
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColossal negative thermal expansion in a cucurbit[8]uril-enabled uranyl-organic polythreading framework via thermally induced relaxation†
Qiu-Yan
Jin
ab,
Yuan-Yuan
Liang
ab,
Zhi-Hui
Zhang
 *c,
Liao
Meng
a,
Jun-Shan
Geng
a,
Kong-Qiu
Hu
a,
Ji-Pan
Yu
a,
Zhi-Fang
Chai
a,
Lei
Mei
*a and
Wei-Qun
Shi
*a
*c,
Liao
Meng
a,
Jun-Shan
Geng
a,
Kong-Qiu
Hu
a,
Ji-Pan
Yu
a,
Zhi-Fang
Chai
a,
Lei
Mei
*a and
Wei-Qun
Shi
*a
aLaboratory of Nuclear Energy Chemistry, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China. E-mail: meil@ihep.ac.cn; shiwq@ihep.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University, Changzhou 213164, China. E-mail: zhangzh@cczu.edu.cn
First published on 10th May 2023
Abstract
It is an ongoing goal to achieve the effective regulation of the thermal expansion properties of materials. In this work, we propose a method for incorporating host–guest complexation into a framework structure and construct a flexible cucurbit[8]uril uranyl-organic polythreading framework, U3(bcbpy)3(CB8). U3(bcbpy)3(CB8) can undergo huge negative thermal expansion (NTE) and has a large volumetric coefficient of −962.9 × 10−6 K−1 within the temperature range of 260 K to 300 K. Crystallographic snapshots of the polythreading framework at various temperatures reveal that, different from the intrinsic transverse vibrations of the subunits of metal–organic frameworks (MOFs) that experience NTE via a well-known hinging model, the remarkable NTE effect observed here is the result of a newly-proposed thermally induced relaxation process. During this process, an extreme spring-like contraction of the flexible CB8-based pseudorotaxane units, with an onset temperature of ∼260 K, follows a period of cumulative expansion. More interestingly, compared with MOFs that commonly have relatively strong coordination bonds, due to the difference in the structural flexibility and adaptivity of the weakly bonded U3(bcbpy)3(CB8) polythreading framework, U3(bcbpy)3(CB8) shows unique time-dependent structural dynamics related to the relaxation process, the first time this has been reported in NTE materials. This work provides a feasible pathway for exploring new NTE mechanisms by using tailored supramolecular host–guest complexes with high structural flexibility and has promise for the design of new kinds of functional metal–organic materials with controllable thermal responsive behaviour.
Introduction
The thermal expansion of solid-state components,1,2 is a ubiquitous and unavoidable effect that occurs upon heating. It is a critical issue in materials with large structures and in precision scientific instruments because it usually leads to problems within these architectures and, as a consequence, the failure of the devices. The control and regulation of the thermal expansion effect helps to improve the reliability of devices and prolong their service life. Negative thermal expansion (NTE)3–8 is a counter-intuitive but intriguing physical phenomenon where the cell volume of a material undergoes contraction upon heating, as observed for the density of water within the temperature range 0 °C to 4 °C. The utilization of NTE provides a promising method to inhibit and eventually eliminate undesired thermal expansion through rational combination of both NTE and positive thermal expansion (PTE) components to achieve near zero thermal expansion (ZTE).9–13The NTE in framework-structured materials4,14 is mainly driven by low-frequency phonons, i.e. the rocking or tilting motions of rigid structural units (metal polyhedra or clusters) induced by transverse vibration of flexible bridging atoms or groups after the temperature increases. However, a different electron-driven mechanism is also observed in functional materials that undergo temperature-related electron changes, such as semiconductors, Mott insulators and superconductors.7 Since the first comprehensive research on the NTE properties of the ZrW2O8 framework by Sleight et al.,15 an increasing number of framework-structured NTE materials have been developed over the past two decades, ranging from oxides to fluorides10,16,17 and molecular framework materials, including Prussian blue analogues18 (PBAs, mostly metal cyanides19–23) and metal–organic frameworks (MOFs).24–34 Compared with well-studied inorganic NTE materials, especially oxides, NTE materials with organic structures have not been fully developed, and more work on the optimization of the NTE properties of these materials needs to be carried out.
