
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFragment expansion with NUDELs – poised DNA-encoded libraries†
Catherine L. A.
Salvini
a,
Benoit
Darlot
bc,
Jack
Davison
a,
Mathew P.
Martin
d,
Susan J.
Tudhope
d,
Shannon
Turberville
d,
Akane
Kawamura
bc,
Martin E. M.
Noble
d,
Stephen R.
Wedge
d,
James J.
Crawford
 e and
Michael J.
Waring
*a
e and
Michael J.
Waring
*a
aCancer Research Horizons Therapeutic Innovation Newcastle Drug Discovery Group, Chemistry, School of Natural and Environmental Sciences, Newcastle University, Bedson Building, NE1 7RU, UK. E-mail: mike.waring@ncl.ac.uk
bChemistry, School of Natural and Environmental Sciences, Newcastle University, Bedson Building, NE1 7RU, UK
cDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
dCancer Research Horizons Therapeutic Innovation Newcastle Drug Discovery Group, Translational and Clinical Research Institute, Newcastle University, Paul O'Gorman Building, NE2 4HH, UK
eGenentech Inc., 1 DNA Way, South San Francisco, California 94080, USA
First published on 12th July 2023
Abstract
Optimisation of the affinity of lead compounds is a critical challenge in the identification of drug candidates and chemical probes and is a process that takes many years. Fragment-based drug discovery has become established as one of the methods of choice for drug discovery starting with small, low affinity compounds. Due to their low affinity, the evolution of fragments to desirable levels of affinity is often a key challenge. The accepted best method for increasing the potency of fragments is by iterative fragment growing, which can be very time consuming and complex. Here, we introduce a paradigm for fragment hit optimisation using poised DNA-encoded chemical libraries (DELs). The synthesis of a poised DEL, a partially constructed library that retains a reactive handle, allows the coupling of any active fragment for a specific target protein, allowing rapid discovery of potent ligands. This is illustrated for bromodomain-containing protein 4 (BRD4), in which a weakly binding fragment was coupled to a 42-member poised DEL via Suzuki–Miyaura cross coupling resulting in the identification of an inhibitor with 51 nM affinity in a single step, representing an increase in potency of several orders of magnitude from an original fragment. The potency of the compound was shown to arise from the synergistic combination of substructures, which would have been very difficult to discover by any other method and was rationalised by X-ray crystallography. The compound showed attractive lead-like properties suitable for further optimisation and demonstrated BRD4-dependent cellular pharmacology. This work demonstrates the power of poised DELs to rapidly optimise fragments, representing an attractive generic approach to drug discovery.
Introduction
Fragment-based lead generation1–3 and DNA-encoded library screening4–6 are two important technologies for identifying ligands for protein targets in chemical biology and medicinal chemistry. The primary rationale for fragment-based approaches is that by screening small (typically <350 Da) compounds, a much greater range of chemical structures can be represented with fewer compounds, making screening more efficient and increasing the chances of finding hits.7,8 Because these hits are small, they typically bind with low affinity often in the millimolar range, necessitating sensitive biophysical techniques (NMR, SPR, X-ray crystallography) to detect their binding. It can take considerable time and effort to develop them into compounds that have high enough affinity to achieve activity in biochemical assays and to carry out target validation studies, which are critical steps in the development of chemical probes and therapeutics.In contrast, DNA-encoded libraries (DELs) consist of collections of compounds covalently linked to unique DNA-oligonucleotides that encode their chemical structure. Through combinatorial synthesis, DELs allow the screening of vast numbers (often billions) of pooled compounds.9 They can be screened very efficiently by affinity selection, subsequent PCR amplification of the bound species and DNA sequencing.5 Synthesis of the molecules identified in the selection without the DNA-tags is then carried out to confirm the hits using traditional assays. Despite the ability to screen such large numbers of compounds, it is still not feasible to significantly cover chemical diversity at the higher molecular weight ranges that are typical of DELs. While the exact size of this chemical space is difficult to estimate, it is believed to be extremely vast, with estimates ranging from 1060 to 10200.10,11 Such libraries should still be focused on biologically relevant chemical space, i.e. consisting mainly of compounds with lead-like physicochemical properties and free of undesirable functionality. This places significant constraints on library design. In addition, subsequent optimisation of hits from DEL screens often requires significant reengineering of their chemical structures, which again can be inefficient and time consuming.
