
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo biodistribution of kinetically stable Pt2L4 nanospheres that show anti-cancer activity†
Eduard O.
Bobylev‡
 a,
Renzo A.
Knol‡
b,
Simon
Mathew
a,
David A.
Poole
III
a,
Ioli
Kotsogianni
c,
Nathaniel I.
Martin
c,
Bas
de Bruin
a,
Alexander
Kros
*b and
Joost N. H.
Reek
*a
a,
Renzo A.
Knol‡
b,
Simon
Mathew
a,
David A.
Poole
III
a,
Ioli
Kotsogianni
c,
Nathaniel I.
Martin
c,
Bas
de Bruin
a,
Alexander
Kros
*b and
Joost N. H.
Reek
*a
avan ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
bDept. of Supramolecular & Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands
cBiological Chemistry Group, Institute of Biology Leiden, Leiden University, 2333 BE Leiden, The Netherlands
First published on 22nd May 2023
Abstract
There is an increasing interest in the application of metal–organic cages (MOCs) in a biomedicinal context, as they can offer non-classical distribution in organisms compared to molecular substrates, while revealing novel cytotoxicity mechanisms. Unfortunately, many MOCs are not sufficiently stable under in vivo conditions, making it difficult to study their structure–activity relationships in living cells. As such, it is currently unclear whether MOC cytotoxicity stems from supramolecular features or their decomposition products. Herein, we describe the toxicity and photophysical properties of highly-stable rhodamine functionalized platinum-based Pt2L4 nanospheres as well as their building blocks under in vitro and in vivo conditions. We show that in both zebrafish and human cancer cell lines, the Pt2L4 nanospheres demonstrate reduced cytotoxicity and altered biodistribution within the body of zebrafish embryos compared to the building blocks. We anticipate that the composition-dependent biodistribution of Pt2L4 spheres together with their cytotoxic and photophysical properties provides the fundament for MOC application in cancer therapy.
Introduction
The biomedical application of metal–organic cages (MOCs) is getting increasing interest. Since the discovery and clinical success of the first inorganic anti-cancer drug cisplatin, research on more potent, selective, and multi-functional metallodrugs progressed.1,2 During the last 10 years, MOCs have emerged as potent alternatives to classical inorganic and organometallic complexes.3–5 MOCs are well-defined nanostructures, which are self-assembled by reversible coordination of multi-topic ligands (linkers) to metal (nodes).6–8 As MOCs are frequently composed of cytotoxic metals, they can act as anticancer metallodrugs. In this context, anticancer activity of many different MOCs in different shapes (spheres, prisms, helicates) and compositions (Ni2L3, Ni4L6, Pd2L4, Pd12L24, Pt8LA2LB4, Ru6LA2LB3) have been explored.9–22 MOCs have characteristic advantages over the use of molecular drugs, such as their large size (usually 1–10 nm), well-defined shape, and confined cavity that can be exploited. These characteristics can improve their therapeutic profiles through alternative activity mechanisms. Amongst the cytotoxic MOC complexes, M2L4 nanospheres (Fig. 1) are especially interesting for reasons provided below.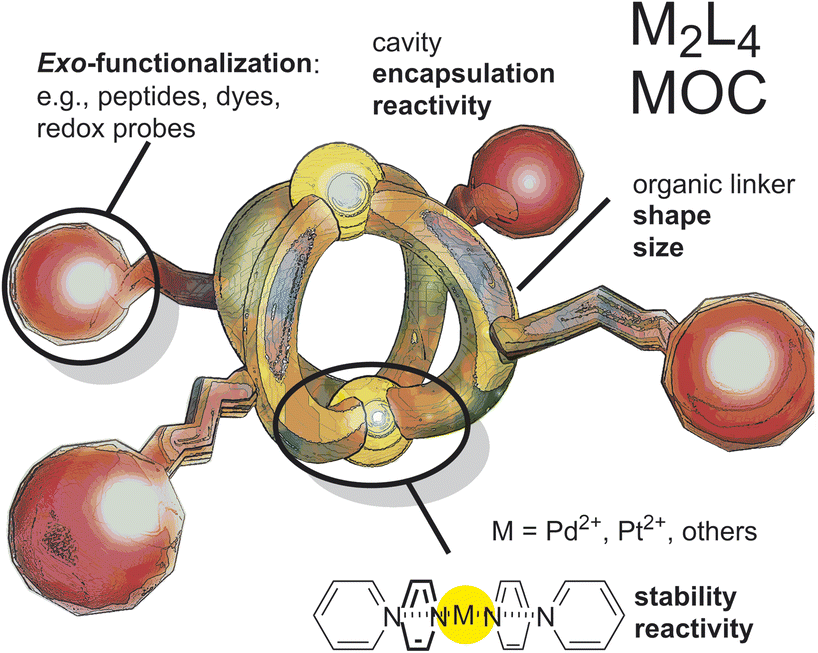 | ||
| Fig. 1 Schematic representation of M2L4 spheres with different modulation possibilities. For example, outer functionalization for solubility; organic linker modification for size and shape modulation and metal modification for modulation of stability. | ||
M2L4 nanospheres are typically prepared by the combination of ditopic pyridine donors together with square planar palladium or platinum precursors (Fig. 1). This type of nanospheres have modulation possibilities by ligand variation or functionalization and can be obtained in a variety of different shapes, which makes them interesting candidates for biological investigation.23–27 Furthermore, a number of reports describe the cytotoxicity of Pd2L4 nanospheres by disruption of cell membranes14 or alternative pathways28–30 or by encapsulation of toxic guest molecules (i.e. cisplatin),12,13,15,31 making further studies into this direction promising for the development of novel anti-cancer drugs. In this context, understanding how a certain shape of the nanosphere influences its cytotoxicity or the activity/toxicity of covalently attached drugs or encapsulated drugs against cancer (herein called structure–activity relationship) is important to design novel, potent and tumor-specific nanospheres. Unfortunately, Pd2L4 nanospheres were shown to decompose quickly when exposed to physiological conditions,12,15 hampering understanding of the cytotoxicity mechanism and development of structure–activity relationships. Despite numerous fascinating examples of in vitro application of M2L4 nanospheres, in vivo applications are rare.32 Imaging of nanospheres in vivo remains, due to the so called “heavy atom effect”, a problem and only a few fluorescent M2L4 spheres have been reported.33–35 Although, some M2L4 nanospheres have shown to be fluorescent, biological application requires a good fluorescent quantum yield and an emission wavelength preferably in the red light. Unfunctionalized nanospheres typically absorb and emit between 250 and 400 nm, which is impractical due to the absorption of living matter (e.g., proteins) in the same region. Furthermore, red fluorescent probes allow for deeper tissue imaging and typically yield a brighter signal to background ratio.36 Motivated by these challenges and by the potential of M2L4 nanospheres for therapeutic treatment, we explore in this contribution kinetically robust and fluorescent Pt2L4 nanospheres in vitro and in vivo. In a proof-of-concept study, we show that the well-defined arrangement of fluorescent drugs around nanospheres changes their biodistribution in vivo (using a zebrafish model), photophysical and cytotoxic properties, while the nanosphere itself (without covalent drug conjugation) acts as a delivery vehicle (for dyes or anti-cancer drugs) with little toxicity.
