
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the extracellular function of HMGB1 N-terminal region acetylation assisted by a protein semi-synthesis approach†‡
Tongyao
Wei
,
Jiamei
Liu
,
Can
Li
,
Yi
Tan
,
Ruohan
Wei
,
Jinzheng
Wang
,
Hongxiang
Wu
,
Qingrong
Li
,
Heng
Liu
,
Yubo
Tang
and
Xuechen
Li
 *
*
Department of Chemistry, State Key Lab of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, SAR, P. R. China. E-mail: xuechenl@hku.hk
First published on 7th September 2023
Abstract
HMGB1 (high-mobility group box 1) is a non-histone chromatin-associated protein that has been widely reported as a representative damage-associated molecular pattern (DAMP) and to play a pivotal role in the proinflammatory process once it is in an extracellular location. Accumulating evidence has shown that HMGB1 undergoes extensive post-translational modifications (PTMs) that actively regulate its conformation, localization, and intermolecular interactions. However, fully characterizing the functional implications of these PTMs has been challenging due to the difficulty in accessing homogeneous HMGB1 with site-specific PTMs of interest. In this study, we developed a streamlined protein semi-synthesis strategy via salicylaldehyde ester-mediated chemical ligations (Ser/Thr ligation and Cys/Pen ligation, STL/CPL). This methodology enabled us to generate a series of N-terminal region acetylated HMGB1 proteins. Further studies revealed that acetylation regulates HMGB1–heparin interaction and modulates HMGB1's stability against thrombin, representing a regulatory switch to control HMGB1's extracellular activity.
Introduction
High mobility group box 1 (HMGB1) is a highly abundant non-histone nuclear protein that plays critical roles both inside and outside the cell. Within the nucleus, HMGB1 acts as a DNA chaperone and a chromosome guardian, interacting with DNA and histones to modify chromatin structures and regulate nuclear processes (e.g., transcription and DNA repair).1–4 Besides, HMGB1 translocation commonly occurs and gains new identities to function in inflammatory or autoimmune diseases. Inside the cytoplasm, HMGB1 acts as an autophagy sustainer to mediate cell autophagy.5 Outside the cell, HMGB1 serves as a key damage-associated molecular pattern (DAMP) and a central mediator of lethal inflammation. It facilitates the migration and activation of inflammatory cells through toll-like receptor 4 (TLR4) and advanced glycation end products (RAGE).1,6 Moreover, the extracellular function of HMGB1 can be extended by interacting with pathogen-associated molecular patterns (PAMPs) (e.g., lipopolysaccharides, LPSs), cytokines and chemokines.7,8 By these means, HMGB1 plays multiple roles in several important processes ranging from inflammation to tissue repair.5,9 It is worth mentioning that the relevance of extracellular HMGB1 as a biomarker or therapeutic target has been demonstrated in various diseases.10,11 For instance, in cases of severe bacterial infections, circulating bacteria can trigger a deleterious systemic inflammatory response (endotoxemia and bacterial sepsis), which can be alleviated by neutralizing extracellular HMGB1.12 In the recent COVID-19 pandemic, elevated serum HMGB1 was observed in severe COVID-19 patents and plasma concentrations of HMGB1 assessed at ICU admission can be used for accurately predicting patient fatality.13 Other studies also showed that exogenous HMGB1 induces the expression of SARS-CoV-2 entry receptor ACE2 in a RAGE-dependent manner, suggesting the potential for innovative therapeutic strategies for COVID-19.14,15Over the past few years, the picture of HMGB1 function has grown more nuanced, with the recognition that HMGB1 undergoes multiple posttranslational modifications (PTMs) that extensively regulate its activity.4 Acetylation is a prominent PTM that plays a crucial role in facilitating HMGB1's shuttling between the nucleus and the cytoplasm. Subsequent accumulation of HMGB1 in the cytoplasm leads to active secretion.16,17 Multiple animal studies have provided substantial evidence that diverse oxidative stresses can trigger the hyperacetylation and release of HMGB1 under certain pathological conditions.18 Besides, other modifications, such as methylation, phosphorylation, and polyADP-ribosylation, have been identified to modulate HMGB1's DNA binding affinity and promote its translocation to the cytoplasm in response to various stress stimuli.19–21 Furthermore, HMGB1 undergoes both O- and N-linked glycosylations, which also impact its function22,23 Recent study revealed that O-GlcNAcylation at Ser100 in HMGB1 alters its DNA binding on different DNA structures and reduces its DNA damage processing activities.23 Moreover, the function and secretion of HMGB1 are intricately dependent on its oxidation status, which is tightly regulated by three redox-sensitive cysteines.24 For instance, fully reduced HMGB1 can interact with CXCL12 and facilitate the migration of immune cells.25 Overall, a diverse array of modifications contributes to the finely tuned subcellular localization and functional versatility of HMGB1. However, the precise interplay between these post-transcriptional modifications and whether they exert competitive, cooperative, or independent effects under different cellular conditions remains an intriguing area warranting further investigation.1,26
As mentioned above, efficient secretion of HMGB1 requires hyper-acetylation on its two NLS regions, while proteomic studies have also identified that in addition to the NLS, significant acetylations occur in the N-terminal regions (Lys2, 6, 7, and 11) of extracellular HMGB1 (Fig. S1‡).17 However, to date, a comprehensive investigation of these acetylation(s) is still lacking.
To dissect the role of the acetylation at specific residues (Lys2, 6, 7, and 11) in HMGB1, it is essential to obtain homogeneous HMGB1 proteins with site-specific lysine acetylation(s). However, current methods such as directed isolation and in vitro enzyme treatment are limited in achieving site-specificity. Unnatural amino acid incorporation, while enabling targeted modifications at desired positions, also faces challenges in flexibility such as installing multiple unnatural amino acids simultaneously. Modern protein chemical synthesis methods combining solid phase peptide synthesis (SPPS) and peptide ligation have evolved to provide a versatile strategy to generate proteins with tailor-made PTMs with precision and flexibility.27–33 To maximize the efficiency of chemical synthesis of the proteins, the semi-synthetic approach facilitated by chemical ligation offers an expedient solution. This method involves ligating short synthetic peptides containing modified residues with long peptide fragments obtained through recombinant expression.34–37 As a result, the size of proteins that can be accessed through semi-synthesis continues to increase in a cost-effective manner. The protein semi-synthesis lies in two manifolds: either C-terminal peptides or N-terminal peptides are recombinantly expressed. As peptide ligation relies on the reacting counterparts at C- and N-termini (e.g., C-terminal thioesters/N-terminal cysteine in native chemical ligation38) for chemoselectivity, the use of natural amino acids as the reactive group is advantageous for protein expression. For example, the protein with an N-terminal cysteine for NCL can be readily obtained. In addition, a significant milestone has been achieved in generating recombinant proteins with C-terminal thioesters using the intein splicing system, known as expressed protein ligation (EPL).34 However, the lowest abundance of cysteine among the 20 natural amino acids limits the choice of the ligation site in certain cases. NCL-desulfurization methods that convert Cys to Ala expand the potential ligation junction to the more abundant alanine, but they are only applicable to non-cysteine containing proteins.39,40 Furthermore, proteins with other thiolated unnatural amino acids at the N-terminus are hard to generate recombinantly yet.29 Previously, we reported that C-terminal salicylaldehyde esters can react with highly abundant (N-terminal) Ser, Thr, and Cys, restoring the natural Xaa-Ser/Thr/Cys peptide linkage at the ligation site upon acidolysis, which we termed Ser/Thr ligation (STL) and Cys/Pen ligation (CPL).41,42 Notably, CPL is found to proceed independently of steric hindrance at C-terminal amino acids, such as Val, Ile, Thr, and Pro, which often severely impairs the efficacy of other ligations.38,42 STL and CPL have been widely used for the synthesis of cyclic peptides and proteins, representing significant advancements in the field.43,44 As a further step forward, the application of STL/CPL-based protein semi-synthesis holds great promise, particularly for the synthesis of large proteins. In this study, we present an efficient Ser/Thr/Cys ligation-mediated semi-synthesis of proteins of interest (HMGB1 proteins with site-specific acetylations). Through this method, we elucidate the regulatory effect of acetylation on HMGB1's polysaccharide binding and stability against enzymatic proteolysis, offering valuable insights for HMGB1's biological functions and therapeutic development.