We proposed that it would be possible to significantly accelerate the process of optimising fragment hits for any given protein by preparing a poised DNA-encoded library in which every member contained a functional handle that allowed the attachment of an active fragment (Fig. 1a). Such a poised library would then be available to couple with active fragments for any selected target in a single step. A subsequent fragment screen would generate fragment hits specific for a chosen target (Fig. 1b). A selected fragment hit derivatised with a complementary reactive group would then be coupled to the poised library, producing a DEL that was focused on the chosen target (Fig. 1c). By employing two synthesis cycles in the poised library synthesis, this would maintain the optimal profile of lead-likeness, library fidelity, synthesis yields and chemical diversity in the final libraries once the active fragment is coupled, as well as maintaining the required signal-to-noise ratios in the selections.12,13
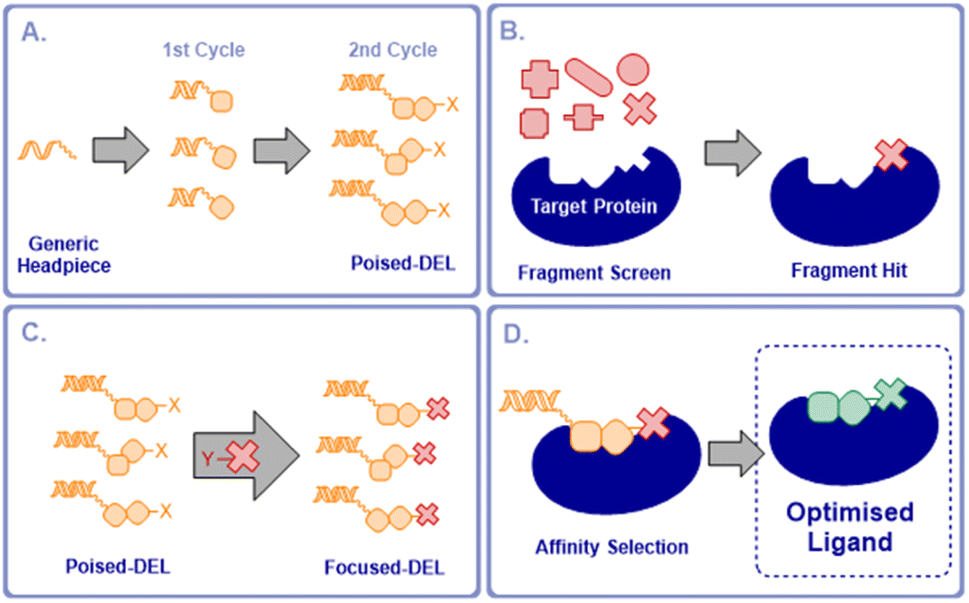 | ||
| Fig. 1 Concept of a poised DEL for fragment elaboration. (a) Synthesis of a 2-cycle poised DEL with a reactive group X; (b) fragment screen to identify active fragments; (c) coupling of the poised DEL to a selective fragment derivatised with complementary reactive group Y to produce a focused DEL; (d) affinity selection of focused DEL to select optimised ligands. | ||
The focused DEL would be screened by affinity selection to identify higher affinity, elaborated ligands (Fig. 1d). This would enable the rapid expansion of fragment hits to compounds with potentially much increased affinity in a single operation without the need for the stepwise fragment growing process, currently the most commonly adopted approach to FBLG.14 Alternatively, this concept could be viewed as a flexible means of focusing a DEL on chemical space that was directly relevant to a selected protein target. These poised libraries we term “NUDELs” because of their origin at Newcastle University.
The ESAC approach15–17 facilitates the discovery of hits for proximal binding pockets on a protein. Similar approaches have been applied to the discovery of ligands with applications in selective delivery of therapeutic radionuclides.18 Generated hits have then been connected by flexible linker to give probe-like compounds, to our knowledge this has yet to be used to yield drug-like molecules. The NUDEL methodology by contrast provides direct covalent connection between the hit fragment and expansion library, resulting directly in lead-like compounds, without the need for further iterative optimisation.
Results and discussion
Synthesis of the poised DNA-encoded library
The first NUDEL was designed based on two sequential amide couplings involving coupling of a set of Fmoc-protected amino acids to an amino-derivatised DNA-head piece in cycle 1, followed by Fmoc-deprotection and second amide coupling with a set of aryl halide-containing acids. Libraries of this design could then incorporate any active fragment for which a derivative was available with a functional group that allowed coupling with the halide. Commonly used reactions of this type, such as Suzuki–Miyaura19,20 and Buchwald–Hartwig21 reactions, have been recently shown to work efficiently on DNA-tagged substrates.The library components were selected based on diversity of shape and chemical functionality such that the resulting test compounds would represent diverse lead-like structures (Fig. 2a). Seven Fmoc amino acids, consisting of canonical amino acids alanine, valine and phenylalanine, pyrrolidine-3 carboxylic acid and aniline derivatives 3- and 4-aminobenzoic acid and (4-methylamino)benzoic acid, were selected for cycle 1. Six aryl halide-containing acids: 3- and 4-iodobenzoic acid, (4-iodophenyl)acetic acid 5-iodonicotinic acid, 2-(6-bromopyridin-3-yl)acetic acid and 2-(4-bromo-1H-pyrazol-1-yl)acetic acid, were selected for cycle 2. Thus, the final library would contain a variety of hydrogen bond donor, acceptor, aromatic and lipophilic functionality, which make up the majority of pharmacophoric interactions, with differing overall shapes that present different relative orientations of the components. Incorporation of additional hydrogen bond donors was avoided in the building block variation due to their necessary presence in the amide linkages used in the assembly of the library.
