
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonoarsine-protected icosahedral cluster [Au13(AsPh3)8Cl4]+: comparative studies on ligand effect and surface reactivity with its stibine analogue†
Jiu-Hong
Yu
a,
Zhi-Rui
Yuan
a,
Jing
Xu
a,
Jin-Gui
Wang
 a,
Mohammad
Azam
c,
Tian-Duo
Li
a,
Ying-Zhou
Li
*a and
Di
Sun
*b
a,
Mohammad
Azam
c,
Tian-Duo
Li
a,
Ying-Zhou
Li
*a and
Di
Sun
*b
aSchool of Chemistry and Chemical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Ji'nan, 250353, P. R. China. E-mail: liyz@qlu.edu.cn
bSchool of Chemistry and Chemical Engineering, State Key Laboratory of Crystal Materials, Shandong University, Ji'nan, 250100, P. R. China. E-mail: dsun@sdu.edu.cn
cDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 29th May 2023
Abstract
Ligand shells of gold nanoclusters play important roles in regulating their molecular and electronic structures. However, the similar but distinct impacts of the homologous analogues of the protecting ligands remain elusive. The C2v symmetric monoarsine-protected cluster [Au13(AsPh3)8Cl4]+ (Au13As8) was facilely prepared by direct reduction of (Ph3As)AuCl with NaBH4. This cluster is isostructural with its previously reported stibine analogue [Au13(SbPh3)8Cl4]+ (Au13Sb8), enabling a comparative study between them. Au13As8 exhibits a blue-shifted electronic absorption band, and this is probably related to the stronger π-back donation interactions between the Au13 core and AsPh3 ligands, which destabilize its superatomic 1P and 1D orbitals. In comparison to the thermodynamically less stable Au13Sb8, Au13As8 achieves a better trade-off between catalytic stability and activity, as demonstrated by its excellent catalytic performance towards the aldehyde–alkyne–amine (A3) coupling reaction. Moreover, the ligand exchange reactions between Au13As8 with phosphines, as exemplified by PPh3 and Ph2P(CH2)2PPh2, suggest that Au13As8 may be a good precursor cluster for further cluster preparation through the “cluster-to-cluster” route.
Introduction
The icosahedral Au13 core is considered a fundamental building block which is commonly observed as the metal core in various structurally well-defined Au nanoclusters.1–17 It was theoretically found that the electronic structures of the Au13 unit might dictate the physicochemical properties of its assembled clusters.18–21 It is therefore of importance to study the influencing factors in depth on the geometric and electronic structures of the individual icosahedral Au13 clusters. In this context, the past 15 years have first witnessed a renewed interest in the typical phosphine-protected Au13 clusters involving development of new synthetic routes (e.g., HCl-induced convergence),6,7,22,23 exploration of formation mechanisms,24,25 tuning of electronic properties (e.g., by heterometal doping26–29), and optical properties (e.g., NIR emission6,7,30 and chiroptical activity31–33), as well as further expansion of their potential applications (e.g., photodynamic catalysis and nano-electronic devices).34,35 More recently, comparative studies on several new types of Au13 analogues protected by other pnictines (e.g., functionalized diphosphine,36 phosphinous acid,37 diarsine,38 and monostibine39–41) or mimics of pnictine (N-heterocyclic carbenes)42–47 have also been extensively made both experimentally and theoretically.The large number of studies on icosahedral Au13 clusters showed that the stereoelectronic factors of the pnictine/NHC ligands have a non-negligible influence on their electronic structures.14,48–54 These ligand-based influencing factors, however, are often intertwined with other factors such as the core geometry and cluster charge state. For example, in 2021, the Konishi group synthesized a pair of dipnictine-protected clusters [Au13(dppe)5Cl2]3+ (dppe = Ph2P(CH2)2PPh2) and [Au13(dpap)5Cl2]3+ (dpap = Ph2As(CH2)3AsPh2), and demonstrated that the Au13 core geometry (deformation), rather than the coordinating P or As atoms, has a more pronounced effect on their absorption and photoluminescence properties.38 In this sense, it is necessary to employ Au13 cluster analogues, with essentially the same metal cores and ligand arrangements, as the comparable cases for better understanding the pnictine ligand effect. The monopnictine-protected Au13 clusters may serve as good candidates for this purpose since the monopnictines are usually more fluxional in the coordination sphere, and the Au13 core geometry is expected to be less affected by the ligands due to the absence of (possibly) strained Au–L′–Au staple/bridge moieties (L′ = bridging ligands). Unfortunately, few has been done with the monopnictine-protected Au13 clusters, largely due to the lack of comparable analogue clusters with known crystal structures.
To our knowledge, only four monophosphine-protected Au13 clusters have been reported, i.e., [Au13(PMe2Ph)10Cl2]3+, [Au13(PMePh2)8Cl4]+, [Au13(POct3)8Cl4]+ (POct3 = trioctylphosphine) and [Au13(L)8Cl4]+ (L = P(CH2CH2CO2CH3)3), and the last of these could further convert to Au25 nanoclusters by reacting with thiols.2,3,7,16 In contrast, much less is known about their heavier analogues. In 2018, we reported the monostibine-protected Au13 cluster [Au13(SbPh3)8Cl4]+ (Au13Sb8),39 which was found more amenable to ligand exchange and also more catalytically active than PPh3-protected Au11 clusters.40,41 Systematic studies on monoarsine-protected Au13 clusters have lagged behind, despite the structurally similar cluster [Au16(AsPh3)8Cl6] published long ago.55 We are thus intrigued by further synthesis of the AsPh3-protected Au13 clusters, so as to better understand the heavier pnictine ligand effect on their structures and reactivities by comparative studies with their heavier analogue Au13Sb8.
Herein, we present the synthesis, characterization and reactivity of the monoarsine-protected cluster [Au13(AsPh3)8Cl4]+ (Au13As8) which is isostructural with its heavier analogue Au13Sb8, in terms of the core geometry and ligand arrangement. Comparative studies between them revealed that the blue-shift electronic absorption band of Au13As8 is probably related to the stronger π-back donation interactions between the Au13 core and AsPh3 ligands, which destabilize its superatomic 1P and 1D orbitals. Moreover, Au13As8 is thermodynamically more stable than its stibine counterparts due to the moderate Au–As coordination bond strength, but its surface arsine ligands are kinetically more labile than phosphines. The unique characteristic of Au13As8 enables a better trade-off between its catalytic stability and activity, and also makes it an ideal candidate for the precursor cluster in the preparation of other clusters via the “cluster-to-cluster” strategy.
