
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-selective duplex formation and template effect in recognition-encoded oligoanilines†
Daniele
Rosa-Gastaldo
 ,
Andrea
Dalla Valle
,
Tommaso
Marchetti
and
Luca
Gabrielli
*
,
Andrea
Dalla Valle
,
Tommaso
Marchetti
and
Luca
Gabrielli
*
Dipartimento di Scienze Chimiche, Università degli studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: luca.gabrielli@unipd.it
First published on 26th July 2023
Abstract
A new family of duplex-forming recognition encoded oligomers, capable of sequence selective duplex formation and template directed synthesis, was developed. Monomers equipped with both amine and aldehyde groups were functionalized with 2-trifluoromethylphenol or phosphine oxide as H-bond recognition units. Duplex formation and assembly properties of homo- and hetero-oligomers were studied by 19F and 1H NMR experiments in chloroform. The designed backbone prevents the undesired 1,2-folding allowing sequence-selective duplex formation, and the stability of the antiparallel duplex is 3-fold higher than the parallel arrangement. Dynamic combinatorial chemistry was exploited for the templated synthesis of complementary oligomers, showing that an aniline dimer can template the formation of the complementary imine. The key role of the H-bond recognition confers to the system the ability to discriminate a mutated donor monomer incapable of H-bonding. Sequence selective duplex formation combined with the template effect makes this system an attractive target for further studies.
Introduction
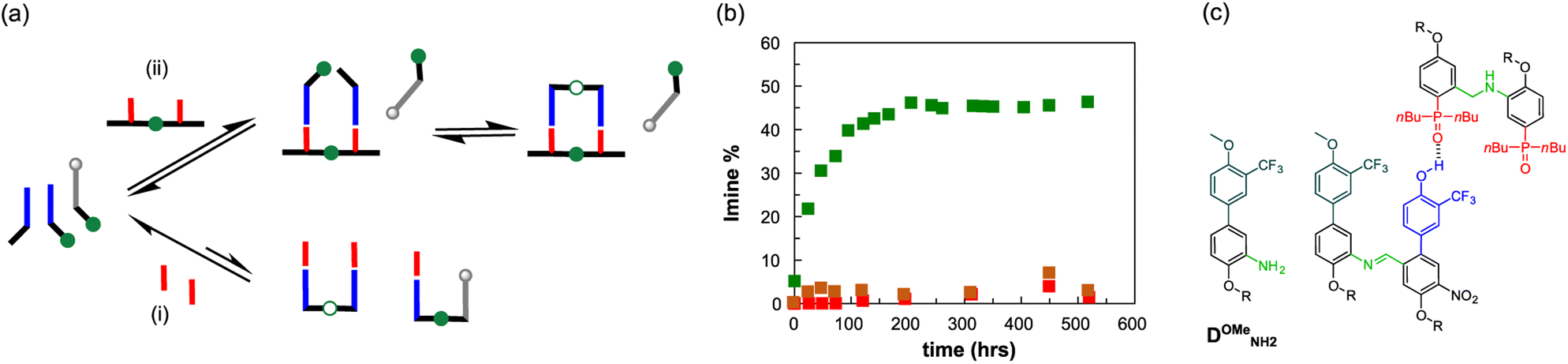
Nature selected duplex formation as the fundamental architecture that allows to replicate, transcript and translate, via template-directed synthesis, the information stored as a sequence of recognition-encoded monomers.1 Currently only nucleic acids possess these properties, hence the first approaches to synthetic sequence-controlled molecules were based on nucleic acids analogues.2–19 Cycles of templated polymerization and selection were used to evolve functional sequences of nucleic acid with recognition or catalytic properties.20–25 Synthetic recognition-encoded oligomers have the potential to display similar properties, which would open the way for directed evolution of function in synthetic polymers. The first steps in this direction have been taken with the development of oligomers that form duplexes in a sequence-selective manner, and a range of such systems have been reported.26–45 DNA directed synthesis of non-natural oligomers has been performed using amide coupling,46,47 and reductive amination.48,49 Templated synthesis of triazole oligomers have been recently reported using H-bond,50 salt bridges51 and covalent ester base-pair.52–56We have recently developed a class of duplex forming oligomers based on the H-bond base pair 2-trifluoromethylphenol–phosphine oxide and an aniline backbone, that can be synthesized using a two-step reductive amination (imine formation and reduction) between aldehyde monomers and aniline linkers.44,57 An interesting feature of this approach is the possibility to exploit dynamic combinatorial chemistry for oligomers' synthesis. Indeed, the addition of a recognition-encoded amine template to a dynamic combinatorial library of informational oligoimines should shift the equilibrium favouring the complementary oligomer, that would form the most stable duplex with the template.58–61
The library could then be trapped by reduction to obtain the complementary sequence of the template as an oligoaniline (Fig. 1a).62 The reported aniline systems were built assembling aldehyde monomers with 1,3-dianiline,44,63 however the geometry of the backbone allowed the folding between adjacent recognition units, thus preventing the formation of stable duplexes between hetero-oligomers (i, Fig. 1b).
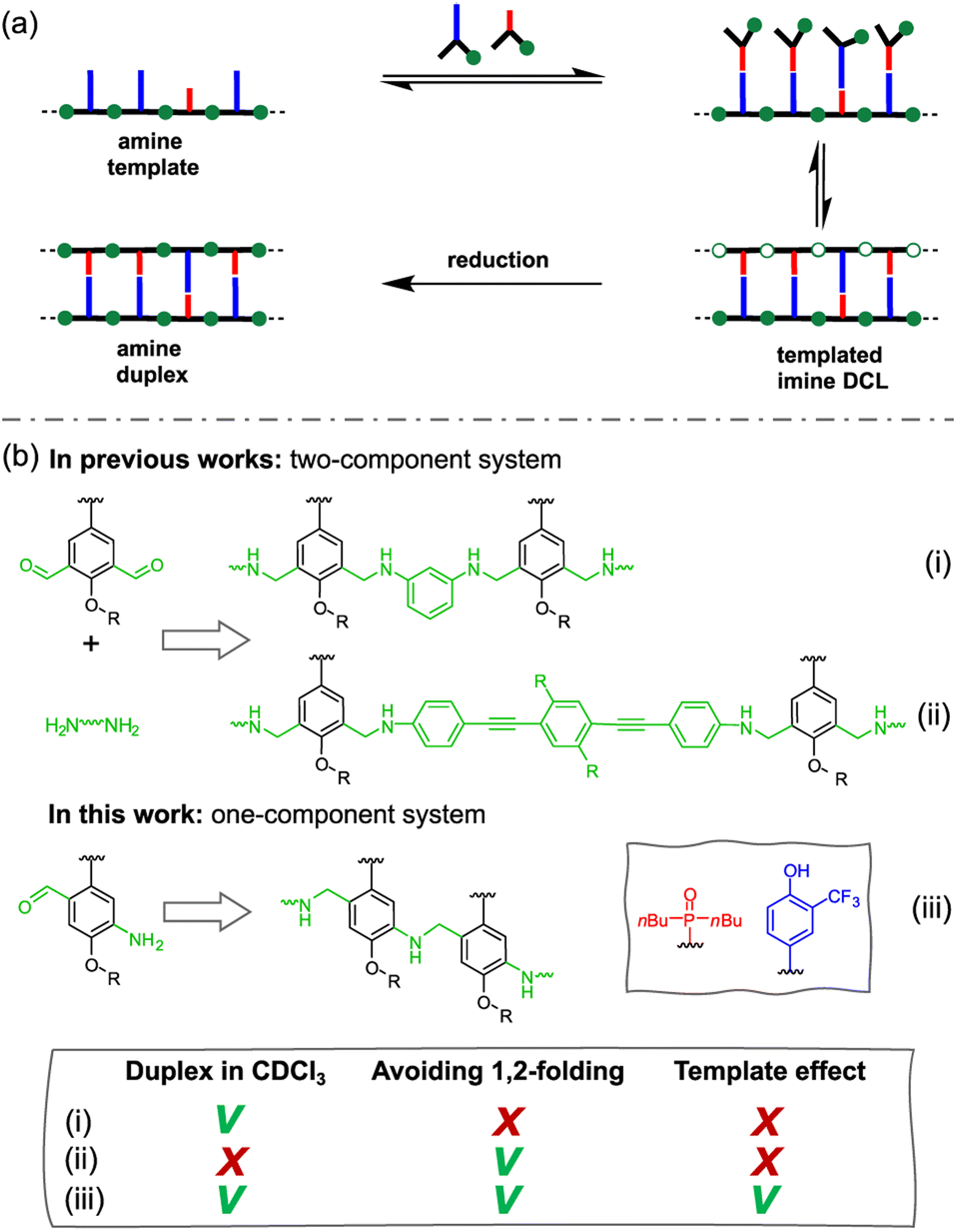 | ||
| Fig. 1 (a) Templated synthesis of a duplex forming molecule through reductive amination. The template sequence organizes the monomers (or codons) favouring the formation of the complementary sequence imine oligomer, which can be finally reduced to the more stable amine. (b) Structure comparison of duplex forming oligoanilines. There are three key design elements that can be independently optimised in a highly modular approach: the coupling chemistry used for the synthesis of oligomers (green), the recognition module responsible for intermolecular binding (blue/red) and the backbone which links these components together (black). The long-short 2-trifluoromethylphenol–phosphine oxide base-pairing system is shown, along with a backbone that is assembled through reductive amination (R = 2-ethylhexyl). Reported approaches are based on the two components assembly of aldehyde monomers and short (i) or long (ii) aniline linkers. The system described here is based on the assembly of recognition encoded monomers equipped with both aniline and aldehyde moieties (iii). This design allows duplex formation in chloroform without the undesired 1,2-folding and shows template effect. | ||
To impede undesired folding, we increased the intermonomer distance, using a long dianiline linker (ii, Fig. 1b).57 Despite this modification succeeded in preventing folding in toluene, homo-oligomers were unable to assemble into duplexes in chloroform solution. Furthermore, when we tested the ability of an aniline oligomer (or dimer) to favour the formation of the complementary sequence, we did not observe any template effect in both the reported systems. We reasoned that in a two-component system the formation of the first imine between monomer and the linker is not directly affected by the presence of the template, since the aniline is not equipped with a H-bonding recognition unit.