Results
Exemplifying STL/CPL in large protein semi-synthesis
To initiate the assembly of HMGB1 protein, our first objective was to establish the working process and optimal conditions for executing the SAL ester-mediated protein semi-synthesis (Fig. 1A). As STL/CPL requires an N-terminal Ser, Thr, or Cys, proteolysis methods were employed to generate proteins with these residues at the N-terminus, as summarized in Fig. S2.‡ For our model study, maltose-binding protein (MBP, ∼40 kDa) was used. MBPs with N-terminal Ser, Thr, or Cys were produced by ubiquitin-like-specific protease 1 (Ulp1) digestion. A number of typical peptide SAL esters of different lengths were prepared via solid phase peptide synthesis (Fig. 1B). We performed preliminary attempts and found that the low solubility of expressed large proteins in pyridine/acetic acid solution was the main issue limiting ligation efficiency. After extensive condition screening (Table S1‡), we found that MBP could be dissolved at a concentration of 2 mM (∼80 mg mL−1) with the inclusion of certain additives. Although the typical concentration for peptide ligation is 10 mM, we identified that ligation could also proceed smoothly at this concentration by increasing the peptide ester concentration. After acidolysis, the reaction products were subjected to analysis using SDS-PAGE and LC/MS (Fig. 1C and S3–S5‡). MBP with an N-terminal Thr consistently exhibited the highest reactivity, except for peptides with C-terminal bulky Pro or Val SAL esters. MBP with an N-terminal Cys effectively reacted with all peptide SAL esters, achieving >60% conversion regardless of the C-terminal amino acids. MBP with an N-terminal Ser showed the lowest ligation efficiency, yet it was still capable of completing the reaction with two short peptide esters (P1 and P2) with >95% conversion. Based on these results, we believe that the reaction features of STL/CPL in large protein semi-synthesis are similar to those observed in short peptide synthesis (Table S2‡).43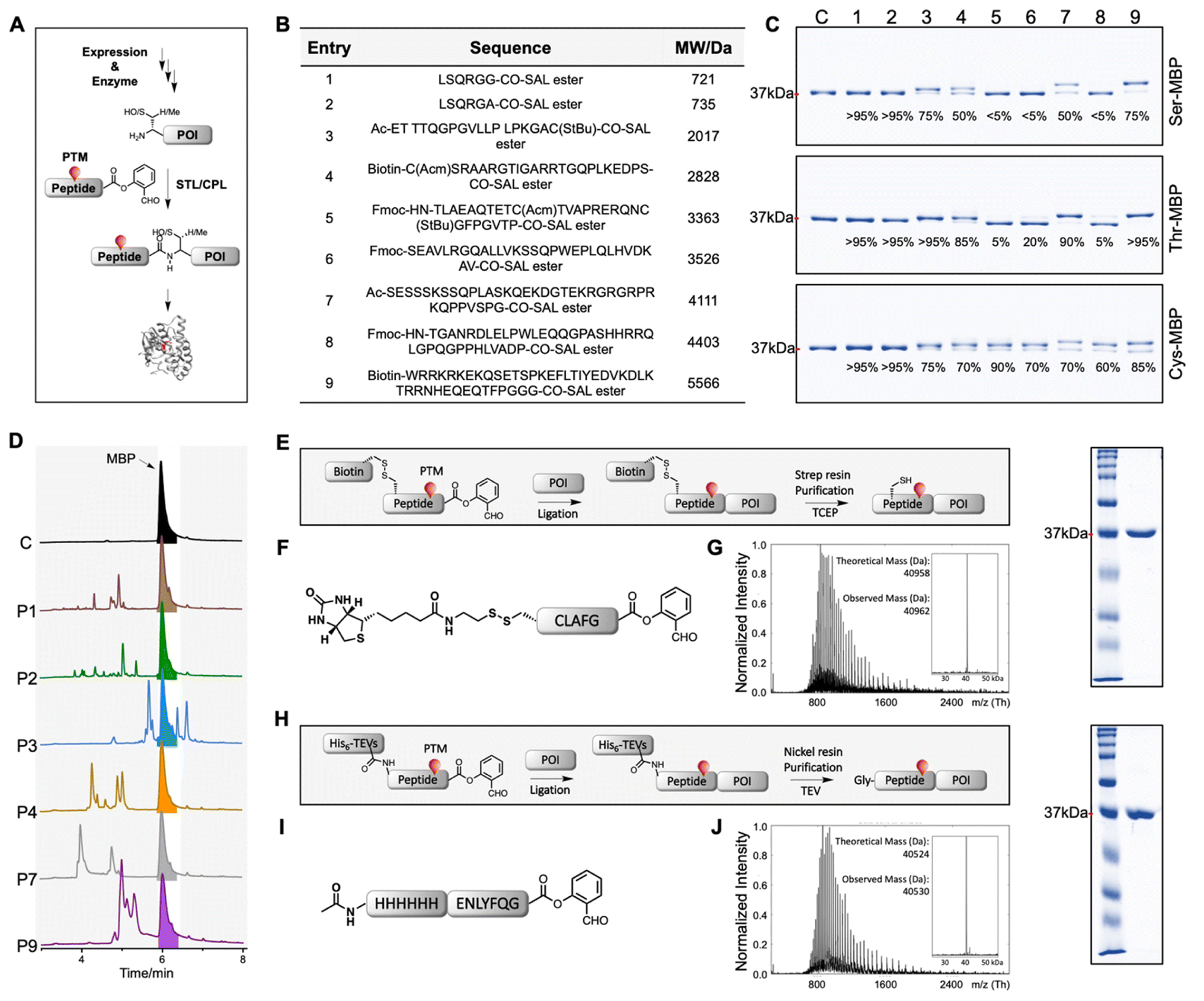 | ||
| Fig. 1 STL/CPL-assisted protein semi-synthesis and purification strategies using MBP (maltose-binding protein) as a model protein. (A) Overview of STL/CPL-mediated protein semi-synthesis. (B) Peptide esters used for demonstrating the ligation. SAL: salicylaldehyde. (C) SDS-PAGE analysis of ligation mixtures. Conversions were calculated according to the peak intensity after deconvolution and indicated below the bands. c: control. See also Fig. S3–S5.‡ (D) UV traces of reaction mixtures of the indicated peptide esters reacting with Ser-MBP. The reactions with more than 50% conversion were chosen ((C) top). No significant shift was observed on the HPLC UV trace after ligation. Except for the desired products, other peaks observed in UV traces were mainly of the unconsumed peptide esters, the hydrolysed peptide esters and the products of SAL esters being displaced by HFIP. (E) Disulfide linker-mediated purification strategy. Strep resin: streptavidin resin. (F) The sequence of a model peptide for the disulfide linker-mediated purification strategy. (G) Left: The mass spectrum of the final product. Right: SDS-PAGE analysis of the final product. See also Fig. S6.‡ (H) His6 tag and TEVs based purification strategy. TEVs: TEV protease recognition site. (I) The sequence of the model peptide for the His6 tag and TEVs based purification strategy. Note: this ligation can be fully converted after 3 h. In order to demonstrate the design, we stopped the reaction after 30 min. (J) Left: The mass spectrum of the final product. Right: SDS-PAGE analysis of the final product. See also Fig. S6.‡ | ||
However, the purification process presents an additional challenge. We noticed that the final products often overlapped with unconsumed expressed parts on high performance liquid chromatography (HPLC) due to the minimal change in polarity after ligation, making the purification difficult (Fig. 1D). To address this issue, we introduced an extra tag, such as His6 or biotin, into the synthetic peptide fragment.45 Additionally, we employed a traceless disulfide linker, which is applicable to cysteine-containing proteins. The tag could be removed by tris(2-carboxyethyl)phosphine (TCEP) treatment after purification (Fig. 1E–G and S6‡). Alternatively, an enzymatically cleavable sequence, such as a tobacco etch virus (TEV) protease recognition site, was used (Fig. 1H–J and S6‡). After purification, the tag was removed by TEV protease digestion, leaving an extra Gly at the N-terminus of the synthetic protein. These HPLC-free purification strategies proved to be highly effective for isolating ligated proteins, offering time-saving benefits and generally giving us good purity and higher recovery rates due to the small purification system (Fig. 1G and J).
Semi-synthesis of acetylated HMGB1 proteins