Results and discussion
Synthesis and characterization
Briefly, Au13As8 was prepared by direct reduction of (Ph3As)AuCl with 0.5 equivalent of NaBH4 in dichloromethane (DCM)–ethanol mixed solvent (Scheme 1). The identity of the precursor (Ph3As)AuCl was first confirmed prior to reduction (Fig. S1†). After reduction, the crude Au13As8 could be easily precipitated from its DCM solution by addition of hexane and is proved to be fairly pure by comparison of its IR spectrum with that of the purer crystalline Au13As8, which was obtained from its DCM–hexane solution by slow evaporation. In addition, the primary byproduct in this reaction, mainly found in the supernatant separated in the step of using hexane to precipitate the crude Au13As8, was identified as (Ph3As)3AuCl by X-ray diffraction analysis (Fig. S2†). This finding provides strong evidence that even minor alterations to the reaction conditions, such as the type of solvent or quantity of reducing agent used, can have a considerable impact on the structure of the resulting product.55 | ||
| Scheme 1 Synthetic route to Au13As8. | ||
Crystallographic data showed that Au13As8 crystallized in a monoclinic lattice with the P21/n space group (Table S1†). Two crystallographically independent molecules of Au13As8, with almost the same bond parameters, are present in the unit cell (Fig. S3†). Each comprises an icosahedral Au13 core, eight AsPh3 ligands and four Cl− ligands (Fig. 1). As is the case with its heavier analogue Au13Sb8,39 the ligands in Au13As8 also arranged in an approximate C2v symmetry. The radial Au–Au bond lengths range from 2.726(2) Å to 2.810(2) Å (2.757(2) Å on average), while the peripheral ones are in the range of 2.808(2)–2.985(2) Å (2.900(2) Å on average). The Au–Cl bond lengths fall in the range of 2.31(1)–2.37(1) Å. The Au–As bond lengths range from 2.392(4) Å to 2.414(6) Å, which is longer than that in its precursor complex (Ph3As)AuCl (2.334(1) Å). As expected, these Au–Au and Au–Cl bond lengths in Au13As8 are also quite close to the corresponding ones in Au13Sb8 (Table S2†).
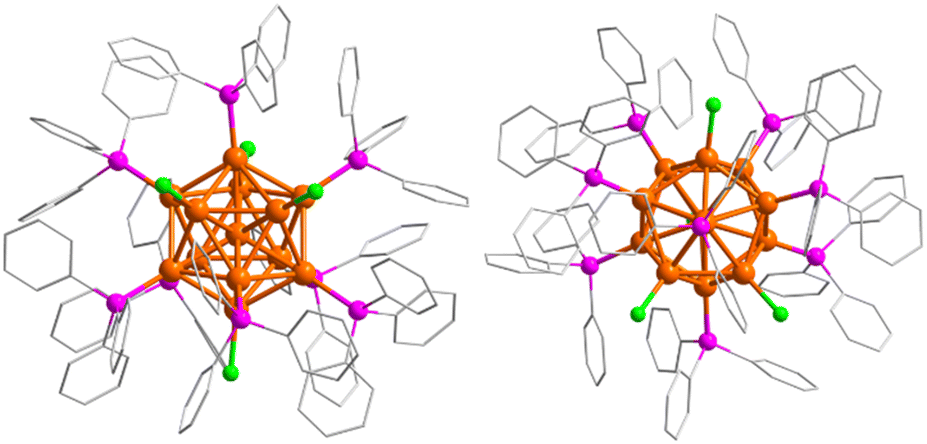 | ||
| Fig. 1 Crystal structure of Au13As8 with all hydrogen atoms omitted (left: side view; right: top view). Au: orange, As: magenta, Cl: green, C: grey. | ||
The mass spectrum of Au13As8 shows a major peak at m/z = 5151.7672, attributed to the molecular ion [Au13(AsPh3)8Cl4]+ (calcd = 5151.7738), which matches well with its calculated one; the isotopic interval of 1.0 amu support its +1 charge (Fig. 2). Besides, the 31P NMR of the product shows a heptet resonance ascribable to PF6− (Fig. S4†), and its IR spectrum also shows the characteristic absorptions of PF6− at 839 and 557 cm−1 (Fig. S5†). All these indicate that Au13As8 bears +1 charge, and the counterion is most probably PF6−, though it fails to be appropriately located crystallographically, probably due to its severe disorder.
