
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free highly chemo-selective bisphosphorylation and deoxyphosphorylation of carboxylic acids†
Liguang
Gan
a,
Tianhao
Xu
a,
Qihang
Tan
a,
Mengjie
Cen
a,
Lingling
Wang
a,
Jingwei
Zhao
a,
Kuang
Liu
a,
Long
Liu
a,
Wen-Hao
Chen
b,
Li-Biao
Han
 *c,
Jacek E.
Nycz
*d and
Tieqiao
Chen
*a
*c,
Jacek E.
Nycz
*d and
Tieqiao
Chen
*a
aKey Laboratory of Ministry of Education for Advanced Materials in Tropical Island Resources, Hainan Provincial Key Lab of Fine Chem, Hainan Provincial Fine Chemical Engineering Research Center, Hainan University, Haikou, 570228, China. E-mail: chentieqiao@hnu.edu.cn
bKey Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education, College of Chemistry and Chemical Engineering, Hainan Normal University, Haikou, 571158, China
cZhejiang Yangfan New Materials Co. Ltd, Shangyu 312369, Zhejiang, China. E-mail: hlb@shoufuchem.com
dInstitute of Chemistry, Faculty of Science and Technology, University of Silesia in Katowice, ul. Szkolna 9, PL-40007 Katowice, Poland. E-mail: jacek.nycz@us.edu.pl
First published on 24th April 2023
Abstract
Carboxylic acids are readily available in both the natural and synthetic world. Their direct utilization for preparing organophosphorus compounds would greatly benefit the development of organophosphorus chemistry. In this manuscript, we describe a novel and practical phosphorylating reaction under transition metal-free reaction conditions that can selectively convert carboxylic acids into the P–C–O–P motif-containing compounds through bisphosphorylation, and the benzyl phosphorus compounds through deoxyphosphorylation. This strategy provides a new route for carboxylic acid conversion as the alkyl source, enabling highly efficient and practical synthesis of the corresponding value-added organophosphorus compounds with high chemo-selectivity and wide substrate scope, including the late modification of complex APIs (active pharmaceutical ingredients). Moreover, this reaction also indicates a new strategy for converting carboxylic acids into alkenes by coupling this work and the subsequent WHE reaction with ketones and aldehydes. We anticipate that this new mode of transforming carboxylic acids will find wide application in chemical synthesis.
Introduction
Carboxylic acids are one of the most ubiquitous functional chemicals found in chemistry, chemical engineering, and related industries.1 Due to their abundance and availability, carboxylic acids are considered the ideal starting substrates in organic synthesis and attract tremendous attention from both academic and industrial researchers. Usually, carboxylic acids are extensively used as the building blocks through a decarboxylative2 or decarbonylative3 process (Scheme 1A(1)). They can also serve well as the acyl source for preparing target functional molecules4 (Scheme 1A(2)) or be reduced by strong reductants5 (Scheme 1A(3)). Herein, we report a new manifold for their conversion under metal-free reaction conditions as the alkyl source (Scheme 1B). It is found that after being activated in situ by Boc2O, both aliphatic and aromatic carboxylic acids, including those bearing functional groups, can be bisphosphorylated by P(O)–H compounds to produce the corresponding phosphorus compounds bearing a P–C–O–P motif. By tuning the reaction conditions, various benzoic acids can also be selectively deoxyphosphorylated by P(O)–H compounds to give the corresponding benzylphosphorus compounds.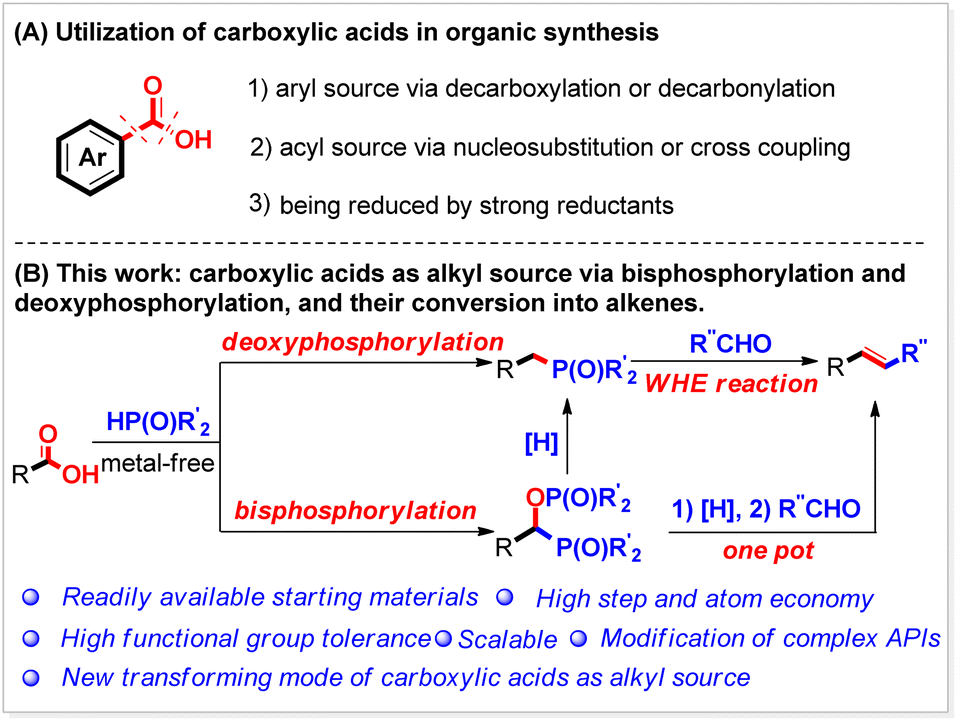 | ||
| Scheme 1 Utilization of carboxylic acids in synthetic chemistry. | ||
Organophosphorus compounds have wide applications in chemical science, medicinal science and materials science.6 However, the methods for their synthesis are limited and highly depend on the transformation of (pseudo)halides.7 The synthesis of organophosphorus compounds, starting from the readily available carboxylic acids, would greatly advance phosphorus chemistry forward. Functional molecules with a P–C–O–P skeleton can be used as ligands or building blocks.8 They can also be used as a dental material and flame retardant material.9 Moreover, some of these compounds have bioactivity exemplified by the PPARg-specific antagonist mifobate (SR-202), which can suppress the differentiation of adipocytes induced by hormones and affect the consumption of glucose in cells and sensitivity to insulin in vivo.10
However, they can only be synthesized through the nucleophilic substitution of alpha-hydroxyl phosphorus compounds with P(O)–X compounds11 or the phosphorylation of acyl derivatives such as acyl phosphorus compounds,12 acyl thioesters,13 acyl chlorides,14etc.15 with P(O)–H compounds. Meanwhile, the benzyl phosphorus compounds are also important phosphorus reagents that can be used as synthetic intermediates, ligands, and bioactive and material functional molecules. For example, they are widely used in Wittig–Horner–Emmons (WHE) reactions and can react with ketones or aldehydes to produce alkenes.16 Benzyl phosphorus compounds can be synthesized through nucleophilic substitution of benzyl metals (Li or Mg) with P(O)–X or the Arbuzov reaction of benzyl halides with phosphites at a high reaction temperature17 (usually >140 °C). They can also be generated via coupling of benzyl halides, aromatic esters, and thiol esters with P(O)–H compounds.18 The reactions described above can produce P–C–O–P skeleton-containing and benzyl phosphorus compounds; however, they obviously suffer from low step economic efficiency since the required intermediates, such as hydroxyl phosphorus compounds, acyl derivatives, and benzyl halides, must be pre-synthesized from the corresponding carboxylic acids.19 Moreover, the conditions of some reactions are very hazardous, limiting their application in organic synthesis. Thus, new facile and efficient methods for their synthesis are still highly desired in the synthetic community.