Following the established protocol from the above study, we proceeded with the semi-synthesis of HMGB1 proteins (Fig. 2A). The synthesis strategy involved disconnection at Met12–Ser13 via Ser ligation. To achieve this, HMGB1 (13-214) was produced through TEVs-mediated proteolysis (Fig. S7‡), while the acetylated peptide (1–12) SAL esters along with a His6 tag and a TEV cleavage site were chemically synthesized. The ligation reaction exhibited 35% conversion after 5 h, as determined by the deconvoluted peak intensity (with the remaining 65% primarily comprising the starting material, and impurities appearing after 5 h). After nickel resin purification, TEV protease cleavage, refolding, and size-exclusion chromatography purification, acetylated full-length HMGB1 proteins were successfully obtained with an overall isolated yield of 10% (Fig. 2B–D and S8‡).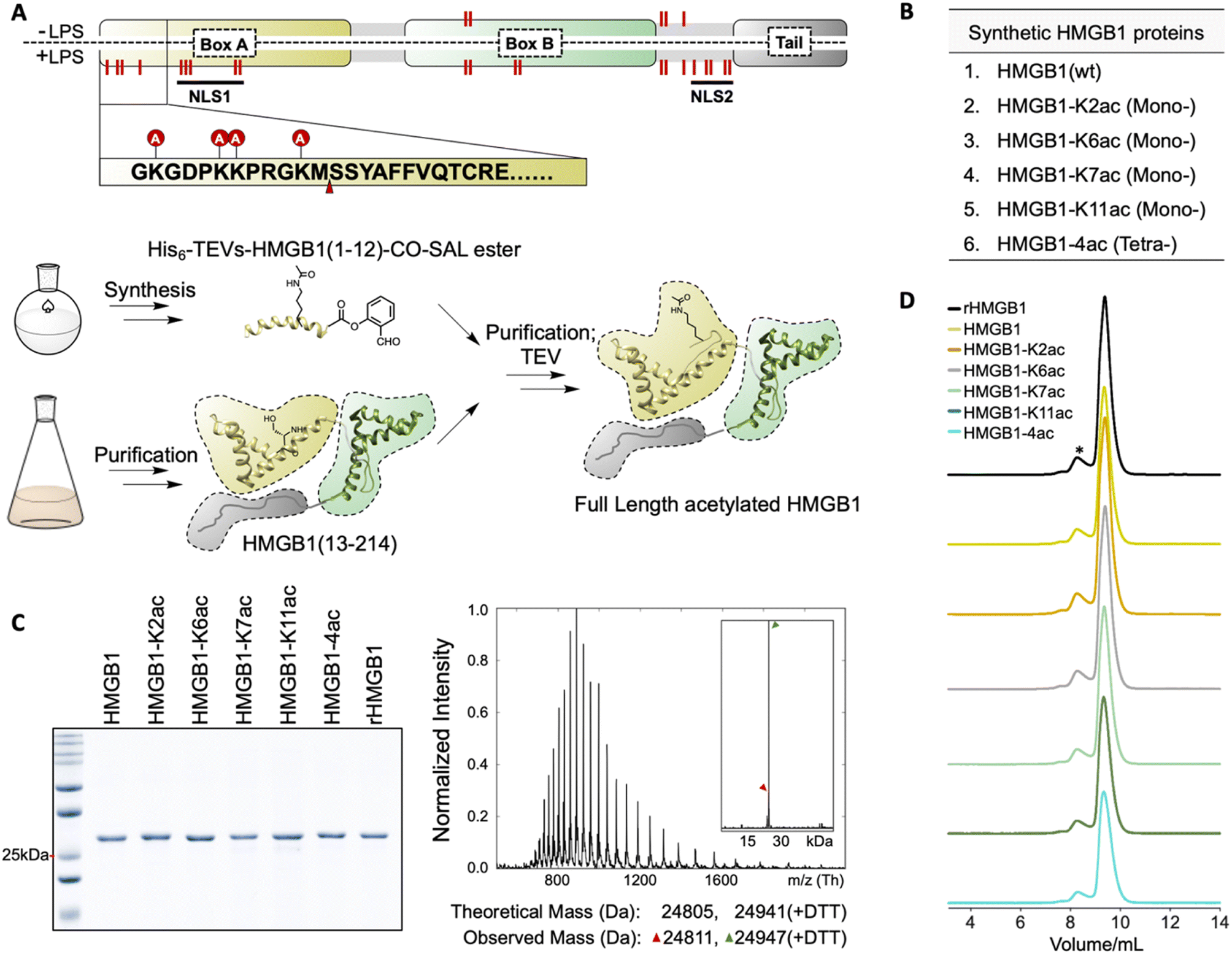 | ||
| Fig. 2 Chemical semi-synthesis of acetylated HMGB1 proteins. (A) Up: HMGB1 is acetylated at Lys2, 6, 7, and 11 after LPS stimulation. The modification sites are highlighted using red lines. The ligation site is indicated by an arrow. NLS1: the first nuclear localization sequence; NLS2: the second nuclear localization sequence. Bottom: Semi-synthesis routes of HMGB1. (B) Full length HMGB1 proteins that were synthesized. (C) Left: SDS-PAGE analysis of semi-synthetic HMGB1 proteins and recombinant full length HMGB1 (rHMGB1). Right: Mass spectra of semi-synthetic HMGB1-K2ac. +DTT: intein tag-based purification will install a DTT ester on the C-terminus of the protein, which will hydrolyze gradually. See also Fig. S7 and S8.‡ (D) Refolding of synthetic HMGB1 proteins. Size-exclusion spectra showed that HMGB1 proteins are folded mainly as monomers; recombinant full length HMGB1 (rHMGB1) purified under native conditions served as a control. *: dimers of HMGB1. | ||
Acetylation inhibits HMGB1–heparin interaction
Heparin, an endogenous sulfated glycosoaminoglycan anticoagulant, has been clinically approved for the treatment of heart attacks and unstable angina.46,47 Recently, it was identified as a potent inhibitor of HMGB1, selectively suppressing the HMGB1-caspase-11 dependent inflammatory response during sepsis.10,11,48,49 The N-terminal sequence of HMGB1 contains a consensus BBXB motif responsible for binding to heparin and other polysaccharides, where B represents any basic residue and X represents any hydrophobic residue.50,51 To investigate whether the acetylation regulates HMGB1–polysaccharide interaction, we employed a heparin affinity column, in which heparin was covalently coupled to highly cross-linked agarose beads. By elution with a continuous gradient of sodium chloride, we observed that all mono-acetylations resulted in decreased heparin-binding, even though Lys2 and Lys11 do not locate in the BBXB motif. This suggests that all four lysine residues contribute to the positive electrostatic potential surface of HMGB1 towards heparin binding (Fig. 3A). As expected, tetra-acetylated HMGB1 was eluted earliest due to a synergistic effect (Fig. 3A). These results were further validated by pull-down experiments using heparin across-linked agarose resin (Fig. 3B) and site mutation mimics at these four lysine residues (Fig. 3C). In addition, methylene blue (M.B.) was reported as an antagonist of heparin and has been used to effectively prevent heparin-mediated bleeding in patients with protamine allergy.52 Upon binding to heparin, the absorbance peak of M.B. will shift from 664 nm to 568 nm.53 However, the addition of HMGB1 restored the absorbance peak at 664 nm, indicating that HMGB1 displaced M.B. from heparin (Fig. 3D). We also utilized the ratio (Abs of 664 nm/Abs of 568 nm) to further evaluate the heparin-binding efficiency of acetylated HMGB1 proteins (Fig. 3E left and S9‡) and mutant HMGB1 proteins (Fig. 3E right). Our results clearly showed that charge neutralization by acetylation or mutation on N-terminal lysine residues impaired the HMGB1–heparin interaction. | ||
| Fig. 3 Acetylation inhibits heparin binding. (A) Heparin column elution spectra of HMGB1 proteins. rHMGB1: full length HMGB1 purified under native conditions. ΔHMGB1: HMGB1 (13-214, N-terminus deleted) purified under native conditions. (B) Pull-down of HMGB1 proteins using heparin resin. First lane: heparin sodium used as the competitor. (C) Heparin column elution spectra of mutated HMGB1 proteins. (D) Absorbance spectra of M.B., M.B.-heparin complex, and M.B.-heparin complex with HMGB1. (E) M.B. displacement assay. Left: Synthetic HMGB1 proteins. Right: Mutated HMGB1 proteins. BSA is used as a negative control. Error bars indicate average ± SEs, n = 3. | ||
HMGB1-directed therapies have been extensively tested in preclinical models and diseases and under preclinical conditions.10 The potential benefits of these therapies have been well supported by numerous previous studies.9–11,54,55 Notably, oligosaccharide-based targeting strategies have emerged as a promising avenue to expand the therapeutic window.10 Moving forward, to further optimize or redefine HMGB1-directed therapies, the intrinsic propensity of HMGB1 for PTMs should be considered, as the targeting efficiency can be significantly affected, potentially leading to HMGB1 escape and therapy failure.
Acetylation regulates the thrombin-mediated degradation