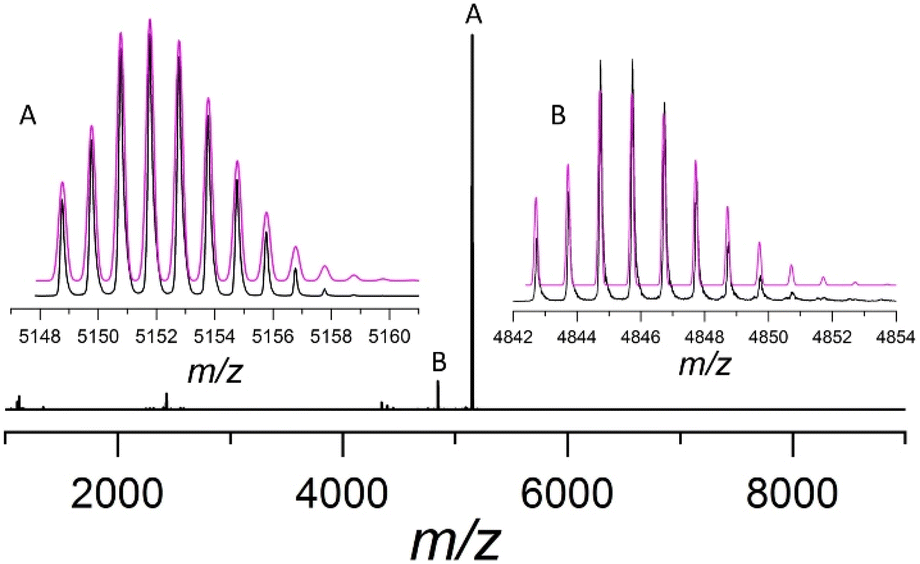 | ||
| Fig. 2 ESI-MS(+) spectrum of Au13As8 in CH2Cl2. Inset: the experimental (black trace) and calculated (magenta trace) isotopic patterns of the Au13As8 ion (A) [Au13(AsPh3)8Cl4]+ and its fragment ion (B) [Au13(AsPh3)7Cl4]+. | ||
The 1H NMR spectrum of Au13As8 shows three doublet resonances in the lower magnetic field (ca. 7.10–7.50 ppm) region in an integration ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, and with a pattern quite akin to that of Au13Sb8 (Fig. S6†). This suggests the presence of three types of chemically non-equivalent AsPh3 ligands, which is consistent with its solid-state structure; this also further confirms that the icosahedral Au13 clusters are stereochemically more rigid than the phosphine-protected Au11 clusters.56,57
:2:1, and with a pattern quite akin to that of Au13Sb8 (Fig. S6†). This suggests the presence of three types of chemically non-equivalent AsPh3 ligands, which is consistent with its solid-state structure; this also further confirms that the icosahedral Au13 clusters are stereochemically more rigid than the phosphine-protected Au11 clusters.56,57
Theoretical calculations
Since the Au13As8 and Au13Sb8 are isostructural either in the solid state or in solution, they constitute a good comparable pair for comparatively studying the heavier pnictine ligand effect on their structures and reactivities. As shown in Fig. 3, the experimental electronic absorptions for Au13As8 appear at 346 and 439 nm, which are blue-shifted by 6 and 15 nm, respectively, as compared to those for Au13Sb8 at 352 and 454 nm. Interestingly, the absorptions for the previously reported monophosphine-protected Au13 clusters are further blue-shifted (Fig. S7†).2,3,7 Given the more similar structures of Au13As8 and Au13Sb8, TD-DFT calculations for them were performed at the same level of theory, to reveal the stereoelectronic influences of the pnictine ligands on the absorptions; the metal cores in both after structure relaxation are more of C2v symmetry, probably transferred from the ligand shells and reflected by the continuous symmetry measure (CSM) value of 0.00 for both.58 The optimized Au–Au bond lengths in Au13As8 are also slightly longer than those in Au13Sb8 (Table S3†), which is consistent with the crystallographic data. The calculated UV-vis spectra of them are well matched with their respective experimental ones, despite a small blue-shift, probably due to the overestimation of the excitation energies for the functional employed (Fig. S8 and S9†). Thus, the calculated peaks at 335 and 417 nm for Au13As8 (340 and 435 nm for Au13Sb8) should correspond to its experimental ones at 346 and 439 nm (352 and 454 nm for Au13Sb8), respectively.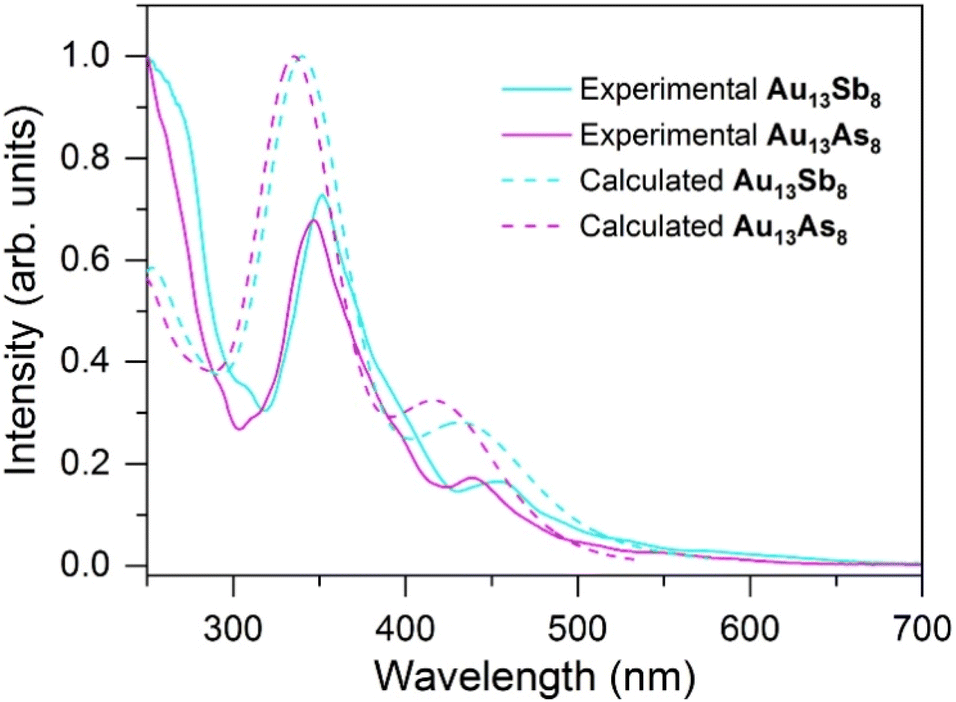 | ||
| Fig. 3 Experimental (solid lines) and TD-DFT calculated (dash lines) UV-vis spectra for Au13As8 (magenta traces) and Au13Sb8 (cyan traces) in CH2Cl2. | ||
Au13As8 and Au13Sb8 possess a similar electronic structure, with a HOMO–LUMO gap of 3.32 and 3.09 eV, respectively. The HOMO, HOMO − 1 and HOMO − 2 orbitals are typical of 1P superatomic orbitals while LUMO through LUMO + 4 show 1D characteristics (Tables S4 and S5†). The long-wavelength absorptions (417 nm for Au13As8 and 435 nm for Au13Sb8) are mainly ascribable to metal-to-metal charge transfer (MMCT) transitions from 1P to 1D shell, while the shorter-wavelength absorptions (335 nm for Au13As8 and 340 nm for Au13Sb8) are mainly attributed to ligand-to-metal charge transfer (LMCT) transitions to 1P orbitals from higher-lying occupied orbitals (HOMO − 3 to ca. HOMO − 10). In addition, the absorptions below 300 nm for both result from the even higher-energy transitions, such as HOMO − n (n > 15) to 1D, or HOMO − n (n < 5) to LUMO + n (n > 10); the phenyl rings in the ligands contributed significantly to HOMO − n (n > 15) orbitals with energy lower than −8.2 eV, and LUMO + n (n > 10) orbitals with energy higher than −2.2 eV (Fig. 4a, b and Tables S6, S7†).