Results and discussion
Ligand design and nanosphere synthesis
Excellent photophysical properties of rhodamine B for in vivo imaging made this dye the subject of our studies leading to the design of two novel building blocks (Fig. 2). Next to the required spectroscopic features for in vivo detection, rhodamine B amide derivatives have shown recently potent cytotoxicity against different human cancer types.37,38 Furthermore, singlet oxygen formation using rhodamine dyes emerged as a tool in photodynamic therapy of tumor cells.39,40 Therefore, rhodamine functionalization of nanospheres can yield systems which are not only suitable for in vivo imaging, but also can have cytotoxic properties by themselves or through light driven singlet oxygen (1O2) formation. The rhodamine unit is attached via two different linker types to a sphere-forming building block (Fig. 2). We chose a long hydrophilic (tetra-ethylene glycol (TEG)) and a short linker (ethyl) to study the effect of the resulting well-defined 3-dimensional nanospheres on (A) photophysical properties, (B) cytotoxicity against cancer lines in vitro and (C) biodistribution within the body of zebrafish embryos. Both building blocks, LRB and LTEGRB are prepared according to standard organic synthesis protocols (summarized in Section SI1†).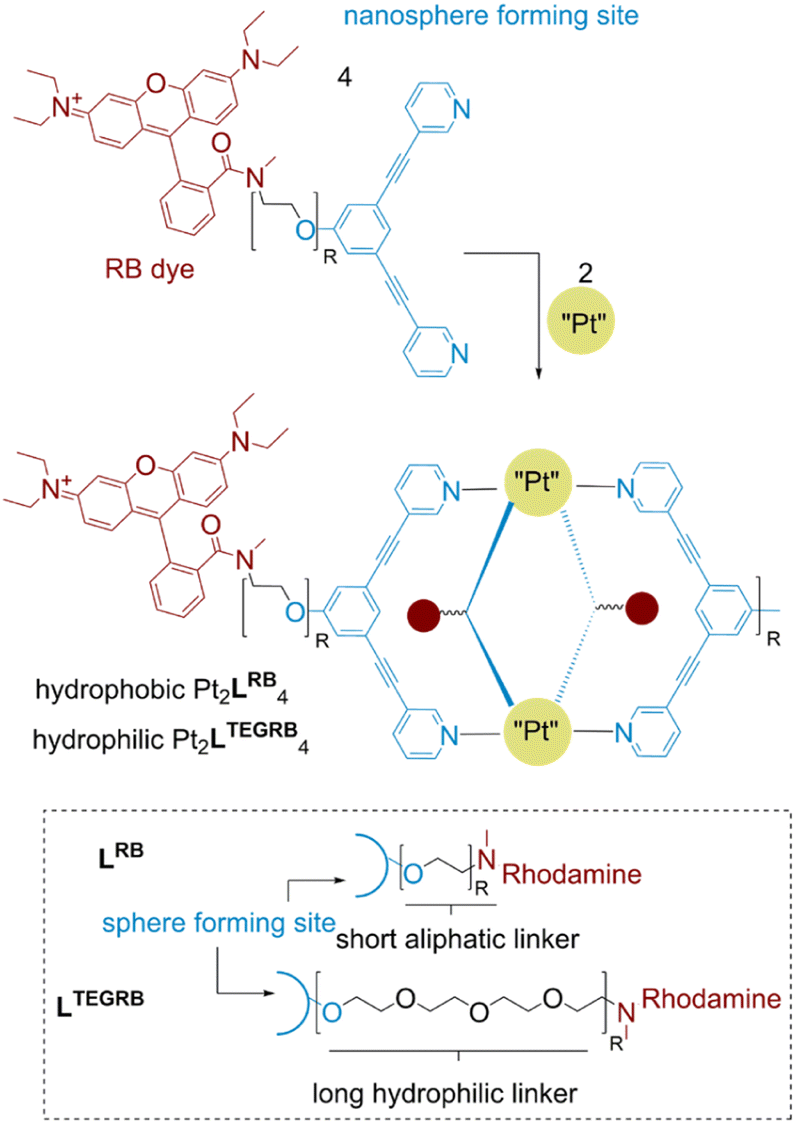 | ||
| Fig. 2 Chemical structure of the two building blocks LRB and LTEGRB and the corresponding nanospheres Pt2LRB4 and Pt2LTEGRB4. | ||
To obtain kinetically robust nanospheres, we used platinum for the nanosphere formation, as the platinum–pyridine bond was demonstrated to be more kinetically inert compared to the conventionally applied palladium analogues.12,41,42 Pt2L4 spheres are prepared according to a recently published procedure.38LRB (1 eq.), and [Pt(BF4)(MeCN)4] (0.6 eq.) were heated to 150 °C in acetonitrile-d3 for 1 day (Fig. 3A). The observed downfield shifts of the pyridine protons in the 1H-NMR spectra are in line with coordination to platinum (Fig. 3C). DOSY NMR spectra show the appearance of a slower diffusing species, with a calculated hydrodynamic radius of 1.1 nm, in line with the formation of Pt2L4 nanosphere (Fig. 3B). ESI-HR-MS analysis confirms selective formation of the desired structure by showing only signals corresponding to different charged states of the assembly with the general formula [Pt2LRB4(BF4)x](8−x)+ and matching isotope pattern for x = 2–8 (Fig. 3D). Crystals suitable for X-ray diffraction were grown from slow vapor diffusion of diethyl ether into a solution of the sphere in acetonitrile. The solid-state structure of Pt2LRB4 confirms formation of the desired nanosphere (Fig. 4). The metal–metal distance and other structural properties are in accordance with similar M2L4 spheres. An interesting feature in the solid state is the bending of the rhodamine dyes towards the windows of the nanosphere. This bending provides the most compact packing and involves four π–π interactions between the rhodamine and the central aromatic ring of the building block with a distance of 3.7 Å (in line with 6 Å limit of other typical cation–π and π–π interactions).43 The displayed densely packed form might occur in water as the hydrophobic surface area is minimized in this conformation.
