 4077–4082 CrossRef CAS PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC.
- D. Zhao, T. van Leeuwen, J. Cheng and B. L. Feringa, Nat. Chem., 2017, 9, 250–256 CrossRef CAS PubMed.
- A. Santoro, J. Holub, M. A. Fik-Jaskółka, G. Vantomme and J.-M. Lehn, Chem.–Eur. J., 2020, 26, 15664–15671 CrossRef CAS PubMed.
- V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. Günther, in NMR Spectroscopy, Wiley-VCH, New York, 3rd edn, 2013, ch. 13 Search PubMed.
- F. A. L. Anet and A. J. R. Bourn, J. Am. Chem. Soc., 1967, 89, 760–768 CrossRef CAS.
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and supplementary figures, including 1H and 13C NMR spectra, crystallographic data, FT-IR spectra, HR-MS and elemental analysis. CCDC 2178526. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00851g |
| This journal is © The Royal Society of Chemistry 2023 |