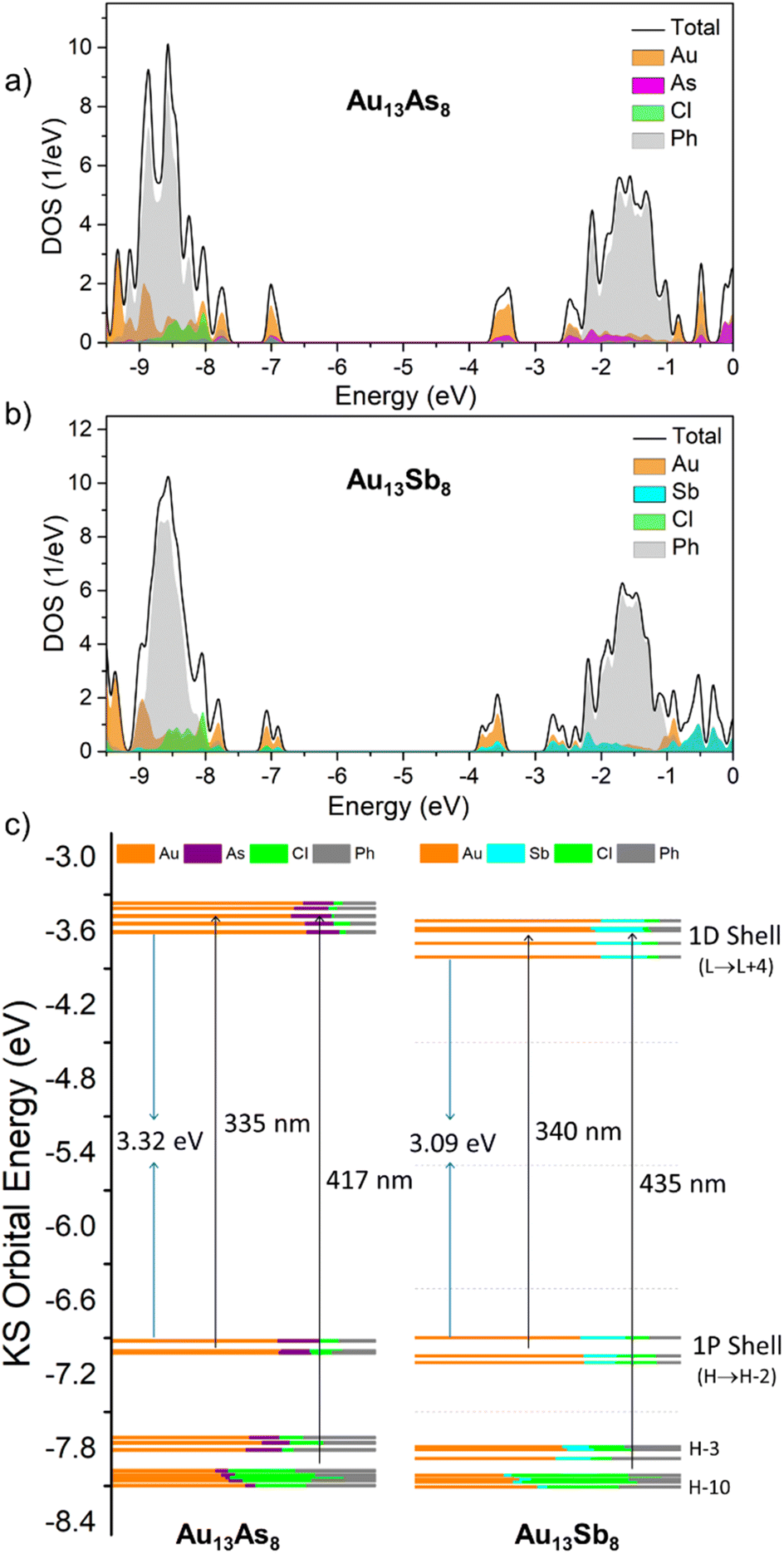 | ||
| Fig. 4 Total density-of-states (TDOS) and partial density-of-states (PDOS) spectra of (a) Au13As8, (b) Au13Sb8, and (c) their selected Kohn–Sham molecular orbitals showing the populations of atomic orbitals. | ||
As shown in Fig. 4c, on average, the 1P orbitals of Au13As8 comprise Au 5d/6sp orbitals (ca. 64%), As 4sp orbitals (ca. 13%), Cl 3sp orbitals (ca. 9%) and C 2sp orbitals (ca. 14%). The 1D orbitals are also mainly composed of Au atomic orbitals (Au:As:Cl:C = 72:13:3:12). The proportions of Au and As in orbitals from HOMO − 3 to HOMO − 10 decreased to a certain extent (48% for Au and 7% for As), but those of Cl and phenyl rings increased (21% for Cl and 24% for C). This means that, for a given type of cluster [Au13(ER3)8Cl4]+ (E = pnictogen), the R substituent of ER3 and the inorganic Cl− may less affect the longer-wavelength absorptions resulting from 1P to 1D transitions, due to their less contributions to 1P/D orbitals; in other words, the shorter-wavelength absorptions are more susceptible to the stereoelectronic property of the ER3 ligand. Consistent with this are the experimental UV-vis spectra of [Au13(L)8Cl4]+ (L = PPh2Me; P(CH2CH2CO2CH3)3),3,16 which show almost the same absorption peaks at 432 nm in the lower-energy region but obviously different peaks at 342 nm and 333 nm, respectively, in the higher-energy region (Fig. S7†).
The orbital composition pattern of Au13Sb8 is akin to that of Au13As8. The 1P orbitals are composed of Au 5d/6sp (ca. 63%), Sb 5sp (ca. 13%), Cl 3sp (ca. 13%) and C 2sp (ca. 12%) orbitals. The 1D orbitals also mainly comprise Au atomic orbitals (Au:Sb:Cl:C = 67:17:4:11). The proportions of Au and As from HOMO − 3 to HOMO − 10 also decreased (44% for Au and 6% for Sb), but those of Cl and phenyl rings increased (29% for Cl and 20% for C). In comparison, the superatomic 1P, 1D and the higher-lying occupied orbitals (e.g. HOMO − 3 to HOMO − 10) in Au13As8 all up-shifted relative to their counterparts in Au13Sb8, except for the HOMO in Au13As8, which is marginally lower in energy by 0.025 eV than that in Au13Sb8.