For the phonon-driven mechanism specifically, it is observed that the NTE properties of a framework-structured material is largely affected by its chemical structure, where larger frameworks and/or more flexible bridging atoms/groups can facilitate stronger NTE.3,4 For example, different from inorganic framework-structured materials that have relatively strong linkages, organic–inorganic hybrid frameworks such as MOFs exhibit more complex vibrational modes and significantly enhanced NTE since they are constructed by organic linkers with higher flexibility.24–34 On the other hand, there is still room for improvement of the NTW properties of organic–inorganic hybrid frameworks when compared with weak-bonding molecule-based NTE materials.35–37 It is expected that the incorporation of weak interactions such as hydrogen bonds, π–π interactions and ion–dipole interactions into a framework could enhance its flexibility greatly and improve the NTE properties. Along these lines, a feasible method to achieve this is to develop weakly bonded supramolecular framework structures through replacement of covalently bonded organic linkers with supramolecular structures capable of forming host–guest complexes. For example, (pseudo)rotaxanes or catenanes38,39 (Scheme 1A) have been already proved to be inherently flexible enough to enhance the structural dynamics of MOFs,40–46 and thus possess the intrinsic attributes required for the development of state-of-the-art NTE materials. Herein, we report a trimeric uranyl-based metal–organic polythreading framework, U3(bcbpy)3(CB8), of which the main framework backbone is built up through the threading of the macrocyclic cucurbit[8]uril (CB8)47–49 based on a three-component host–guest complex, (bcbpy)2@CB8, (Scheme 1B, bcbpy is the deprotonated viologen-derived monocarboxylic acid, 1-(4-carboxybenzyl)-[4,4′-bipyridin]-1-ium, see ESI† for details). Notably, the U3(bcbpy)3(CB8) polythreading framework exhibits, via a rare relaxation process, a huge NTE as the temperature increases (Scheme 1C). The NTE occurs via a drastic contraction of the framework structure following a process of slow thermal expansion.
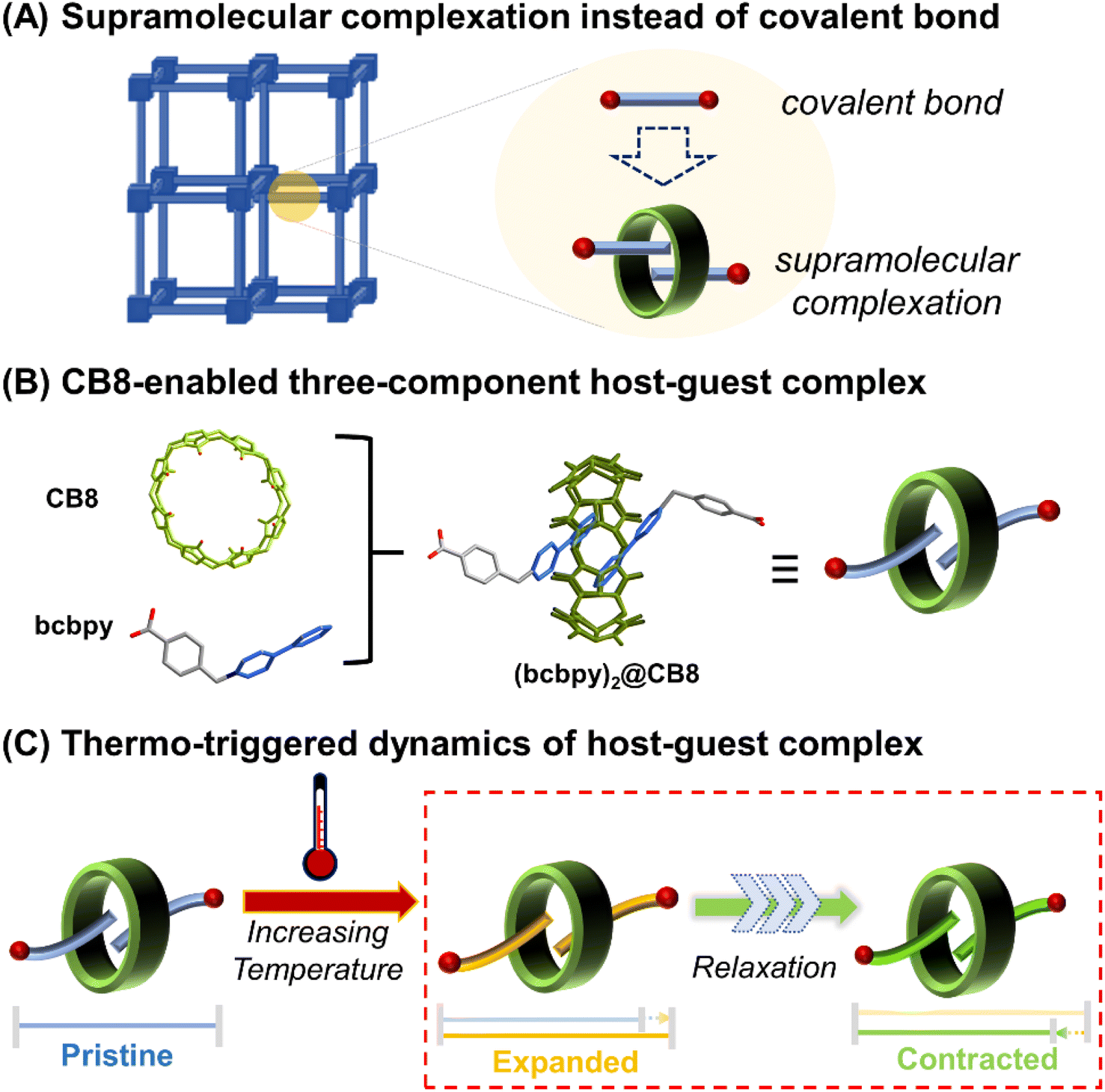 | ||
| Scheme 1 A macrocyclic CB8 three-component host–guest supramolecular complex as a flexible linkage for the development of NTE materials: (A) replacement of covalent bonds with supramolecular complexes in MOFs to produce metal–organic polythreading frameworks; (B) construction of a three-component host–guest complex, (bcbpy)2@CB8; and (C) thermally driven expansion and relaxation-induced contraction of the flexible host–guest complex (bcbpy)2@CB8. | ||