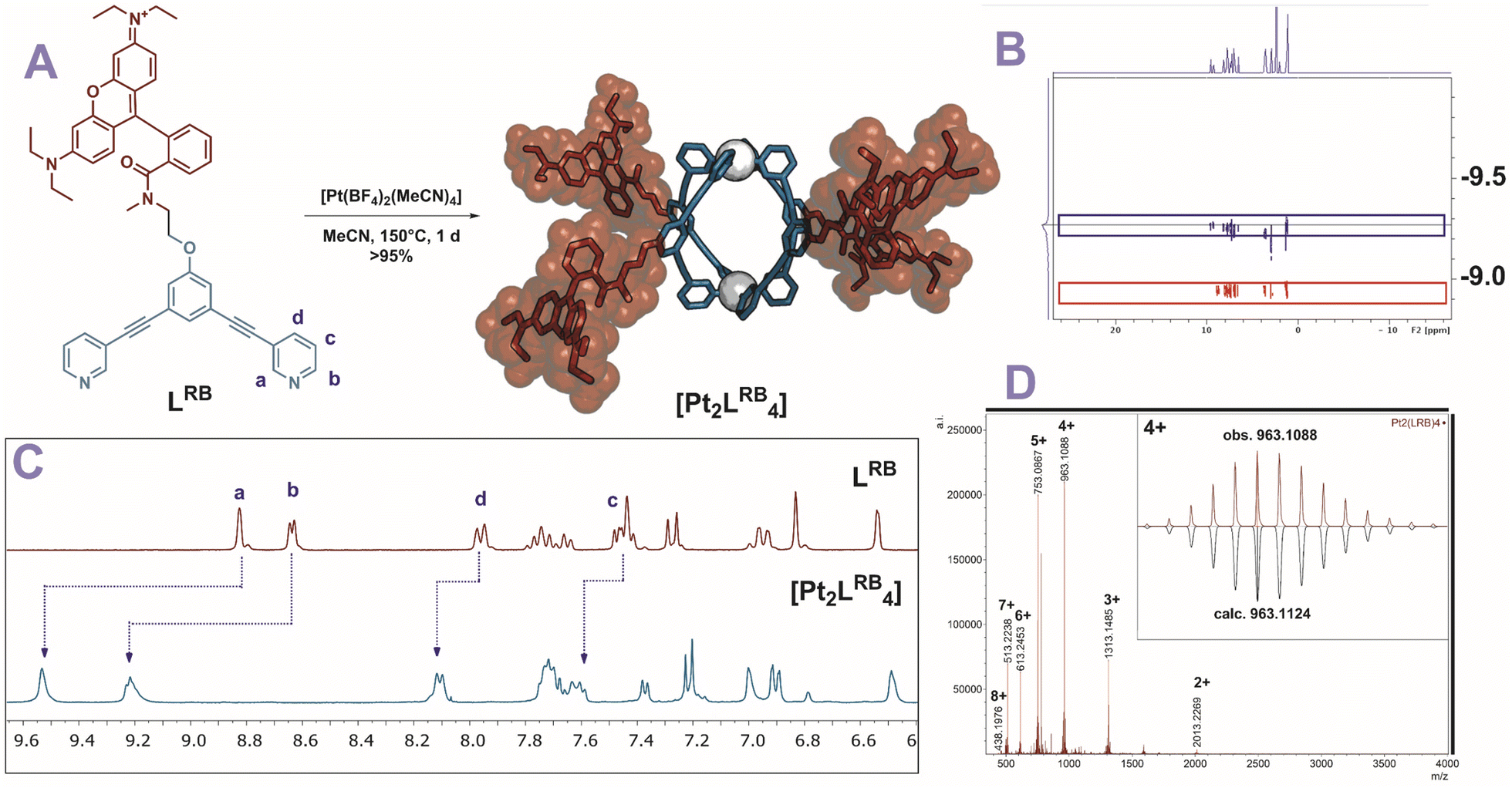 | ||
| Fig. 3 Characterization of [Pt2LRB4]8+. (A) Reaction conditions for formation of nanospheres. Molecular structure of the displayed sphere was minimized on PM3 level. Carbon and nitrogen of nanosphere framework displayed in blue, rhodamine dye in red, platinum as white spheres. (B) Overlayed DOSY NMR of the Pt2LRB4 nanosphere (blue) and the building block (red). (C) 1H-NMR spectra of [Pt2LRB4] nanosphere and the corresponding building block. (D) ESI-MS spectrum of Pt2LRB4. | ||
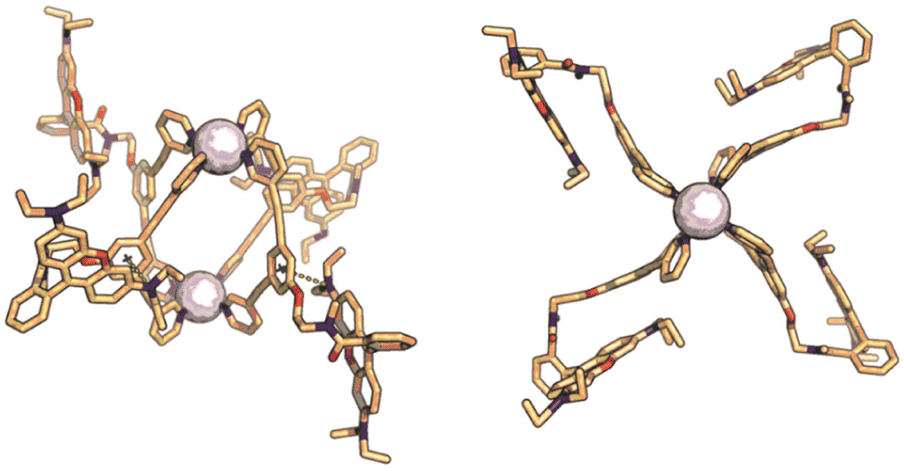 | ||
| Fig. 4 Solid state structure of Pt2LRB4, sphere framework displayed in stick form; carbon in beige, nitrogen in blue, oxygen in red, platinum as white spheres. Side view indicating cation–π interaction between rhodamine and central benzene group. Top view showing the densely packed arrangement of the sphere. | ||
Pt2LTEGRB4 is obtained via an analogous experimental procedure to Pt2LRB4 with similar characteristic spectroscopic features (Fig. S12–15†). Both spheres were precipitated in diethyl ether and redissolved in DMSO-d6 for further application.
Stability and protein binding
A requirement for the development of structure–activity relationships in bio-medicinal application of MOCs is their stability under physiological conditions. Therefore, the kinetic stability of Pt2LRB4 and Pt2LTEGRB4 in the presence of common biological nucleophiles (Cl−, imidazole) and a reducing agent/transition-metal ligand (glutathione) was investigated using 1H-NMR and UV/vis spectroscopy. 1H-NMR experiments were carried out in DMSO-d6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O 1:2 where 0.83 mM nanospheres were treated with 80 eq. NaCl, 30 eq. imidazole and 60 eq. of glutathione at 37 °C. Under these conditions, neither of the two nanospheres show any decomposition within 12 h (Fig. S16†). UV/vis absorbance spectra of the spheres were measured at 37 °C in phosphate-buffered saline (PBS, 1 M, pH 7.4). Absorbance spectra in water show characteristic absorbance at 570 nm and 535 nm (Fig. S16†). The Pt2LTEGRB4 nanosphere shows no decrease in absorption after 12 h at 37 °C in PBS (Fig. S16†), providing support for excellent halide and buffer stability of this Pt2L4 nanosphere. In contrast, Pt2LRB4 shows a decrease in absorbance after 12 h (approx. 20%). As the characteristic sphere absorption features (almost equivalent absorption at 535 and 570 nm) remain the same, we anticipate the decrease of Pt2LRB4 absorption due to the observed adsorption of the sphere to the surface of the UV/vis cuvette.
