
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe promotional role of Mn in CO2 hydrogenation over Rh-based catalysts from a surface organometallic chemistry approach†
Wei
Zhou
,
Scott R.
Docherty
,
Christian
Ehinger
 ,
Xiaoyu
Zhou
and
Christophe
Copéret
*
,
Xiaoyu
Zhou
and
Christophe
Copéret
*
Department of Chemistry and Applied Bioscience, ETH Zürich, Vladimir Prelog Weg2, CH-8093 Zürich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch
First published on 18th April 2023
Abstract
Rh-based catalysts modified by transition metals have been intensively studied for CO2 hydrogenation due to their high activity. However, understanding the role of promoters at the molecular level remains challenging due to the ill-defined structure of heterogeneous catalysts. Here, we constructed well-defined RhMn@SiO2 and Rh@SiO2 model catalysts via surface organometallic chemistry combined with thermolytic molecular precursor (SOMC/TMP) approach to rationalize the promotional effect of Mn in CO2 hydrogenation. We show that the addition of Mn shifts the products from almost pure CH4 to a mixture of methane and oxygenates (CO, CH3OH, and CH3CH2OH) upon going from Rh@SiO2 to RhMn@SiO2. In situ X-ray absorption spectroscopy (XAS) confirms that the MnII is atomically dispersed in the vicinity of metallic Rh nanoparticles and enables to induce the oxidation of Rh to form the Mn–O–Rh interface under reaction conditions. The formed interface is proposed to be key to maintaining Rh+ sites, which is related to suppressing the methanation reaction and stabilizing the formate species as evidenced by in situ DRIFTS to promote the formation of CO and alcohols.
Introduction
The continuous increase in the concentration of atmospheric CO2 has been raising serious concerns due to its visible environmental impact associated with the greenhouse effect and ocean acidification.1,2 CO2 hydrogenation using renewable H2 represents one of the most promising strategies to mitigate CO2 emission and realize carbon neutrality.3,4 Among numerous catalysts employed for CO2 hydrogenation, rhodium-based catalysts have drawn continued interest due to their high activity.5,6 In general, methanation dominates over Rh-based catalysts, yielding methane as the main product. However, to steer product selectivity towards oxygenates, promoters such as Mn and Fe have been intensively investigated.7–11 However, understanding the role of these promoters at the molecular level has been challenging due to the ill-defined structure of these catalysts. To address the complex structure of heterogeneous catalysts, surface organometallic chemistry combined with thermolytic molecular precursors (SOMC/TMP) has emerged as a promising approach to construct well-defined model catalysts with tailored composition and interfaces.12,13 This approach results in materials that are more amenable to molecular-level spectroscopic characterization, hence they can be used to bridge the gap in understanding between complex heterogeneous systems and model catalysts, by enabling the development of detailed structure–activity relationships.Our group has successfully applied this approach to various heterogeneous reactions such as the selective oxidation of methane14 and propane dehydrogenation15,16 as well as the selective CO2 hydrogenation to methanol.17 For instance, a series of Cu-based catalysts doped with various metals (Cu/M@SiO2, M = Ti, Zr, Hf, Nb, Ta, Zn, Ga) were synthesized via the SOMC/TMP approach.18–23 Metal nanoparticles with a narrow particle size distribution, similar metal dispersion, tailored interfaces, and compositions enabled the investigation of the influence of discrete physicochemical properties (Lewis acidity and alloying) on CO2 hydrogenation. These studies have shown that the presence of Lewis acid sites, originating from early transition-metal metal dopants, boosts the CH3OH formation rate via the stabilization of formate and methoxy intermediates.21 For the Cu–Ga@SiO2 and Cu–Zn@SiO2 model catalysts, alloy phases have been observed upon the reduction treatment.19,20 The oxidation of Zn or Ga to form small oxide domains is however observed from these alloys under reaction conditions, thus maximizing the interface with metallic Cu. The high methanol selectivity in a wider range of CO2 conversion is probably associated with such a dynamic process. Overall, the SOMC approach has opened new avenues to shed light on complex systems and to develop detailed structure–activity relationships, that can be transferred to other heterogeneous catalysts.
With this in mind, we synthesized well-defined RhMn@SiO2 and Rh@SiO2 catalysts using the SOMC/TMP approach to elucidate the promotional role of Mn in CO2 hydrogenation. We found that the presence of Mn significantly suppresses methanation, while the product distribution shifts from almost pure CH4 to a mixture of CH4, CO, and CH3OH along with small amounts of CH3CH2OH upon going from Rh@SiO2 to RhMn@SiO2. Detailed in situ studies show that the isolated MnII in the vicinity of Rh nanoparticles is key to suppressing methanation by inducing the oxidation of Rh.
Results and discussion
Catalyst preparation and characterization
We first developed a Rh precursor, amenable for grafting with easy removal of organic ligands during the nanoparticle formation, namely (N,N′-diisopropylacetamidinate)(1,5-cyclooctadiene)rhodium, abbreviated as Rh(COD)(DIA). This compound is prepared by reacting Li(DIA)(THF) (Fig. S1–S3†) with chloro(1,5-cyclooctadiene)rhodium(I) to produce Rh(COD)(DIA) in high yield (90%) (Fig. S4–S7†). Rh(COD)(DIA) reacts with SiO2-700 (silica dehydroxylated at 700 °C) as evidenced by solution NMR, with complete consumption of the molecular precursor upon contacting with SiO2-700 (Fig. S8†). IR spectroscopy of the resulting materials shows the disappearance of Si–OH at 3747 cm−1 and the concomitant appearance of C–H stretching (3100–2700 cm−1) and bending (1500–1300 cm−1) bands, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching bands (1600–1700 cm−1), and N–H stretching band from secondary amine (a low-intensity peak at ca. 3250 cm−1) (Fig. S9†). These results confirm that Rh(COD)(DIA) was grafted on SiO2via protonolysis with the surface silanols to generate Rh(COD)(DIA)@SiO2 (Fig. S10†). Furthermore, 13C cross-polarization magic-angle spinning (CP-MAS) NMR displays signals at 163, 72, 45, 30, 22, and 12 ppm, consistent with the grafting of the molecular precursor (Fig. S11†). Notably the signal for C-sp2 carbon of amidinate shifts from 179 ppm to 163 ppm after grafting due to the cleavage of the Rh–N bond via protonolysis. Reduction at 400 °C under a H2 atmosphere removes all the ligands according to IR spectroscopy (Fig. S9†). The resulting dark solid (denoted as Rh@SiO2) suggests particle formation; transmission electron microscopy (TEM) analysis of Rh@SiO2 shows a narrow distribution of Rh nanoparticles centered at 3.3 nm (Fig. S12†).