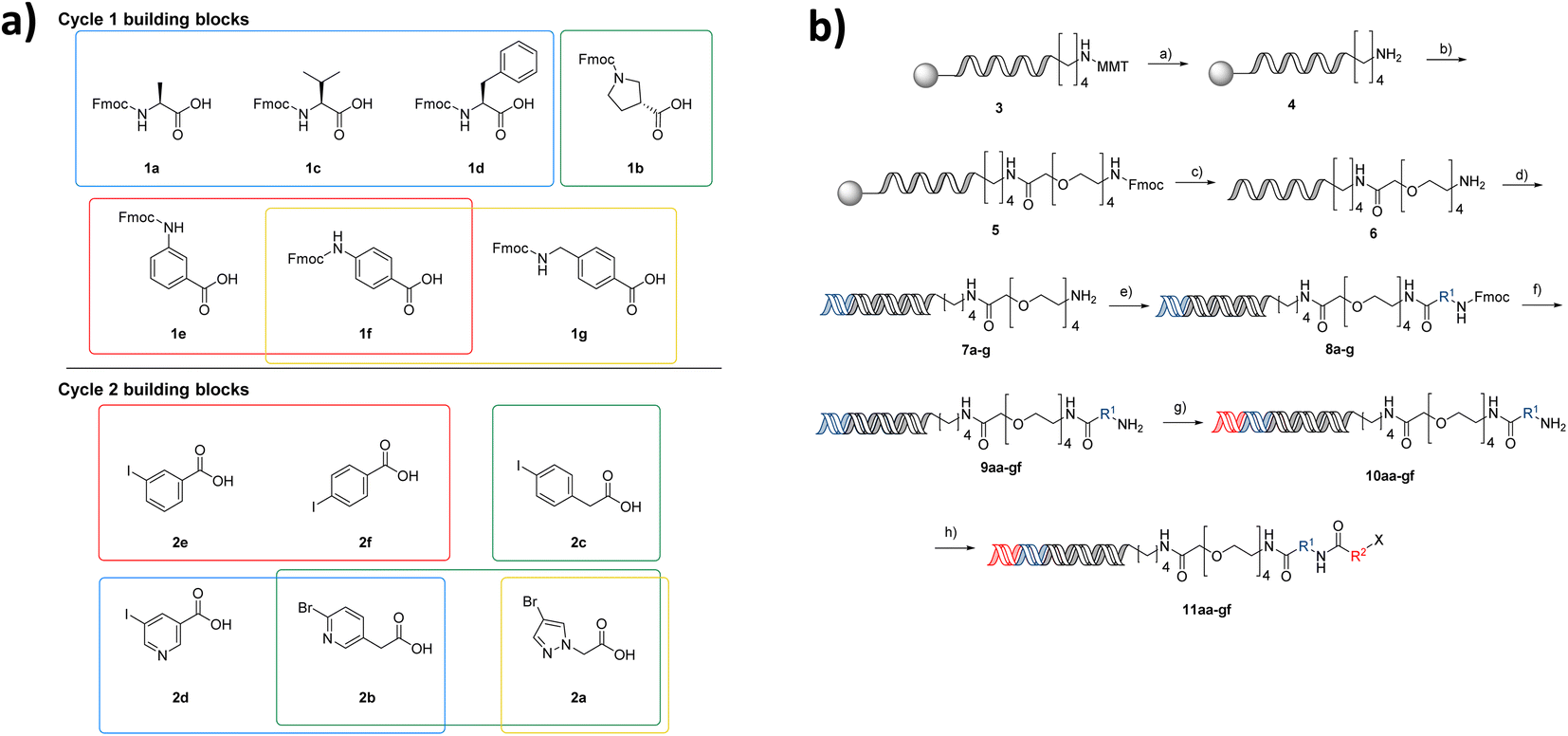 | ||
| Fig. 2 Poised NUDEL library design and synthesis. (a) Building block selection, cycle 1: blue, canonical amino acids; green, 2° amine; red, aniline derivatives and positional isomers; yellow, increased spacer length; cycle 2: red, positional isomers; green, increased spacer; blue, 6 membered heterocycles; yellow, 5-membered heterocycles; (b) NUDEL synthesis (first letter referring to BB1 a = 1a, second letter referring to BB2 a = 2a etc.), reagents and conditions: (a) TCA, DCM; (b) DMF, DIPEA, HATU, Fmoc-PEG acid; (c) MeNH2, NH3, H2O; (d) (i) PNK, PNK buffer (ii) ligase, ligase buffer; (e) DMTMM, Fmoc-amino acid borate buffer pH 9.4, H2O/DMF; (f) 10% piperidine, H2O; (g) (i) PNK, PNK buffer (ii) ligase, ligase buffer; (h) DMT-MM, aryl-acid, H2O/DMF. | ||
Through the DEL methodology, large numbers of compounds become synthetically achievable in parallel. However, one of the major disadvantages of working with DNA-conjugated compounds is that the removal of side products is difficult. Underpinning the fidelity of the library is the requirement that reactions used in its synthesis proceed in high conversion to the intended product. This ensures that the compound structure associated with each label is correct, as side products or incomplete reactions give rise to multiple products attached to the same sequence. To ensure the NUDEL was of high fidelity, examples of amide coupling conditions for compounds 1a–c with headpiece 6 were optimised to the highest conversions, before further expanding for the selected library building blocks. Though this proposed library may be small when compared to DELs in the published literature, it contains numbers of analogues that are comparable to those used in generation of SAR in a fragment growing campaign and allows them to be accessed far more rapidly than via traditional medicinal chemistry synthesis strategies. Working with a small-scale library afforded the ability to conduct thorough validation experiments for each individual coupling and stage of library construction. The purity and conversion of the library synthesis were prioritised over scale in this first proof of concept, to ensure the fidelity of the library.