:D2O 1:2 where 0.83 mM nanospheres were treated with 80 eq. NaCl, 30 eq. imidazole and 60 eq. of glutathione at 37 °C. Under these conditions, neither of the two nanospheres show any decomposition within 12 h (Fig. S16†). UV/vis absorbance spectra of the spheres were measured at 37 °C in phosphate-buffered saline (PBS, 1 M, pH 7.4). Absorbance spectra in water show characteristic absorbance at 570 nm and 535 nm (Fig. S16†). The Pt2LTEGRB4 nanosphere shows no decrease in absorption after 12 h at 37 °C in PBS (Fig. S16†), providing support for excellent halide and buffer stability of this Pt2L4 nanosphere. In contrast, Pt2LRB4 shows a decrease in absorbance after 12 h (approx. 20%). As the characteristic sphere absorption features (almost equivalent absorption at 535 and 570 nm) remain the same, we anticipate the decrease of Pt2LRB4 absorption due to the observed adsorption of the sphere to the surface of the UV/vis cuvette.
Because these MOC's represent multi-charged species with different hydrophobic character (from the organic linkers), we also decided to study the stability of the two nanospheres in the presence of protein, which is typically overlooked in stability assays.44 Although there are many proteins present in vivo, a very prominent protein is albumin. This protein is present in all mammals (e.g., bovine albumin or human albumin) in relatively high concentrations (60% of all plasma proteins) and has the main function to transport apolar and positively charged molecules. As these two functions (apolar and positively charged) are present in our structures (e.g., the positive Pt corners and the organic linker or the positive charged aromatic rhodamine units), we anticipated a strong interaction of our nanospheres with albumin and studied these (Fig. 5B). When we mixed Pt2LRB4 with bovine serum albumin (BSA), a change in rhodamine absorption was observed. The signal at 520 nm showed a decrease in intensity, whereas that at 570 nm was increased. This characteristic change can be attributed to an interaction between the sphere and BSA (Fig. 6). In order to investigate this further, we performed isothermal titration calorimetry (ITC) by addition of BSA to nanosphere solutions in HEPES (10 mM, pH = 7.2) (Fig. 5A). Analysis of the ITC thermogram of Pt2LRB4 provides a high binding constant of the nanosphere to BSA (KD = 2.41 ± 1.2 × 10−7 M). The TEG extended sphere Pt2LTEGRB4 is less hydrophobic relative to Pt2LRB4 and exhibits a lower binding affinity to BSA (KD = 2.59 ± 0.9 × 10−6 M). In both scenarios binding is dominated by hydrophobic interactions (as judged by the signature plots, Fig. S19†). The binding affinities between the nanospheres and BSA are unexpectedly high compared to molecular rhodamine B (KD = 10−4 M). These high binding affinities suggest that albumin binding might contribute to the biodistribution of nanospheres in the body and influence their in vivo activity. We therefore included the BSA complexes in our subsequent biodistribution investigations (vide infra).§
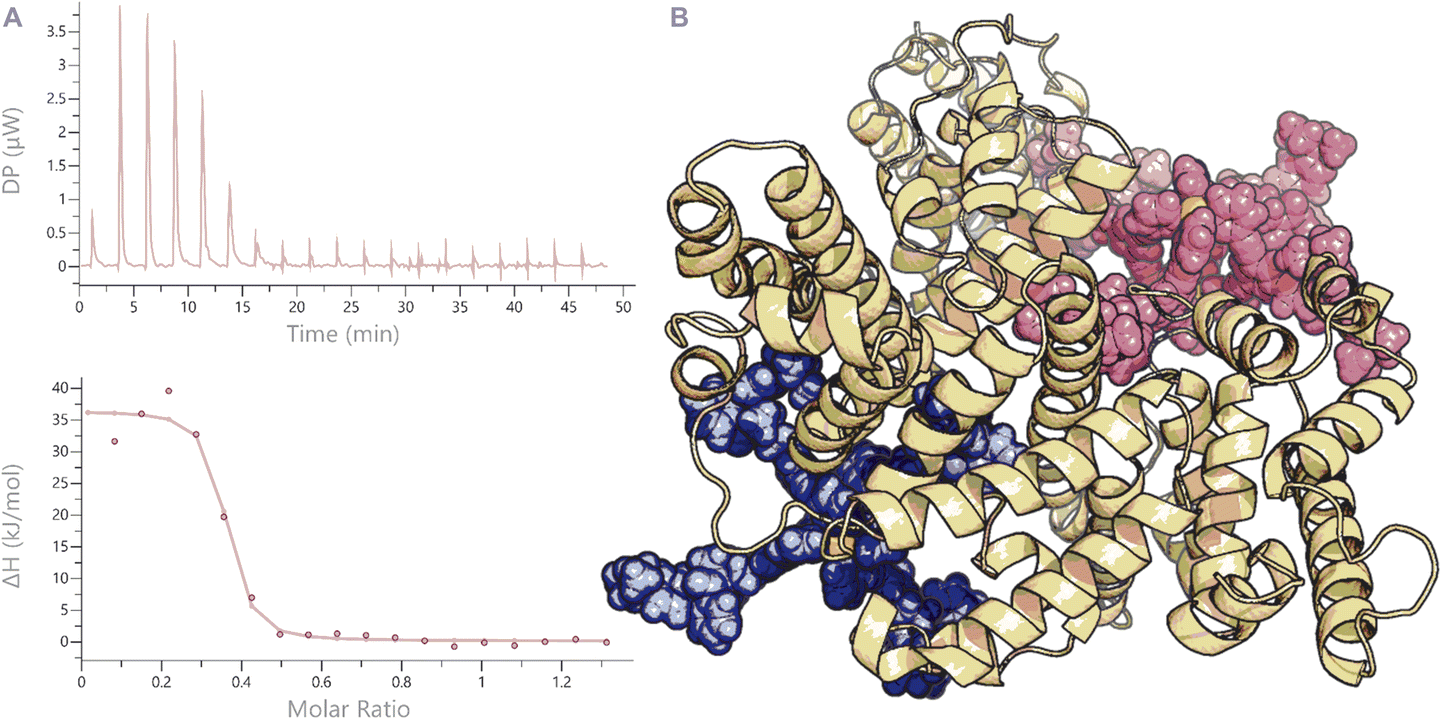 | ||
| Fig. 5 ITC thermogram of the addition of BSA to Pt2LRB4 (A). Below: fitted function to obtain the corresponding binding constants. (B) MD minimized model showing binding of Pt2LRB4 into different hydrophobic pockets of BSA. | ||
Fluorescence studies
With stable nanospheres in hand, we continued to investigate their photophysical properties. The absorption profiles of ligands LRB and LTEGRB and nanospheres Pt2LRB4 and Pt2LTEGRB4 in PBS (1 M, pH = 7.4, 0.2% DMSO) are shown in Fig. 6. Both ligands LRB and LTEGRB display similar broad absorption bands centered at 570 nm with molar absorption coefficients of ε = 18 × 103 M−1 cm−1 and 60 × 103 M−1 cm−1 respectively. After nanosphere formation, higher molar extinction coefficients of ε = 33 × 103 M−1 cm−1 (Pt2LRB4) and 88 × 103 M−1 cm−1 (Pt2LTEGRB4) and an extra absorption located around 535 nm appears for Pt2LRB4. The increase in this absorption is an indication of rhodamine–rhodamine stacking, consistent with previous reports (e.g., rhodamine 6G).45 Rhodamine-dimer formation is less pronounced for Pt2LTEGRB4. We anticipate that the hydrophilic TEG linkers reduce the hydrophobicity of the nanosphere in a significant manner, which results in less inter- and intramolecular rhodamine-stacking. | ||
| Fig. 6 Normalized (maximum absorption = 1) absorption spectra of the two building blocks LRB and LTEGRB and the corresponding nanospheres Pt2LRB4 and Pt2LTEGRB4 in PBS (left). Emission spectra of LRB, LTEGRB, Pt2LRB4 and Pt2LTEGRB4 in PBS (right). Insert: picture of the two nanospheres under UV irradiation (left: Pt2LRB4; right: Pt2LTEGRB4). | ||
After exploring the absorption features of the two rhodamine containing building blocks and of the corresponding spheres, we studied the emission of the ligands and nanospheres (Fig. 6B). All investigated structures emit orange light with a center around 600 nm. LRB displays a moderate quantum yield (QY) of 14%. LTEGRB is four times more efficient with a QY of 55%. Following the same trend as the building blocks, Pt2LRB4 and Pt2LTEGRB4 displayed a QY of 4% and 29% respectively. The lower QY after nanosphere formation can be attributed to rhodamine-stacking, cation–π interactions and the heavy atom effect.