C and CN stretching bands (1600–1700 cm−1), and N–H stretching band from secondary amine (a low-intensity peak at ca. 3250 cm−1) (Fig. S9†). These results confirm that Rh(COD)(DIA) was grafted on SiO2via protonolysis with the surface silanols to generate Rh(COD)(DIA)@SiO2 (Fig. S10†). Furthermore, 13C cross-polarization magic-angle spinning (CP-MAS) NMR displays signals at 163, 72, 45, 30, 22, and 12 ppm, consistent with the grafting of the molecular precursor (Fig. S11†). Notably the signal for C-sp2 carbon of amidinate shifts from 179 ppm to 163 ppm after grafting due to the cleavage of the Rh–N bond via protonolysis. Reduction at 400 °C under a H2 atmosphere removes all the ligands according to IR spectroscopy (Fig. S9†). The resulting dark solid (denoted as Rh@SiO2) suggests particle formation; transmission electron microscopy (TEM) analysis of Rh@SiO2 shows a narrow distribution of Rh nanoparticles centered at 3.3 nm (Fig. S12†).
Based on these encouraging results, the corresponding RhMn bimetallic catalyst was prepared in a two-step SOMC/TMP process, involving first the generation of the well-defined isolated MnII sites dispersed on silica prepared via the combined SOMC/TMP approach.24 This material, denoted as MnII@SiO2 (1.45 wt% Mn, 0.79 Mn/nm2), was first synthesized by grafting the [Mn(OSi(OtBu)3)2]2 on SiO2-700, followed by thermal treatment at 400 °C under vacuum (10−5 mbar, Fig. S13†). The presence of –OH in MnII@SiO2 allows the introduction of Rh through a second grafting step using Rh(COD)(DIA) (Fig. 1a). IR spectroscopy was used to monitor the grafting process (Fig. 1b): grafting of Rh(COD)(DIA) on MnII@SiO2 shows a decrease in intensity of the surface silanols and the appearance of similar IR bands as observed during grafting on silica (vide supra). Upon treatment under H2 at 400 °C, all organic ligands are removed and the surface silanols are regenerated (Fig. 1b). This material is henceforth denoted as RhMn@SiO2. TEM shows the formation of significantly smaller nanoparticles for RhMn@SiO2 centered at 1.9 nm (Fig. 1c) when compared to monometallic Rh@SiO2, which already suggests a substantial influence of Mn on the nanoparticle structure.
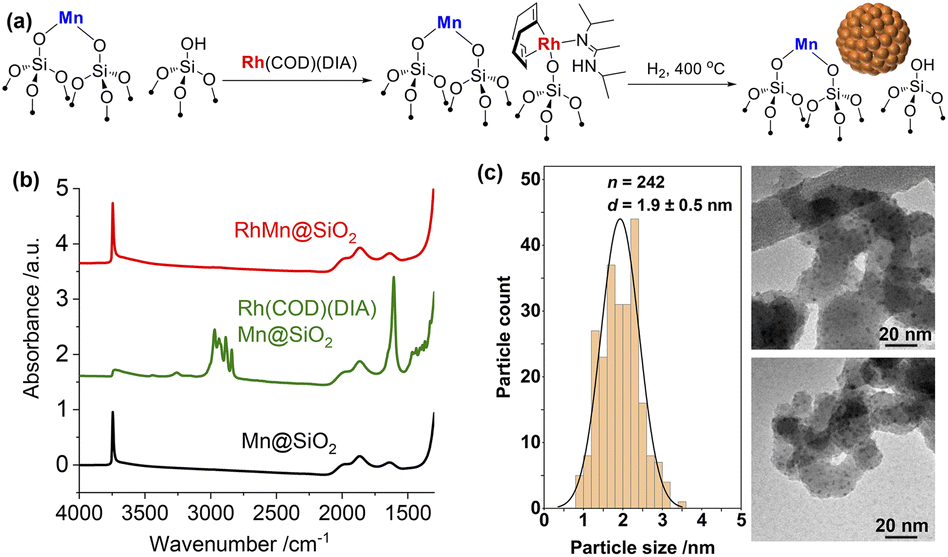 | ||
| Fig. 1 (a) Schematic procedure for grafting of Rh(COD)(DIA) on MnII@SiO2 followed by reduction under H2 at 400 °C. (b) IR spectra throughout the synthesis of RhMn@SiO2 starting from the second grafting. (c) Particle size distribution and TEM images of RhMn@SiO2. | ||
The metal loadings in RhMn@SiO2 and Rh@SiO2, analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES), are similar with ca. 2.3 wt% Rh. The RhMn@SiO2 also contains 1.45 wt% Mn, which corresponds to a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of Rh:Mn (Table S1†). We also carried out H2 and CO chemisorption to better characterize the surface state of Rh (Table S1, Fig. S14 and S15†). The H2 and CO uptake found RhMn@SiO2 are both higher than that for Rh@SiO2, implying that the Rh nanoparticles are smaller in the presence of the Mn promoter. Note that H2/CO chemisorption is not effective for estimating the metal dispersion in the case of Rh due to the multiple adsorption of hydrogen and the complicated CO adsorption modes.25,26
:1 molar ratio of Rh:Mn (Table S1†). We also carried out H2 and CO chemisorption to better characterize the surface state of Rh (Table S1, Fig. S14 and S15†). The H2 and CO uptake found RhMn@SiO2 are both higher than that for Rh@SiO2, implying that the Rh nanoparticles are smaller in the presence of the Mn promoter. Note that H2/CO chemisorption is not effective for estimating the metal dispersion in the case of Rh due to the multiple adsorption of hydrogen and the complicated CO adsorption modes.25,26
Furthermore, FTIR spectra of adsorbed CO under different CO pressures reveal that the adsorption of CO on both Rh@SiO2 and RhMn@SiO2 is irreversible, indicating a strong CO adsorption, consistent with previous reports (Fig. S16†).26 In addition, in both cases, the IR bands are red-shifted by ca. 10 cm−1 after the evacuation likely indicating a decrease in dipole–dipole interaction at lower CO coverage.26 At a CO pressure of 2.4 mbar, two bands at around 2070 and 1890 cm−1 are observed upon subtraction of the background for both materials; they are attributed to linearly bound CO and bridged CO adsorbed on metallic Rh, respectively (Fig. S17†).27,28 Note that bands at 2080 and 2010 cm−1, commonly attributed to the geminal dicarbonyl on Rh+ sites (Rh-(CO)2), are not observed, indicating that the grafted RhI species are completely reduced to Rh(0) metal after H2 treatment.29,30
Catalyst evaluation for CO2 hydrogenation
The catalytic performances of Rh@SiO2 and RhMn@SiO2 were tested at 230 °C under 40 bar (H2/CO2/Ar = 3:1:1). Following exposure to air, the catalyst was reduced at 400 °C under H2 (1 bar, 50 mL min−1) prior to CO2 hydrogenation. The CO2 conversion was maintained below 10% for kinetic consideration. Under these conditions, CH4 is the main product for Rh@SiO2 along with a small fraction of CO across a wide range of CO2 conversion (Fig. 2a and S18†), consistent with earlier reports on monometallic Rh catalysts during COx (CO and CO2) hydrogenation in the absence of promoter.6,31,32 Upon the introduction of Mn as a promoter, the product selectivity significantly shifts to a mixture of CH4, CO, CH3OH, and CH3CH2OH (Fig. 2b and S19†). Note that Mn@SiO2 was also tested under identical conditions and showed no activity in CO2 hydrogenation. This rules out that the products such as CO and CH3OH are formed directly over the Mn promoter.