Thrombin, a serine protease in the vascular endothelium, is capable of recognizing and cleaving HMGB1 at the Arg9-Gly10 site, especially in the presence of the thrombin cofactor (thrombomodulin), producing a less proinflammatory form (Fig. S10‡).56 This process has been known as an important mechanism for the rapid clearance of HMGB1 from circulation.57 Given that the N-terminal acetylation sites are adjacent to the cleavage site, we hypothesized that these modifications might directly influence thrombin-mediated degradation. To investigate the potential role, we reconstituted the extracellular environment using a physiological concentration of thrombin (∼1 U mL−1), a relatively low concentration of HMGB1 (5 μM), and phosphate-buffered saline (PBS). As depicted in Fig. 4A, acetylation of Lys2, which is not located within the conserved thrombin recognition sequence (Fig. S11‡), did not affect the digestion. Remarkably, Lys6 acetylation significantly enhanced thrombin-mediated cleavage. As a result, tetra-acetylation also facilitated the thrombin digestion. These results were further supported by quantitative Michaelis–Menten kinetics analysis (Fig. 4A and S12‡). The analysis revealed that the acceleration effect of Lys6 acetylation was primarily attributable to an increase in Kcat (Vmax), indicating that Lys6 acetylation does not markedly alter the binding affinity towards thrombin but promotes better positioning for proteolytic cleavage. It is worth noting that we also observed a moderate inhibitory effect from Lys7 acetylation, while Lys11 acetylation had no effect on the thrombin digestion process. | ||
| Fig. 4 Acetylation affects thrombin-mediated degradation. (A) HMGB1 digestion assay. HMGB1 proteins were treated with thrombin and analyzed by SDS-PAGE. The digestion rate curve generated from SDS-PAGE analysis (middle). The red arrow indicates cleaved products. Right: Kinetic parameters for thrombin on HMGB1 proteins. Values for Vmax and Km represent 95% profile likelihood. (B) Left: HMGB1 N-terminal peptides that were synthesized. Middle: Kinetic parameters for thrombin on HMGB1 N-terminal peptides. Values for Vmax and Km represent 95% profile likelihood. Right: Representative UV traces of the digested HMGB1 N-terminal peptide mixtures after treatment with thrombin. (a): HMGB1; (b): HMGB1-K2ac; (c): HMGB1-K6ac; (d): HMGB1-K7ac; (e): HMGB1-K11ac; (f): HMGB1-4ac. Starting materials are labelled with red arrows; cleaved products are labelled with black arrows. (C) Michaelis–Menten kinetics analysis of thrombin on HMGB1 proteins and N-terminal peptides. Due to the protein solubility limits, the highest concentration that we can use is 400 μM. (D) The structures of HMGB1 and its mutations were predicted using AlphaFold2 and were coloured using the predicted local distance difference test (pLDDT) score (per-residue confidence score). Only the top-ranking (rank1) models are shown. | ||
A previous study demonstrated that aliphatic residues (e.g., Leu) at P4 are preferred by thrombin (Fig. S11‡).58 As the acetylation reduces the hydrophilicity of Lys, it is plausible that Lys6 acetylated HMGB1 may become a more favourable substrate for thrombin. To test our hypothesis, various HMGB1 N-terminal peptides were therefore synthesized (Fig. 4B) and then subjected to the thrombin digestion assay. The cleaved products were analyzed by HPLC (Fig. 4B). Consistent with the protein digestion results, acetylation on Lys6 substantially enhanced thrombin digestion, while Lys7 acetylation inhibited it (Fig. 4B and S13‡). Interestingly, our findings also revealed that acetylation of Lys11 hindered thrombin's activity, and in the case of tetra-acetylation, the overall effect was inhibitory on peptide digestion, which contradicted the results obtained from protein digestion (Fig. 4A–C). This discrepancy between the thrombin digestion results of full-length HMGB1 proteins and HMGB1 N-terminal peptides prompted us to consider that conformational hindrance may affect the enzymatic activity. Indeed, based on the resolved structure of HMGB1 boxA by different studies, the N-terminus, especially the region from Pro8 to Met12, is consistently tethered to the third α-helix through several putative hydrogen bonding interactions (Fig. S14‡).59,60 Thus, it is highly probable that the N-terminus is twisted by these intramolecular interactions and is unable to be optimally positioned for thrombin cleavage, as observed in flexible short N-terminal peptides. To validate this, we first examined the intramolecular interaction between the N-terminus and third α-helix. AlphaFold2 was employed to predict the structure of HMGB1 containing corresponding mutations that were expected to interrupt the putative hydrogen bonds.61 As shown in Fig. 4E, S16A, and B,‡ although the predicted models are structurally conserved and do not show significant conformational differences between mutations and wild type HMGB1, we noticed that the prediction confidence for the N-terminus was decreased, implying that hydrogen bond interruption resulted in a structural destabilization of the N-terminus (low confidence represents a flexible region).62 By combining computational modelling and biochemistry characterization, we identified that the three-dimensional structure of HMGB1 protein indeed prevents Lys11 from being involved in thrombin-mediated degradation (ESI Notes and Fig. S15–S22‡). Consequently, the predominant promoting effect of Lys6 acetylation, together with the absence of an inhibitory effect of Lys11 acetylation, ultimately resulted in enhanced digestion of tetra-acetylated HMGB1 protein.