Previous studies with thiol/alkynyl-protected Au25 clusters based on the jellium model assumed that the more electronegative (usually X-type) ligands bonded to the Au13 core might lead to the reduction of potential volume for the confinement of Au13 core valence electrons, and result in the destabilization of the superatomic 1P orbitals.15 In this work, the L-type two-electron-donor pnictine ligands are more suitable to be classified by overall electron-donating ability. AsPh3 is a stronger σ-donating and also a better π-accepting ligand than SbPh3;59 it is thus envisioned that both interactions may influence the superatomic cluster orbitals. The π-back donation from the Au core to the π-acid ligand is not uncommon, for instance, Au(5d) → CO(π*) in the carbonyl-protected Au cluster,60 and Au(5d) → N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(π*) in the guanine-protected Au13 cluster.61
C(π*) in the guanine-protected Au13 cluster.61
To get more insight into this, the natural bond orbital (NBO) analyses were performed. The qualitative natural population analyses (NPA) show that the Au13 core in Au13As8 is less negatively charged than that in Au13Sb8 (−0.50 vs. −1.43, Table S9†), as is also the case with the Mulliken charge analysis (−1.95 vs. −2.27, Table S10†). This implies that, given the stronger σ-donating ability of AsPh3, a stronger electron back donation from the Au13 core to AsPh3 may contribute to the relatively lower electron density for the Au13 core in the case of Au13As8. As expected, the second-order perturbation theory analyses in NBO basis for Au13As8 show strong interactions between the central Au lone pair orbitals and the radial Au–As σ* orbitals, as reflected by the large second-order perturbation stabilization energies E(2) (Table S11†). In comparison, this type of interaction in Au13Sb8 is much weaker (Table S12†). This is probably associated with the fact that the As 4sp orbitals involved in the Au–As σ* orbitals are less diffuse, lower in energy, and thus more electrophilic, compared to the Sb 5sp orbitals in Au–Sb σ* orbitals. Since the stronger π-back donation interactions between Au and Au–As σ* orbitals would reduce the Au–As bond orders more significantly, the Wiberg bond indices for the Au–As bonds in Au13As8 are indeed smaller (ca. 0.46) than those for the Au–Sb bonds in Au13Sb8 (ca. 0.54), whereas those for the innocent Au–Cl bonds in both are almost the same (ca. 0.42) (Table S13†). In addition, other types of π-back donation interactions between surface Au lone pairs and As/Sb–C σ* orbitals are rather weak and comparable for both Au13As8 and Au13Sb8 (Tables S11 and S12†). Based on these, the upshift of 1P orbitals in Au13As8 relative to that in Au13Sb8 is tentatively accounted for by the stronger π-back donation interactions between central Au and radial Au–As σ* orbitals, by which the better π-accepting L-type ligand AsPh3 exerts a similar impact on the Au13 core valence electrons to that for the case of the more electronegative X-type ligand.
On the other hand, the correlations of 1D orbitals with the electronegativity of ligands are rather complicated, though they shift to the same direction as 1P orbitals in most cases but lack an obvious trend in the shift amount.15 In this work, the 1D orbitals in Au13As8 up-shifted more (by 0.156 eV on average) than 1P orbitals (by 0.031 eV on average). That is to say, the 1D orbitals are more susceptible to the electronic factors of the pnictine ligands. In view of this, more comparative studies, including with the lighter phosphine-protected analogue clusters, may be of interest to better explain the behavior of 1D orbitals with respect to the integrated σ-donating/π-accepting parameters of the L-type pnictine ligands.
Optical properties
The solution photoluminescence (PL) of Au13As8 emits at ca. 802 nm, which is red-shifted in wavelength and also higher in intensity, as compared to the main emission of Au13Sb8 at ca. 740 nm (Fig. 5). This is presumably due to the heavy metal effect of Sb, which facilitates the reverse intersystem crossing (RISC) process from the T1 state to the S1 state and therefore leads to the blue-shift and lower intensity of the emission for Au13Sb8.62,63 The previously reported two phosphine-protected clusters [Au13(LP)nCl4]+ (LP = trioctylphosphine (POct3), n = 8; LP = Ph2P(CH2)3PPh2, n = 4) both emit at ca. 775 nm.7 Considering the likely dual emissions of Au13Sb8, if the shoulder at ca. 825 nm is of the same phosphorescence character as the emissions of Au13As8 and [Au13(LP)nCl4]+, the emission wavelengths would be in the order of Au13Sb8 > Au13As8 > [Au13(LP)nCl4]+.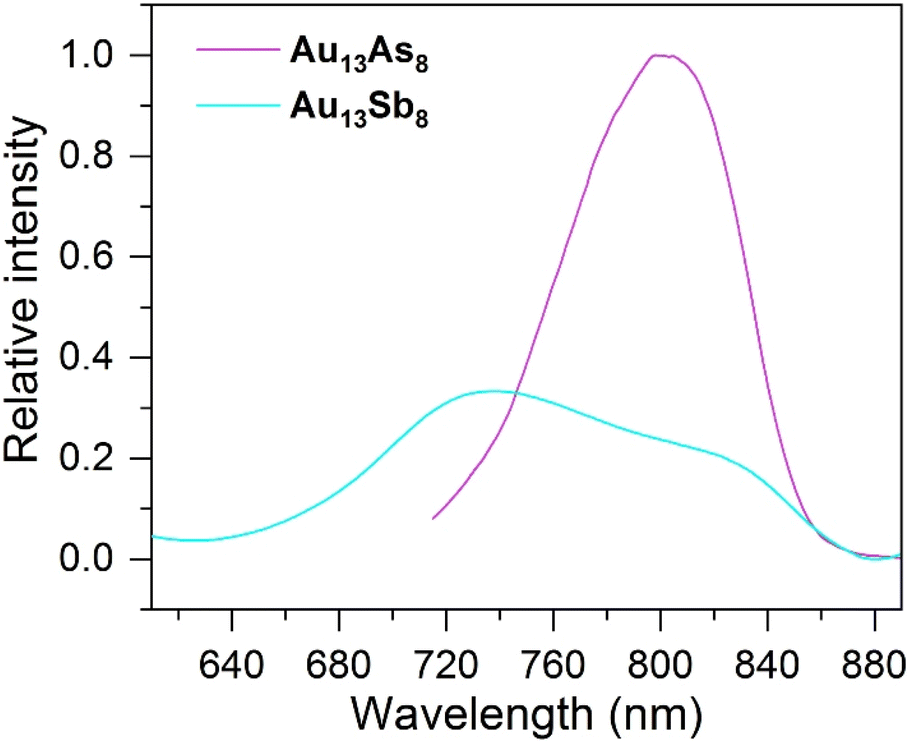 | ||
| Fig. 5 Photoluminescence spectra for Au13As8 (magenta line, λex = 460 nm) and Au13Sb8 (cyan line, λex = 470 nm) in CH2Cl2 (ca. 2.0 mg mL−1) at room temperature. | ||
Au13As8 in its crystalline state exhibits a PL emission over a wider range of wavelengths (from ca. 650 nm to ca. 850 nm), and the emission maximum at ca. 740 nm is blue-shifted by ca. 60 nm compared to that in solution. The PL emission of Au13Sb8 in the crystalline state displays a similar but also blue-shifted weak dual-emission-like pattern to that in solution, with an emission peak centered at ca. 805 nm and a shoulder at ca. 710 nm (Fig. S10†). In general, the PL blue-shift for both can be partially attributed to the rigidity enhancement of the structures in the solid state. In addition, the largely broadened and blue-shifted solid PL spectrum of Au13As8, somewhat resembling that of Au13Sb8, also suggest that a similar dual-emission phenomenon to that of Au13Sb8 may be present in its solid/crystalline state.