Results and discussion
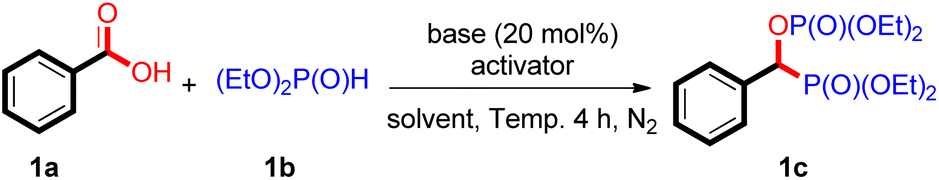
Decarbonylative coupling of benzoic acids emerged as a powerful method in organic synthesis.20 Our group also made a contribution to this research field.21 During the study on the mechanism of decarbonylative phosphorylation of carboxylic acids, a trace amount of bisphosphorus compound 1c was detected by GC-MS. Regarding the importance of such compounds and their synthetic difficulty, we think this scientific finding deserves further investigation. We started this work by investigating the reactivity of benzoic acid (1a) with diethyl phosphite (2a), and the obtained results are compiled in Table 1. By heating a mixture of benzoic acid 1a (0.2 mmol), (EtO)2P(O)H (1b, 2 equiv.), Boc2O (2 equiv.) and Na2CO3 (20 mol%) in toluene at 80 °C for 4 h, the corresponding bisphosphorylating product 1c was produced in 97% yield (Table 1, entry 1). The base is important for this reaction. In its absence, no 1c was detected (Table 1, entry 2). NaHCO3 could also mediate this reaction (Table 1, entry 3), while the weaker base NaOAc showed no reactivity (Table 1, entry 4). The organic base Et3N could also mediate this reaction, but with a relatively low yield (Table 1, entry 5). By increasing the loading to 1 equiv, 95% yield of 1c was obtained (Table 1, entry 6). A similar yield was obtained with 1 equiv. Na2CO3 (Table 1, entry 7). The activator anhydride of a carboxylic acid is also essential to this reaction. Without the addition of anhydride, the reaction did not progress (Table 1, entry 8). Other selected anhydrides, such as Ac2O, Piv2O, and TFAA, did not work either (Table 1, entries 9–11). This reaction could also take place in cyclohexane, THF, and dioxane, but poorly in polar CH3CN and NMP (Table 1, entries 12–16). To our delight, when the reaction was conducted under solvent-free conditions, a high yield was obtained (Table 1, entry 17). Lowering the reaction temperature decreased the yield of 1c (Table 1, entry 18). Shortening the reaction time to 2 h also decreases the yield (Table 1, entry 19). Obviously, the reaction conditions are rather facile and mild. In addition, the atom utilization efficiency of the starting material carboxylic acid and (EtO)2P(O)H is over 90%. This reaction would undoubtedly be a simple and efficient method for preparing bisphosphorus compound 1c.|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Activator | Solvent | Temp. (°C) | Yieldb/% |
| a Reaction conditions: 1a (0.2 mmol), 1b (0.4 mmol), base (20 mol%), activator (0.4 mmol), solvent (1 mL), 4 h, N2. b GC yield using dodecane as an internal standard. c Base (1 equiv.). d Isolated yield. e 2 h. | |||||
| 1 | Na2CO3 | Boc2O | Toluene | 80 | 97 |
| 2 | — | Boc2O | Toluene | 80 | None |
| 3 | NaHCO3 | Boc2O | Toluene | 80 | 95 |
| 4 | NaOAc | Boc2O | Toluene | 80 | Trace |
| 5 | Et3N | Boc2O | Toluene | 80 | 49 |
| 6c | Et3N | Boc2O | Toluene | 80 | 95 |
| 7 | Na2CO3 | Boc2O | Toluene | 80 | 93 |
| 8 | Na2CO3 | — | Toluene | 80 | None |
| 9 | Na2CO3 | Ac20 | Toluene | 80 | None |
| 10 | Na2CO3 | Piv2O | Toluene | 80 | None |
| 11 | Na2CO3 | TFAA | Toluene | 80 | Trace |
| 12 | Na2CO3 | Boc2O | Cyclohexane | 80 | 98 |
| 13 | Na2CO3 | Boc2O | THE | 80 | 95 |
| 14 | Na2CO3 | Boc2O | Dioxane | 80 | 98 |
| 15 | Na2CO3 | Boc2O | CH3CN | 80 | 30 |
| 16 | Na2CO3 | Boc2O | NMP | 80 | 49 |
| 17 | Na2CO3 | Boc2O | 80 | 98 (95d) | |
| 18 | Na2CO3 | Boc2O | 60 | 25 | |
| 19e | Na2CO3 | Boc2O | 80 | 53 | |
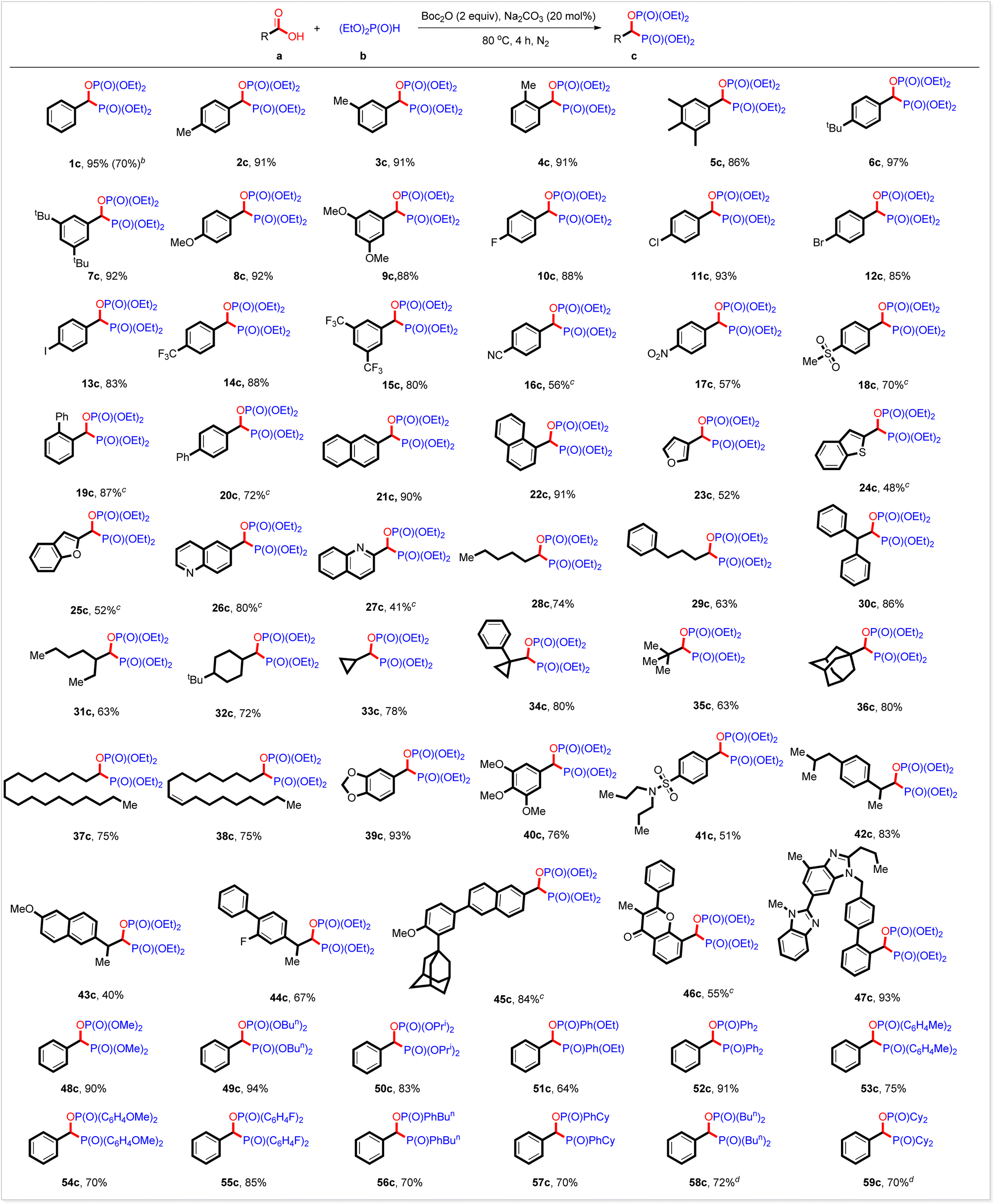