In summary, our study indicates that acetylation of Lys2 does not affect the thrombin digestion, while acetylation of Lys6 greatly improves the thrombin digestion, and acetylation of Lys7 inhibits the thrombin digestion; Lys11 is sequestered by the tertiary structure, and its acetylation does not regulate the thrombin-mediated degradation. In cases of multiple acetylations, Lys6 acetylation always facilitates the digestion regardless of the status of the other three lysine residues (e.g., tetra-acetylation). In recent decades, extensive signalling crosstalk between phosphorylation and proteolysis has been reported as a fine-tuned mechanism to tilt the cell's fate towards apoptosis.63,64 However, acetylation, as another highly abundant PTM in cells, remains largely unexplored in terms of this important regulation mechanism. Here, Lys6 acetylation-induced thrombin cleavage on HMGB1 is likely the first example of acetylation-directed proteolysis, representing a new multidimensional regulation mechanism in cell activities. Additionally, our studies on Lys11 showed that the structure and function of a short peptide may not necessarily be correlated with those of the same sequence in the context of a protein. This finding highlights the importance of protein structural morphism, which is capable of sequestering PTM-mediated phenotypes and raises a cautionary note on the validity of studies that use modified peptides to investigate PTMs. It is also worth noting that K7Q mutation on HMGB1, mimicking K7ac, showed an opposite effect to K7ac (Fig. S18 and S19‡). Nowadays, protein site mutation mimics are widely used to study the PTM effects in cells (e.g., Gln mimicking Lys acetylation and Glu mimicking Ser/Thr/Tyr phosphorylation). Nevertheless, the validity of such a strategy also remains to be further tested and validated.
Conclusions
As a sentinel for the immune system, HMGB1 plays a critical role in cell survival/death pathways. In the meantime, HMGB1 isoforms resulting from PTMs are also responsible for its diverse activities. To systematically elucidate the role of HMGB1 in the immune system, full characterization of posttranslationally modified isoforms is important. One key to explore how PTMs contribute to multifarious cellular activities at the molecular level increasingly relies on our ability to access the homogeneous proteins with site-specific PTMs of interest. Notably, a variety of proteins with complicated modifications (e.g., glycosylation) are becoming available by means of chemical synthesis.65–71 However, the advantages of chemical protein synthesis have not been fully realized since the process can be relatively burdensome and is often difficult to initiate in a biological laboratory. Alternatively, chemical ligation enabled protein semi-synthesis, which harnesses the advantages of both organic synthesis and recombinant protein technology, presents a more feasible approach for biologists (small synthetic peptide parts will be more affordable or synthetically accessible by themselves). In this study, we established a streamlined method for SAL ester-mediated protein semi-synthesis. The success of stitching various peptide SAL esters with MBP, a 40 kDa-protein, exemplified the operability of STL and CPL for large protein synthesis. Additionally, two efficient purification strategies were introduced to isolate the ligated products. Our results demonstrated that STL/CPL-mediated protein semi-synthesis, in tandem with HPLC-free purification, offers a straightforward yet effective approach to address the challenge of synthesizing large proteins with specific PTMs of interest.We have successfully used this method to generate multiple acetylated full-length HMGB1 proteins, which enable us to illustrate that the N-terminal region acetylation represents a regulatory switch to control the HMGB1–heparin interaction and HMGB1's stability. The critical interaction between the HMGB1 N-terminus and polysaccharides (e.g., heparin) has likely guided the development of sugar-based inhibitors for HMGB1, which is one of the most popular strategies currently employed.10 Therefore, a comprehensive investigation into the N-terminal modification holds significant promise for advancing the development of extracellular HMGB1-targeted drugs. In addition, our thrombin digestion studies revealed a crosstalk between acetylation and circular enzyme-mediated proteolysis. Specifically, acetylation at Lys6 may subvert the proinflammatory function of HMGB1 by accelerating its degradation, potentially serving as a rescue mechanism for cells against pyroptosis and lethality upon infectious injury (Fig. 5). Further in vivo studies should be conducted to confirm this.