Hence, we describe here a new family of recognition-encoded oligoaniline, based on monomers that are equipped with both aldehyde and amine moieties (iii, Fig. 1b). A 4-amino-benzaldehyde scaffold was functionalised with a recognition unit in position 2 and with a solubility group in position 5. In this single component approach the template should be able to directly affect the formation of every intermonomer imine linkage. In addition, we speculated that nearing the monomers would make more effective the long-short phenol-phosphine oxide base-pair in reducing the probability of undesired 1,2-folding interactions. The properties of the backbone are fundamental because the duplex formation is in competition with other self-assembly channels (Fig. 2).
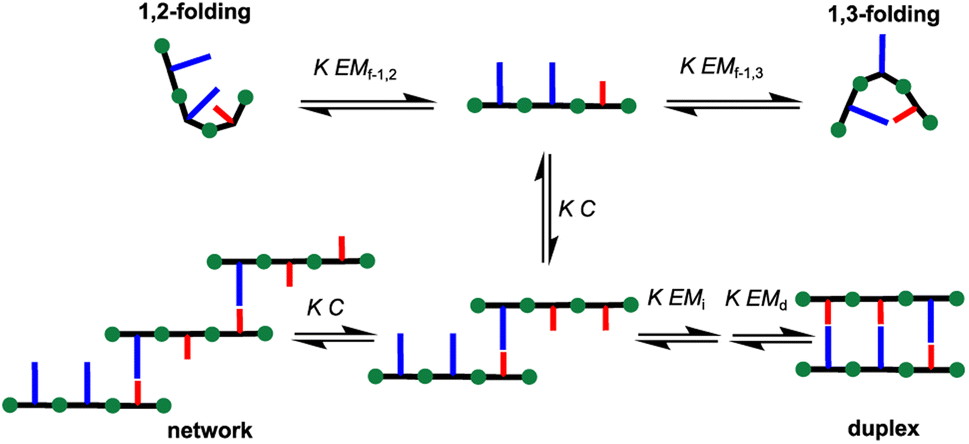 | ||
| Fig. 2 Self-assembly channels for recognition-encoded trimers. The first base-pairing interaction can take place in an intermolecular or intramolecular fashion, leading to the 1,2 or 1,3 folding channels. The second base-pairing interaction can be intramolecular, initiating duplex formation or intermolecular, leading to the networks channel. The outcome depends on the concentration, C, the association constant for the intermolecular base-pairing interaction, K, and the effective molarities for folding, (EMf-1,2, EMf-1,3), duplex initiation and propagation (EMi, EMd). | ||
The formation of networks can be inhibited working at concentrations (C) lower than the effective molarity for duplex propagation EMd. The competition with the folding channels is highly dependent on the conformational properties of the backbone, thus it is more difficult to treat. In particular if two adjacent recognition units can form a base-pair, the folding pathway will be favoured over the duplex assembly channel. Longer range intramolecular interactions, such as 1,3-folding, will compete with duplex formation, decreasing the observed association constant for duplex formation, but the duplex assembly can be favoured.
In this paper we will demonstrate that the designed system is able to perform sequence selective duplex formation via H-bonding interactions and that an amine template can favour the formation of the complementary imine oligomer in a dynamic combinatorial library.
Results and discussion
Synthetic procedures
Three sets of acceptor and donor monomers equipped with aldehyde, amine or both the groups have been synthesized as shown in Scheme 1 (see Sections 1–4 of the ESI† for detailed procedures). Nitration of 2-bromo-5-hydroxybenzaldehyde with copper nitrate supported on claycop and acetic anhydride in DCM gave intermediate 1, which was then alkylated with racemic 2-ethylhexyl bromide to give 2. Selective reduction of the nitro group was performed using SnCl2 in ethanol, to give compound 3.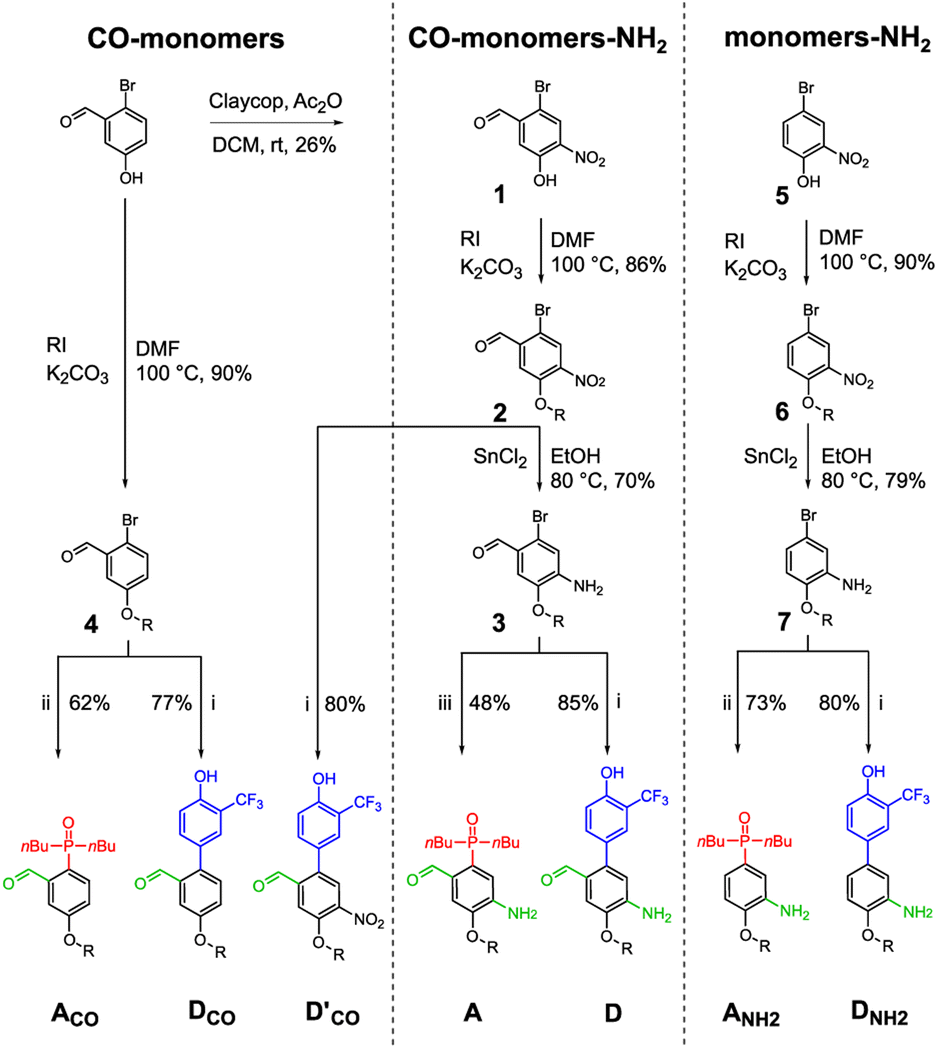 | ||
| Scheme 1 Synthesis of acceptor and donor monomers (R = 2-ethylhexyl). (i) Pd2(dba)3, KF, HP(tBu)3BF4, 4-pinacolborane-2-(trifluoromethyl)phenol, THF/H2O; (ii) Pd2(dba)3, XantPhos, TEA, PHOBu2, dioxane; (iii) CuI, PHOBu2, Na2CO3, toluene. | ||
This key intermediate was coupled with dibuthylphosphine oxide using CuI and potassium carbonate to give the acceptor monomer A, or it was coupled with 4-pinacolborane-2-(trifluoromethyl)phenol under Suzuki–Miyaura conditions to give the donor monomer D. Direct alkylation of 2-bromo-5-hydroxybenzaldehyde with racemic 2-ethylhexyl bromide gave the aldehyde intermediate 4, which was coupled with dibuthylphosphine oxide using palladium and XantPhos to give acceptor monomer ACO; Suzuki–Miyaura coupling of intermediates 4 and 2 gave respectively the aldehyde monomers DCO and D′CO. Amine monomers were synthesized alkylating 4-bromo-2-nitrophenol with racemic 2-ethylhexyl bromide to give 6, which was then reduced with SnCl2 giving intermediate 7. Palladium catalysed coupling of 7 with dibuthylphosphine oxide and XantPhos gave acceptor monomer ANH2 while donor monomer DNH2 was obtained reacting 7 with 4-pinacolborane-2-(trifluoromethyl) phenol under Suzuki–Miyaura conditions. Homo- and heterodimers were synthesized via two-step reductive amination between the appropriate aldehyde and amine monomers, using NaBH4 as reducing agent (Scheme 2).