Addition of BSA enhanced the QY of Pt2LRB4 by 3-fold from 4% to 12%, which can be attributed to rhodamine–rhodamine stack dissociation as supported by a decrease in absorbance at 535 nm (which is characteristic for rhodamine–rhodamine stacks). In a similar manner, but to a lesser extent, also the QY of Pt2LTEGRB4 was enhanced by 20% (from 29% to 35%). The QY of the applied building blocks remained unchanged upon addition of BSA. In summary, a hydrophilic TEG linker between a dye and the nanosphere allows to maintain similar emissive properties of dyes in PBS buffer as before sphere formation. When the dye is placed close to the sphere, inter-/intra-molecular dye stacking and the heavy atom effect led to a significant reduction of QY. In this context, BSA has shown to be a suitable candidate to break up rhodamine-dimer formation of sphere aggregates in PBS, improving their QY by a factor of 1.2 and 3.
Cytotoxicity
While some specific palladium-based Pd2L4 nanospheres are highly toxic,14,28–30 other structures, especially with similar design to our framework have shown little toxicity.12,15,31 Recent studies revealed the high toxicity of rhodamine derivatives,37,38 making our systems, with the well-defined nanosphere and linked toxic units, interesting candidates for structure-cytotoxicity assays. We anticipate differences between the molecular building blocks and their nanosphere analogues. We studied the building block LTEGRB, the nanospheres Pt2LRB4 and Pt2LTEGRB4 as well as a dummy sphere Pt2LOMe4 without rhodamine functionalization against two cell lines: PC3M-Pro4 (prostate carcinoma) and cisplatin-resistant MDA-MB-231 (breast cancer). The dummy sphere Pt2LOMe4 shows no significant cytotoxicity within 24 h (IC50 > 20 μM, Table 1). The rhodamine-functionalized building block shows very high cytotoxicity against PC3M-Pro4 (IC50 = 1.5 μM for LTEGRB) and against MDA-MB-231 (IC50 = 0.5 μM for LTEGRB). The corresponding nanospheres show reduced toxicity compared to their molecular building blocks against both cell lines. The cytotoxicity of the rhodamine incorporated into the nanospheres is 4 to 13-times lower against both cell lines compared to the corresponding building blocks (IC50 = 6.8–10 μM for Pt2LRB4 and IC50 = 6.8 μM for Pt2LTEGRB4). Overall, the building block and the nanospheres display similar cytotoxicity for both cancer cell lines, regardless of the linker type between the nanosphere forming part of the building block and the dye. Although rhodamines have emerged very recently as cytotoxic anti-cancer agents, currently the mechanism of action is not fully understood. Rhodamine-based dyes can possess cytotoxicity due to DNA complexation46 by π-stacking and electrostatic attraction. More recent work describes rhodamine derivatives as cytotoxic agents which can disrupt/destroy mitochondrial membranes, causing apoptosis.37,38,47 Although, our study was performed thus far on only two different cell lines, it demonstrates how reduction of toxicity to an applicable level can be achieved by the formation of supramolecular nanospheres.| IC50 (μM) | ||
|---|---|---|
| Compound | PC3M-Pro4 (prostate) | MDA-MB-231 (breast) |
| a The cells were incubated for 24 hours at 37 °C, 5% CO2. Then 9 μL WST-1 reagent (Sigma) was added to each well mixed, and incubated for 2 hours at 37 °C, 5% CO2. Then the absorbance at 400 nm was measured for each well, using the Infinite M1000Pro plate reader (Tecan). | ||
| LTEGRB | 0.5 (0.5) | 1.5 (1.5) |
| Pt2LRB4 | 1.7 (6.8) | 2.5 (10) |
| Pt2LTEGRB4 | 1.7 (6.8) | 1.7 (6.8) |
| Pt2LOMe4 | 41.7 (n.a.) | >20 (n.a.) |
In vivo biodistribution
To investigate the influence of self-assembly of rhodamine building blocks to a nanosphere on the biodistribution of the rhodamine dyes, we used the zebrafish model using a transgenic kdrl:EGFP and mpeg1:EGFP zebrafish line (with green emissive endothelium of the vasculature and EGFP expressing macrophages, respectively, for schematic overview of zebrafish, see Fig. 8B).48,49 Acute toxicity studies of Pt2LTEGRB4 in wild type zebrafish embryos over the course of 4 days revealed no apparent toxicity, abnormalities, or premature death in any of the injected embryos intravenously injected with concentrations up to 200 μM in 10 mM HEPES pH 7.4. Zebrafish do not express albumin, but instead a protein with similar function: vitamin D binding protein (VDBP),50 making them also a good platform to study the biodistribution of albumin-bound spheres. The nanospheres were heated at 80 °C in DMSO (to break up possible aggregation), then diluted with HEPES (10 mM, pH 7.4) prior injection. 1 nL of 5 μM probe samples were directly injected intravenously into the duct of Cuvier in 48–80 hours post fertilization (hpf) zebrafish embryos. Initially, BSA-nanosphere complexes were prepared prior to injection by mixing 10 mM stock solutions of nanosphere in DMSO with PBS or HEPES buffer containing 150 μM BSA (15 eq.) to obtain a final concentration of 5 μM nanosphere bound to BSA. The BSA-nanosphere samples were directly injected without prior heating. The biodistribution of the nanospheres and BSA-nanosphere complexes within the body of the zebrafish embryos was analyzed by confocal imaging at 90 minutes post injection (mpi) (Fig. S23†). After ITC studies revealed stoichiometry of roughly two spheres bound to one BSA-molecule (Fig. 5A), and with therapeutic application in mind, the complexes were prepared by injecting a higher 160 μM Pt2LTEGRB4 concentration into a 160 μM BSA solution in 10 mM HEPES, pH 7.4. The biodistribution of this higher concentration of the complex was studied in 72 hpf transgenic kdrl:EGFP zebrafish embryos.A non-injected control showed red autofluorescence emission only in the yolk sac and eyes (Fig. S22†). Pt2LRB4 shows rapid non-selective uptake within 30 min throughout the body by endothelial cells (Fig. 7B). The relatively high uptake in the scavenger endothelial cells (SECs) in the tail compared to endothelial cells in the arteries can be explained by the majority of spheres interacting with serum proteins (negatively charged) directly after entering the bloodstream. These sphere–protein complexes likely have a net negative charge, directing them to the SECs. The fluorescent intensity of Pt2LRB4 is qualitatively much lower (Fig. 7B is 5 times less bright under identical laser and detector settings compared to Fig. 7A). Similar endothelial cell uptake was observed when Pt2LTEGRB4 was injected (Fig. 7A). Pt2LTEGRB4 showed more intense fluorescence, comparable to free building block LTEGRB (Fig. 7C compared to Fig. 7A, same settings were used when taking the pictures), which we attribute to the molecular behavior of dyes due to the long TEG linker between dye and nanosphere. As opposed to the spheres, building block LTEGRB remained primarily in circulation, besides some accumulation in the pronephric ducts (analogous to kidneys in mammals) at 1.5 hpi (Fig. 7C).