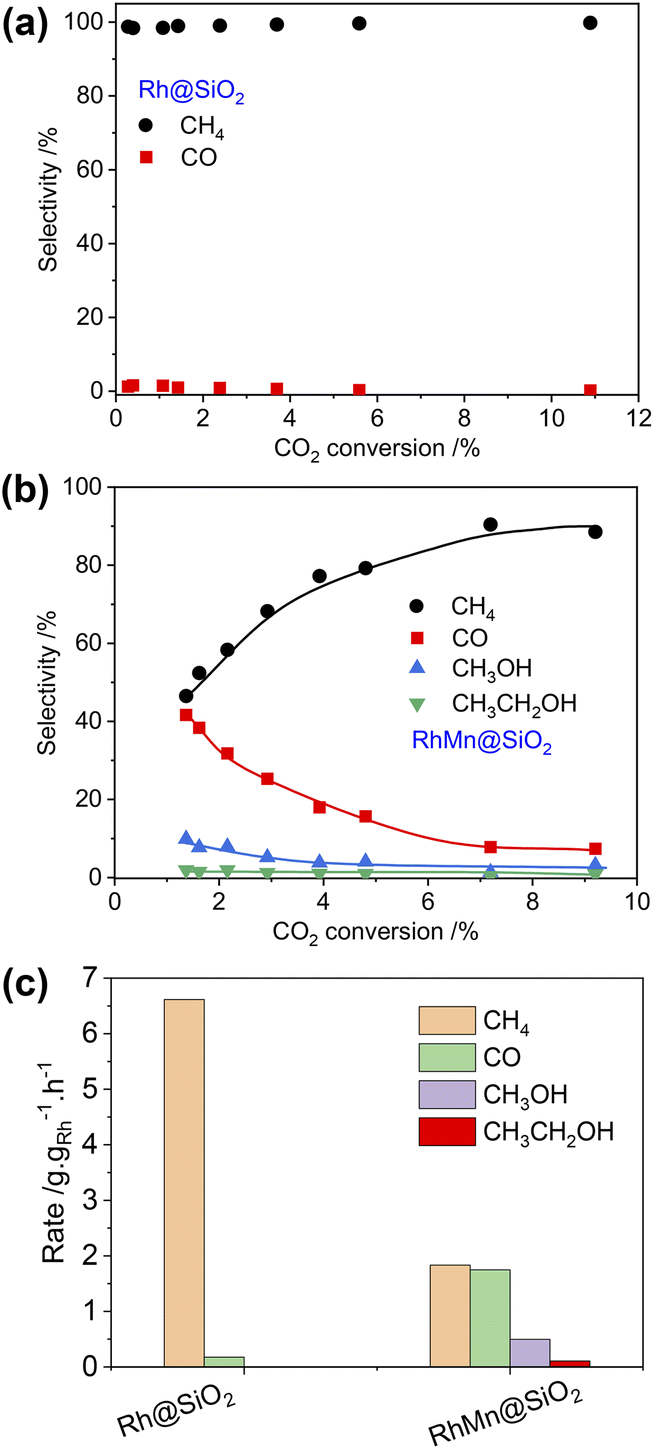 | ||
| Fig. 2 Product selectivity vs. CO2 conversion for Rh@SiO2 (a) and RhMn@SiO2 (b). Reaction conditions: 30 mg for Rh@SiO2, 125 mg for RhMn@SiO2, F = 6–100 mL min−1, T = 230 °C, P = 40 bar. (c) Product formation rate on Rh@SiO2 and RhMn@SiO2 at a flow rate of 50 mL min−1. | ||
In addition, the CO selectivity and methanol selectivity on the RhMn@SiO2 catalyst gradually decrease with the increase in CO2 conversion, while the CH4 selectivity shows the opposite trend. This indicates that CO and CH3OH are further hydrogenated to CH4 with increased contact time. When the flow rate was set at 50 mL min−1, we compared the formation rate of products of Rh@SiO2 and RhMn@SiO2. As shown in Fig. 2c, the formation rate of CH4 on Rh@SiO2 (6.62 g gRh−1 h−1) is higher than that of RhMn@SiO2 (1.83 g gRh−1 h−1) by a factor of around 3.5. Moreover, the formation rate of CO, CH3OH, and CH3CH2OH on RhMn@SiO2 is 1.75 g gRh−1 h−1, 0.50 g gRh−1 h−1 and 0.10 g gRh−1 h−1 respectively. Therefore, these catalytic results clearly show that the introduction of the Mn promoter significantly affects the methanation reaction, while promoting the formation of CO, CH3OH, and CH3CH2OH.