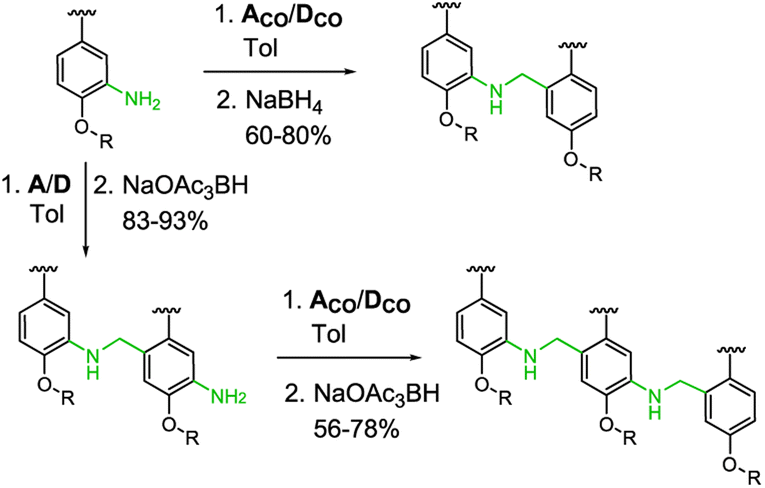 | ||
| Scheme 2 Synthesis of dimers and trimers (R = 2-ethylhexyl). | ||
Homo- and mixed trimers were synthesized reacting the selected amine monomer (DNH2 or ANH2) with a bifunctional monomer (A or D), in the presence of sodium triacetoxyborohydride, since the poor reactivity of the conjugated aniline allows a protective groups-free approach. The obtained intermediate dimers were then capped with the appropriate aldehyde monomer, using NaBH(OAc)3 in a one-step reductive amination. To avoid the stability issues observed only in the electron rich trimer DDD, instead of DCO we used the nitrated monomer D′CO as capping aldehyde monomer.
NMR binding studies of homo-oligomers
To assess whether the designed backbone was suitable for duplex formation, we studied the complexation of length-complementary homo-oligomers using 1H and 19F NMR titration experiments (see Section 5 in the ESI†). The association constant K for A·D complex formation was measured by titrating monomer ACO into DCO in toluene-d8. Addition of ACO caused an upfield shift in the 19F NMR signal due to the donor CF3 group and a downfield change in the 1H NMR chemical shift of the OH group signal. The titration data fit well to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm, giving an association constant of 4 × 103 M−1 (Table 1). Despite the aldehyde groups on ACO and DCO differ from the reductive amination products, these substituents have no effect on the H-bonding properties of the phosphine oxide and phenol recognition units. Indeed, the measured K is identical to the value previously observed for the corresponding monomers with different substituents.42,44 Titration of AA into DD caused an upfield shift of the 19F NMR signal due to the CF3 groups of DD and a downfield shift in the 1H NMR signal of the OH groups. The data fit well to a 1:1 binding isotherm, giving an association constant of 1 × 106 M−1. The K measured for AA·DD is two orders of magnitude higher than the value measured for monomers ACO·DCO, indicating that AA·DD forms a fully assembled duplex with cooperative formation of two H-bonds. The effective molarity (EM) for the second intramolecular H-bond formation in AA·DD duplex was determined as shown in eqn (1), where K1 is the association constant for formation a single intermolecular H-bond in A·D and KN is the association constant for duplex formation between two oligomers with N interaction sites.
:1 binding isotherm, giving an association constant of 4 × 103 M−1 (Table 1). Despite the aldehyde groups on ACO and DCO differ from the reductive amination products, these substituents have no effect on the H-bonding properties of the phosphine oxide and phenol recognition units. Indeed, the measured K is identical to the value previously observed for the corresponding monomers with different substituents.42,44 Titration of AA into DD caused an upfield shift of the 19F NMR signal due to the CF3 groups of DD and a downfield shift in the 1H NMR signal of the OH groups. The data fit well to a 1:1 binding isotherm, giving an association constant of 1 × 106 M−1. The K measured for AA·DD is two orders of magnitude higher than the value measured for monomers ACO·DCO, indicating that AA·DD forms a fully assembled duplex with cooperative formation of two H-bonds. The effective molarity (EM) for the second intramolecular H-bond formation in AA·DD duplex was determined as shown in eqn (1), where K1 is the association constant for formation a single intermolecular H-bond in A·D and KN is the association constant for duplex formation between two oligomers with N interaction sites.| KN = 2K1NEMN−1 | (1) |
K, effective molarities (EM), limiting NMR chemical shifts (δfree and δbound) and limiting complexation-induced changes in chemical shift (Δδ) measured by NMR titrations in toluene-d8 and chloroform-d at 298 Ka
| Solvent | Complex | logK (M−1) |
EM (mM) | 1H NMRb (ppm) | 19F NMRb (ppm) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H (1) | 1H (2) | 19F (1) | 19F (2) | 19F (3) | ||||||||||||||
| δ free | δ bound | Δδ | δ free | δ bound | Δδ | δ free | δ bound | Δδ | δ free | δ bound | Δδ | δ free | δ bound | Δδ | ||||
| a Each titration was repeated twice, and the average value is reported with errors at the 95% confidence limit. b Data for the signals due to the OH and CF3 groups on the phenol recognition units. c Signals due to the OH protons were too broad to be detected. n.d. = not detected. | ||||||||||||||||||
| CDCl3 | D·A | 2.4 ± 0.1 | — | 5.7 | 11.8 | 6.1 | — | — | — | −61.3 | −63.0 | −1.7 | — | — | — | — | — | — |
| DD·AA | 3.9 ± 0.1 | 47 | 5.5 | 10.9 | 5.5 | 5.4 | 10.8 | 5.4 | −60.7 | −62.1 | −1.4 | −60.7 | −62.2 | −1.5 | — | — | — | |
| DDD·AAA | 5.3 ± 0.1 | 66 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | −60.7 | −62.0 | −1.3 | −60.7 | −62.2 | −1.5 | −61.0 | −62.4 | −1.4 | |
| Toluene | D·A | 3.6 ± 0.1 | — | 4.9 | 12.1 | 7.2 | — | — | — | −61.4 | −61.9 | −0.5 | — | — | — | — | — | — |
| DD·AA | 6.1 ± 0.1 | 32 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | −61.1 | −61.5 | −0.4 | −61.2 | −61.6 | −0.4 | — | — | — | |
The effective molarity for the AA·DD is 32 mM, therefore the chelate cooperativity associated with duplex formation (expressed as K1 EM) is equal to 130, implying that the doubly H-bonded closed duplex is almost exclusively populated (99%).42,44 The complex AAA·DDD was too stable in toluene for measuring the association constant via NMR titration, so the binding studies of length-complementary oligomers were repeated in chloroform-d. The measured association constants are sensibly lower than in toluene, however the association constant (KN) increases by an order of magnitude for every recognition module added to the oligomer, and for all three complexes the values of Δδ for the CF3 and OH groups are similar. These observations confirm that the donor recognition units are fully bound in all the complexes (Table 1). The association constant for AAA·DDD formation was investigated also using isothermal titration calorimetry (ITC) in chloroform. The data fit well to a 1:1 binding isotherm (see Section 9 in the ESI†) and confirm the association constant obtained via NMR titrations (105 M−1). The effective molarities for AA·DD and AAA·DDD are slightly higher than in toluene (respectively 47 and 66 mM); however, due to the reduction of the association constants in chloroform, the chelate cooperativity for duplex formation is approximately reduced by an order of magnitude. It is important to observe the effective molarity for formation of the AAA·DDD duplex is higher that the value obtained for the AA·DD. This suggests that the backbone is suitable for assembling stable duplexes using longer oligomers. Indeed, although duplex initiation (EM1) is relatively insensitive to the conformational properties of the backbone, the subsequent values of effective molarities for duplex propagation (EM2, EM3etc.) can be much lower in systems incompatible with formation of an extended duplex.37,64 Hence, the results in Table 1 indicate that this backbone will support formation of longer duplexes.35–37,64
Geometry is more critical in a rigid one-component backbone compared to a more flexible two-component system (i, Fig. 1b).
In fact, conformational flexibility allows the backbone to adapt to a geometry compatible with extended duplex, while more rigid backbones are difficult to design.42
Hence, the fact that the increase in association constant (KN) as a function of the recognition module number (N) is higher in this system, than in the more flexible two-component backbone, confirms that the developed system fulfils the geometrical requirements for duplex formation (Fig. 3).