After a period of optimisation, the library synthesis protocol involved on-resin monomethoxytrityl deprotection of a hexylamino-tagged DNA adapter sequence, followed by HATU-mediated coupling of a PEG-4-linker bearing an Fmoc protected amine (Fig. 2b). Concomitant Fmoc deprotection and cleavage from the resin was achieved by methylamine treatment. The resulting headpiece was annealed with a complementary DNA strand and the building block 1 codons attached by enzymatic ligation. The Fmoc amino acids in cycle 1 were incorporated by DMTMM coupling in borate buffer, followed by deprotection to yield the 7 intermediate amines. These intermediates were pooled, and the resulting mixture split into 6 wells. Codon 2 ligation was then carried out followed by a second DMTMM-mediated acylation of the cycle 2 halide-containing acids, resulting in a fully encoded library of 42 aryl halides (11aa–gf), poised for coupling of active fragments. Although this library is small by DELs standards, synthesis of matrices of compounds in such a manner in a traditional fragment growing campaign would be far more resource intensive. Additionally, working on a small scale initially provided the opportunity to confirm all transformation combinations employed in the NUDEL construction.
Coupling of an active fragment
To demonstrate the use of the NUDEL to generate a focused library for fragment expansion, we selected bromodomain-containing protein 4 (BRD4) as a test case. 3,5-Dimethylisoxazole is a known active fragment binder of BRD4.22 We proposed that the NUDEL aryl halides could be derivatised by coupling to the corresponding boronic acid of dimethyl isoxazole via on-DNA Suzuki–Miyaura cross-coupling,19,20 thus providing a BRD4 focused library.To establish the coupling of dimethylisoxazole boronic acid to the library, a potentially challenging coupling due to the presence of the heterocyclic system and sterically hindering bis-ortho-methyl substitution, a model DNA substrate containing the adapter sequence, was used as a trial reaction (Fig. 3a). Micellar-mediated Suzuki–Miyaura conditions performed very well with this substrate, furnishing the coupled dimethylisoxazole with 100% conversion, giving confidence that the BRD4 fragment could be coupled efficiently on-DNA. A sample of the NUDEL was subjected to the same coupling conditions resulting in the BRD4 targeted library.
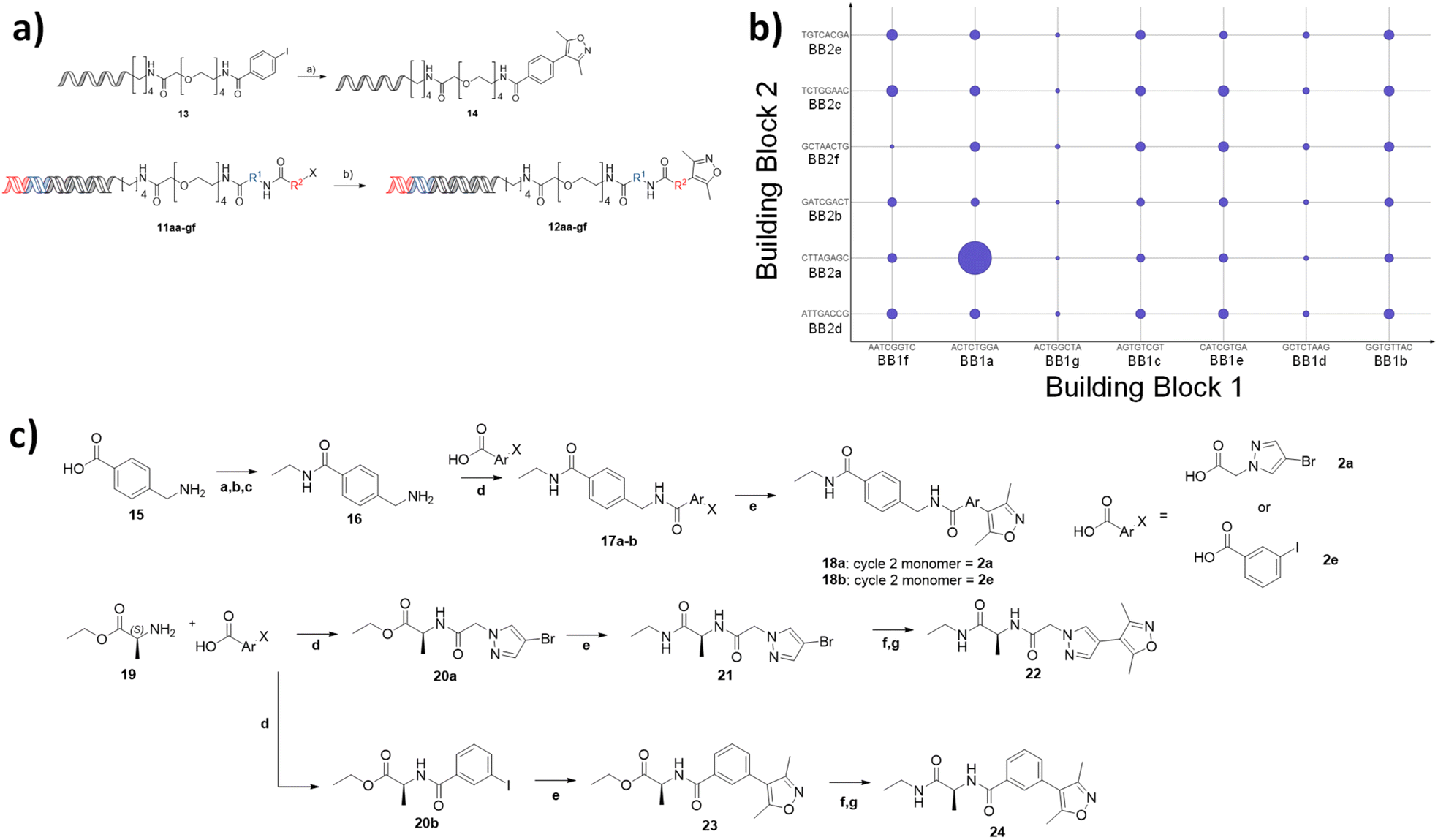 | ||
Fig. 3 (a) Coupling of the isoxazole fragment to a model substrate, showing confirmation of Suzuki coupling on DNA, and coupling to the full NUDEL, reagents: (a) Pd(dtbpf)Cl2, K3PO4, H2O/THF, TPGS-750M 2.5%, 3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoxazole, (b); (j) Pd(dtbpf)Cl2, 2% TPGS-750-M, K3PO4, H2O/THF; (b) Selection of targeted library against BRD4 showing enrichment of sequences by size of the points, broken down by building block 1 (X-axis) and building block 2 (Y-axis) codons; (c) Off-DNA synthesis of 4 compounds. 