We subsequently investigated the substrate scope with the optimized reaction conditions. As shown in Table 2, the substrate scope is rather general. Electron-rich and electron-deficient aromatic carboxylic acids and aliphatic ones served well. All the three kinds of hydrogen phosphoryl compounds, H-phosphonates, H-phosphinates, and secondary phosphine oxides, were suitable for this reaction. Thus, when the reaction of benzoic acid with diethyl phosphonate was conducted at a 5 mmol scale, 1c was generated in 70% yield. In addition to benzoic acid, substrates bearing 4-methyl, 3-methyl, 2-methyl, 3,4,5-trimethyl, 4-tert-butyl, 3,5-di(tert-butyl), 4-methoxyl and 3,5-dimethoxyl groups at the benzene ring all coupled readily with diethyl phosphonate 1b to produce the corresponding products in high yields (Table 2, 1c–9c). Halo groups (F, Cl, Br, and I) survived well under the reaction conditions, greatly facilitating further derivation of those products (Table 2, 10c–13c). Benzoic acids with electron-withdrawing groups such as 4-trifluoromethyl, 3,5-ditrifluoromethyl, 4-nitrile, 4-nitro, and 4-methylsulfonyl groups also worked well and were transformed into the expected bisphosphorus compounds in high yields (Table 2, 14c–18c). The steric hindrance seemed not to affect the yield of this reaction. For example, 2-phenyl and 4-phenyl benzoic acid acted as the right substrate to give the desired products 19c and 20c in 87% and 74% yield, respectively. High yields were also obtained from the π-extended and heterocyclic derivatives (Table 2, 21c–27c). By using this strategy, aliphatic carboxylic acids were also smoothly bisphosphorylated. It should be noted that primary, secondary, and tertiary carboxylic acids all were workable under the reaction conditions, furnishing the expected products in high yields (Table 2, 28c–35c). Notably, this bisphosphorylation strategy enables the late modification of bioactive carboxylic acids, including some drugs used in the clinic, showing the potential application of this new reaction in the design and development of drugs. For instance, bioactive adamantanyl carboxylic acid, stearic acid, oleic acid, piperic acid, and eudesmic acid all reacted with diethyl phosphonate to produce the expected bisphosphorus compounds in high yields (Table 2, 36c–40c). Probenecid, ibuprofen, naproxen, flurbiprofen, adapalene, 3-methylflavone-8-carboxylic acid and telmisartan are widely used in clinics. By using this strategy, they are also converted smoothly into the corresponding coupling products in high yields (Table 2, 41c–47c). The substrate scope of hydrogen phosphoryl compounds is also investigated. In addition to diethyl phosphonate, other selected H-phosphonates such as dimethyl phosphonate, dibutyl phosphonate, and diisopropyl phosphonate also reacted readily with benzoic acid to produce the corresponding products in high yields (Table 2, 48c–50c). H-phosphinates exemplified by ethyl phenylphosphinate also acted well (Table 2, 51c). To our delight, both aromatic and aliphatic secondary phosphine oxides worked well under the present reaction conditions. Thus, various biaryl phosphine oxides are coupled with benzoic acids (Table 2, 52c–55c). Phenyl butyl phosphine oxide and phenyl cyclohexyl phosphine oxide served as the right substrates (Table 2, 56c and 57c). Aliphatic dibutyl phosphine oxides and the sterically hindrant dicyclohexyl phosphine oxide also reacted with benzoic acid and were transformed smoothly into the expected products in high yields (Table 2, 58c and 59c). These results outlined in Table 2 demonstrated well that this reaction would be a facile, general, and efficient method for preparing bisphosphorus compound c.