Thermal stability
The Au13As8 is rather stable in the solid state, but gradually decomposes with time in solution at room temperature, although such a process is much slower than for the case of Au13Sb8. Lower temperature could effectively inhibit the decomposition of Au13As8; its UV-vis monitoring spectra showed no essential decomposition even after 12 days when its DCM solution was stored at 4 °C (Fig. S11†). Thermogravimetric analysis (TGA) of Au13As8 at a heating rate of 10 °C min−1 under N2 purge (60 mL min−1) showed that the rapid weight loss started at ca. 200 °C, whereas that for Au13Sb8 was observed at 155 °C (Fig. S12†).39 These results suggest that Au13As8 is more stable than Au13Sb8 either in solution or in the solid state but most probably still less stable than the phosphine-protected analogue clusters; this is believed to result from the moderate Au–As bond strength.Surface reactivity-catalysis
Since the electron-rich Au13 core was speculated to activate the CO bond,36,64,65 the pnictine-protected Au13 clusters may find uses in the aldehyde/ketone-involved catalytic reactions. Au13Sb8 was found to be able to efficiently catalyze the aldehyde–alkyne–amine (A3) coupling reaction, but the drawback of lower catalytic stability could also not be neglected.40 To better realize the trade-off between stability and activity, the catalytic performance of Au13As8 towards the same A3 coupling reaction was tested (Table 1). It was found that, despite its relatively stronger Au–As coordination bonds, Au13As8 could also achieve a comparable conversion ratio to Au13Sb8 after 12 h, probably associated with different reaction kinetics. More importantly, Au13As8 is catalytically more stable than Au13Sb8; it can be simply recovered (and also purified) from the reaction mixture by precipitation with hexane (Fig. S13†), and can be reused for five cycles with only slight decrease in catalytic activity (Fig. S14 and S15†). This finding further underlines the important role of ligands in tuning the catalytic performance and may provide some implications for the design of more practically useful ligand-protected metal nanocluster catalysts.
|
|
|||||
|---|---|---|---|---|---|
| Catalystb | NMR yield (%) | ||||
| Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 5 | |
| a Benzaldehyde (1.0 mmol), piperidine (1.2 mmol), phenylacetylene (1.3 mmol), solvent-free reaction. b With respect to the amount of benzaldehyde. c Given the obvious decomposition in each cycle for Au13Sb8, no further trial was made after the second cycle due to the marked decrease in yield. | |||||
| Au13As8 | 73 | 74 | 69 | 64 | 62 |
| Au13Sb8 | 81 | 32 | — | — | — |
Surface reactivity-ligand exchange
The exquisite “cluster-to-cluster” synthetic strategy based on ligand exchange has recently become increasingly prevailing.47,66,67 Taking Au13 clusters as examples, reactivity studies on Au13Sb8 or its bromo-analogue [Au13(SbPh3)8Br4]+ had shown that they could readily react with glutathione (GSH) or 1-adamantanethiol (S-Adm) to give the achiral Au25(SG)18 or chiral Au18(SAdm)8(SbPh3)4Br2, respectively.39,41 The Wang group also developed a new method of synthesizing thiol-protected Au25 clusters through reactions of [Au13(L′′)8Cl4]+ (L′′ = P(CH2CH2CO2CH3)3) with appropriate thiols.16 In analogy to these stibine or phosphine-protected Au13 clusters, Au13As8 may also possess the potential as a precursor cluster in the syntheses of other new clusters by reacting with stronger coordinating ligands. Thus, the reactions of Au13As8 with monophosphine PPh3 or diphosphine Ph2P(CH2)2PPh2 (dppe) at room temperature were carried out as a proof-of-concept in this work. As shown in Scheme 2, two well-known clusters [Au11(PPh3)8Cl2]+ and [Au13(dppe)5Cl2]3+ were obtained in the cases of PPh3 and dppe, respectively, and their structures were determined by comparison of their UV-vis, 1H NMR and MS spectra with those from literature (Fig. S16–S20†).7,68 These preliminary trials confirm the potential use of Au13As8 in the “cluster-to-cluster” syntheses. In comparison, the reactions with Au13As8 proceed more slowly than with Au13Sb8, indicating that it is more likely for the former to realize more controlled derivatization or functionalization by ligand engineering.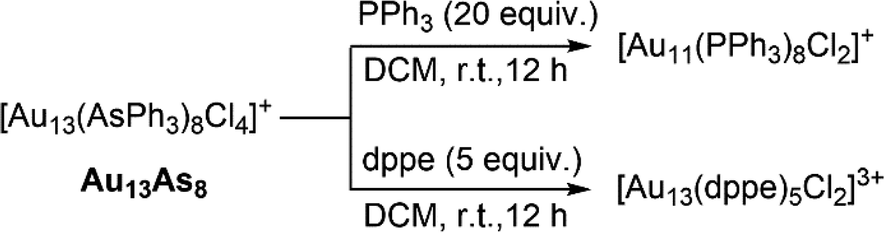 | ||
| Scheme 2 Reactions of Au13As8 with phosphines. | ||
Conclusions