18a synthesised from 2a, 18b synthesised from 2e. Reagents: (a) TFAA; (b) SOCl2; (c) NH2Et in THF 2M; (d) HATU, DIPEA, DCM (e) Pd(dtbpf)Cl2, Cs2CO3, Diox./H2O 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoxazole; (f) LiOH, THF/H2O 1:1; (g) HATU, DIPEA, DCM, NH2Et in THF 2 M. :1, 3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoxazole; (f) LiOH, THF/H2O 1:1; (g) HATU, DIPEA, DCM, NH2Et in THF 2 M. | ||
Affinity selection against BRD4
The targeted library was incubated with immobilised BRD4 (first bromodomain, BD1) protein in the presence of DNA-linked version of the established BRD4 ligand JQ1 (ref. 23) as a positive control. After washing to remove non-binders, the protein was denatured, and the sample subjected to 40 cycles of PCR amplification and next generation sequencing (>150000 reads). The resulting output was analysed to assess the frequency of occurrence of each building block codon and their enrichment relative to the starting library. Remarkably, this highlighted a specific combination of cycle 1 codon (ACTATGGA) with cycle 2 codon (CTTAGAGC), corresponding to alanine and methylamidopyrazole respectively, which occurred significantly more times (11-fold) than any other sequence and was comparable to the enrichment of the JQ1 derived control, spiked into the library at the concentration of a representative library member (Fig. 3b). Interestingly, neither codon appeared significantly enriched in any other combination. Together, this suggested that compound 22 would be a potent BRD4 ligand and that the combination of 1a and 2a, rather than either motif individually, resulted in a synergistic and multiplicative gain in affinity.
Off-DNA hit confirmation
To confirm the synergistic finding, a ‘matched square’ of compounds consisting of compound 22, which corresponded to the hit, 24, which contained 1a (active cycle 1 monomer) and 2e (representative non-enriched cycle 2 monomer), 18a containing 1g (representative non-enriched cycle 1 monomer) with 2a (enriched cycle 2 monomer) and finally 18b (both non-enriched monomers) were selected for off-DNA synthesis and evaluation (Fig. 3c).Compounds 18a,b were synthesised from 4-(aminomethyl)benzoic acid 15 by formation of the primary ethyl amide via a protection and deprotection of the amine as the trifluoroacetamide. Subsequent amide coupling to the appropriate cycle 2 monomer and incorporation of the dimethyl isoxazole warhead via Suzuki cross-coupling resulted in the desired off-DNA compounds 18a,b (Fig. 3c). 22 and 24 were obtained in a similar manner, first coupling alanine ethyl ester 19, with either 2a, 2e, followed by Suzuki cross coupling to install the isoxazole. Ester hydrolysis and amide coupling gave the final ethyl amide products 22 and 24.