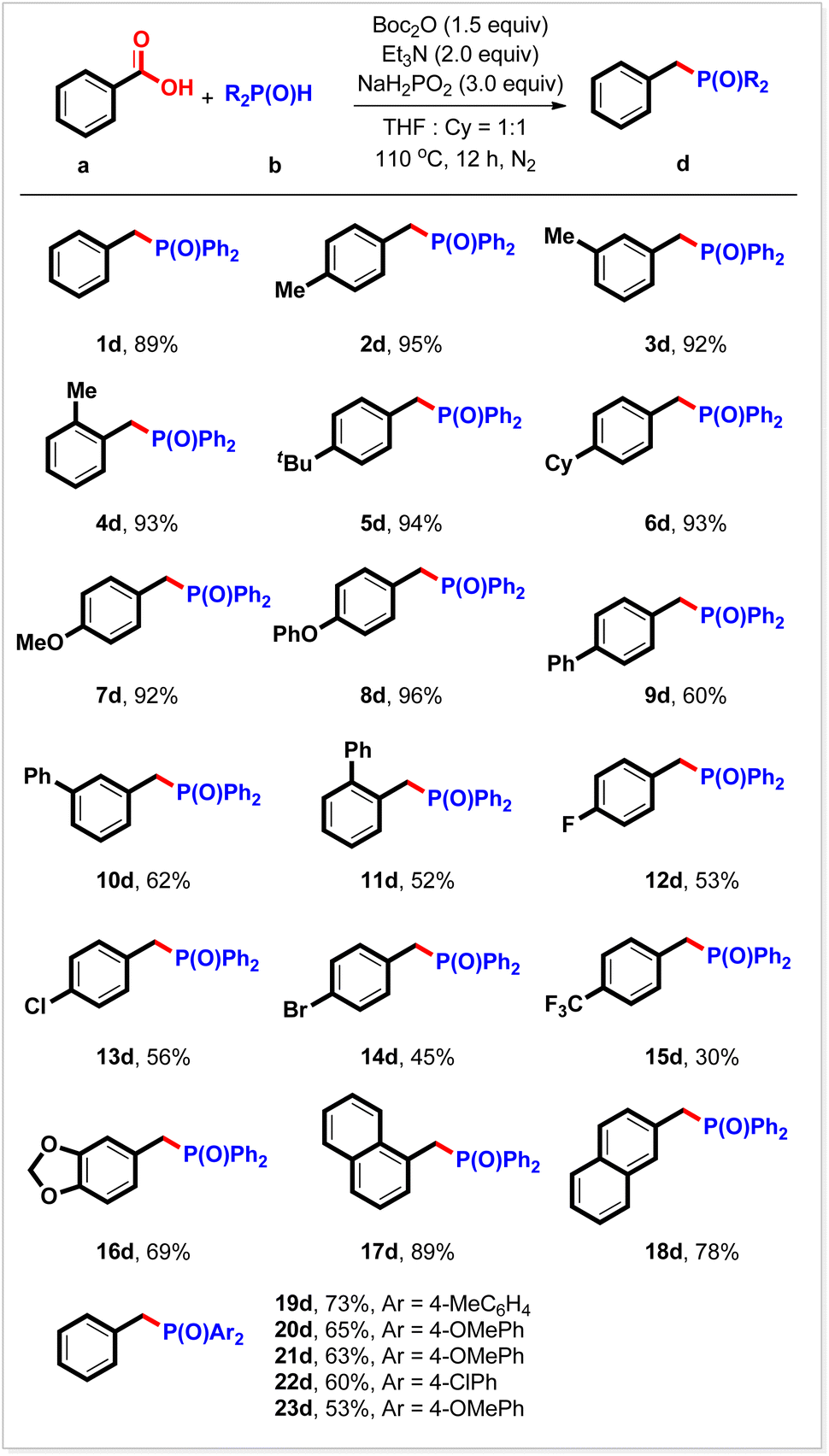
During the reaction of diphenyl phosphine oxide with benzoic acid, it was found that when over-stoichiometric Ph2P(O)H was used, a new benzyl phosphorus compound 1dvia deoxyphosphorylation was detected by GC-MS. Boc2O (1.5 equiv.), Et3N (2 equiv.), and NaH2PO2 (3 equiv.)20 in a mixed solvent (THF/cyclohexane) at 110 °C for 12 h produced the corresponding benzyl phosphorous compounds in 89% yield (Table 3, 1d). This reaction was also suitable for other substituted benzoic acids. Thus, high yields were obtained from derivatives bearing 4-methyl, 3-methyl, 2-methyl, 4-tert-butyl, 4-cyclohexyl, 4-methoxyl, and 4-phenoxyl groups at the benzene ring (Table 3, 2d–8d). All three kinds of phenyl group-substituted benzoic acids were readily deoxyphosphorylated, furnishing the expected products in moderate yields (Table 3, 9d–11d). The halogen groups (F, Cl, and Br) survived under the reaction conditions (Table 3, 12d–14d). The electron-deficient benzoic acid exemplified by 4-trifluoromethyl benzoic acid showed relatively low reactivity, giving the desired product 15d in 30% yield. By using this strategy, heterocyclic and π-extensive substrates were also transformed readily into the corresponding benzyl phosphorus compounds in good yields (Table 3, 16d, 17d, and 18d). Other selected secondary phosphine oxides can also be transformed into the expected benzyl phosphorus compounds in good yields under the current reaction conditions through deoxyphosphorylation (Table 3, 19d–23d).
a Reaction conditions: carboxylic acid (0.2 mmol), R2P(O)H (0.4 mmol), Boc2O (0.3 mmol), NaH2PO2 (0.6 mmol), mixed solvent (VTHF/Vcyclohexane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.5 mL), 110 °C, 12 h, N2. Isolated yield. :1, 0.5 mL), 110 °C, 12 h, N2. Isolated yield.
|
|---|
|
It should be noted that when diethyl phosphonate was used, only a trace amount of the desired product (24d) was produced. To our delight, 24d could be obtained in 85% yield after treatment of the isolated bisphosphorus compound 1c in the presence of AgOAc and Ph2SiH2 in cyclohexane at 100 °C for 2 h (Scheme 2, eqn (1)). Meanwhile, 52c could be converted into 1d in 44% yield under the conditions of a deoxyphosphorylation reaction (Scheme 2, eqn (2)). These results would be a good complement to the deoxyphosphorylation reaction described above. Worth noting is that benzyl phosphorus compounds are widely used in Wittig–Horner–Emmons (WHE) reactions. Indeed, both 1d and 24d could react with benzaldehyde to produce the corresponding alkene 1e in 85% and 87% yield, respectively (Scheme 2, eqn (3)). In particular, we found that the alkene 1e could also be obtained in 70% yield by adding benzaldehyde and a base to the reaction mixture after completing eqn (1) and stirring at 60 °C for 12 h (Scheme 2, eqn (4)). By using this strategy, a variety of alkenes were synthesized in good to high yields (Scheme 2, eqn (4)). These results indicated that carboxylic acids could be easily transformed into alkenes by coupling the current reaction and subsequent WHE reaction, and would find wide applications in organic synthesis.