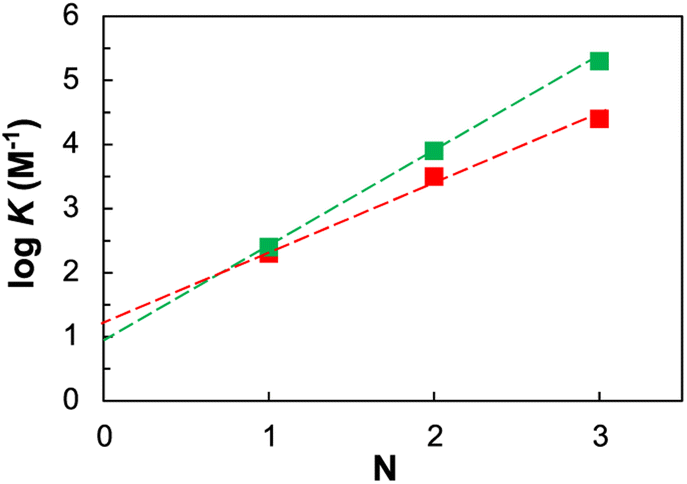 | ||
| Fig. 3 Relationship between the association constants for duplex formation between length-complementary homo-oligomers measured in chloroform at 298 K (K) and the number of intermolecular H-bonds formed (N) the described single-component system (green squares) is compared with the reported (Fig. 1b (i)) two-components system (red squares). The lines of best fit shown are respectively logK = 1.5N + 1 (green) and logK = 1.0N + 1.3 (red). | ||
The duplex assembly was further explored via thermal denaturation experiments, carried out using 19F NMR. Spectra of equimolar solutions of length-complementary oligomers at 2 mM in 1,1,2,2-tetrachloroethane-d2 were recorded at different temperatures ranging between 253 and 363 K. The changes in the 19F NMR chemical shifts of the donor's CF3 groups indicate that the population of H-bonded complexes increases at lower temperatures while the duplex is denatured at higher temperatures (Fig. 4).
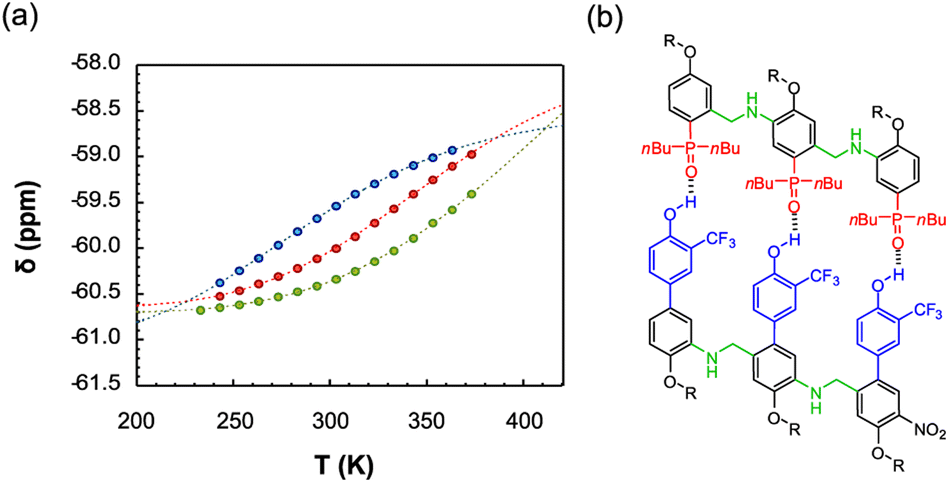 | ||
Fig. 4 (a) Experimental 19F NMR chemical shift plotted as a function of temperature for 1:1 mixtures (2 mM) of A·D (blue), AA·DD (red), and AAA·DDD (green) in 1,1,2,2-tetrachloroethane-d2. The fitting equation is reported in the ESI (eqn (S6.1)†) and the dashed lines are the best fit of the data (total rmsd < 0.01 ppm) the values of Tm,N, and 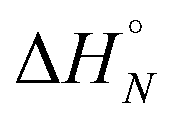 are 289 K and −23 kJ mol−1 for A·D, 354 K and −33 kJ mol−1 for AA·DD, and 422 K and −34 kJ mol−1 for AAA·DDD. (b) Structure of the AAA·DDD duplex. are 289 K and −23 kJ mol−1 for A·D, 354 K and −33 kJ mol−1 for AA·DD, and 422 K and −34 kJ mol−1 for AAA·DDD. (b) Structure of the AAA·DDD duplex. | ||
Assuming that only single strands and duplex are present, the experimental melting data were fit to a two-state model (see Section 6 of the ESI†). The values of the transition melting temperatures increase from 289 to 422 K as the length of the duplex increases, and the enthalpy change on duplex formation becomes progressively more favourable with increasing numbers of H-bonds. These observations are consistent with the titration experiments and confirm the cooperative H-bonding interactions along the duplex.
Folding and self-association of hetero-oligomers
The data obtained with homo-oligomers indicate that the backbone is suitable for extended duplex formation. However, in mixed-sequence oligomers the duplex channels might compete with the folding pathways, which can limit or prevent the formation of reliable duplexes. Thus, we initially assessed the presence of 1,2-folding, which is the most detrimental for duplex formation, because when the number of H-bonds formed in the folded state and in the duplex state are identical, the intramolecular process will dominate.The approach used to determine the folding properties assumes that DD·AA and DA·DA duplexes have similar effective molarities (EMdimer). Hence, we can use the value of Kduplex measured for the homo-dimer (KDD·AA) to determine the equilibrium constant for folding (Kfold) in the heterodimer (eqn (2)). The factor of 4 considers the degeneracies of the hetero- and homoduplexes.57
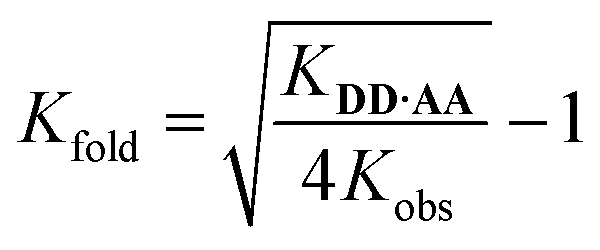 | (2) |
Self-association of the DA hetero-dimer was studied in chloroform-d through 1H and 19F NMR titration and dilution experiments (Table 2). The observed association constant is similar to the value measured for the complex DD·AA and is one order of magnitude higher than the value measured for a single H-bond in D·A. Furthermore, the limiting 19F NMR chemical shift of monomeric DA shows that δfree is equal to δfree of DD, confirming that there is no intramolecular H-bonding between adjacent recognition units. This result indicates that bringing the H-bonding groups closer together along the covalent backbone for maximizing the effect of long-short recognition units is a successful strategy for preventing the undesired 1,2-folding.
K, effective molarities (EM), limiting NMR chemical shifts (δfree and δbound) and limiting complexation-induced changes in chemical shift (Δδ) measured by NMR titrations in chloroform-d at 298 Ka
| Complex | log K (M−1) |
K fold | EMf (mM) | X fold | 19F NMRb (ppm) | 1H NMRb (ppm) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 19F (1) | 19F (2) | 1H (1) | |||||||||||
| δ free | δ bound | Δδ | δ free | δ bound | Δδ | δ free | δ bound | Δδ | |||||
| a Each titration was repeated twice, and the average value is reported with errors at the 95% confidence limit. b Data for the signals due to the OH and CF3 groups on the phenol recognition units. c Signals due to the OH protons were too broad to be detected. | |||||||||||||
| DA·DA | 3.3 ± 0.1 | 0.0 | 0.0 | 0.0 | −60.6 | −62.0 | −1.4 | — | — | — | 5.0 | 10.3 | 5.3 |
| DAA·DAA | 3.8 ± 0.1 | 0.0 | 0.0 | 0.0 | −60.7 | −62.1 | −1.4 | — | — | — | — | — | — |
| DDA·DDA | 3.4 ± 0.1 | 1.1 | 0.2 | 0.5 | −60.7 | −61.9 | −1.2 | −60.9 | −61.5 | −0.6 | — | — | — |
| ADD·ADD | 2.6 ± 0.1 | 3.9 | 0.6 | 0.9 | −60.7 | −61.9 | −1.2 | −62.0 | −61.9 | 0.1 | — | — | — |
Trimers DDA, ADD and DAA were used to investigate the 1,3-folding (Fig. 2). The signals of internal and external donor units have been assigned using a combination of 1H–1H, 1H–19F and 1H–13C 2D-NMR experiments, as summarised in the ESI (Section 5.1†). 19F NMR dilution data fit well to a dimerization isotherm in all cases, and the results are reported in Table 2. For trimer DAA the limiting chemical shift for the single-stranded free state is equal to the free chemical shift of unfoldable DD or DDD. Furthermore, the observed association constants are similar to those measured for the formation of DD·AA and DA·DA complexes.
These results suggest that DAA does not experience 1,3-folding. On the other hand, the behaviour of trimers equipped with a central biphenyl donor unit (ADD and DDA) is different. In fact, the observed self-association constant of ADD is about one order of magnitude smaller that the values obtained for DD·AA and DA·DA complexes formation. Moreover, the chemical shift due to the external donor unit for the single-stranded monomeric state is very similar to the value typical of the bound state. These results indicate that 1,3-folding is present in ADD. Trimer DDA has a behaviour that is intermediate between the unfolded DAA and the 1,3-folded ADD, in terms of both self-association constant and limiting chemical shift for the external donor unit. Indeed, analysing both Kfold and the limiting complexation-induced change in chemical shift (Δδ = δbound − δfree)57 ≈ 90% of ADD, ≈ 55% of DDA and ≈ 0% of DAA are 1,3-folded in the monomeric state.
Duplexes formation in hetero-oligomers
Interactions between all pairwise combinations of the trimer sequences were investigated by 19F NMR titration experiments in chloroform-d. The titration data fit well to 1:1 binding isotherm, and the resulting association constants are reported in Table 3. The most stable complex is the sequence-complementary DDD·AAA duplex, which has an association constant of 105 M−1, but the stabilities of the complexes span 3 orders of magnitude and other complexes are significantly less stable.