In situ X-ray absorption spectroscopy
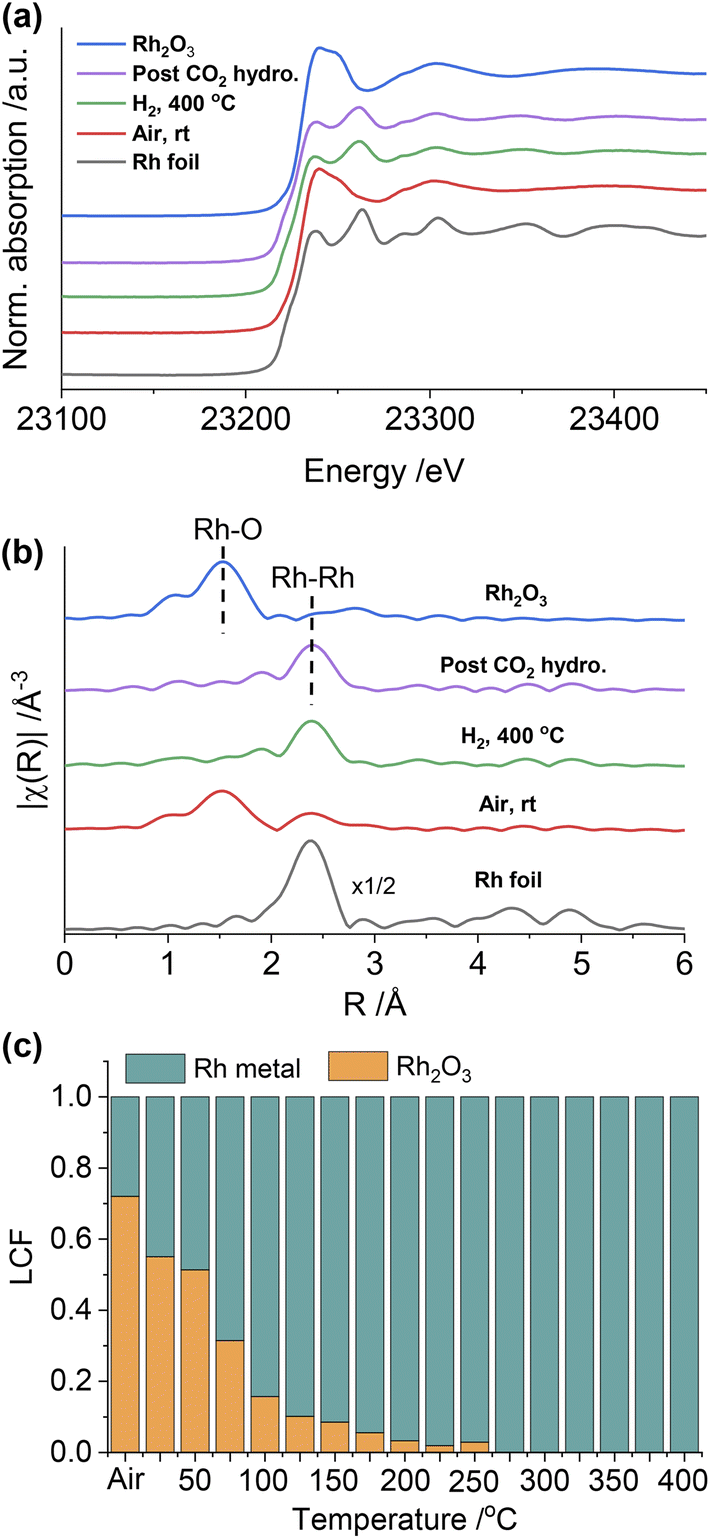
To gain further insight into the structure of the catalysts under different conditions, in situ XAS was performed on both Rh@SiO2 and RhMn@SiO2, at both the Rh and Mn K-edges (Fig. S20†). The catalyst structures of the material as prepared (exposed to air), reduced and post-CO2 hydrogenation were studied by extended X-ray absorption fine structure (EXAFS), while any changes in the chemical state in the duration of H2 reduction and CO2 hydrogenation were studied by X-ray absorption near edge structure (XANES). We first investigated the evolution of the structure of Rh@SiO2 and RhMn@SiO2 catalysts under different conditions from the Rh K-edge (Fig. 3 and 4). The corresponding best-fit details are shown in Fig. S21–S27, Tables S2 and S3.† Compared to the XANES spectra of Rh and Rh2O3 references, the XANES spectra of Rh@SiO2 after exposure to air closely match that of metallic Rh (Fig. 3a). Furthermore, only one peak at 2.4 Å corresponding to the contribution of Rh–Rh scattering path appears in the Fourier transforms of EXAFS spectra combined with the fitting results (Fig. 3b, and S22–S24†). These results indicate that Rh in the as-synthesized Rh@SiO2 catalyst is exclusively present in a metallic state, even upon exposure to air. After H2 reduction, the average coordination number (CN) in the first Rh–Rh shell increases from 8.9 to 10.1 (Table S3†), suggesting that the size of Rh particles grows in the reduced sample. Of interest, the CN was reduced to 8.7 after the CO2 hydrogenation, indicating that a re-dispersion of Rh nanoparticles may occur during the reaction. This is consistent with earlier reports that show that CO species formed from CO2 hydrogenation can generate carbonyl species and induce the re-dispersion of Rh particles.33 Therefore, we consider that the same re-dispersion of Rh particles likely occurs on Rh@SiO2 due to the formation of Rh carbonyl species during the reaction.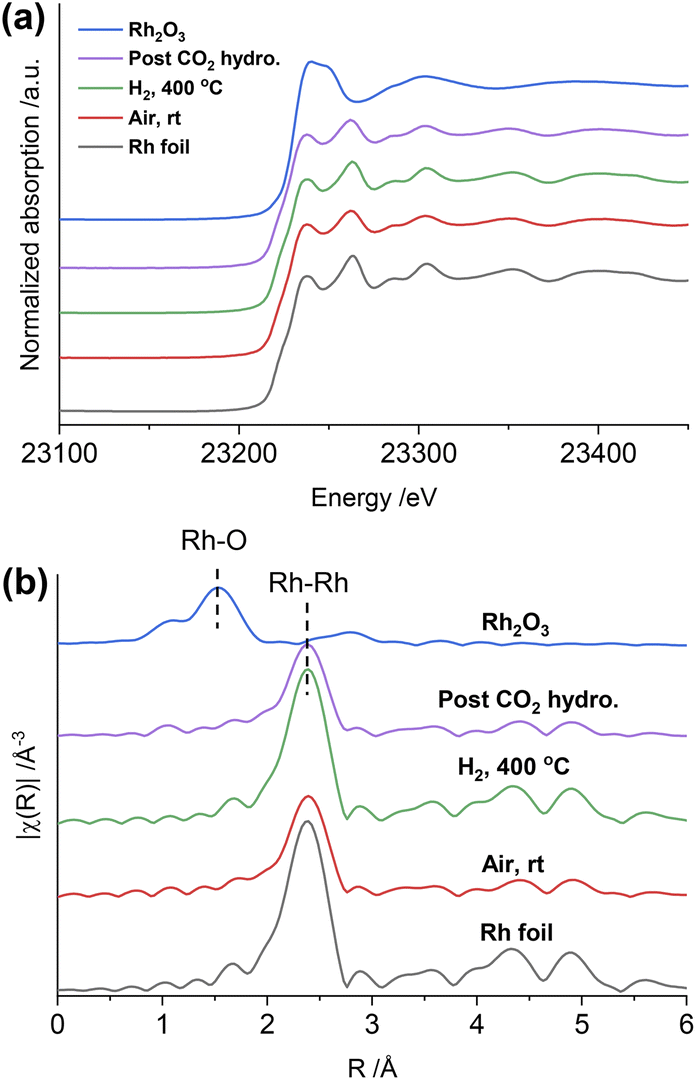 | ||
| Fig. 3 In situ Rh K-edge XANES spectra (a) and the k2-weighted Fourier transforms of EXAFS spectra (b) of the Rh@SiO2 catalyst under different conditions and reference samples. | ||
 | ||
| Fig. 4 In situ Rh K-edge XANES spectra (a) and the k2-weighted Fourier transforms of EXAFS spectra (b) of the RhMn@SiO2 catalyst under different conditions and reference samples. (c) Results of linear combination fitting (LCF) of Rh K-edge XANES. | ||
Regarding RhMn@SiO2, the edge position and white line intensity of air-exposed XANES spectra are more similar to that of the Rh2O3 reference, indicating that Rh is oxidized in such materials (Fig. 4a). The main peak in the Fourier transform of EXAFS spectra is at 1.5 Å, attributed to the contribution of the Rh–O scattering path (Fig. 4b). The EXAFS spectra were modeled using Rh–O (2.03 Å) and Rh–Rh (2.68 Å) scattering paths, showing a CN of 4.5 and 2.2 for Rh–O and Rh–Rh, respectively (Table S3, Fig. S25†). Therefore, the air-exposed RhMn@SiO2 mainly contains oxidized Rh species with a small amount of metallic Rh. After H2 reduction at 400 °C, the edge position shifts to lower energy, the white line intensity decreases, and the characteristic peak at 23262 eV belonging to metallic