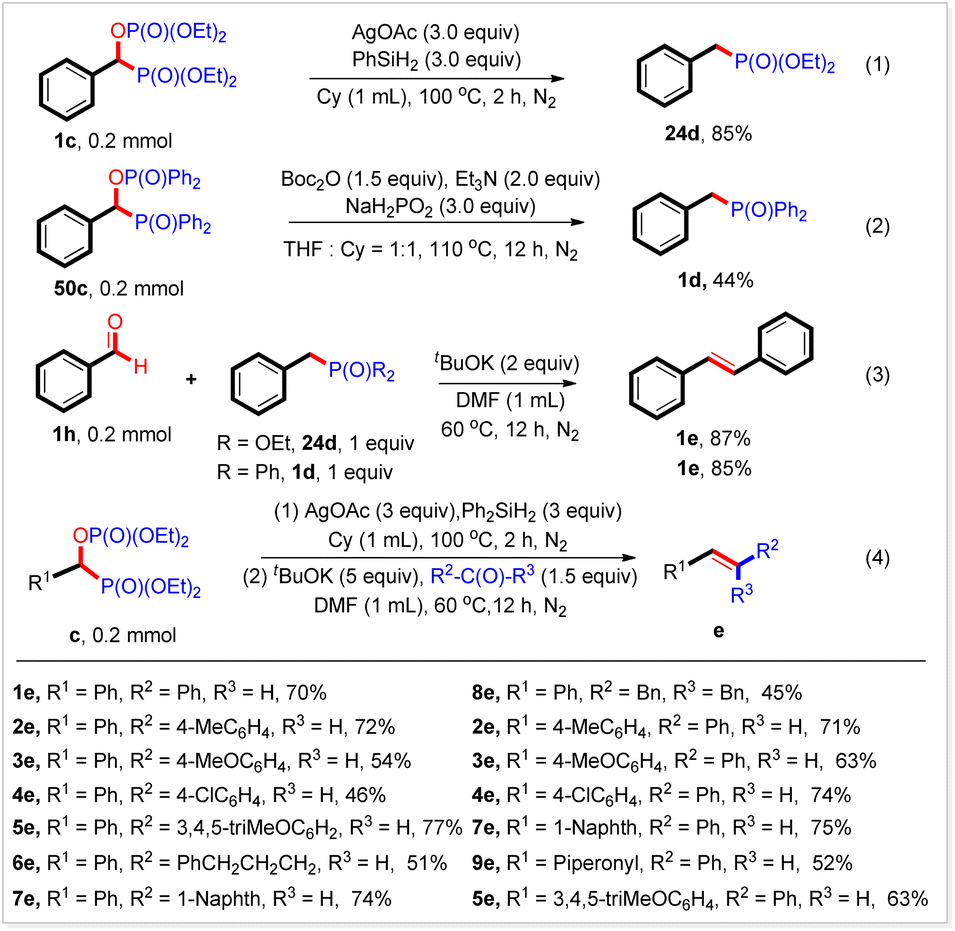 | ||
| Scheme 2 Application of this new transformation. | ||
To probe the mechanism of the reaction, more control experiments were performed. It is known that Boc2O can activate carboxylic acids to form a mixed anhydride. We think anhydride might be the key intermediate in this reaction.21,22 We found that benzoic anhydride could react with diethyl phosphonate to produce 1c in 40% yield (Scheme 3A, eqn (1)). It is deduced that anhydride can react with the P(O)–H compound to give an acyl phosphorus compound, which might also be an intermediate in this reaction. We synthesized acyl phosphorus compound 5g and found that it indeed could couple with diphenyl phosphine oxide to generate 5c in 45% yield (Scheme 3A, eqn (2)). A Hammett analysis was subsequently performed (Scheme 3B). Under the conditions of a bisphosphorylation reaction, a negative slope (ρ = −0.22) was observed for the reaction of substituted aromatic carboxylic acids with diethyl phosphonate. This result indicated that the release of the Boc fragment might be the rate-determining step in the bisphosphorylation reaction.
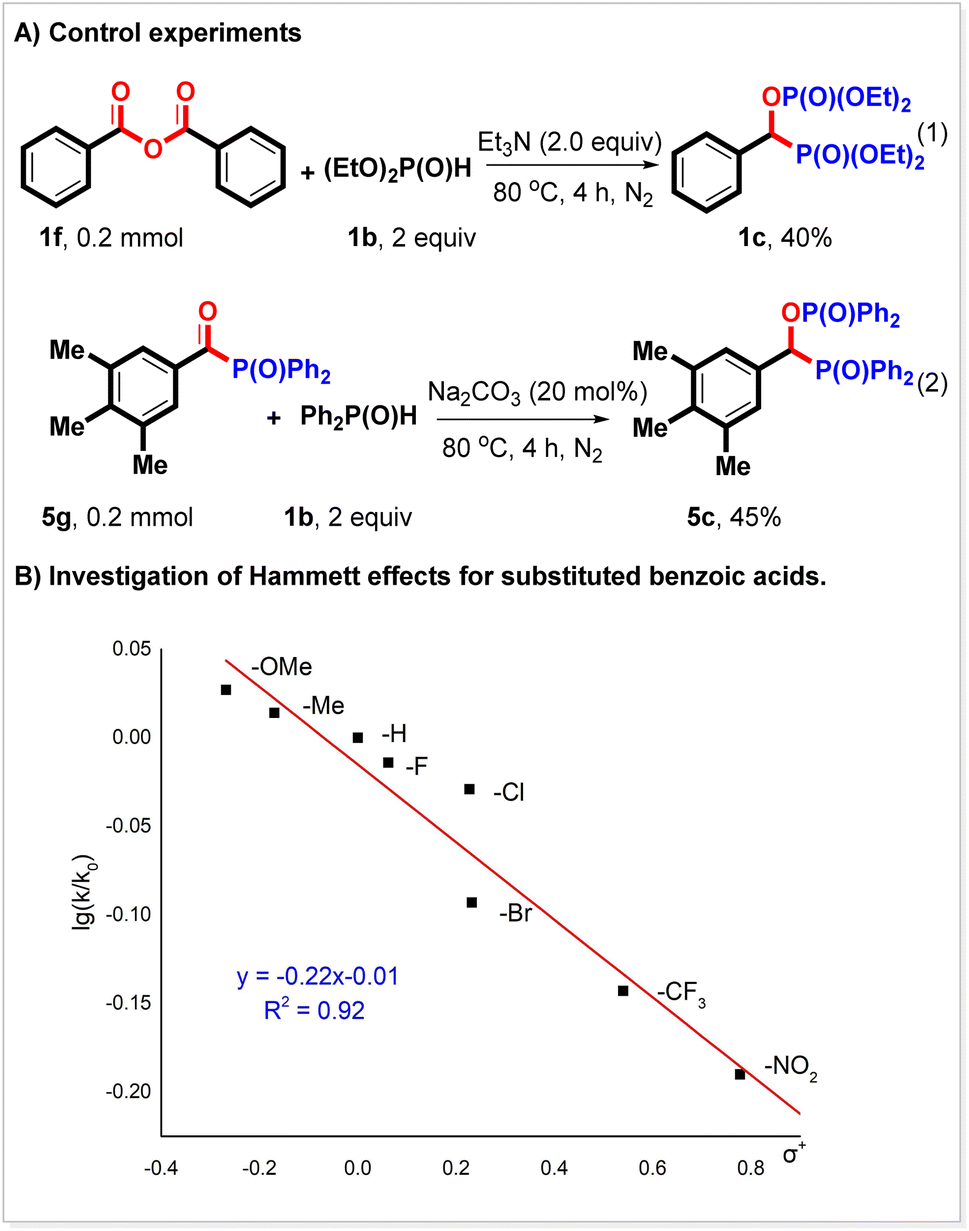 | ||
| Scheme 3 Control experiments and Hammett analysis. | ||
Several inter-molecular competing experiments were conducted to investigate the chemo-selectivity of the bisphosphorylation reaction (Scheme 4A). When benzoic acid 1a was allowed to compete with electron-rich 4-methoxyl benzoic acid 8a to react with diethyl phosphonate 1b under the conditions of the bisphosphorylation reaction, the expected products 1c and 8c were produced in 37% and 65% yield, respectively (Scheme 4A, eqn (1)). Similar results were also observed with the competing reactions between benzoic acid 1a and 4-trifluoromethyl benzoic acid 14a and 4-methoxyl benzoic acid 8a and 4-trifluoromethyl benzoic acid 14a (Scheme 4A, eqn (2) and (3)), indicating that this reaction favored electron-richer carboxylic acids. It seems that the steric hindrance of the carboxylic acid substrate did not affect the yield as described above, but it would reduce the reaction rate. For example, when 2-phenyl benzoic acid 19a competed with 4-phenyl benzoic acid 20a, 20c was produced in 74% yield, while 19c was only obtained in 21% yield (Scheme 4A, eqn (4)). The chemo-selectivity was also studied with a P(O)H substrate. It was found that diphenyl phosphine oxide reacted with benzoic acid more quickly than diethyl phosphonate under the reaction conditions (Scheme 4, eqn (5)). Its reaction rate was also faster than dibutyl phosphine oxide's (Scheme 4A, eqn (6)). However, when diethyl phosphonate competed with dibutyl phosphine oxide in one pot, the expected 1c was generated in 90% yield, while 58c was only given in 4% yield (Scheme 4A, eqn (7)). These results would be ascribed to the stability, and not the nucleophilicity of the conjugated base of P(O)H compounds. The more stable the conjugated base, the faster the rate of this reaction. The steric hindrance of the P(O)H substrate was also investigated and found not to affect the reaction. For instance, similar yields of the corresponding products were obtained with the competing experiment of dibutyl phosphine oxide and dicyclohexyl phosphine oxide (Scheme 4A, eqn (8)). It should be noted that these results herein were consistent with the Hammett analysis described above.