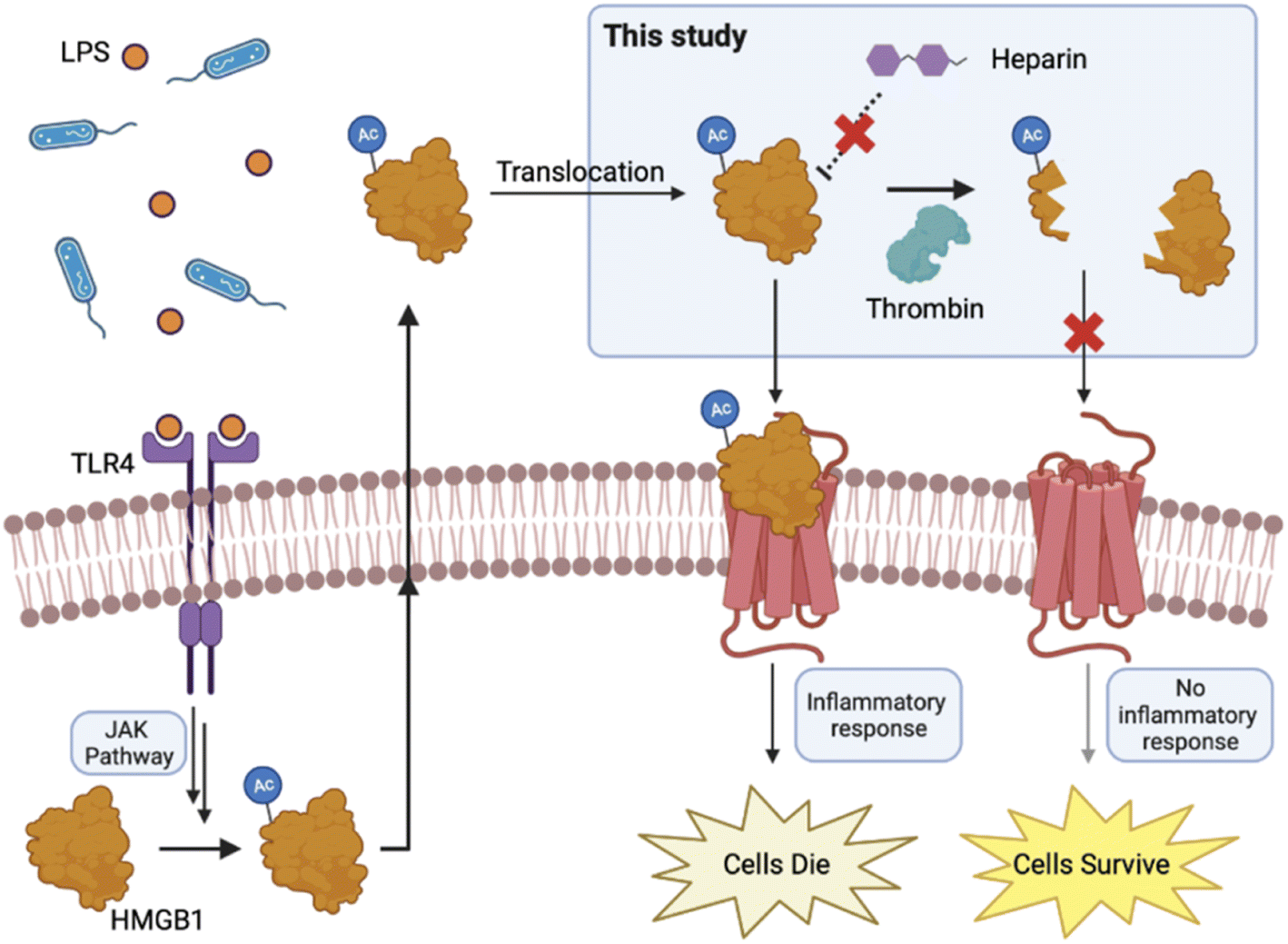 | ||
| Fig. 5 The proposed outcome for N-terminal acetylation of HMGB1. Circulating bacteria stimulate the JAK/STAT1 signalling pathway. Afterwards, HMGB1 undergoes hyper acetylation, leading to its secretion. In the extracellular environment, N-terminal region acetylation impairs the targeting efficiency of heparin towards HMGB1. On the other hand, acetylation of Lys6 promotes rapid degradation of HMGB1 by thrombin, resulting in cleaved HMGB1 that fails to stimulate more severe inflammatory responses. BioRender was used to generate this figure. | ||
Data availability
The data supporting this article have been uploaded as part of the ESI.‡Author contributions
X. L., T. W., and Y. T. designed this project and experiments, analyzed the data, and wrote the paper with input from other authors. T. W. and C. L. performed biochemistry experiments. T. W., J. L., Y. T., J. W., and H. W. performed synthetic experiments. R. W., Q. L., H. L. and Y. T. provided discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grants Research Council of the University Grants Committee of Hong Kong (C7147-20G, 17303920, AoE/P-706/16). We thank Dr Han Liu for proofreading the manuscript.Notes and references
- R. Kang, R. Chen, Q. Zhang, W. Hou, S. Wu, L. Cao, J. Huang, Y. Yu, X. G. Fan, Z. Yan, X. Sun, H. Wang, Q. Wang, A. Tsung, T. R. Billiar, H. J. Zeh, M. T. Lotze and D. Tang, HMGB1 in health and disease, Mol. Aspects Med., 2014, 40, 111–116 CrossRef PubMed.
- S. A. Richard, L. H. Xiang, J. X. Yun, S. S. Zhou, Y. Y. Jiang, J. Wang, Z. L. Su and H. X. Xu, Carcinogenic and therapeutic role of high-mobility group Box 1 in cancer: is it a cancer facilitator, a cancer inhibitor or both?, World Cancer Res. J., 2017, 4, 919 Search PubMed.
- S. A. Richard, M. Sackey, Z. Su and H. Xu, Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke, Biosci. Rep., 2017, 37, BSR20171104 CrossRef CAS PubMed.
- S. A. Richard, Y. Jiang, L. H. Xiang, S. Zhou, J. Wang, Z. Su and H. Xu, Post-translational modifications of high mobility group box 1 and cancer, Am. J. Transl. Res., 2017, 9, 5181–5196 CAS.
- M. Magna and D. S. Pisetsky, The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases, Mol. Med., 2014, 20, 138–146 Search PubMed.
- G. P. Sims, D. C. Rowe, S. T. Rietdijk, R. Herbst and A. J. Coyle, HMGB1 and RAGE in inflammation and cancer, Annu. Rev. Immunol., 2019, 28, 367–388 CrossRef.
- H. Yang, D. J. Antoine, U. Andersson and K. J. Tracey, The many faces of HMGB1: a molecular structure-functional activity in inflammation, apoptosis, and chemotaxis, J. Leukocyte Biol., 2013, 93, 856–873 Search PubMed.
- U. Andersson, H. Yang and H. Harris, High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells, Semin. Immunol., 2018, 38, 40–48 CrossRef CAS.
- U. Andersson and H. E. Harris, The role of HMGB1 in the pathogenesis of rheumatic disease, Biochim. Biophys. Acta, 2010, 1799, 141–148 CrossRef CAS PubMed.
- S. VanPatten and Y. Al-Abed, High mobility group box-1 (HMGb1): current wisdom and advancement as a potential drug target: miniperspective, J. Med. Chem., 2017, 61, 5093–5107 CrossRef PubMed.
- K. Arnold, Y. Xu, E. M. Sparkenbaugh, M. Li, X. Han, X. Zhang, K. Xia, M. Piegore, F. Zhang, X. Zhang and M. Henderson, Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure, Sci. Transl. Med., 2020, 12, eaav8075 CrossRef CAS PubMed.