In this work, a monoarsine-protected icosahedral cluster [Au13(AsPh3)8Cl4]+ (Au13As8) was prepared. It is isostructural with its previously reported stibine analogue [Au13(SbPh3)8Cl4]+ (Au13Sb8), which provides an opportunity to better comparatively study the heavier pnictine ligand effect on their electronic structures, optical properties, and surface reactivities. The influencing mechanism of the L-type pnictine ligands on the Au13 superatomic orbitals may be different from those of the X-type ligands (e.g., organic thiol, selenol, alkynyl). The superatomic 1P and 1D orbitals of Au13As8 are up-shifted relative to those of Au13As8, which is tentatively ascribed to the stronger π-back donation interactions between the Au13 core and AsPh3 ligands. In addition, compared to the thermodynamically less stable Au13Sb8, a better trade-off between stability and activity was realized with Au13As8, as reflected by its good catalytic performance towards the A3 coupling reaction. Finally, the ligand exchange reactions between Au13As8 with phosphines, as exemplified by PPh3 and Ph2P(CH2)2PPh2, suggest that Au13As8 may be a good precursor cluster and may find more use in the preparation of other clusters via the prevailing “cluster-to-cluster” strategy. Taken together, the heavier organometalloidal arsines or organometallic stibines, especially the latter, are not simply “weaker organic phosphines”, and they are more likely to exert distinct ligand effects on the structures and properties of metal nanoclusters. Since there has been only a limited number of heavier pnictine-protected metal nanoclusters, more diverse syntheses of such type of clusters, as well as explorations of their physicochemical properties and applications, are of much interest and deserve deeper scrutiny.Data availability
All experimental and computational data associated with this article have been included in the main text and ESI.†Author contributions
Y.-Z. L. and D. S. conceived the manuscript; J. H. Y. performed the experiments; J.-H. Y., Z.-R. Y., J. X., J.-G. W., M. A., T.-D. L. and Y.-Z. L. analyzed data, prepared figures and provided conceptual contributions; J. H. Y., Y. Z. L. and D. S. wrote the manuscript with contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Natural Science Foundation of China (no. 22001139, 22178184, and 52072190), Natural Science Foundation of Shandong Province (no. ZR2022MB099), the Taishan Scholar Project of Shandong Province of China (no. tsqn201812003 and ts20190908), the Fok Ying Tong Education Foundation (171009), the National Postdoctoral Innovative Talents Support Program (No. BX2021171), the China Postdoctoral Science Foundation (No. 2021M700081), and the Science, Education and Industry Integration Pilot Project Program from Qilu University of Technology (Shandong Academy of Science) (no. 2022PY066 and 2022JBZ02-04). Dr M. Azam also acknowledges the financial support through the Researchers Supporting Project (RSP2023R147) at King Saud University, Riyadh, Saudi Arabia.Notes and references
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334 RSC.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- R. C. B. Copley and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1996, 491–500 RSC.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- Y. Shichibu, K. Suzuki and K. Konishi, Nanoscale, 2012, 4, 4125–4129 RSC.
- J.-i. Nishigaki, S. Yamazoe, S. Kohara, A. Fujiwara, W. Kurashige, Y. Negishi and T. Tsukuda, Chem. Commun., 2014, 50, 839–841 RSC.
- X. K. Wan, Q. Tang, S. F. Yuan, D. E. Jiang and Q. M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef PubMed.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS PubMed.
- L. Liao, S. Zhuang, C. Yao, N. Yan, J. Chen, C. Wang, N. Xia, X. Liu, M. B. Li, L. Li, X. Bao and Z. Wu, J. Am. Chem. Soc., 2016, 138, 10425–10428 CrossRef CAS PubMed.
- K. Weerawardene, P. Pandeya, M. Zhou, Y. Chen, R. Jin and C. M. Aikens, J. Am. Chem. Soc., 2019, 141, 18715–18726 CrossRef CAS PubMed.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed.
- Z. Lei, J. J. Li, Z. A. Nan, Z. G. Jiang and Q. M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14415–14419 CrossRef CAS PubMed.
- T. Wang, W.-H. Zhang, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Chem. Commun., 2018, 54, 10367–10370 RSC.
- M. Zhou, R. Jin, M. Y. Sfeir, Y. Chen, Y. Song and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4697–E4705 CrossRef CAS PubMed.
- K. Nobusada and T. Iwasa, J. Phys. Chem. C, 2007, 111, 14279–14282 CrossRef CAS.
- D.-e. Jiang, K. Nobusada, W. Luo and R. L. Whetten, ACS Nano, 2009, 3, 2351–2357 CrossRef CAS PubMed.
- C. Xu, Y. Zhou, J. Yi, D. Li, L. Shi and L. Cheng, J. Phys. Chem. Lett., 2022, 13, 1931–1939 CrossRef CAS PubMed.
- S. Jin, W. Du, S. Wang, X. Kang, M. Chen, D. Hu, S. Chen, X. Zou, G. Sun and M. Zhu, Inorg. Chem., 2017, 56, 11151–11159 CrossRef CAS PubMed.
- S. Zhang, L. Feng, R. Senanayake, C. M. Aikens, X.-P. Wang, Q. Zhao, C.-H. Tung and D. Sun, Chem. Sci., 2018, 9, 1251–1258 RSC.
- L. Yang, H. Cheng, Y. Jiang, T. Huang, J. Bao, Z. Sun, Z. Jiang, J. Ma, F. Sun, Q. Liu, T. Yao, H. Deng, S. Wang, M. Zhu and S. Wei, Nanoscale, 2015, 7, 14452–14459 RSC.
- L. Yang, L. Huang, X. Song, W. He, Y. Jiang, Z. Sun and S. Wei, Acta Phys.-Chim. Sin., 2018, 34, 762–769 CAS.
- Y. Lv, X. Kang, S. Yang, T. Chen, A. Liu, H. Yu and M. Zhu, RSC Adv., 2017, 7, 51538–51545 RSC.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem., 2020, 59, 17889–17895 CrossRef CAS PubMed.
- N. Fedik, A. I. Boldyrev and A. Munoz-Castro, Phys. Chem. Chem. Phys., 2019, 21, 25215–25219 RSC.
- J. Wei, P. L. Rodriguez-Kessler, J. F. Halet, S. Kahlal, J. Y. Saillard and A. Munoz-Castro, Inorg. Chem., 2021, 60, 8173–8180 CrossRef CAS PubMed.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS PubMed.