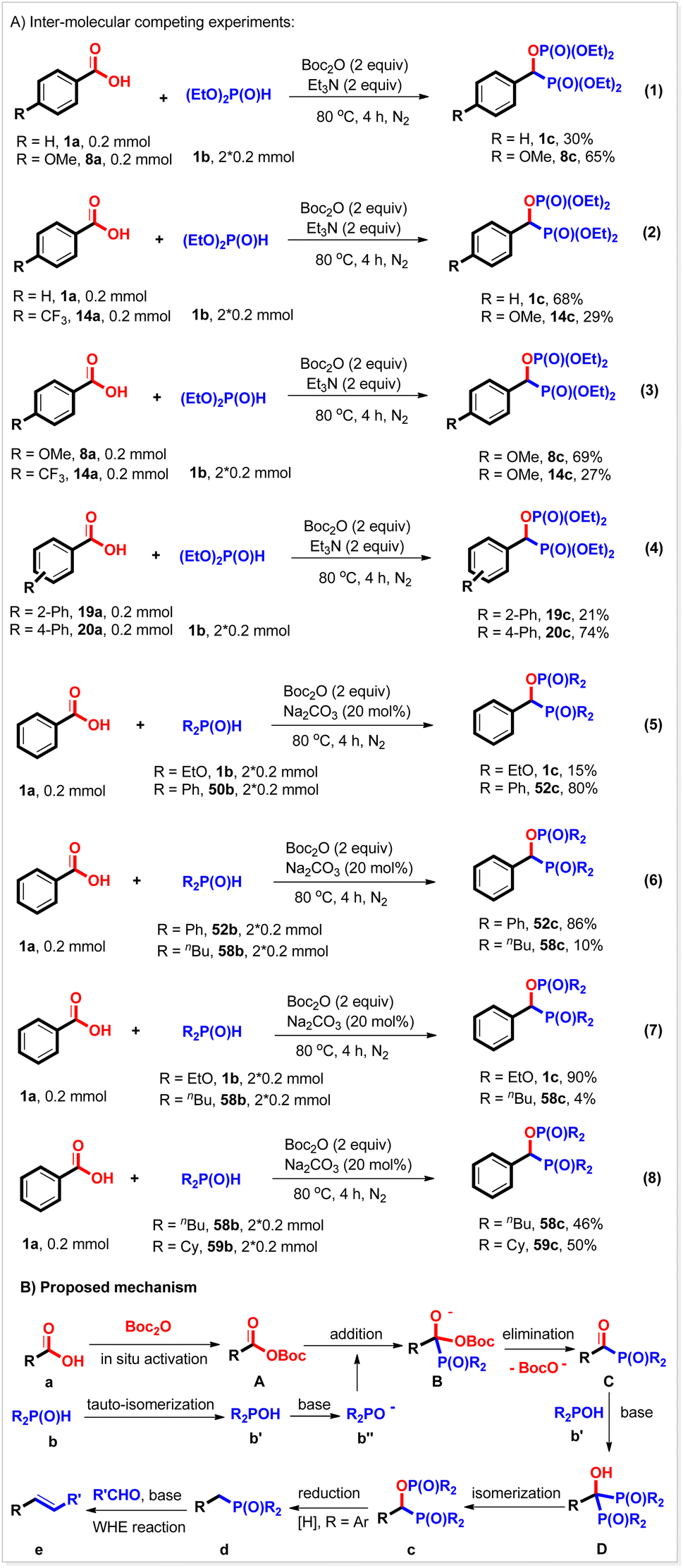 | ||
| Scheme 4 Inter-molecular competing experiments and the proposed mechanism. | ||
On the basis of the mechanistic studies described above and previous references, a probable mechanism was proposed for this reaction. As shown in Scheme 4B, carboxylic acid a was first activated by Boc2O to generate a mixed anhydride A,21,22 which then reacted with hydrogen phosphoryl compound b to give acyl phosphorus compound C and released a Boc fragment. The resulting acyl species C subsequently underwent addition with another hydrogen phosphoryl compound b, followed by [1, 2]-phospha-Brook isomerization23 to produce the target bisphosphorus compound c. When diaryl phosphine oxides were used, product c could be further reduced by the external reductant NaH2PO2 to afford the corresponding benzyl phosphorus compound d and therefore, complete deoxyphosphorylation in one pot. By further addition of aldehydes (ketones) and base, this reaction could efficiently couple with the WHE reaction, thus converting carboxylic acids into value-added alkenes.
Conclusions
In summary, we have achieved both bisphosphorylation and deoxyphosphorylation of carboxylic acids under metal-free and mild reaction conditions, generating the corresponding P–C–O–P motif-containing compounds and benzyl phosphorus compounds selectively. Specifically, this reaction can efficiently couple with the WHE reaction and thus also provides an efficient method for converting carboxylic acids into valuable alkenes. Wide substrate scope for both carboxylic acids and P(O)H compounds was demonstrated with high functional group tolerance. The scale-up experiments and application to modification of complex APIs will also show this new reaction's practicality. This work extends the application of carboxylic acids and provides efficient methods for preparing related organophosphorus compounds and alkenes. We think these new reactions would find wide application in the synthetic community.Author contributions
L. G., T. X., Q. T., M. C., L. W., J. Z. and K. L. did the experiments. L. L. and W. C. discussed the mechanism and revised the manuscript. L. H., J. N. and T. C. designed the experiment project, discussed the mechanism and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. C. thanks the National Nature and Science Foundation of China (Grant No. 22261015 and 21871070) and the Key R&D project of Hainan province (No. ZDYF2020168) for financial support. L. H. thanks Zhejiang Province (2022R01021) and Zhejiang Yangfan New Materials Co. Ltd for financial support.Notes and references
- (a) G.-I. Badea and G. L. Radu, Carboxylic Acid-Key Role in Life Sciences, InTech, 2018 CrossRef; (b) C. Lamberth and J. Dinges, Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals, Wiley-VCH, Weinheim, Germany, 2016 Search PubMed; (c) C. Ballatore, D. M. Huryn and A. B. Smith III, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed; (d) S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed; (e) D. Seidel and C. Min, Chem. Soc. Rev., 2017, 46, 5889–5902 RSC.