- N. E. Stevens, M. J. Chapman, C. K. Fraser, T. R. Kuchel, J. D. Hayball and K. R. Diener, Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes, Sci. Rep., 2017, 7, 1–14 CrossRef CAS PubMed.
- C. Sivakorn, J. Dechsanga, L. Jamjumrus, K. Boonnak, M. J. Schultz, A. M. Dondorp and T. Techarang, High mobility group box 1 and interleukin 6 at intensive care unit admission as biomarkers in critically ill COVID-19 Patients, Am. J. Trop. Med. Hyg., 2021, 105, 73 CAS.
- R. Chen, Y. Huang, J. Quan, J. Liu, H. Wang, T. R. Billiar, M. T. Lotze, H. J. Zeh, R. Kang and D. Tang, HMGB1 as a potential biomarker and therapeutic target for severe COVID-19, Heliyon, 2020, 6, e05672 CrossRef CAS PubMed.
- U. Andersson, W. Ottestad and K. J. Tracey, Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19?, Mol. Med., 2020, 6, 1–13 Search PubMed.
- T. Bonaldi, F. Talamo, P. Scaffidi, D. Ferrera, A. Porto, A. Bachi, A. Rubartelli, A. Agresti and M. E. Bianchi, Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion, EMBO J., 2003, 22, 5551–5560 CrossRef CAS PubMed.
- B. Lu, D. J. Antoine, K. Kwan, P. Lundbäck, H. Wähämaa, H. Schierbeck, M. Robinson, M. A. Van Zoelen, H. Yang, J. Li and H. Erlandsson-Harris, JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3068–3073 CrossRef CAS PubMed.
- S. H. Davé, J. S. Tilstra, K. Matsuoka, F. Li, R. A. DeMarco, D. Beer-Stolz, A. R. Sepulveda, M. P. Fink, M. T. Lotze and S. E. Plevy, Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis, J. Leukocyte Biol., 2009, 86, 633–643 CrossRef PubMed.
- I. Ito, J. Fukazawa and M. Yoshida, Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils, J. Biol. Chem., 2007, 282, 16336–16344 CrossRef CAS PubMed.
- J. H. Youn and J. S. Shin, Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion, J. Immunol., 2006, 177, 7889–7897 CrossRef CAS PubMed.
- D. Ditsworth, W. X. Zong and C. B. Thompson, Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus, J. Biol. Chem., 2007, 282, 17845–17854 CrossRef CAS PubMed.
- Y. H. Kim, M. S. Kwak, J. B. Park, S. A. Lee, J. E. Choi, H. S. Cho and J. S. Shin, N-linked glycosylation plays a crucial role in the secretion of HMGB1, J. Cell Sci., 2016, 129, 29–38 CrossRef CAS PubMed.
- A. T. Balana, A. Mukherjee, H. Nagpal, S. P. Moon, B. Fierz, K. M. Vasquez and M. R. Pratt, O-GlcNAcylation of High Mobility Group Box 1 (HMGB1) Alters Its DNA Binding and DNA Damage Processing Activities, J. Am. Chem. Soc., 2021, 143, 16030–16040 CrossRef CAS PubMed.
- D. Tang, T. R. Billiar and M. T. Lotze, A Janus tale of two active high mobility group box 1 (HMGB1) redox states, Mol. Med., 2012, 18, 1360–1362 CAS.
- M. Schiraldi, A. Raucci, L. M. Munoz, E. Livoti, B. Celona, E. Venereau, T. Apuzzo, F. De Marchis, M. Pedotti, A. Bachi and M. Thelen, HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4, J. Exp. Med., 2012, 209, 551–563 CrossRef CAS PubMed.
- R. Chen, R. Kang and D. Tang, The mechanism of HMGB1 secretion and release, Exp. Mol. Med., 2022, 54, 91–102 CrossRef CAS PubMed.
- S. Bondalapati, M. Jbara and A. Brik, Expanding the chemical toolbox for the synthesis of large and uniquely modified proteins, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- J. W. Bode, Chemical protein synthesis with the α-ketoacid–hydroxylamine ligation, Acc. Chem. Res., 2017, 50, 2104–2115 CrossRef CAS PubMed.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Rapid and efficient protein synthesis through expansion of the native chemical ligation concept, Nat. Rev. Chem, 2018, 2, 1–17 CrossRef.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. C. M. Monbaliu and O. Melnyk, Native chemical ligation and extended methods: mechanisms, catalysis, scope, and limitations, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- L. Liu, Chemical synthesis of proteins that cannot be obtained recombinantly, Isr. J. Chem., 2019, 59, 64–70 CrossRef CAS.
- Y. Tan, H. Wu, T. Wei and X. Li, Chemical Protein Synthesis: Advances, Challenges, and Outlooks, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed.
- T. Wei, H. Liu, B. Chu, P. Blasco, Z. Liu, R. Tian, D. X. Li and X. Li, Phosphorylation-regulated HMGA1a-P53 interaction unveils the function of HMGA1a acidic tail phosphorylations via synthetic proteins, Cell Chem. Biol., 2021, 28, 722–732 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Expressed protein ligation: a general method for protein engineering, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS PubMed.
- T. W. Muir, Semisynthesis of proteins by expressed protein ligation, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. Becker, Native chemical ligation in protein synthesis and semi-synthesis, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- R. E. Thompson and T. W. Muir, Chemoenzymatic semisynthesis of proteins, Chem. Rev., 2019, 120, 3051–3126 CrossRef PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wang and S. J. Danishefsky, Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef PubMed.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Protein chemical synthesis by serine and threonine ligation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS PubMed.
- Y. Tan, J. Li, K. Jin, J. Liu, Z. Chen, J. Yang and X. Li, Cysteine/Penicillamine Ligation Independent of Terminal Steric Demands for Chemical Protein Synthesis, Angew. Chem., Int. Ed., 2020, 132, 12841–12845 CrossRef.
- H. Liu and X. Li, Serine/threonine ligation: origin, mechanistic aspects, and applications, Acc. Chem. Res., 2018, 51, 1643–1655 CrossRef CAS PubMed.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Ligation Technologies for the Synthesis of Cyclic Peptides, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- (a) D. Bang and S. B. Kent, His6 tag-assisted chemical protein synthesis, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5014–5019 CrossRef CAS PubMed; (b) S. F. Loibl, Z. Harpaz, R. Zitterbart and O. Seitz, Total chemical synthesis of proteins without HPLC purification, Chem. Sci., 2016, 7, 6753–6759 RSC; (c) X. Liu, J. Liu, Z. Wu, L. Chen, S. Wang and P. Wang, Photo-cleavable purification/protection handle assisted synthesis of giant modified proteins with tandem repeats, Chem. Sci., 2019, 10, 8694–8700 RSC.
- V. Menon, S. D. Berkowitz, E. M. Antman, R. M. Fuchs and J. S. Hochman, New heparin dosing recommendations for patients with acute coronary syndromes, Am. J. Med., 2001, 110, 641–650 CrossRef CAS PubMed.