SAR analysis
The Kd of 18a, 18b and 22 and 24 were evaluated via SPR analysis for binding to BRD4 BD1. Gratifyingly, hit compound 22, had a Kd of 51 nM (Fig. 4a). In contrast the affinities of 18a,18b and 24 were 35 μM, 2.5 μM, and 15 μM, respectively, i.e. two to three orders of magnitude less potent (Fig. 4b). This result is consistent with the enrichment observed during the selection and provides evidence that the enrichment from the on-DNA selection depends on the affinity of the compounds. It shows that it is the combination of the alanine and the pyrazolylacetamide moieties, rather than either of them in isolation that is responsible for the dramatic gain in affinity. This highlights a distinct advantage of this approach over traditional fragment growing. It would not be possible to identify this unique, synergistic combination of substructures without synthesising every combination, something that is essentially prohibitive for analogues synthesised off-DNA. Moreover, using fragment growing approaches, this combination could not be identified because the pyrazoleacetamide species would not be identified as beneficial when lacking the cycle 2 alanine and so would not have been selected for subsequent elaboration. The use of the NUDEL, which contains every possible combination of the selected substructures, makes the discovery of such synergistic combinations of groups facile.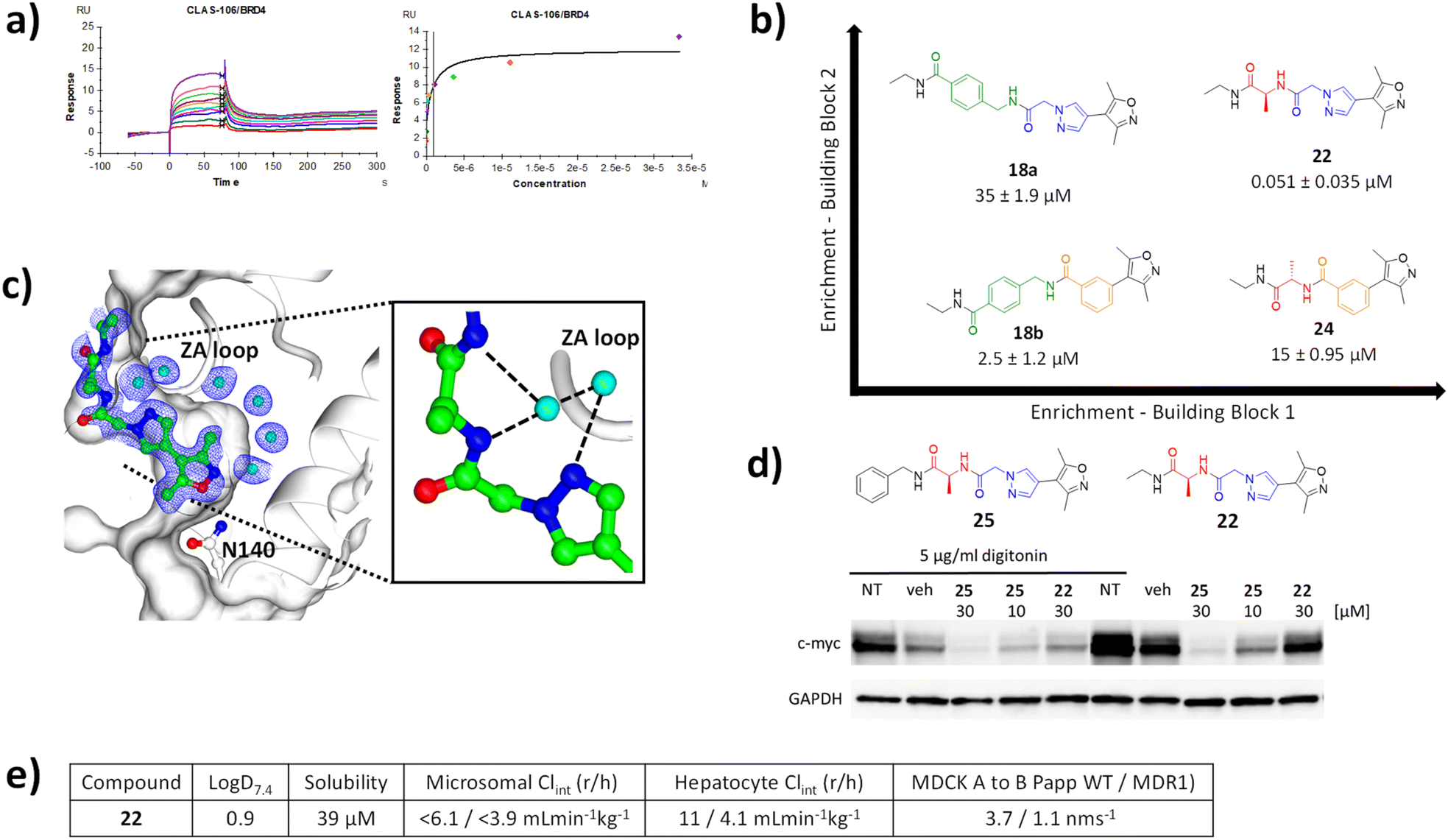 | ||
| Fig. 4 (a) SPR sensograms for 22 against BRD4; (b) matched square of compounds showing increased potency with increased selection enrichment; (c) X-ray crystal structure of 22 bound to BRD4 BD1 (pdb code 8C11); (d) western blot analysis of downregulation of c-Myc in MM.1S cells by compounds 22 and 25 in the presence and absence of digitonin, compared to non-treated (NT) and vehicle (veh) treated cells relative to GAPDH loading control; (e) ADMET profile of 22. | ||
Characterisation of the binding mode of 22 (CLAS-106) was achieved by solving the crystal structure in complex with BRD4 (Fig. 4c). The crystal structure of the BRD4 complex was determined at 1.8 Å resolution and, as expected, 22 was observed to interact with the KAc recognition site. The oxygen of the isoxazole moiety forms a hydrogen bond with the conserved Asn-140, and a conserved water mediated a hydrogen bond between the nitrogen of the isoxazole and Tyr-97. The pyrazole core is sandwiched between the Pro-82 of the WPF shelf and Leu-92 of the ZA-loop, with a key hydrogen bond formed between a conserved structural water and the main chain of Pro-86 and Gln-85 of the ZA-loop. The extended pyrazole core, a product of cycle 2 synthesis, is further stabilised through additional water mediated hydrogen bonding network involving the main chain of Asp-86. This provides an explanation for the synergistic increase in potency from the combination of the pyrazole and alanine, in that it provides a unique relative orientation of the three critical groups to form complementary hydrogen bonds to the two adjacent structural water molecules.