K) measured by NMR titrations in chloroform-d at 298 Ka
| DDD | DDA | ADD | DAA | AAA | |
|---|---|---|---|---|---|
| a Each titration was repeated twice, and the average value is reported with errors at the 95% confidence limit. n.d. = not detected. For some complexes with low association constants, reliable determination of the association constant was not possible. | |||||
| DDD | n.d. | — | — | — | — |
| DDA | n.d. | 3.43 | — | — | — |
| ADD | n.d. | n.d | 2.60 | — | — |
| DAA | 4.10 | 4.14 | 3.44 | 3.73 | — |
| AAA | 5.26 | 3.97 | 3.16 | n.d | n.d |
In complexes where the structure of the duplex is dictated by the sequence of the recognition units, it should be possible to distinguish antiparallel (DDA·DAA) and parallel (ADD·DAA) arrangements of the backbone, while in symmetric sequences (e.g.DDD·AAA) both the arrangements can coexist in equilibrium. Since parallel and antiparallel complexes can be composed of oligomers with different folding properties, we will initially consider the arrangement of the recognition units and later we will return to the directionality of the backbone.
Table 3 shows that the common feature of the less stable complexes is the possibility to form intramolecular H-bonding interactions between the terminal recognition units, since folding would compete with duplex formation. Indeed, the association constant for formation of the sequence-complementary duplexes between foldable oligomers are from 10 to 100 times lower than the Kduplex for DDD·AAA formation. In particular trimer ADD, which is the most affected by 1,3-folding, forms the complexes with the lowest Kobs. The undesired consequence of the 1,3-folding is that mismatch sequences might have similar association constants of complementary sequences. The Kobs for the formation of DDA·DAA complex is very close to the association constant of DDD·DAA, in fact contrary to DDD, about 55% of DDA is folded in the monomeric state.
It is important to notice that differently from the 1,2-folding, the presence of 1,3-folding should not abolish the formation of duplex in the system. Furthermore, even though complementary and mismatch complexes might have similar association constants, the system is still able to perform sequence selective duplex formation. However, before discussing the sequence selective assembly it is fundamental to highlight that the selectivity depends on what the competitors are. In fact, if trimer DAA is in competition with both DDA and DDD (100 mM each), at the equilibrium the concentration of the single mismatch complex DDD·DAA will be 4 times higher than DDA·DAA. However, if AAA is present in the same solution, it will strongly compete with DAA in favour of the most stable DDD·AAA complex formation, thus promoting the formation of DDA·DAA duplex. Self-sorting in a complex mixture can indeed be driven by the formation of a complex with very high thermodynamic stability, which drives the hierarchical reconfiguration of the mixture composition, thus leading to a self-sorting process.40,65–67
Fig. 5 illustrates the populations of all possible duplexes calculated for an equimolar mixture of DDD, DDA, DAA and AAA at 100 mM, based on the association constants reported in Table 3. Titration experiments on the complexes for which the association constants are not reported in Table 3 suggest that these are weak binding systems (K ≈ 102 M−1), hence they would have no significant effect on the speciation shown in Fig. 5. The trimers are organised so that all the sequence-complementary duplexes lie on the plot's diagonal, and these duplexes are the most populated complexes (dark blue columns). This result confirms that in a system of competing sequences affected by 1,3-folding high-fidelity duplex formation is prevented,45 thus the presence of the optimal partner for each component is required for minimizing the mismatch effect and leading to the domination of sequence-complementary duplexes.40
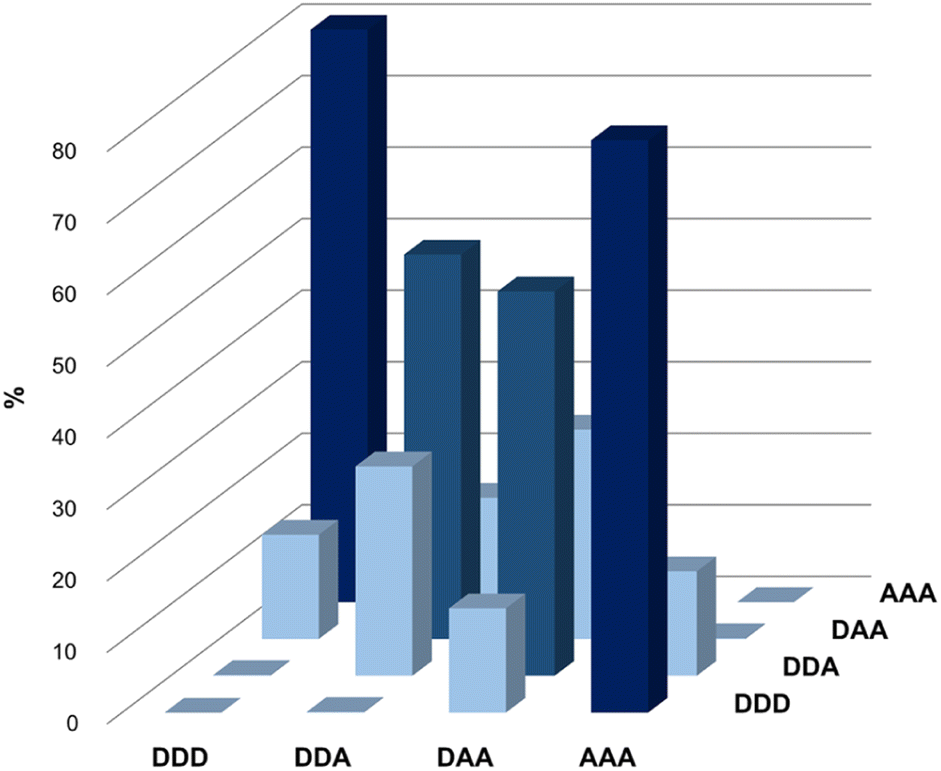 | ||
| Fig. 5 Calculated populations of duplexes formed in a 100 mM equimolar mixture of 3-mers in chloroform-d. | ||
Backbone arrangement
The use of monomers equipped with both amine and aldehyde groups confers directionality to the oligomer's backbone, so we describe the sequence of recognition units in the direction of the synthesis, starting from the aniline to the benzaldehyde monomer. In symmetric oligomers, where the assembly is not dictated by the sequence, the system could be in equilibrium between parallel and antiparallel duplexes. To assess any preferences in the backbone arrangement, we focused on a minimal set of trimers that differ in the orientation of the backbone with respect to the sequence of the recognition units. Hence, the formation of the parallel ADD·DAA and the antiparallel DDA·DAA duplexes was investigated by 19F NMR titration experiments in chloroform-d (binding constants in Table 3 and duplexes structure in Fig. 6).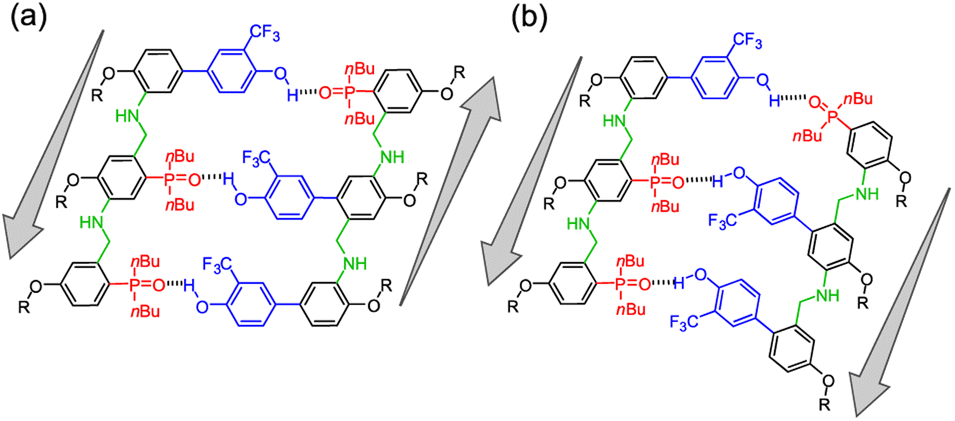 | ||
| Fig. 6 (a) Structures of antiparallel DDA·DAA and (b) ADD·DAA parallel duplexes. | ||
However, since ADD and DDA have different folding properties, the evaluation of the preference in backbone arrangement cannot be done simply by comparing the two observed association constants. Folding between terminal recognition units is not present in DAA, thus the observed association constant for duplex formation where only one partner folds is given by eqn (3).40
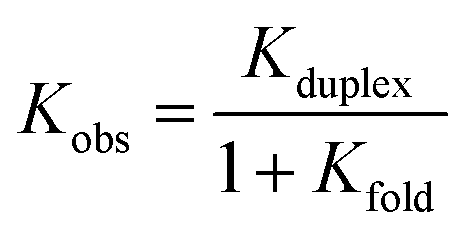 | (3) |
Hence, it is possible to calculate the association constant for the formation of antiparallel and parallel duplexes, as shown respectively in eqn (4) and (5).
| Kantipduplex = KobsDDA·DAA(1 + KfoldDDA) | (4) |
| Kparaduplex = KobsADD·DAA(1 + KfoldADD) | (5) |
The obtained association constant for the formation of antiparallel duplex (3 × 104 M−1) is higher than the one for parallel duplex formation (1 × 104 M−1). These results suggest that in symmetric complexes, such as DDD·AAA, the duplex structure is in equilibrium between a 70% of antiparallel and a 30% of parallel duplexes.