 | ||
| Fig. 7 Confocal images showing biodistribution of nanospheres and building block in 54 hours post fertilization (hpf) transgenic kdrl:EGFP zebrafish embryos injected intravenously in the duct of Cuvier with (A) Pt2LTEGRB4 (shown in red), which is rapidly cleared from circulation into the venous and arterial vasculature (shown in green) within 30 minutes post injection (mpi); (A′) nanocages (shown in white), showing the uptake pattern throughout the vascular endothelium; (B) Pt2LRB4 showing lower fluorescence intensity was, similar to Pt2LTEGRB4, rapidly taken up in venous and arterial vessel walls within 30 mpi; (C) LTEGRB remained primarily in circulation besides accumulation in the pronephric duct at 1.5 hours post injection (hpi). Scale bar represents 250 μm. | ||
The presence of BSA changed the biodistribution of both nanospheres in a similar fashion (e.g., Fig. 8 and S23†). Both nanospheres show better fluorescence (especially Pt2LRB4⊃BSA) compared to the free nanospheres in the absence of BSA, in line with the enhanced fluorescent quantum yield under physiological conditions. Furthermore, the circulation of both nanospheres was significantly enhanced (Fig. 8 and S23†). In line with anionic charged molecules, Pt2LTEGRB4⊃BSA is taken up by scavenger endothelial cells in the tail (Fig. 8, likely due to the net negative charge of BSA (18−)). In addition, the sphere–BSA complexes showed major accumulation in a different cell type identified as ionocytes located superficially on the skin (Fig. 8). Before full development of gills, kidneys, and intestine of fish embryos, both gas exchange and ion exchange take place through specialized cells in the skin. Ionocytes in the skin of fish embryos regulate body fluid pH and ionic composition.51 We assume that the very small size of the spheres allows them to diffuse relatively easily through the tissue of the fish, far beyond the bloodstream and the vascular endothelium allowing accumulation in ionocytes. Furthermore, the chemistry of rhodamine B might explain the bright staining of these cells. Under acidic conditions, the dye's fluorescence is bright.52 A certain type of ionocytes in embryonic zebrafish is involved in acid and ammonia secretion, providing an acidic environment in which rhodamine B fluorescence is brighter red compared to the intensity in physiological pH (in circulation).51 Likely, due to the high degree of H+ present in these cells and the lipophilic nature of rhodamine B the (mitochondrial) membranes of these mitochondria-rich cells are brightly stained.
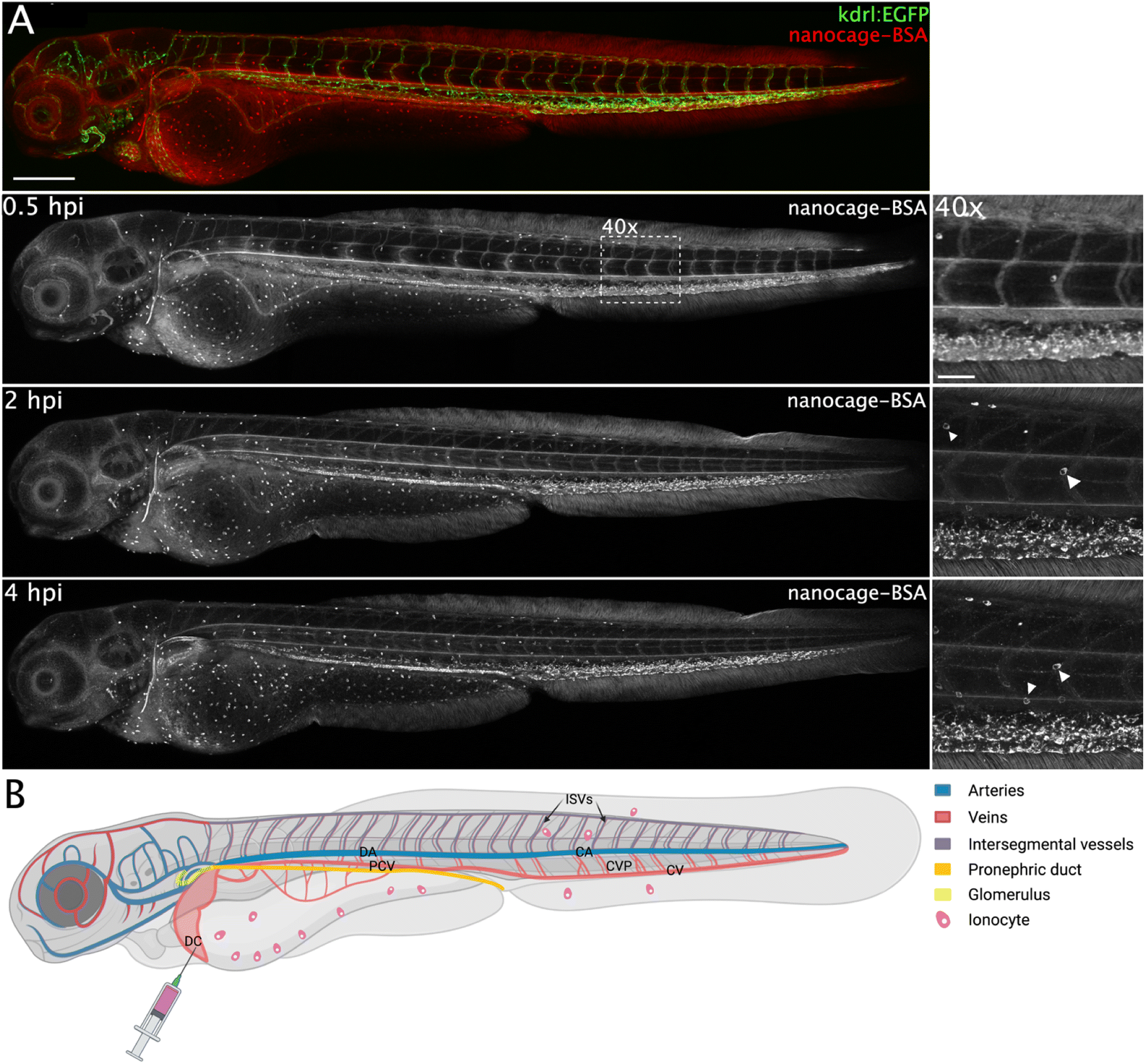 | ||
| Fig. 8 (A) Confocal images showing biodistribution of 160 μM Pt2LTEGRB4⊃BSA intravenously injected (1 nL) in 72 hpf transgenic kdrl:EGFP zebrafish embryos. At 30 minutes post injection (mpi) the complexes remained primarily in circulation, although accumulation in ionocytes, mainly around the yolk sac, and slight uptake in scavenger endothelial cells (SECs), as well as accumulation in the pronephric duct was already visible. At 2 hpi and 4 hpi this accumulation became increasingly pronounced, with clear accumulation in the glomerulus (schematically depicted on the right) visible at 4 hpi, although a (small) fraction remained in circulation. Accumulation in ionocytes is indicated in 2 hpi (40×) and 4 hpi (40×) by white arrow heads. Scale bars represent 250 μm (0.5 hpi) and 50 μm (0.5 hpi, 40×). (B) Schematic representation of accumulation of nanosphere materials in different tissues of zebrafish embryos. Intravenous injection of 1 nL nanosphere solution for biodistribution studies was performed in the Duct of Cuvier (DC). Upon injection, the nanospheres enter the heart, are pumped into the arterial vessels, among others the dorsal aorta (DA), followed by the caudal aorta (CA), intersegmental vessels (ISVs), caudal vein (CV) and caudal vein plexus (CVP) containing scavenger endothelial cells (SECs). Finally, the circulation continues back to the heart via the posterior cardinal vein (PCV) and the DC. | ||
Additionally, we studied the possible uptake of nanosphere-BSA complexes by macrophages using transgenic zebrafish line mpeg1:EGFP carrying macrophages expressing EGFP. Pt2LRB4⊃BSA showed no apparent uptake by macrophages (as the green and red emissions do not overlap, Fig. S23†). For both nanosphere-BSA adducts, as well as for the free building block, accumulation in the pronephric duct (part of the developing kidney) was observed, starting at around 45 mpi (Fig. 8 and S23†). Additionally, in case of nanosphere-BSA complexes, accumulation in the glomerulus (part of developing kidney) was observed starting at 2 hpi (Fig. 8). The developing kidney plays an important role in the clearance of various molecules, notably carrier proteins such as albumin and VDBP, likely explaining the accumulation of the BSA-bound spheres in this tissue. As for the building blocks, their positive charge might cause adsorption to (carrier) proteins in circulation, possibly including VDBP, which likely points at an accumulation mechanism similar to that of the nanosphere-BSA complexes. For a schematic representation of the tissues in which the injected samples accumulated refer to Fig. 8B.