ADME and cellular pharmacology of 22
To confirm the activity of 22 in a cellular context, human multiple myeloma MM.1S cells were treated with compound. Consistent with the high polarity of the compound, and hence expected low permeability and significant efflux, cells required permeabilisation with digitonin to show activity. In the presence of digitonin, 22 showed downregulation of c-Myc, an established downstream pharmacodynamic marker of BRD4 inhibition24 at a concentration of 30 μM (Fig. 4d). A more lipophilic, cell permeable analogue 25 (benzyl amide derivative) showed downregulation of c-Myc in a concentration dependent manner in both the presence and absence of digitonin.The logD value for compound 22 was 0.90, thus a lipophilic ligand efficiency of 6.4, which is consistent with a high quality lead compound (Fig. 4e).25 It had high solubility and low in vitro clearance in human and rat microsomes and hepatocytes. As expected, and consistent with the cellular activity, permeability in MDCK cell was low without evidence of MDR1-mediated efflux. Overall, compound 22 is a high-quality technical profile with scope for facile further optimisation.
Methods
Synthesis of the NUDEL
Ligations contained DNA (100 μM, 9000 pmol in overall reaction media of 90 μL), phosphorylated DNA strands (Max: 20 μL, 9000 pmol), 10X T4 DNA ligase buffer (9 μL, 400 mM Tris-HCl, 100 mM MgCl2, 100 mM DTT, 5 mM ATP9), water (up to 90 μL) and T4 DNA Ligase (3 μL, 30 Weiss U μL−1). The ligations were carried out at 25 °C for 16 hours, followed by heating to 75 °C for 10 min. Each ligation was purified by ethanol precipitation prior to the subsequent organic reaction taking place.
Conclusions
The identification of an inhibitor with nanomolar potency in a single step from a weakly binding fragment demonstrates the power of the NUDEL poised library approach to fragment growing. Due to the careful design of the library, this also led to compounds with desirable drug like properties and cellular activity. With iterative fragment growing approaches, currently still best practice in fragment-based lead generation, reaching such an endpoint can take years and is beset with challenges associated with traversing form weakly active fragments to compounds of lead-like affinity.26 With the availability of a NUDEL, such a goal can be achieved in mere weeks. The identification of synergistic combinations of sub-groups is enabled by the combinatorial nature of the library, the synthesis of which is often resource prohibitive with traditional off-DNA analogue exploration. Hence, as illustrated in this case, combinations of groups can be discovered that otherwise would have been highly unlikely to have been identified. This approach should be applicable to any traditional fragment library including specialised binding site mapping libraries such as MiniFrags27 and FragLites28,29 that return high hit rates but relatively low affinity hits. Whilst this study demonstrates the value of a small NUDEL, the same approach will be scalable to much larger libraries covering a greater degree of chemical space. We believe this approach will provide a solution to the problem of rapidly identifying potent ligands from fragment start points and become a highly valuable approach in drug discovery and chemical biology.Data availability
Crystallographic data for 22 has been deposited at the PBD under 8C11. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
CLAS carried out the synthesis of the NUDEL, developed the coupling reaction and carried out the selections, analysed the data and carried out subsequent chemical synthesis. BD carried out selections. JD carried out HPLC purification. MPM carried out SPR and crystallography. SJT carried out the cellular experiments. ST synthesised BRD4 protein. AK supervised BD. MEMN supervised MPM and ST. SRW supervised SJT. JC supervised CLAS. MJW conceived of the project, supervised CLAS and carried out data analysis. CLAS and MJW wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of Genentech, Inc. (PhD studentship award to CLAS), EPSRC MoSMed CDT (centre funding for PhD training for CLAS, grant reference EP/S022791/1), Cancer Research UK (programme funding of the Drug Discovery Group, grant references C2115/A21421 and DRCDDRPGMApr2020\100002; Centre Network Accelerator Award, grant reference A20263); EPSRC (PhD and Doctoral Prize finding for BD, ref. EP/T517811/1); EU Horizon 2020 (fellowship awards to AK, ref. 679479 and 101003111). We thank Newcastle Structural Biology Facility and Dr. Arnaud Basle for X-ray crystallography support. We thank Emile Plise, Ivy Chen and Ruth Dorel (Genentech) for generation of ADME profiling data.Notes and references
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS PubMed.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. V Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS.