Template effect
An interesting feature of a system based on monomers equipped with aniline and aldehyde groups is the possibility to exploit dynamic combinatorial chemistry. To explore whether the system is compatible with the templated synthesis of complementary-sequence oligomers, we focused on a minimal set of homodimers, studying the ability of the aniline template AA to favour the synthesis of the complementary imine dimer DD′ (Fig. 7d). Template molecules pre-organise the monomers in a defined spatial arrangement, which permits to transfer the structural information from the molecular template to the molecular copy.68,69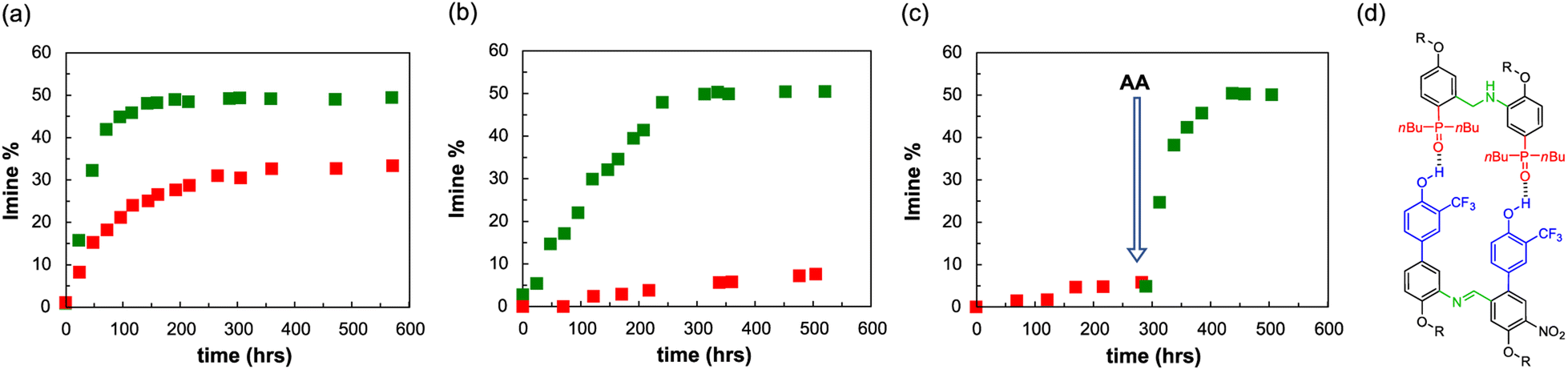 | ||
| Fig. 7 (a) Imine formation in toluene-d8 at 70 °C between D′CO and DNH2 (10 mM each), in the presence of POOct3 (20 mM, red squares) or AA (10 mM, green squares). (b) Imine formation (toluene-d8, 70 °C) between D′CO and DNH2 (1 mM each), in the presence of POOct3 (2 mM, red squares) or AA (1 mM, green squares). (c) Imine formation (toluene-d8, 70 °C) between D′CO and DNH2 (1 mM each) before (red squares) and after the addition of the dimer template AA (1 mM, green squares). (d) Structure of the dimeric complex between acceptor template AA and the imine donor product. | ||
In order to favour the templated intramolecular imine formation, it is fundamental to operate at concentrations lower than the effective molarity, which is about 32 mM for dimers in toluene. Hence, we initially studied the imine formation at 70 °C in a 10 mM solution of aldehyde (D′CO) and aniline (DNH2) monomers in toluene, in the presence of the aniline template AA (5 mM) or of an equimolar amount of monomeric phosphine oxide as a control experiment (trioctylphosphine oxide, POOct3 10 mM). Toluene was preferred to chloroform for maximizing the binding constants. The consumption of the aldehyde and the imine formation were monitored via1H NMR and, as it is shown in Fig. 7a, the presence of AA has two effects: the first is kinetic and it is related to the preorganization of the monomers on the template, favouring the formation of imine, which reaches its maximum within 100 hours, compared to the 300 hours required by the control experiment (complete NMR data are reported in the Section 7 of the ESI†).70 The second effect is thermodynamic, and it is associated to the increased imine concentration at the equilibrium, which is about 50% in the templated experiment and around 30% in non-templated conditions. In these conditions the imine dimer could be formed through two different pathways: an intramolecular bond formation between two monomers pre-assembled on the template and an intermolecular non-templated process.
To further prove the observed template effect, we repeated the experiments decreasing by a factor of 10 the concentration of all the components (Fig. 7b). Indeed, if the association constant is enough to guarantee the assembly of the monomers on the template, the decrease of concentration would greatly affect the non-templated pathway, with no or little influence on the templated process. Fig. 7b shows that in the presence of the template dimer AA the imine reaches the equilibrium concentration of 50% in about 200 hours, while the non-templated system experiences a sensible decrease in the imine formation rate and after 500 hours only ≈7% of DD′ dimer is formed.
To additionally highlight the effect of the template oligomer we followed the imine formation at 70 °C in a 1 mM toluene solution of aldehyde (D′CO) and aniline (DNH2) monomers, in the absence of any phosphine oxide counterpart. After about 290 hours only less than 5% of imine was formed, however the addition of 0.5 mM of AA sensibly increased the imine formation rate, which reached the equilibrium concentration of 50% about 160 hours after the template addition (Fig. 7c).
Next, we explored the effect of the template concentration on the initial reaction rate. Equimolar toluene solutions of D′CO and DNH2 (1 mM each) were reacted at 70 °C in the presence of variable amounts of the aniline template dimer AA (0–1.5 mM) and the proceeding of the reactions was monitored via1H NMR. For all the tested conditions, initial rates of template synthesis Vinit, were determined from concentration–time profiles at early reaction times and plotted as a function of the square root of the template concentration (Fig. 8).
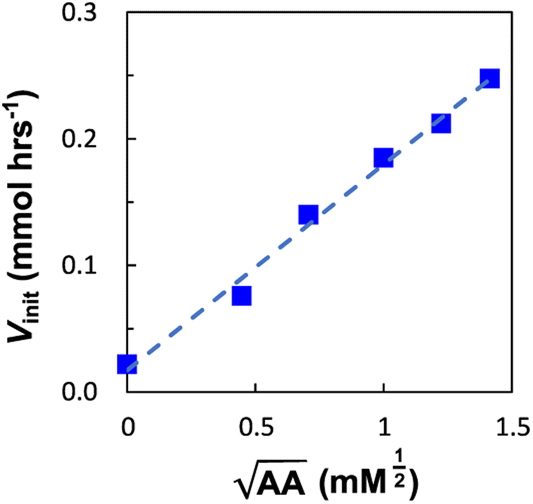 | ||
| Fig. 8 The initial reaction rate (Vinit) of imine formation between D′CO and DNH2 (1 mM each, toluene-d8 70 °C) is plotted as a function of the square root of AA concentration. The line of best fit shown is Vinit = 0.2[AA]1/2 + 0.02. | ||
The plot shown in Fig. 8 reveals a linear relationship between the Vinit and the square root of the AA concentration. This allows to establish the empirical relationship reported in eqn (6), which indicates that the complementary imine is formed from monomers D′CO and DNH2via two pathways: a template-catalysed route, characterised by the empirical constant a, and a template-independent bimolecular process which is measured with the empirical constant b, that accounts for the initial rate in the absence of AA.
The rate of templated synthesis follows the square-root of the initial template concentration. This peculiar rate law was initially observed in self-replicant systems, where the product remains bound to the template causing its inactivation.71–73
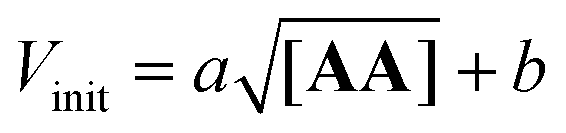 | (6) |
In order to test the key role of the H-bonding recognition event in the observed template effect, we synthesised a mutated analogue of DNH2, in which the phenol recognition unit has been methylated (DOMeNH2), thus hampering its ability to act as H-bonding donor (Fig. 9a and c). We then focused on a dynamic combinatorial library composed of D′CO, DNH2 and DOMeNH2 (1 mM each) and we studied its composition at 70 °C via1H NMR in the presence of template AA (1 mM) or POOct3 (2 mM). With monomeric phosphine oxide we can detect only traces of the possible imine products DD′ and the methylated DOMeD′, both at similar concentrations (Fig. 9b). However, in the presence of the template acceptor dimer AA the DD′ imine product reached its equilibrium concentration (≈50%) within 150 hours. It is important to observe that in these conditions no methylated dimer was detected, hence confirming the fundamental role of the H-bonding recognition unit in the observed template effect.