Rh appears. In addition, the peak at 1.5 Å completely disappears while the intensity of the peak at 2.4 Å increases markedly. No Rh–O and Rh–Mn features were observed in either XANES and Fourier transforms of EXAFS spectra, which indicates that the oxidized Rh was fully reduced to metallic Rh with no alloy formation. No significant change in the CN of the RhMn@SiO2 catalyst is observed post-CO2 hydrogenation compared to that of the reduced RhMn@SiO2. Notably, the CN of reduced RhMn@SiO2 (6.4) is less than that found in reduced Rh@SiO2 (10.1) consistent with the presence of smaller metal particles, resulting in lower average CN.34 This is consistent with the presence of smaller particle size in RhMn@SiO2 than in Rh@SiO2 as observed by TEM. Compared to the pure Rh@SiO2 the chemical state of Rh in air-exposed RhMn@SiO2 is quite distinct, suggesting that the oxidation of Rh is induced by the presence of Mn.
As described above, the chemical state of Rh in the RhMn@SiO2 sample was dramatically changed after H2 reduction. Therefore, the XANES spectra were further collected to monitor the subtle changes during H2 temperature programmed reduction (H2-TPR) (Fig. S28†). The XANES spectra were analyzed by linear combination fitting (LCF). To minimize the influence of particle size, we employ Rh@SiO2 as the standard Rh metal spectra (Fig. 4c and S29†). The air-exposed RhMn@SiO2 sample contains approximately a 70:30 mixture of Rh2O3 and Rh metal phases. When the temperature is increased to 200 °C, the spectra match well with the Rh metal spectra, and there are no obvious changes on further increasing the temperature. Therefore, the Rh oxide in RhMn@SiO2 can be completely reduced at around 200 °C. In contrast, there is no chemical state change observed for Rh in the Rh@SiO2 sample during the H2-TPR (Fig. S30†).
We also collected the in situ Mn K-edge XAS spectra under identical conditions. As shown in Fig. 5a, compared to the MnII@SiO2, the edge position of air-exposed RhMn@SiO2 shifts to a higher position, suggesting that the pristine MnII is oxidized upon exposure to air. We further quantify the average oxidation state of the Mn by establishing a calibration curve describing the relationship between the oxidation state and the difference in edge energy (Fig. 5b and S31†).35 The evaluated average oxidation state of Mn in air-exposed RhMn@SiO2 is +2.6. Due to the proximity between Mn sites and Rh nanoparticles, we speculate that the oxidation of Mn enables to induce the oxidation of Rh (confirmed from the Rh K-edge spectra), probably resulting in the formation of Mn–O–Rh interfaces. After the H2 reduction at 400 °C, the edge position shifts back to lower energy, which is similar to that of the MnII@SiO2 standard, indicating that the oxidized Mn is reduced back to MnII. The Fourier transforms of Mn K-edge EXAFS spectra were analyzed, and references (Mn@SiO2, MnO, and Mn foil) are employed for comparison (Fig. S32†). We can see that the peak of RhMn@SiO2 post-H2 reduction or CO2 hydrogenation is similar to that of Mn@SiO2-air free, and there is no Mn–Mn peak observed. It is suggested that Mn in RhMn@SiO2 catalyst remains atomically dispersed on SiO2, while no sintering or alloying occurs during the H2 reduction and CO2 hydrogenation.