With this set of experiments, we demonstrate a high degree of tunability of the biodistribution properties of Pt2L4 nanospheres by (A) their hydrophobic character and (B) by protein complexation. Depending on the probe formulation, we were able to achieve fast uptake by endothelial cells (plain nanospheres), longer circulation, uptake by scavenger endothelial cells in the tail (nanosphere-BSA complexes, anionic charged small particle) and accumulation in ionocytes and the pronephric duct (building blocks and nanosphere-BSA complexes). We anticipate that the high degree of tunable particle delivery together with their cytotoxic properties provides the fundament for new avenues of coordination-based spherical assembly application in cancer therapy. Depending on the cancer type which should be treated, sticky spheres (such as Pt2LRB4) can be useful when the drugs can be directly injected into the tumor. For other applications (e.g., intravenous administration) more circulating materials are required (such as Pt2LTEGRB4⊃BSA). We have shown that simple ligand modification of the nanospheres has a huge influence on their properties in vivo.
Conclusions
While clinical use of coordination-based nanospheres is still in its infancy, they have promising properties, some of which are described in this paper. We have shown a clear step forward by designing two new exo-functionalized self-assembled Pt2L4 nanospheres with rhodamine groups, as was shown by NMR, mass spectroscopy, and single-crystal X-ray diffraction. The nanospheres were shown to be stable under physiological conditions for 12 h and exhibit good fluorescence, making Pt2L4 nanospheres suitable candidates for toxicity and the biodistribution study in the body of zebrafish embryos. Previous studies on Pd2L4 nanospheres displayed decomposition under in vivo conditions, making it unclear what the effect of the supramolecular structure is on toxicity and biodistribution. With the first set of robust and fluorescent Pt2L4 nanospheres, we made some important observations on the effects of the supramolecular structure for in vitro and in vivo applications. We have demonstrated that albumin binding occurs with a very high binding constant in comparison to molecular analogues, which enhances the fluorescent properties and in turn, can play a role in the distribution of typically rather hydrophobic nanospheres in vivo. The toxicity of nano-assembled rhodamine drugs is downregulated towards specific cancer types in comparison to the molecular analogue which stems from the well-defined three-dimensional composition of the nanospheres, a handle that may come useful for specific drug design when too toxic materials are delt with. The nanospheres show faster cell uptake than the building blocks they are assembled of and display alternated distributions in the presence of albumin. We foresee that Pt2L4 or similar systems could act as a universal platform for imaging purposes, which can be expanded to the delivery of therapeutically active drugs by encapsulation strategies (e.g., ref. 12, 13, 15, and 31) or covalent attachment.Data availability
All relevant data is provided in the ESI† of the paper. If other data is needed, it will be made available on reasonable request.Author contributions
Conceptualization: EOB, RK, AK, JNHR; formal analysis: EOB, RK; funding acquisition: JNHR, BdB, AK, NIM; investigation: EOB, RK, SM, DAP, IK; supervision: JNHR, BdB, AK, NIM; validation: EOB, RK, SM, DAP, IK, JNHR, BdB, AK, NIM; visualization: EOB, RK; writing – original draft: EOB; writing – review & editing: EOB, RK, SM, DAP, IK, JNHR, BdB, AK, NIM. EOB and RK contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge the University of Amsterdam for financial support to RPA sustainable chemistry. G. Zhou and Prof. E. Snaar-Jagalska (IBL, Leiden University) are acknowledged for their support with the cancer cell lines and zebrafish studies.Notes and references
- K. J. Franz and N. Metzler-Nolte, Chem. Rev., 2019, 119, 727–729 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed.
- A. Pöthig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef PubMed.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734–777 CrossRef PubMed.
- N. P. E. Barry, N. H. Abd Karim, R. Vilar and B. Therrien, Dalton Trans., 2009, 10717 RSC.
- A. Dubey, Y. J. Jeong, J. H. Jo, S. Woo, D. H. Kim, H. Kim, S. C. Kang, P. J. Stang and K.-W. Chi, Organometallics, 2015, 34, 4507–4514 CrossRef CAS.
- A. Dubey, J. W. Min, H. J. Koo, H. Kim, T. R. Cook, S. C. Kang, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2013, 19, 11622–11628 CrossRef CAS PubMed.