 | ||
| Fig. 9 (a) Cartoon showing the template effect of AA (ii) compared to monomeric phosphine oxide (i), on a dynamic combinatorial library formed by D′CO, DNH2 and the methylated donor DOMeNH2. (b) Imines formation (toluene-d8, 70 °C) between D′CO, DNH2 and DOMeNH2 (1 mM each): in the presence of AA (1 mM) only the DNH2 based imine is formed (green squares), while in the presence of POOct3 (2 mM) traces of both DNH2 (red) and DOMeNH2 (orange) based imines are detected. (c) Structure of DOMeNH2: the corresponding imine obtained with reaction with D′CO is unable to form duplex with the template AA. | ||
Conclusions
A new family of recognition-encoded oligoanilines has been developed. The H-bond base pair 2-trifluoromethylphenol–phosphine oxide permits duplex assembly through the formation of intermolecular H-bonds in both toluene and chloroform. The backbone is assembled via reductive amination of monomers equipped with both aniline and aldehyde groups. The self-assembly properties of the oligomers were characterize using 1H and 19F NMR spectroscopy. For length-complementary homo-oligomers, duplex formation in toluene is characterised by an increase in stability of two orders of magnitude for every base-pair added to the chain, while in chloroform the association constant increases of one order of magnitude. The long-short base-pairing system together with the backbone rigidity prevent the undesired folding between adjacent monomers, as it is shown by NMR spectra of DA dimer dilute solutions. On the other hand, similar experiments on mixed sequence trimers show that 1,3-folding takes place in trimers with an internal donor unit, such as ADD and DDA, but not in DAA. These folding processes compete with duplex formation and reduce the stability of the corresponding duplex. The result is that some of the mismatch complexes (DDD·DAA) have association constant similar to those measured for some of the sequence-complementary duplexes (DDA·DAA), thus hampering the possibility of high-fidelity sequence-selective duplex formation.45 However, the affinity of DDD for AAA is much higher than for any other sequence, thus in a mixture of trimers the presence of the optimal partner for each component minimizes the mismatch effects, leading to the domination of the sequence-complementary duplexes in solution. The use of asymmetric monomers confers directionality to the oligomer's backbone hence, to characterize the relative stabilities of parallel and antiparallel duplexes we studied the formation of ADD·DAA and DDA·DAA complexes. These systems show that the antiparallel arrangement is more stable than the parallel arrangement by a factor of 3, suggesting that symmetric complexes, such as DDD·AAA, exist as a 70:30 mixture of the two backbone arrangements.
An interesting feature of this system is the possibility to exploit imine based dynamic combinatorial chemistry for the templated synthesis of complementary oligomers. This approach, which has been previously proposed on DNA based oligomers,48,49 is now applied to a fully synthetic system capable of sequence-selective extended duplex formation. To assess the template effect, we used dimer AA as a template for the synthesis of the complementary donor imine. 1H NMR kinetics showed that the aniline template successfully pre-organises the donor monomers, accelerating the complementary imine formation. A linear relationship between the initial rate and the square root of the template concentration was found. This indicates that the complementary imine is formed via two pathways: a template-catalysed route and a template-independent bimolecular process. Since the non-templated bimolecular route is highly influenced by the monomers concentration, the observed effect increases diluting the system from 10 mM to 1 mM. The presence of a template oligomer affects not only the kinetic but also the imine population at the equilibrium, which is higher in the template experiment (50%) compared to non-templated conditions (30%). Finally, the key role of the H-bonding recognition was highlighted adding a methylated donor monomer to the dynamic combinatorial library. The system can discriminate the mutated base, indeed the presence of AA dimer favours only the formation of the complementary imine dimer, to the disadvantage of the mutated monomer.
The sequence-selective duplex formation together with the possibility to exploit the template effect in dynamic combinatorial chemistry make this system an attractive target for the investigation of templated synthesis in longer mixed sequence oligomers.
Data availability
The ESI† contains all the experimental details, including the synthetic procedures, the characterization and the 1H, 19F, 31P, 13C-NMR spectra of all products reported in this study. ITC experiments and NMR spectra of titrations, dilutions, variable temperature and templated experiments are included as well.Author contributions
The manuscript was written through contributions of all authors. D. R.-G. performed the synthesis and the NMR and ITC experiments for the duplex characterization. A. D. V. replicated the synthesis of monomers and oligomers. T. M. performed the NMR templated experiments. L. G. designed the project, analysed the data, and supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LG thanks Cristopher A. Hunter for the helpful discussions and UNIPD STARS-StG (DyNAseq) for funding.Notes and references
- J. D. Watson and F. H. Crick, Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- D. H. Appella, Non-natural nucleic acids for synthetic biology, Curr. Opin. Chem. Biol., 2009, 13, 687–696 CrossRef CAS PubMed.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Triazole-linked analogue of deoxyribonucleic acid ((TL)DNA): design, synthesis, and double-strand formation with natural DNA, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed.
- K. Burgess, R. A. Gibbs, M. L. Metzker and R. Raghavachari, Synthesis of an oxyamide linked nucleotide dimer and incorporation into antisense oligonucleotide sequences, J. Chem. Soc., Chem. Commun., 1994, 915–916 RSC.
- L. Zhang, Z. Yang, K. Sefah, K. M. Bradley, S. Hoshika, M. J. Kim, H. J. Kim, G. Zhu, E. Jimenez, S. Cansiz, I. T. Teng, C. Champanhac, C. McLendon, C. Liu, W. Zhang, D. L. Gerloff, Z. Huang, W. Tan and S. A. Benner, Evolution of functional six-nucleotide DNA, J. Am. Chem. Soc., 2015, 137, 6734–6737 CrossRef CAS PubMed.
- J. A. Piccirilli, T. Krauch, S. E. Moroney and S. A. Benner, Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet, Nature, 1990, 343, 33–37 CrossRef CAS PubMed.
- B. A. Schweitzer and E. T. Kool, Aromatic Nonpolar Nucleosides as Hydrophobic Isosteres of Pyrimidine and Purine Nucleosides, J. Org. Chem., 1994, 59, 7238–7242 CrossRef CAS PubMed.
- H. Liu, J. Gao, S. R. Lynch, Y. D. Saito, L. Maynard and E. T. Kool, A four-base paired genetic helix with expanded size, Science, 2003, 302, 868–871 CrossRef CAS PubMed.
- J. C. Delaney, J. Gao, H. Liu, N. Shrivastav, J. M. Essigmann and E. T. Kool, Efficient replication bypass of size-expanded DNA base pairs in bacterial cells, Angew. Chem., Int. Ed. Engl., 2009, 48, 4524–4527 CrossRef CAS PubMed.
- A. Eschenmoser, Towards a chemical etiology of nucleic acid structure, Origins Life Evol. Biospheres, 1997, 27, 535–553 CrossRef CAS PubMed.
- S. Obika, D. Nanbu, Y. Hari, K.-i. Morio, Y. In, T. Ishida and T. Imanishi, Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3, -endo sugar puckering, Tetrahedron Lett., 1997, 38, 8735–8738 CrossRef CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- A. Aerschot Van, I. Verheggen, C. Hendrix and P. Herdewijn, 1,5-Anhydrohexitol Nucleic Acids, a New Promising Antisense Construct, Angew. Chem., Int. Ed. Engl., 1995, 34, 1338–1339 CrossRef.
- K. Schoning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Chemical etiology of nucleic acid structure: the alpha-threofuranosyl-(3′→2′) oligonucleotide system, Science, 2000, 290, 1347–1351 CrossRef CAS PubMed.
- L. Zhang, A. Peritz and E. Meggers, A simple glycol nucleic acid, J. Am. Chem. Soc., 2005, 127, 4174–4175 CrossRef CAS PubMed.
- A. B. Gerber and C. J. Leumann, Synthesis and properties of isobicyclo-DNA, Chem. Eur. J., 2013, 19, 6990–7006 CrossRef CAS PubMed.
- S. C. Zimmerman and T. J. Murray, Hydrogen bonded complexes with the AA·DD, AA·DDD, and AAA·DD motifs: The role of three centered (bifurcated) hydrogen bonding, Tetrahedron Lett., 1994, 35, 4077–4080 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, Strong Dimerization of Ureidopyrimidones via Quadruple Hydrogen Bonding, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- J. L. Sessler and R. Wang, Self-Assembly of an “Artificial Dinucleotide Duplex”, J. Am. Chem. Soc., 1996, 118, 9808–9809 CrossRef CAS.
- C. Tuerk and L. Gold, Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, In vitro selection of RNA molecules that bind specific ligands, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA, Nature, 1990, 344, 467–468 CrossRef CAS PubMed.
- Z. Chen, P. A. Lichtor, A. P. Berliner, J. C. Chen and D. R. Liu, Evolution of sequence-defined highly functionalized nucleic acid polymers, Nat. Chem., 2018, 10, 420–427 CrossRef CAS PubMed.
- D. L. Usanov, A. I. Chan, J. P. Maianti and D. R. Liu, Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules, Nat. Chem., 2018, 10, 704–714 CrossRef CAS PubMed.
- J. C. Chen, J. P. Chen, M. W. Shen, M. Wornow, M. Bae, W. H. Yeh, A. Hsu and D. R. Liu, Generating experimentally unrelated target molecule-binding highly functionalized nucleic-acid polymers using machine learning, Nat. Commun., 2022, 13, 4541 CrossRef CAS PubMed.
- A. P. Bisson, F. J. Carver, C. A. Hunter and J. P. Waltho, Molecular Zippers, J. Am. Chem. Soc., 2002, 116, 10292–10293 CrossRef.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, Synthesis and Recognition Properties of Aromatic Amide Oligomers: Molecular Zippers, J. Am. Chem. Soc., 2000, 122, 8856–8868 CrossRef CAS.
- H. Gong and M. J. Krische, Duplex molecular strands based on the 3,6-diaminopyridazine hydrogen bonding motif: amplifying small-molecule self-assembly preferences through preorganization and iterative arrangement of binding residues, J. Am. Chem. Soc., 2005, 127, 1719–1725 CrossRef CAS.