- R. A. Goodnow, C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2016, 16, 131–147 CrossRef PubMed.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS PubMed.
- A. R. Leach and M. M. Hann, Curr. Opin. Chem. Biol., 2011, 15, 489–496 CrossRef CAS PubMed.
- I. F. S. F. Castan, J. S. Graham, C. L. A. Salvini, H. A. Stanway-Gordon and M. J. Waring, Bioorg. Med. Chem., 2021, 43, 116273 CrossRef CAS PubMed.
- J.-L. Reymond, L. Ruddigkeit, L. Blum and R. van Deursen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 717–733 Search PubMed.
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649–663 CrossRef CAS.
- A. L. Satz, ACS Chem. Biol., 2015, 10, 2237–2245 CrossRef CAS.
- A. L. Satz, R. Hochstrasser and A. C. Petersen, ACS Comb. Sci., 2017, 19, 234–238 CrossRef CAS.
- I. J. P. de Esch, D. A. Erlanson, W. Jahnke, C. N. Johnson and L. Walsh, J. Med. Chem., 2022, 65, 84–99 CrossRef CAS.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- G. Bassi, N. Favalli, M. Vuk, M. Catalano, A. Martinelli, A. Trenner, A. Porro, S. Yang, C. L. Tham, M. Moroglu, W. W. Yue, S. J. Conway, P. K. Vogt, A. A. Sartori, J. Scheuermann and D. Neri, Adv. Sci., 2020, 7, 2001970 CrossRef CAS PubMed.
- M. Catalano, M. Moroglu, P. Balbi, F. Mazzieri, J. Clayton, K. H. Andrews, M. Bigatti, J. Scheuermann, S. J. Conway and D. Neri, ChemMedChem, 2020, 15, 1752–1756 CrossRef CAS PubMed.
- S. Puglioli, E. Schmidt, C. Pellegrino, L. Prati, S. Oehler, R. De Luca, A. Galbiati, C. Comacchio, L. Nadal, J. Scheuermann, M. G. Manz, D. Neri, S. Cazzamalli, G. Bassi and N. Favalli, Chem, 2023, 9, 411–429 CAS.
- J. H. Hunter, L. Prendergast, L. F. Valente, A. Madin, G. Pairaudeau and M. J. Waring, Bioconjugate Chem., 2020, 31, 149–155 CrossRef CAS PubMed.
- J. H. Hunter, M. Potowski, H. A. Stanway-Gordon, A. Madin, G. Pairaudeau, A. Brunschweiger and M. J. Waring, J. Org. Chem., 2021, 86, 17930–17935 CrossRef CAS PubMed.
- J. S. Graham, J. H. Hunter and M. J. Waring, J. Org. Chem., 2021, 86, 17257–17264 CrossRef CAS PubMed.
- D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, S. Knapp, S. J. Conway and T. D. Heightman, J. Med. Chem., 2011, 54, 6761–6770 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- J. E. Delmore, G. C. Issa, M. E. Lemieux, P. B. Rahl, J. Shi, H. M. Jacobs, E. Kastritis, T. Gilpatrick, R. M. Paranal, J. Qi, M. Chesi, A. C. Schinzel, M. R. McKeown, T. P. Heffernan, C. R. Vakoc, P. L. Bergsagel, I. M. Ghobrial, P. G. Richardson, R. A. Young, W. C. Hahn, K. C. Anderson, A. L. Kung, J. E. Bradner and C. S. Mitsiades, Cell, 2011, 146, 904–917 CrossRef CAS PubMed.
- R. J. Young and P. D. Leeson, J. Med. Chem., 2018, 61, 6421–6467 CrossRef CAS PubMed.
- M. P. Martin and M. E. M. Noble, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 1294–1302 CrossRef CAS PubMed.
- M. O'Reilly, A. Cleasby, T. G. Davies, R. J. Hall, R. F. Ludlow, C. W. Murray, D. Tisi and H. Jhoti, Drug Discovery Today, 2019, 24, 1081–1086 CrossRef.
- D. J. Wood, J. D. Lopez-Fernandez, L. E. Knight, I. Al-Khawaldeh, C. Gai, S. Lin, M. P. Martin, D. C. Miller, C. Cano, J. A. Endicott, I. R. Hardcastle, M. E. M. Noble and M. J. Waring, J. Med. Chem., 2019, 62(7), 3741–3752 CrossRef CAS.
- G. Davison, M. P. Martin, S. Turberville, S. Dormen, R. Heath, A. B. Heptinstall, M. Lawson, D. C. Miller, Y. M. Ng, J. N. Sanderson, I. Hope, D. J. Wood, C. Cano, J. A. Endicott, I. R. Hardcastle, M. E. M. Noble and M. J. Waring, J. Med. Chem., 2022, 65(22), 15416–15432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for synthesis and selection of the libraries and off-DNA synthesis, protein production SPR testing, crystallography and cellular pharmacology. See DOI: https://doi.org/10.1039/d3sc01171b |
| This journal is © The Royal Society of Chemistry 2023 |