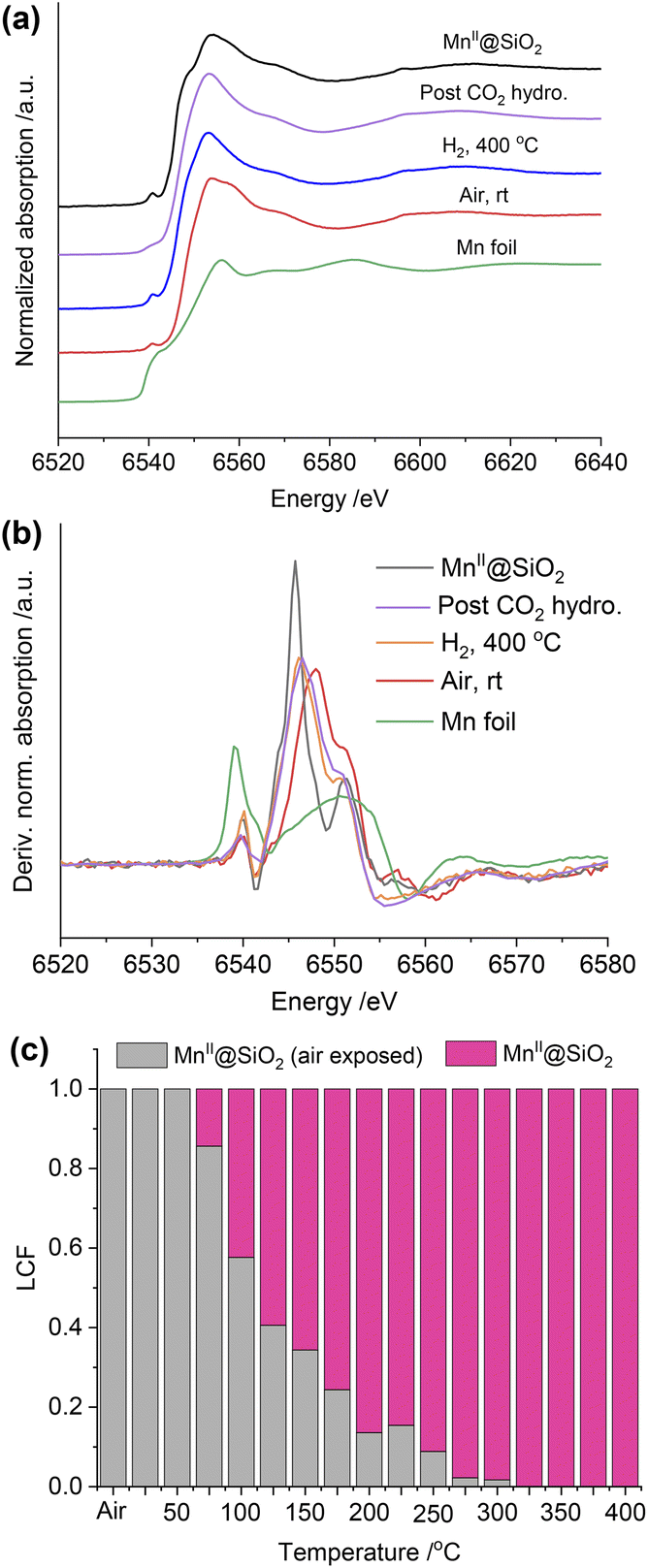 | ||
| Fig. 5 (a) In situ Mn K-edge XANES spectra of RhMn@SiO2 under different conditions and reference. (b) The first derivative of the EXAFS spectra at the Mn K-edge of RhMn@SiO2 under different conditions and reference samples. (c) Results of linear combination fitting (LCF) of Mn K-edge XANES. | ||
The Mn K-edge XANES spectra were also recorded to monitor the chemical changes of Mn during the H2 temperature programmed reduction (Fig. S33†). Because Mn in RhMn@SiO2 is mostly isolated (vide supra), we employed the air-free and air-exposed MnIISiO2 samples as the references for the linear combination fitting, which enables to obtain a good fitting (Fig. S34†). As shown in Fig. 5c, the oxidized Mn starts to resemble MnII@SiO2 at around 75 °C and was completely transformed to MnII at around 300 °C. The reduced temperature is about 100 °C higher than that of Rh (200 °C). This is because the oxygen affinity of Mn is higher than that of Rh, making Mn more difficult to reduce.
Upon exposing the catalyst to CO2 hydrogenation reaction conditions at 20 bar (H2/CO2/Ar = 1:3:1), we observed the alterations in both Mn K-edge and Rh K-edge XANES spectra with time on stream (Fig. S35† and 6a and b). The edge position gradually shifts to higher energy for both Mn K-edge and Rh K-edge XANES spectra during CO2 hydrogenation, with slight changes in the white line intensity of both (Fig. S35†). These transformations are consistent with the partial oxidation of Mn and Rh under CO2 hydrogenation conditions. However, we did not observe any changes when comparing the XANES spectra before and post-CO2 hydrogenation for the pure Rh@SiO2 catalyst (Fig. S36†). These changes in Rh are thereby induced by the Mn promoter during CO2 hydrogenation and correlate with suppressing the methanation reaction and promoting the formation of CO and alcohols.
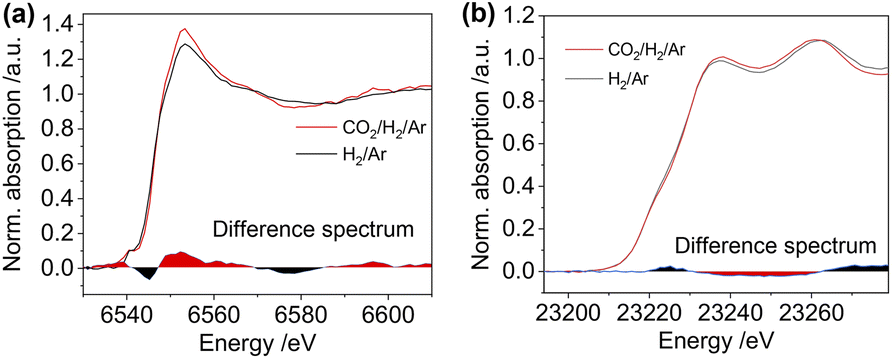 | ||
| Fig. 6 The in situ Mn K-edge XANES spectra (a) and Rh K-edge XANES spectra (b) of the RhMn@SiO2 catalyst collected under H2/Ar (3:2, 20 bar) (black line) and under CO2/H2/Ar (1:3:1, 20 bar) after the reaction of 2 h (red line). The difference spectra between these two spectra are shown at the bottom of the respective spectra. | ||
To gain insight into the surface-adsorbed species, in situ DRIFTS studies were carried out over Rh@SiO2 and RhMn@SiO2 at 20 bar and 230 °C. Upon switching to CO2/H2/Ar (1:3:1) gas, IR bands at 1294, 2050, and 2170 cm−1 were observed over the Rh@SiO2 catalyst, these bands are attributed to adsorbed bidentate carbonate 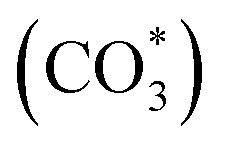 , absorbed CO* and gaseous CO, respectively (Fig. 7a).36,37 In parallel, adsorbed CHx species and gaseous CH4 were detected, as evidenced by the signals at 2856/2956 cm−1 and 3015 cm−1, respectively.6,38