- Y. Yang, Z. Y. Yang, Y. P. Yi, J. F. Xiang, C. F. Chen, L. J. Wan and Z. G. Shuai, Helical molecular duplex strands: multiple hydrogen-bond-mediated assembly of self-complementary oligomeric hydrazide derivatives, J. Org. Chem., 2007, 72, 4936–4946 CrossRef CAS.
- P. N. Taylor and H. L. Anderson, Cooperative Self-Assembly of Double-Strand Conjugated Porphyrin Ladders, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS.
- B. Gong, Y. Yan, H. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, A New Approach for the Design of Supramolecular Recognition Units: Hydrogen-Bonded Molecular Duplexes, J. Am. Chem. Soc., 1999, 121, 5607–5608 CrossRef CAS.
- H. Zeng, R. S. Miller, R. A. Flowers and B. Gong, A Highly Stable, Six-Hydrogen-Bonded Molecular Duplex, J. Am. Chem. Soc., 2000, 122, 2635–2644 CrossRef CAS.
- H. Zeng, H. Ickes, R. A. Flowers 2nd and B. Gong, Sequence specificity of hydrogen-bonded molecular duplexes, J. Org. Chem., 2001, 66, 3574–3583 CrossRef CAS PubMed.
- B. Gong, Molecular duplexes with encoded sequences and stabilities, Acc. Chem. Res., 2012, 45, 2077–2087 CrossRef CAS PubMed.
- G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Mix and match backbones for the formation of H-bonded duplexes, Chem. Sci., 2016, 7, 1760–1767 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Mix and match recognition modules for the formation of H-bonded duplexes, Chem. Sci., 2016, 7, 5686–5691 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Cooperative duplex formation by synthetic H-bonding oligomers, Chem. Sci., 2016, 7, 94–101 RSC.
- D. Nunez-Villanueva and C. A. Hunter, Homochiral oligomers with highly flexible backbones form stable H-bonded duplexes, Chem. Sci., 2017, 8, 206–213 RSC.
- D. Nunez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. A. Swain and C. A. Hunter, H-Bond Self-Assembly: Folding versus Duplex Formation, J. Am. Chem. Soc., 2017, 139, 6654–6662 CrossRef CAS PubMed.
- A. E. Stross, G. Iadevaia, D. Nunez-Villanueva and C. A. Hunter, Sequence-Selective Formation of Synthetic H-Bonded Duplexes, J. Am. Chem. Soc., 2017, 139, 12655–12663 CrossRef CAS PubMed.
- J. A. Swain, G. Iadevaia and C. A. Hunter, H-Bonded Duplexes based on a Phenylacetylene Backbone, J. Am. Chem. Soc., 2018, 140, 11526–11536 CrossRef CAS PubMed.
- F. T. Szczypinski and C. A. Hunter, Building blocks for recognition-encoded oligoesters that form H-bonded duplexes, Chem. Sci., 2019, 10, 2444–2451 RSC.
- F. T. Szczypiński, L. Gabrielli and C. A. Hunter, Emergent supramolecular assembly properties of a recognition-encoded oligoester, Chem. Sci., 2019, 10, 5397–5404 RSC.
- L. Gabrielli, D. Nunez-Villanueva and C. A. Hunter, Two-component assembly of recognition-encoded oligomers that form stable H-bonded duplexes, Chem. Sci., 2020, 11, 561–566 RSC.
- P. Troselj, P. Bolgar, P. Ballester and C. A. Hunter, High-Fidelity Sequence-Selective Duplex Formation by Recognition-Encoded Melamine Oligomers, J. Am. Chem. Soc., 2021, 143, 8669–8678 CrossRef CAS PubMed.
- C. Bohler, P. E. Nielsen and L. E. Orgel, Template switching between PNA and RNA oligonucleotides, Nature, 1995, 376, 578–581 CrossRef CAS.
- D. M. Rosenbaum and D. R. Liu, Efficient and sequence-specific DNA-templated polymerization of peptide nucleic acid aldehydes, J. Am. Chem. Soc., 2003, 125, 13924–13925 CrossRef CAS PubMed.
- X. Li, Z. Y. Zhan, R. Knipe and D. G. Lynn, DNA-catalyzed polymerization, J. Am. Chem. Soc., 2002, 124, 746–747 CrossRef CAS PubMed.
- J. T. Goodwin and D. G. Lynn, Template-directed synthesis: use of a reversible reaction, J. Am. Chem. Soc., 1992, 114, 9197–9198 CrossRef CAS.
- D. Nunez-Villanueva and C. A. Hunter, H-Bond Templated Oligomer Synthesis Using a Covalent Primer, J. Am. Chem. Soc., 2022, 144, 17307–17316 CrossRef CAS PubMed.
- D. Nunez-Villanueva and C. A. Hunter, Replication of a synthetic oligomer using chameleon base-pairs, Chem. Commun., 2022, 58, 11005–11008 RSC.
- D. Nunez-Villanueva, M. Ciaccia, G. Iadevaia, E. Sanna and C. A. Hunter, Sequence information transfer using covalent template-directed synthesis, Chem. Sci., 2019, 10, 5258–5266 RSC.
- D. Nunez-Villanueva, M. Ciaccia and C. A. Hunter, Cap control: cyclic versus linear oligomerisation in covalent template-directed synthesis, RSC Adv., 2019, 9, 29566–29569 RSC.
- D. Nunez-Villanueva and C. A. Hunter, Molecular replication using covalent base-pairs with traceless linkers, Org. Biomol. Chem., 2019, 17, 9660–9665 RSC.
- D. Nunez-Villanueva and C. A. Hunter, Controlled mutation in the replication of synthetic oligomers, Chem. Sci., 2021, 12, 4063–4068 RSC.
- D. Nunez-Villanueva and C. A. Hunter, Effect of backbone flexibility on covalent template-directed synthesis of linear oligomers, Org. Biomol. Chem., 2022, 20, 8285–8292 RSC.
- D. Rosa-Gastaldo, V. Peciukenas, C. A. Hunter and L. Gabrielli, Duplex vs. folding: tuning the self-assembly of synthetic recognition-encoded aniline oligomers, Org. Biomol. Chem., 2021, 19, 8947–8954 RSC.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Fast transimination in organic solvents in the absence of proton and metal catalysts. A key to imine metathesis catalyzed by primary amines under mild conditions, Chem. Sci., 2013, 4, 2253–2261 RSC.
- J.-M. Lehn, Dynamic Combinatorial Chemistry and Virtual Combinatorial Libraries, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. Sanders and S. Otto, Dynamic combinatorial chemistry, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Dynamic covalent chemistry, Angew. Chem., Int. Ed. Engl., 2002, 41, 898–952 CrossRef PubMed.
- I. Huc and J. M. Lehn, Virtual combinatorial libraries: dynamic generation of molecular and supramolecular diversity by self-assembly, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- L. Gabrielli and C. A. Hunter, Supramolecular catalysis by recognition-encoded oligomers: discovery of a synthetic imine polymerase, Chem. Sci., 2020, 11, 7408–7414 RSC.
- G. Iadevaia, D. Nunez-Villanueva, A. E. Stross and C. A. Hunter, Backbone conformation affects duplex initiation and duplex propagation in hybridisation of synthetic H-bonding oligomers, Org. Biomol. Chem., 2018, 16, 4183–4190 RSC.
- A. Wu and L. Isaacs, Self-sorting: the exception or the rule?, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- G. Men and J. M. Lehn, Higher Order Constitutional Dynamic Networks: [2 × 3] and [3 × 3] Networks Displaying Multiple, Synergistic and Competitive Hierarchical Adaptation, J. Am. Chem. Soc., 2017, 139, 2474–2483 CrossRef CAS PubMed.
- C. Pezzato, P. Scrimin and L. J. Prins, Zn2+-regulated self-sorting and mixing of phosphates and carboxylates on the surface of functionalized gold nanoparticles, Angew. Chem., Int. Ed. Engl., 2014, 53, 2104–2109 CrossRef CAS PubMed.
- G. von Kiedrowski, 40 Jahre Fonds der Chemischen Industrie 1950–1990, 1990, pp. 197–218 Search PubMed.
- L. E. Orgel, Molecular replication, Nature, 1992, 358, 203–209 CrossRef CAS PubMed.
- The monomer DCO was used as well in some preliminary experiments showing a similar behaviour (see ESI†). However the imine formation rate and the equilibrium population of imine were slightly lower, requiring longer experimental times and resulting in a lower S/N ratio and higher uncertainity in the signal integration of the first points. Therfore we preferred to proceed with the D′CO monomer instead.
- G. von Kiedrowski, A Self-Replicating Hexadeoxynucleotide, Angew. Chem., Int. Ed. Engl., 1986, 25, 932–935 CrossRef.
- W. S. Zielinski and L. E. Orgel, Autocatalytic synthesis of a tetranucleotide analogue, Nature, 1987, 327, 346–347 CrossRef CAS PubMed.
- E. Szathmary, Simple growth laws and selection consequences, Trends Ecol. Evol., 1991, 6, 366–370 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis detail, complete product characterizations, NMR titrations, extra NMR experiments and templated experiments. See DOI: https://doi.org/10.1039/d3sc00880k |
| This journal is © The Royal Society of Chemistry 2023 |