- For selected reviews, see: (a) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS PubMed; (b) N. Rodríguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (c) P. Xiao, X. Pannecoucke, J.-P. Bouillon and S. Couve-Bonnaire, Chem. Soc. Rev., 2021, 50, 6094–6151 RSC; (d) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (e) L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef PubMed; (f) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed.
- For selected reviews, see: (a) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed; (b) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671–2678 RSC; (c) Q. Zhao and M. Szostak, ChemSusChem, 2019, 12, 2983–2987 CrossRef CAS PubMed; (d) C. Liu and M. Szostak, Org. Chem. Front., 2022, 9, 216–222 RSC; (e) C. Liu and M. Szostak, ChemCatChem, 2021, 13, 4878–4881 CrossRef CAS PubMed; (f) X. Zhang, F. Jordan and M. Szostak, Org. Chem. Front., 2018, 5, 2515–2521 RSC.
- For selected examples, see: (a) G. Bergonzini, C. Cassani and C. J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed; (b) X. Jiang, F.-T. Sheng, Y. Zhang, G. Deng and S. Zhu, J. Am. Chem. Soc., 2022, 144, 21448–21456 CrossRef CAS PubMed; (c) T. Scattolin, K. Deckers and F. Schoenebeck, Org. Lett., 2017, 19, 5740–5743 CrossRef CAS PubMed; (d) R. G. Kinney and B. A. Arndtsen, Angew. Chem., Int. Ed., 2019, 58, 5085–5089 CrossRef CAS PubMed; (e) E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- For selected examples, see: (a) T. J. Korstanje, V. I. Vlugt, C. J. Elsevier and B. Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed; (b) D. Chen, L. Xu, Y. Yu, Q. Mo, X. Qi and C. Liu, Angew. Chem., Int. Ed., 2022, e202215168 Search PubMed; (c) X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 36, 10596–10599 CrossRef PubMed; (d) M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 9042–9046 CrossRef CAS PubMed.
- (a) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley Interscience, New York, 2000 Search PubMed; (b) Organophosphorus Reagents, ed. P. J. Murphy, Oxford University Press, Oxford, UK, 2004 Search PubMed; (c) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (d) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed; (e) C. Q. ffelec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed.
- (a) S. V. Jeught and C. V. Stevens, Chem. Rev., 2009, 109, 2672–2702 CrossRef PubMed; (b) A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS; (c) A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC; (d) New Aspects in Phosphorus Chemistry, ed. J.-P. Majoral, Springer, Berlin, 2003, vol. 1–5 Search PubMed.
- (a) X. Wang, Y. Hu, L. Song, H. Yang, W. Xing and H. Lu, Prog. Org. Coat., 2011, 71, 72–82 CrossRef CAS; (b) J. Rieusset, F. Touri, L. Michalik, P. Escher, B. Desvergne, E. Niesor and W. Wahli, Mol. Endocrinol., 2022, 16, 2628–2644 CrossRef PubMed; (c) D. Rejman, M. Olesiak, L. Chen, S. E. Patterson, D. Wilson, H. N. Jayaram, L. Hedstrom and K. W. Pankiewicz, J. Med. Chem., 2006, 49, 5018–5022 CrossRef CAS PubMed; (d) W. Li, Y. Niu, D. C. Xiong, X. Cao and X. S. Ye, J. Med. Chem., 2015, 58, 7972–7990 CrossRef CAS PubMed; (e) J. Guo, W. Li, W. Xue and X. S. Ye, J. Med. Chem., 2017, 60, 2135–2141 CrossRef CAS PubMed.
- (a) W. Peng, S. Nie, Y. Xu and W. Yang, Polym. Degrad. Stab., 2021, 193, 109715 CrossRef CAS; (b) J. Wang, M. Qiu, X. Wang and Y. Li, CN 112940034A, 2021; (c) Z. Wang, X. Zhao and H. Ding, WO 2020103529A1, 2020; (d) N. Moszner, Y. Catel and C. Dellsperger, EP 3225228A1, 2017.
- H. Wu, J. Yu, Y. Li, Y. Yang, Q. He, Y. Lou and R. Ji, Acta Pharmacol. Sin., 2007, 28, 417–422 CrossRef CAS PubMed.
- (a) Z. Rádaia, T. Windtb, V. Nagyb, A. Füredib, N. Z. Kissa, I. Ranđelovićd, J. Tóvárid, G. Keglevicha, G. Szakácsb and S. Tóth, New J. Chem., 2019, 43, 14028–14035 RSC; (b) Z. Rádai, V. Hodula, N. Z. Kiss, J. Kótib and G. Keglevich, Mendeleev Commun., 2019, 29, 153–154 CrossRef; (c) N. Z. Kiss, Z. Rádai and G. Keglevich, Phosphorus Sulfur, 2019, 194, 1003–1006 CrossRef CAS.
- (a) M. Isbera, B. Bognár, C. Sár, J. Jekő and T. Kálai, Synth. Commun., 2021, 51, 1353–1362 CrossRef CAS; (b) S. Eymur, M. Gollu and A. S. Demir, Turk. J. Chem., 2014, 38, 164–171 CrossRef CAS; (c) Y. W. Sun, P. L. Zhu, Q. Xu and M. Shi, Tetrahedron, 2012, 68, 9924–9929 CrossRef CAS; (d) A. Grun, I. G. Molńar, B. Greiner, I. Berťok and G. Keglevich, Heteroat. Chem., 2009, 20, 350–354 CrossRef; (e) A. S. Demir, A. Aybey and M. Emrullahoglu, Synthesis, 2009, 10, 1655–1658 CrossRef; (f) D. V. Griffiths, H. A. R. Jamali and J. C. Tebby, Phosphorus Sulfur, 2006, 25, 173–175 CrossRef; (g) M. Sekine, M. Satoh, H. Yamagata and T. Hata, J. Org. Chem., 1980, 45, 4162–4167 CrossRef CAS.
- K. Pachamuthu and R. R. Schmidt, Chem. Commun., 2004, 1078–1079 RSC.
- (a) A. Hosseini, M. A. Khalilzadeh, S. Hallajian and M. A. Tajbakhsh, Phosphorus Sulfur, 2011, 186, 225–232 CrossRef CAS; (b) R. Ruel, J. P. Bouvier and R. N. Young, J. Org. Chem., 1995, 60, 5209–5213 CrossRef CAS; (c) T. Ishihara, T. Maekawa, Y. Yamasaki and T. Ando, J. Fluorine Chem., 1987, 34, 323–335 CrossRef CAS; (d) V. Griffiths, H. A. R. Jamali and J. C. Tebby, Phosphorus Sulfur, 1981, 11, 95–99 CrossRef.
- (a) X. Wang, Y. Hu, L. Song, W. Guo and W. Xing, CN110229190A, 2019; (b) X. Y. Zhang, Q. W. Li, H. Q. Yue, Z. Q. Wu, J. Li, M. Li, L. Lu, S. D. Yang and B. Yang, Chem. Commun., 2022, 58, 6665–6668 RSC.