, absorbed CO* and gaseous CO, respectively (Fig. 7a).36,37 In parallel, adsorbed CHx species and gaseous CH4 were detected, as evidenced by the signals at 2856/2956 cm−1 and 3015 cm−1, respectively.6,38
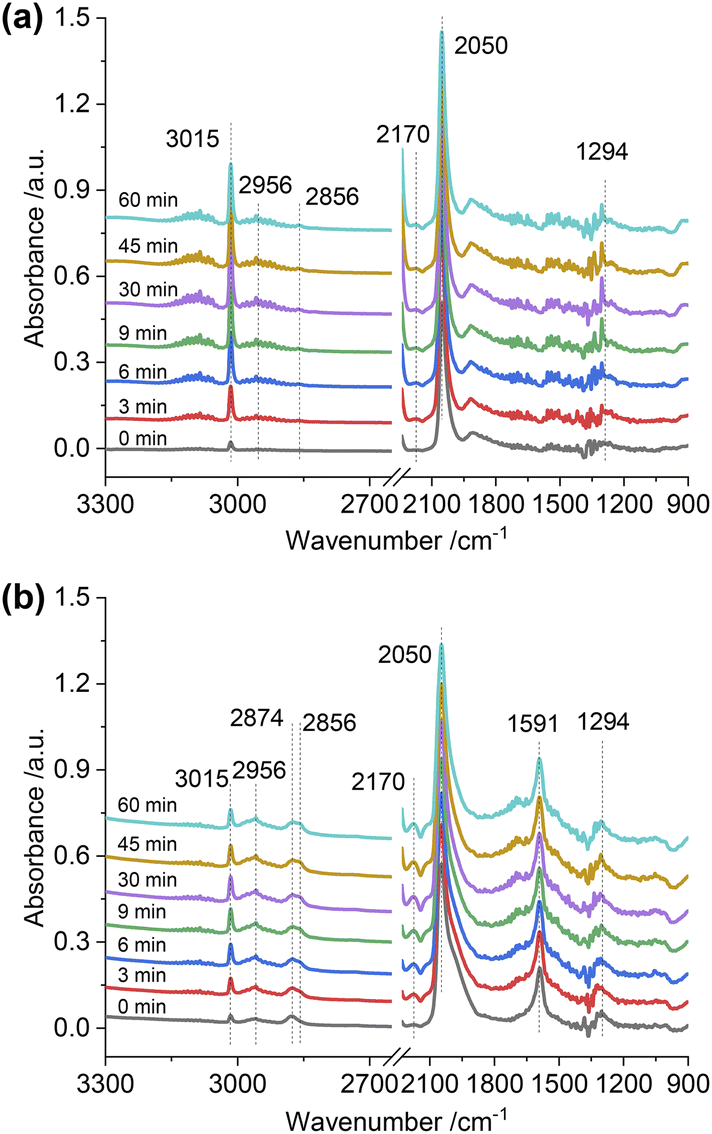 | ||
| Fig. 7
In situ DRIFTS spectra of adsorbed species on Rh@SiO2 (a) and RhMn@SiO2 (b) collected under CO2/H2/Ar (1:3:1, 20 bar). | ||
The same in situ DRIFTS studies for RhMn@SiO2 also demonstrated the presence of adsorbed bidentate 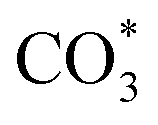 (band at 1294 cm−1), CO* (band at 2050 cm−1), and
(band at 1294 cm−1), CO* (band at 2050 cm−1), and 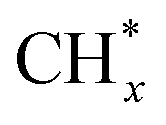 (bands at 2856 and 2956 cm−1) species (Fig. 7b). However, in contrast to Rh@SiO2, the bands for formate species (1591 and 2874 cm−1), a key intermediate for methanol formation, were observed over RhMn@SiO2.39 Moreover, the intensity of the band for gaseous CH4 over RhMn@SiO2 is significantly lower than that of RhMn@SiO2, while the intensity of the band for gaseous CO is much higher. These observations are in line with our CO2 hydrogenation results, where the presence of Mn enables to minimize the methanation reaction and promote the formation of CO and CH3OH.
(bands at 2856 and 2956 cm−1) species (Fig. 7b). However, in contrast to Rh@SiO2, the bands for formate species (1591 and 2874 cm−1), a key intermediate for methanol formation, were observed over RhMn@SiO2.39 Moreover, the intensity of the band for gaseous CH4 over RhMn@SiO2 is significantly lower than that of RhMn@SiO2, while the intensity of the band for gaseous CO is much higher. These observations are in line with our CO2 hydrogenation results, where the presence of Mn enables to minimize the methanation reaction and promote the formation of CO and CH3OH.
Based on the in situ XAS and DRIFTS experiments, we propose a possible mechanism to illustrate the promotional effect of Mn for Rh-based catalysts. After being exposed to air, the MnII is oxidized (average oxidation state is +2.6) in the vicinity of Rh and promotes Rh oxidation. This likely leads to the formation of Mn–O–Rh interfaces due to the proximity between the isolated Mn and Rh nanoparticles, driving the partial oxidation of Rh. It is accepted that the CH4 comes from the dissociative activation of CO on the Rh0 site, while the CO and methanol are produced from the non-dissociative activation of CO on the Rh+ site.5 Therefore, the pure Rh@SiO2 presenting only the metallic state of Rh under CO2 hydrogenation is a methanation catalyst, However, the presence of Mn induces the formation of the Rh–O–Mn interface to maintain the Rh+ site, which is in parallel with the stabilization of formate species as evidenced by in situ DRIFTS, thus promoting the formation of CO and alcohols and the suppression of methanation activity (Fig. 8).