- Y. Yang, Q. Zhang, Z.-J. Guan, Z.-A. Nan, J.-Q. Wang, T. Jia and W.-W. Zhan, Inorg. Chem., 2019, 58, 3670–3675 CrossRef CAS PubMed.
- Y. Shichibu, Y. Ogawa, M. Sugiuchi and K. Konishi, Nanoscale Adv., 2021, 3, 1005–1011 RSC.
- H. Hirai, T. Nakashima, S. Takano, Y. Shichibu, K. Konishi, T. Kawai and T. Tsukuda, J. Mater. Chem. C, 2023, 11, 3095–3100 RSC.
- J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787–5798 CrossRef CAS.
- Z. Wei, W. Jiang, Z. Bai, Z. Lian, Z. Wang and F. Song, Eur. Phys. J. D, 2017, 71, 237 CrossRef.
- J.-Q. Fan, Y. Yang, C.-B. Tao and M.-B. Li, Angew. Chem., Int. Ed., 2023, 62, e202215741 CAS.
- S. Zhuang, D. Chen, W.-P. Ng, D. Liu, L.-J. Liu, M.-Y. Sun, T. Nawaz, X. Wu, Y. Zhang, Z. Li, Y.-L. Huang, J. Yang, J. Yang and J. He, JACS Au, 2022, 2, 2617–2626 CrossRef CAS PubMed.
- Y. Shichibu, F. Zhang, Y. Chen, M. Konishi, S. Tanaka, H. Imoto, K. Naka and K. Konishi, J. Chem. Phys., 2021, 155, 054301 CrossRef CAS PubMed.
- Y.-Z. Li, R. Ganguly, K. Y. Hong, Y. Li, M. E. Tessensohn, R. Webster and W. K. Leong, Chem. Sci., 2018, 9, 8723–8730 RSC.
- Y.-Z. Li and W. K. Leong, RSC Adv., 2019, 9, 5475–5479 RSC.
- J. B. Patty, S. Havenridge, D. Tietje-Mckinney, M. A. Siegler, K. K. Singh, R. Hajy Hosseini, M. Ghabin, C. M. Aikens and A. Das, J. Am. Chem. Soc., 2022, 144, 478–484 CrossRef CAS PubMed.
- Q. Tang and D.-e. Jiang, Chem. Mater., 2017, 29, 6908–6915 CrossRef CAS.
- A. Muñoz-Castro, Inorg. Chem. Front., 2019, 6, 2349–2358 RSC.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed.
- H. Shen, S. Xiang, Z. Xu, C. Liu, X. Li, C. Sun, S. Lin, B. K. Teo and N. Zheng, Nano Res., 2020, 13, 1908–1911 CrossRef CAS.
- H. Yi, K. M. Osten, T. I. Levchenko, A. J. Veinot, Y. Aramaki, T. Ooi, M. Nambo and C. M. Crudden, Chem. Sci., 2021, 12, 10436–10440 RSC.
- M. Bevilacqua, M. Roverso, S. Bogialli, C. Graiff and A. Biffis, Inorg. Chem., 2023, 62, 1383–1393 CrossRef CAS PubMed.
- A. Wing-Bocanegra and A. Tlahuice-Flores, Phys. Chem. Chem. Phys., 2019, 21, 23855–23864 RSC.
- Z.-H. Gao, J. Dong, Q.-F. Zhang and L.-S. Wang, Nanoscale Adv., 2020, 2, 4902–4907 RSC.
- P. L. Rodríguez-Kessler, M. Rojas-Poblete and A. Muñoz-Castro, Phys. Chem. Chem. Phys., 2021, 23, 18035–18043 RSC.
- A. Munoz-Castro, Phys. Chem. Chem. Phys., 2022, 24, 17233–17241 RSC.
- J. Wei, S. Kahlal, J. F. Halet, J. Y. Saillard and A. Munoz-Castro, J. Phys. Chem. A, 2022, 126, 536–545 CrossRef CAS PubMed.
- G. E. Johnson, A. Olivares, D. Hill and J. Laskin, Phys. Chem. Chem. Phys., 2015, 17, 14636–14646 RSC.
- G. Shafai, S. Hong, M. Bertino and T. S. Rahman, J. Phys. Chem. C, 2009, 113, 12072–12078 CrossRef CAS.
- M. Richter and J. Strähle, Z. Anorg. Allg. Chem., 2001, 627, 918–920 CrossRef CAS.
- B. S. Gutrath, U. Englert, Y. Wang and U. Simon, Eur. J. Inorg. Chem., 2013, 2013, 2002–2006 CrossRef CAS.
- S. Mukherjee, A. Das, A. K. Das, A. Sheriff, K. Sunny, A. S. Nair, S. Bhandary, R. Bhowal, D. Chopra, B. Pathak, S. Yamazoe and S. Mandal, Chem. Mater., 2023, 35, 1659–1666 CrossRef CAS.
- I. Tuvi-Arad, G. Alon and D. Avnir, CoSyM, http://csm.ouproj.org.il Search PubMed.
- N. J. Holmes, W. Levason and M. Webster, J. Chem. Soc., Dalton Trans., 1998, 3457–3462 RSC.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Am. Chem. Soc., 2005, 127, 8416–8423 CrossRef CAS PubMed.
- X. Zhang, C. Q. Sun and H. Hirao, Phys. Chem. Chem. Phys., 2013, 15, 19284–19292 RSC.
- X. Cao, K. Pan, J. Miao, X. Lv, Z. Huang, F. Ni, X. Yin, Y. Wei and C. Yang, J. Am. Chem. Soc., 2022, 144, 22976–22984 CrossRef CAS PubMed.
- X. Jiang, T. Zhang, C. Sun, Y. Meng and L. Xiao, Chin. Chem. Lett., 2019, 30, 1055–1058 CrossRef CAS.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS PubMed.
- Y. Pei, N. Shao, Y. Gao and X. C. Zeng, ACS Nano, 2010, 4, 2009–2020 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- W. Suzuki, R. Takahata, Y. Chiga, S. Kikkawa, S. Yamazoe, Y. Mizuhata, N. Tokitoh and T. Teranishi, J. Am. Chem. Soc., 2022, 144, 12310–12320 CrossRef CAS PubMed.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational and crystallographic details, and spectroscopic characterization including IR, ESI-MS, UV-vis, and NMR. CCDC 2244318 and 2244690. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01311a |
| This journal is © The Royal Society of Chemistry 2023 |