- P. Theroux, D. Waters, S. Qiu, J. McCans, P. De Guise and M. Juneau, Aspirin versus heparin to prevent myocardial infarction during the acute phase of unstable angina, Circulation, 1993, 88, 2045–2048 CrossRef CAS PubMed.
- Y. Ling, Z. Y. Yang, T. Yin, L. Li, W. W. Yuan, H. S. Wu and C. Y. Wang, Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro, Int. Immunopharmacol., 2011, 11, 187–193 CrossRef CAS PubMed.
- L. Li, Y. Ling, M. Huang, T. Yin, S. M. Gou, N. Y. Zhan, J. X. Xiong, H. S. Wu, Z. Y. Yang and C. Y. Wang, Heparin inhibits the inflammatory response induced by lps and hmgb1 by blocking the binding of hmgb1 to the surface of macrophages, Cytokine, 2015, 72, 36–42 CrossRef CAS PubMed.
- E. Elass-Rochard, D. Legrand, V. Salmon, A. Roseanu, M. Trif, P. S. Tobias, J. Mazurier and G. Spik, Lactoferrin inhibits the endotoxin interaction with CD14 by competition with the lipopolysaccharide-binding protein, Infect. Immun., 1998, 66, 486–491 CrossRef CAS PubMed.
- J. H. Youn, M. S. Kwak, J. Wu, E. S. Kim, Y. Ji, H. J. Min, J. H. Yoo, J. E. Choi, H. S. Cho and J. S. Shin, Identification of lipopolysaccharide-binding peptide regions within HMGB1 and their effects on subclinical endotoxemia in a mouse model, Eur. J. Immunol., 2011, 41, 2753–2762 CrossRef CAS PubMed.
- E. M. Sloand, C. M. Kessler, C. L. McIntosh and H. G. Klein, Methylene blue for neutralization of heparin, Thromb. Res., 1989, 54, 677–686 CrossRef CAS PubMed.
- Q. Liu, S. Välimäki, A. Shaukat, B. Shen, V. Linko and M. A. Kostiainen, Serum albumin–peptide conjugates for simultaneous heparin binding and detection, ACS Omega, 2019, 4, 21891–21899 CrossRef CAS PubMed.
- D. Musumeci, G. N. Roviello and D. Montesarchio, An overview on hmgb1 inhibitors as potential therapeutic agents in hmgb1-related pathologies, Pharmacol. Ther., 2014, 141, 347–357 CrossRef CAS PubMed.
- H. Wang, M. F. Ward and A. E. Sama, Targeting hmgb1 in the treatment of sepsis, Expert Opin. Ther. Targets, 2014, 18, 257–268 CrossRef CAS PubMed.
- T. Ito, K. I. Kawahara, K. Okamoto, S. Yamada, M. Yasuda, H. Imaizumi, Y. Nawa, X. Meng, B. Shrestha, T. Hashiguchi and I. Maruyama, Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes, Arterioscler., Thromb., Vasc. Biol., 2008, 18, 1825–1830 CrossRef PubMed.
- T. Ito, K. Kawahara, T. Nakamura, S. Yamada, T. Nakamura, K. Abeyama, T. Abeyama and I. Maruyama, High-mobility group box 1 protein promotes development of microvascular thrombosis in rats, J. Thromb. Haemostasis, 2007, 5, 109–116 CrossRef CAS PubMed.
- M. Gallwitz, M. Enoksson, M. Thorpe and L. Hellman, The extended cleavage specificity of human thrombin, PLoS One, 2012, 7, e31756 CrossRef CAS PubMed.
- J. P. Rowell, K. L. Simpson, K. Stott, M. Watson and J. O. Thomas, HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail, Structure, 2012, 20, 2014–2024 CrossRef CAS PubMed.
- J. Wang, N. Tochio, A. Takeuchi, J. I. Uewaki, N. Kobayashi and S. I. Tate, Redox-sensitive structural change in the A-domain of HMGB1 and its implication for the binding to cisplatin modified DNA, Biochem. Biophys. Res. Commun., 2013, 44, 701–706 CrossRef.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- K. M. Ruff and R. V. Pappu, AlphaFold and implications for intrinsically disordered proteins, J. Mol. Biol., 2021, 433, 167208 CrossRef CAS PubMed.
- I. Maluch, J. Grzymska, S. J. Snipas, G. S. Salvesen and M. Drag, Evaluation of the effects of phosphorylation of synthetic peptide substrates on their cleavage by caspase-3 and-7, Biochem. J., 2021, 478, 2233–2245 CrossRef CAS PubMed.
- A. M. Weeks, Kinases leave their mark on caspase substrates, Biochem. J., 2021, 478, 3179–3184 CrossRef CAS PubMed.
- C. Unverzagt and Y. Kajihara, Chemical assembly of N-glycoproteins: a refined toolbox to address a ubiquitous posttranslational modification, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC.
- H. Sun and A. Brik, The journey for the total chemical synthesis of a 53 kDa protein, Acc. Chem. Res., 2019, 52, 3361–3371 CrossRef CAS PubMed.
- F. Ye, J. Zhao, P. Xu, X. Liu, J. Yu, W. Shanguan, J. Liu, X. Luo, C. Li, T. Ying and J. Wang, Synthetic homogeneous glycoforms of the SARS-CoV-2 spike receptor-binding domain reveals different binding profiles of monoclonal antibodies, Angew. Chem., Int. Ed., 2021, 60, 12904–12910 CrossRef CAS PubMed.
- H. Wu, Y. Zhang, Y. Li, J. Xu, Y. Wang and X. Li, Chemical synthesis and biological evaluations of adiponectin collagenous domain glycoforms, J. Am. Chem. Soc., 2021, 143, 7808–7818 CrossRef CAS PubMed.
- C. C. Hanna, J. Kriegesmann, L. J. Dowman, C. F. Becker and R. J. Payne, Chemical synthesis and semisynthesis of lipidated proteins, Angew. Chem., Int. Ed., 2021, 61, e202111266 CrossRef PubMed.
- Q. Zheng, Z. Su, Y. Yu and L. Liu, Recent progress in dissecting ubiquitin signals with chemical biology tools, Curr. Opin. Chem. Biol., 2022, 70, 102187 CrossRef CAS PubMed.
- W. W. Shi, C. Shi, T. Y. Wang, Y. L. Li, Y. K. Zhou, X. H. Zhang, D. Bierer, J. S. Zheng and L. Liu, Total chemical synthesis of correctly folded disulfide-rich proteins using a removable O-linked β-N-acetylglucosamine strategy, J. Am. Chem. Soc., 2022, 144, 349–357 CrossRef CAS PubMed.
Footnotes |
| † Please note that this manuscript has been preprinted at bioRxiv (https://www.biorxiv.org/content/10.1101/2021.10.05.463167v2) since 2021 and has been cited by a published article in Chemical Science (Chem. Sci., 2022, 13, 1367–1374). This study presents a new approach for semi-synthesis of HMGB1 protein, which is based on the method described in Fig. 1F of this study and a solubilizing tag. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01109g |
| This journal is © The Royal Society of Chemistry 2023 |