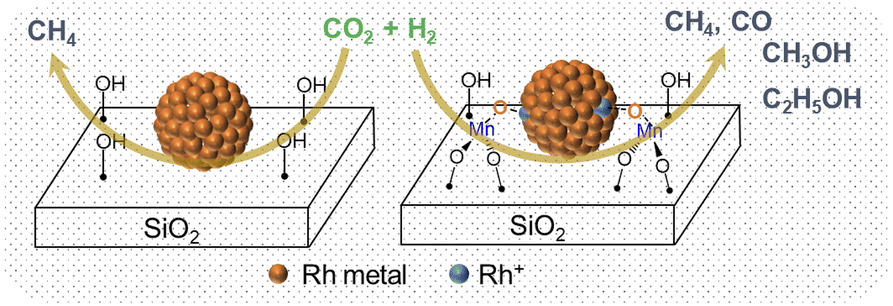 | ||
| Fig. 8 The scheme for CO2 hydrogenation over Rh@SiO2 and RhMn@SiO2 catalysts. | ||
Conclusions
In summary, we employed the SOMC/TMP approach to construct well-defined Rh-based catalysts (RhMn@SiO2 and Rh@SiO2) in order to study the promotional effect of Mn for CO2 hydrogenation. We find that the presence of Mn not only decreases particle size but also dramatically suppresses methanation. The product can be steered from CH4 on non-Mn-promoted Rh@SiO2 to a mixture of methane and oxygenates (CO and CH3OH along with little CH3CH2OH) on RhMn@SiO2. In situ XAS reveals that the isolated Mn remains atomically dispersed in the vicinity of Rh nanoparticles during either the H2 pretreatment or CO2 hydrogenation. This excludes the formation of mixed oxides or alloys as the major contributors to the activity of these catalysts. The presence of MnII in the vicinity of Rh nanoparticles enables to drive the oxidation of Rh, likely generating Mn–O–Rh interfaces under reaction conditions. The formed interface is proposed to be key to maintaining Rh+ sites, which is responsible for suppressing the methanation reaction and stabilizing the formate species as evidenced by in situ DRIFTS to promote the formation of CO and alcohols. These observations reveal how the Mn promoter dynamically influences Rh nanoparticles, thereby steering the product selectivity. This study provides insights into the rational design of catalysts for CO2 hydrogenation.Data availability
We do not have more experimental and computational data associated with this article.Author contributions
Z. W. and C. C. conceived and designed the study. C. C. supervised the project. Z. W. performed the catalyst preparation, characterization, and catalytic reactions. C. E. developed the Rh precursor, S. D., X. Z. and C. E. assisted in in situ XAS experiment. W. Z. and C. C. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C., W. Z. and X. Z. acknowledge the National Centre of Competence in Research (NCCR) Catalysis (grant 180544) for financial support. C. C. and S. R. D. acknowledge the Swiss National Science Foundation (grants 200021_169134, and 200020B_192050), while C. E. acknowledges the Swiss National Science Foundation (grant 200020B_192050) and the Scholarship Fund of the Swiss Chemical Industry (SSCI). The authors acknowledge Dr W. V. Beek and Dr D. Stoian at the Swiss Norwegian Beamlines (SNBL, ESRF) for the provision of beamtime and support with in situ XAS experiments via proposal A31-1-168. Furthermore, ScopeM is gratefully acknowledged for their support and assistance in this work through project No. 2460. The authors thank Dr Y. Wang for assistance with in situ DRIFTS. The authors also thank C. Hansen and C. J. Kaul for assistance with X-ray crystallography and ss-NMR, respectively. Crystal Data for Li(DIA)(THF) and Rh(COD)(DIA) can be obtained free of charge from the Cambridge Crystallographic Data Centre (CCDC number: 2237587 and 2237586†).Notes and references
- G. A. Olah, G. K. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS.
- F. Zhang, W. Zhou, X. Xiong, Y. Wang, K. Cheng, J. Kang, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2021, 125, 24429–24439 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Energy, 1997, 22, 343–348 CrossRef CAS.
- M. R. Gogate and R. J. Davis, Catal. Commun., 2010, 11, 901–906 CrossRef CAS.
- G. Wang, R. Luo, C. Yang, J. Song, C. Xiong, H. Tian, Z.-J. Zhao, R. Mu and J. Gong, Sci. China: Chem., 2019, 62, 1710–1719 CrossRef CAS.
- C. Yang, R. Mu, G. Wang, J. Song, H. Tian, Z. J. Zhao and J. Gong, Chem. Sci., 2019, 10, 3161–3167 RSC.
- M. Suvarna, P. Preikschas and J. Pérez-Ramírez, ACS Catal., 2022, 12, 15373–15385 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Nunez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed.
- J. Meyet, A. Ashuiev, G. Noh, M. A. Newton, D. Klose, K. Searles, A. P. van Bavel, A. D. Horton, G. Jeschke, J. A. van Bokhoven and C. Copéret, Angew. Chem., Int. Ed., 2021, 60, 16200–16207 CrossRef CAS PubMed.
- S. R. Docherty, L. Rochlitz, P.-A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- L. Rochlitz, Q. Pessemesse, J. W. A. Fischer, D. Klose, A. H. Clark, M. Plodinec, G. Jeschke, P.-A. Payard and C. Copéret, J. Am. Chem. Soc., 2022, 144, 13384–13393 CrossRef CAS PubMed.
- S. R. Docherty and C. Copéret, J. Am. Chem. Soc., 2021, 143, 6767–6780 CrossRef CAS PubMed.
- E. Lam, J. J. Corral-Pérez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives, A. Urakawa and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 13989–13996 CrossRef CAS PubMed.
- E. Lam, G. Noh, K. Larmier, O. V. Safonova and C. Copéret, J. Catal., 2021, 394, 266–272 CrossRef CAS.
- E. Lam, G. Noh, K. W. Chan, K. Larmier, D. Lebedev, K. Searles, P. Wolf, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 7593–7598 RSC.
- G. Noh, E. Lam, D. T. Bregante, J. Meyet, P. Šot, D. W. Flaherty and C. Copéret, Angew. Chem., Int. Ed., 2021, 60, 9650–9659 CrossRef CAS PubMed.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS PubMed.
- S. R. Docherty, N. Phongprueksathat, E. Lam, G. Noh, O. V. Safonova, A. Urakawa and C. Copéret, JACS Au, 2021, 1, 450–458 CrossRef CAS PubMed.
- B. Ghaffari, J. Mendes-Burak, K. W. Chan and C. Copéret, Chem.–Eur. J., 2019, 25, 13869–13873 CrossRef CAS PubMed.
- B. J. Kip, F. B. M. Duivenvoorden, D. C. Koningsberger and R. Prins, J. Catal., 1987, 105, 26–38 CrossRef CAS.
- G. Prieto, P. Concepción, A. Martínez and E. Mendoza, J. Catal., 2011, 280, 274–288 CrossRef CAS.
- S. Chuang, R. Stevens and R. Khatri, Top. Catal., 2005, 32, 225–232 CrossRef CAS.
- N. Yang, J. S. Yoo, J. Schumann, P. Bothra, J. A. Singh, E. Valle, F. Abild-Pedersen, J. K. Nørskov and S. F. Bent, ACS Catal., 2017, 7, 5746–5757 CrossRef CAS.
- V. Schwartz, A. Campos, A. Egbebi, J. J. Spivey and S. H. Overbury, ACS Catal., 2011, 1, 1298–1306 CrossRef CAS.
- F. C. Meunier, J. Phys. Chem. C, 2021, 125, 21810–21823 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, Appl. Catal., A, 2000, 197, 255–268 CrossRef CAS.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- A. Campos, N. Lohitharn, A. Roy, E. Lotero, J. G. Goodwin and J. J. Spivey, Appl. Catal., A, 2010, 375, 12–16 CrossRef CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CrossRef CAS.
- H. Du, C. T. Williams, A. D. Ebner and J. A. Ritter, Chem. Mater., 2010, 22, 3519–3526 CrossRef CAS.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2237586 and 2237587. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01163a |
| This journal is © The Royal Society of Chemistry 2023 |