- (a) W. S. Wadsworth, Org. React., 1977, 25, 73–253 CAS; (b) R. Baker and R. J. A Sims, Synthesis, 1981, 2, 117 CrossRef; (c) J. Petrova, N. G. Vassilev and M. Kirilov, Phosphorus Sulfur, 1989, 47, 457–463 CrossRef; (d) Q. Zhao, L. Yang and Y. Shen, Ind. Eng. Chem. Res., 2016, 55, 7604–7611 CrossRef CAS; (e) Q. Zhao, J. Sun, J. Li and J. He, Catal. Commun., 2013, 36, 98–103 CrossRef CAS; (f) S. C. Dakdouki, D. Villemin and N. Bar, Eur. J. Org. Chem., 2011, 23, 4448–4454 CrossRef.
- (a) Y. Nishiyama, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 5771–5774 RSC; (b) A. Jasiak, G. Mielniczak, K. Owsianik, M. Koprowski, D. Krasowska and J. Drabowicz, J. Org. Chem., 2019, 84, 2619–2625 CrossRef CAS PubMed; (c) E. V. Matveeva, I. L. Odinets, V. A. Kozlov, A. S. Shaplov and T. A. Mastryukova, Tetrahedron Lett., 2006, 47, 7645–7648 CrossRef CAS; (d) J. J. Kiddle and A. F. Gurley, Phosphorus Sulfur, 1999, 160, 195–205 CrossRef; (e) K. D. Berlin, D. H. Burpo, R. U. Pagilagan and D. A Bude, Chem. Commun., 1967, 1060–1061 RSC.
- (a) G. Lavén and J. Stawinski, Synlett, 2009, 2, 225–228 Search PubMed; (b) M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed; (c) K. Xu, L. Liu, Z. Li, T. Huang, K. Xiang and T. Chen, J. Org. Chem., 2020, 85, 14653–14663 CrossRef CAS PubMed.
- It should be noted that in Prof. Yamaguchi's work on the palladium-catalyzed deoxyphosphorylation of aromatic esters, two examples converting carboxylic acids into benzyl phosphorus compounds with the use of Boc2O as an in situ activator and HCOONa as an reductant were reported. It now appears that palladium catalysts might be unnecessary. For details, see Ref. 18b.
- For selected examples using NaH2PO2 and its derivatives as the reductant, see: (a) C. Guyon, E. Métay, F. Popowycz and M. Lemairea, Org. Biomol. Chem., 2015, 13, 7879–7906 RSC; (b) G. G. Wu, F. X. Chen, D. LaFrance, Z. Liu, S. G. Greene, Y.-S. Wong and J. Xie, Org. Lett., 2011, 13(19), 5220–5223 CrossRef CAS PubMed; (c) J. E. Milne, T. Storz, J. T. Colyer, O. R. Thiel, M. D. Seran, R. D. Larsen and J. A. Murry, J. Org. Chem., 2011, 76(22), 9519–9524 CrossRef CAS PubMed; (d) C. McMaster, R. N. Breamb and R. S. Grainger, Org. Biomol. Chem., 2012, 10, 4752–4758 RSC; (e) S. R. Graham, J. A. Murphy and D. Coates, Tetrahedron Lett., 1999, 40, 2415–2416 CrossRef CAS; (f) T. Chen, J. Xiao, Y. Zhou, S. Yin and L.-B. Han, J. Organomet. Chem., 2014, 749, 51–54 CrossRef CAS.
- For selected examples, see: (a) K. Nagayama, I. Shimizu and A. Yamamoto, Chem. Lett., 1998, 27, 1143–1144 CrossRef; (b) C. Liu, C. L. Ji, X. Hong and M. Szostak, Angew. Chem., Int. Ed., 2018, 57, 16721–16726 CrossRef CAS PubMed; (c) F. Pan, Z. Lei, H. Wang, H. Li, J. Sun and Z. Shi, Angew. Chem., Int. Ed., 2013, 52, 2063–2067 CrossRef CAS PubMed; (d) B. Feng, G. Zhang, X. Feng and Y. Chen, Org. Chem. Front., 2022, 9, 1085–1089 RSC; (e) C. Liu, C. Ji, Z. Qin, X. Hong and M. Szostak, iScience, 2019, 19, 749–759 CrossRef CAS PubMed; (f) C. Liu, Z. Qin, C. Ji, X. Hong and M. Szostak, Chem. Sci., 2019, 10, 5736–5742 RSC; (g) H. Zhao, X. Xu, H. Yu, B. Li, X. Xu, H. Li, L. Xu and Q. Fan, Org. Lett., 2020, 22, 4228–4234 CrossRef CAS PubMed; (h) C. Liu, C.-L. Ji, T. Zhou, X. Hong and M. Szostak, Angew. Chem., Int. Ed., 2021, 60, 10690–10699 CrossRef CAS PubMed.
- (a) X. Li, L. Liu, T. Huang, Z. Tang, C. Li, W. Li, T. Zhang, Z. Li and T. Chen, Org. Lett., 2021, 23, 3304–3309 CrossRef CAS PubMed; (b) K. Xiang, S. Zhang, L. Liu, T. Huang, Z. Tang, C. Li, K. Xu and T. Chen, Org. Chem. Front., 2021, 8, 2543–2550 RSC; (c) T. Xu, X. Zhou, X. Xiao, Y. Yuan, L. Liu, T. Huang, C. Li, Z. Tang and T. Chen, J. Org. Chem., 2022, 87, 8672–8684 CrossRef CAS PubMed; (d) W. Yu, L. Liu, T. Huang, X. Zhou and T. Chen, Org. Lett., 2020, 22, 7123–7128 CrossRef CAS PubMed; (e) T. Xu, W. Li, K. Zhang, Y. Han, L. Liu, T. Huang, C. Li, Z. Tang and T. Chen, J. Org. Chem., 2022, 87, 11871–11879 CrossRef CAS PubMed; (f) J. Zhang, T. Chen and L.-B. Han, Eur. J. Org. Chem., 2020, 2020, 1148–1153 CrossRef CAS.
- For selected examples, on [1,2]-phospha-Brook isomerization, see: (a) A. Kondoh, R. Ojima and M. Terada, Org. Lett., 2021, 23, 7894–7899 CrossRef CAS PubMed; (b) A. Kondoh, T. Aoki and M. Terada, Chem.–Eur. J., 2017, 23, 2769–2773 CrossRef CAS PubMed; (c) M. A. Horwitz, N. Tanaka, T. Yokosaka, D. Uraguchi, J. S. Johnson and T. Ooi, Chem. Sci., 2015, 6, 6086–6090 RSC; (d) A. Kondoh, T. Aoki and M. Terada, Org. Lett., 2014, 16, 3528–3531 CrossRef CAS PubMed; (e) C. Cheibas, N. N. Fincias, J. Garrec and L. E. Kaïm, Angew. Chem., Int. Ed., 2022, 61, e202116249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures, copies of 1H, 13C and 31P NMR spectra for products. See DOI: https://doi.org/10.1039/d3sc01148H |
| This journal is © The Royal Society of Chemistry 2023 |