- F. Kaiser, A. Schmidt, W. Heydenreuter, P. J. Altmann, A. Casini, S. A. Sieber and F. E. Kühn, Eur. J. Inorg. Chem., 2016, 2016, 5189–5196 CrossRef CAS.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- S. M. McNeill, D. Preston, J. E. M. Lewis, A. Robert, K. Knerr-Rupp, D. O. Graham, J. R. Wright, G. I. Giles and J. D. Crowley, Dalton Trans., 2015, 44, 11129–11136 RSC.
- D. Preston, S. M. McNeill, J. E. M. Lewis, G. I. Giles and J. D. Crowley, Dalton Trans., 2016, 45, 8050–8060 RSC.
- S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS PubMed.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184 RSC.
- S.-F. Xi, L.-Y. Bao, J.-G. Lin, Q.-Z. Liu, L. Qiu, F.-L. Zhang, Y.-X. Wang, Z.-D. Ding, K. Li and Z.-G. Gu, Chem. Commun., 2016, 52, 10261–10264 RSC.
- G. Yu, S. Yu, M. L. Saha, J. Zhou, T. R. Cook, B. C. Yung, J. Chen, Z. Mao, F. Zhang, Z. Zhou, Y. Liu, L. Shao, S. Wang, C. Gao, F. Huang, P. J. Stang and X. Chen, Nat. Commun., 2018, 9, 4335 CrossRef PubMed.
- Y.-R. Zheng, K. Suntharalingam, P. M. Bruno, W. Lin, W. Wang, M. T. Hemann and S. J. Lippard, Inorg. Chim. Acta, 2016, 452, 125–129 CrossRef CAS PubMed.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts, I. Renard, R. Hargreaves, C. Cawthorne, S. J. Archibald and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 16877–16881 CrossRef CAS PubMed.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- B. Woods, M. N. Wenzel, T. Williams, S. R. Thomas, R. L. Jenkins and A. Casini, Front. Chem., 2019, 7, 68 CrossRef CAS PubMed.
- A. Ahmedova, R. Mihaylova, D. Momekova, P. Shestakova, S. Stoykova, J. Zaharieva, M. Yamashina, G. Momekov, M. Akita and M. Yoshizawa, Dalton Trans., 2016, 45, 13214–13221 RSC.
- R. A. S. Vasdev, L. F. Gaudin, D. Preston, J. P. Jogy, G. I. Giles and J. D. Crowley, Front. Chem., 2018, 6, 563 CrossRef CAS PubMed.
- A. Ahmedova, D. Momekova, M. Yamashina, P. Shestakova, G. Momekov, M. Akita and M. Yoshizawa, Chem.–Asian J., 2015, 11, 474–477 CrossRef PubMed.
- A. Schmidt, M. Hollering, J. Han, A. Casini and F. E. Kühn, Dalton Trans., 2016, 45, 12297–12300 RSC.
- B. Woods, R. D. M. Silva, C. Schmidt, D. Wragg, M. Cavaco, V. Neves, V. F. C. Ferreira, L. Gano, T. S. Morais, F. Mendes, J. D. G. Correia and A. Casini, Bioconjugate Chem., 2021, 32, 1399–1408 CrossRef CAS PubMed.
- C. Li, B. Zhang, Y. Dong, Y. Li, P. Wang, Y. Yu, L. Cheng and L. Cao, Dalton Trans., 2020, 49, 8051–8055 RSC.
- Z. Li, N. Kishi, K. Hasegawa, M. Akita and M. Yoshizawa, Chem. Commun., 2011, 47, 8605 RSC.
- B. Aikman, R. Bonsignore, B. Woods, D. Doellerer, R. Scotti, C. Schmidt, A. A. Heidecker, A. Pöthig, E. J. Sayers, A. T. Jones and A. Casini, Dalton Trans., 2022, 51, 7476–7490 RSC.
- C. Li, A. G. Tebo, M. Thauvin, M. Plamont, M. Volovitch, X. Morin, S. Vriz and A. Gautier, Angew. Chem., Int. Ed., 2020, 59, 17917–17923 CrossRef CAS PubMed.
- H. Battula, S. Bommi, Y. Bobde, T. Patel, B. Ghosh and S. Jayanty, J. Photochem. Photobiol., 2021, 6, 100026 CrossRef.
- I. Serbian, S. Hoenke, O. Kraft and R. Csuk, Med. Chem. Res., 2020, 29, 1655–1661 CrossRef CAS.
- R. Alford, H. M. Simpson, J. Duberman, G. C. Hill, M. Ogawa, C. Regino, H. Kobayashi and P. L. Choyke, Mol. Imaging, 2009, 8, 7290 CrossRef.
- G. Gunaydin, M. E. Gedik and S. Ayan, Front. Chem., 2021, 9, 686303 CrossRef CAS PubMed.
- E. O. Bobylev, D. A. Poole III, B. Bruin and J. N. H. Reek, Chem. – Eur. J., 2021, 27, 12667–12674 CrossRef CAS PubMed.
- E. O. Bobylev, D. A. Poole III, B. de Bruin and J. N. H. Reek, Chem. Sci., 2021, 12, 7696–7705 RSC.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- L. E. H. Paul, B. Therrien and J. Furrer, Org. Biomol. Chem., 2015, 13, 946–953 RSC.
- M. Chapman and W. B. Euler, J. Fluoresc., 2018, 28, 1431–1437 CrossRef CAS PubMed.
- Md. M. Islam, M. Chakraborty, P. Pandya, A. Al Masum, N. Gupta and S. Mukhopadhyay, Dyes Pigm., 2013, 99, 412–422 CrossRef CAS.
- R. Guo, J. Yin, Y. Ma, Q. Wang and W. Lin, J. Mater. Chem. B, 2018, 6, 2894–2900 RSC.
- E. Moro, G. Ozhan-Kizil, A. Mongera, D. Beis, C. Wierzbicki, R. M. Young, D. Bournele, A. Domenichini, L. E. Valdivia, L. Lum, C. Chen, J. F. Amatruda, N. Tiso, G. Weidinger and F. Argenton, Dev. Biol., 2012, 366, 327–340 CrossRef CAS PubMed.
- F. Ellett, L. Pase, J. W. Hayman, A. Andrianopoulos and G. J. Lieschke, Blood, 2011, 117, e49–e56 CrossRef CAS PubMed.
- E. S. Noël, M. dos Reis, Z. Arain and E. A. Ober, Gene Expression Patterns, 2010, 10, 237–243 CrossRef PubMed.
- L.-Y. Lin, J.-L. Horng, J. G. Kunkel and P.-P. Hwang, Am. J. Physiol.: Cell Physiol., 2006, 290, C371–C378 CrossRef CAS PubMed.
- N. O. Mchedlov-Petrossyan, V. I. Kukhtik and V. I. Alekseeva, Dyes Pigm., 1994, 24, 11–35 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2216167. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01086d |
| ‡ These authors contributed equally to this work. |
| § Although BSA and other albumins are biologically speaking not identical (ca. 75% identical), all of the albumins share the same function and possess pockets for positively charged and hydrophobic molecules. BSA as such acts chemically as a good estimation for other albumin forms which are less available for research (e.g., human serum albumin HSA). |
| This journal is © The Royal Society of Chemistry 2023 |