Results and discussion
The most important structural feature of the as-synthesized U3(bcbpy)3(CB8) framework is the three-component host–guest complex motif, (bcbpy)2@CB8. As a starting point, the supramolecular assembly of (bcbpy)2@CB8 from bcbpy (using its carboxylic acid form, [Hbcbpy]Cl, Fig. S1, S2†) and CB8 (Fig. S3†) was investigated. A combination of mass spectrometry (Fig. S4†), 1H NMR (Fig. S5†) and single-crystal X-ray analysis (Fig. S6 and Table S1†) proved that the (bcbpy)2@CB8 motif in its chloride form, [(Hbcbpy)2@CB8]Cl2, had been successfully formed when CB8 was mixed with the corresponding guest molecule [Hbcbpy]Cl.Next, when uranyl ions were introduced into a mixture of CB8 and [Hbcbpy]Cl, the metal–organic polythreading framework U3(bcbpy)3(CB8) was synthesized by a one-pot method. Crystallographic analysis (Fig. 1, Tables S1 and S2†) reveals that its asymmetric unit contains a trimeric uranyl unit, three bcbpy motifs with different coordination modes (monodentate, bidentate and bridged bidentate) and the halves of two different CB8 macrocycles (Fig. S7†). Since one of these two CB8 macrocycles acts as a ditopic linker from both ends in the coordination with trinuclear uranyl nodes, the final uranyl complex observed in the crystal lattice is actually a CB8-bridged hexanuclear unit with six bipyridinium ‘arms’ on the outside (Fig. S8†). It is interesting to see that, akin to previously reported cases involving other cucurbituril uranyl-rotaxane compounds,40,50–53 the coordination with uranyl ions does not reduce CB8's ability to capture guest molecules (Fig. S9†). This uranyl-coordinated CB8, together with another non-coordinated CB8, contribute to the formation of the whole supramolecular U3(bcbpy)3(CB8) framework via polythreading between different moieties. Specifically, based on the propensity of the CB8 host and bipyridium guest components to form the host–guest complex [(Hbcbpy)2@CB8]Cl2, as shown above, the CB8-bridged hexanuclear uranyl unit can extend in different directions in three-dimensional (3D) lattice by forming a (bcbpy)2@CB8 supramolecular complex at six different sites (Fig. 1A). Of these sites, 1–4 are of ‘type A’ based on uranyl-tagged CB8, while sites 5 and 6 are of ‘type B’ based on non-coordinated CB8 (Fig. 1B). As a result, an interwoven two-dimensional (2D) network extending along the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) plane is formed through ‘type A’ host–guest complexation between arrays of hexanuclear units along axis b (Fig. 1C and D). These 2D supramolecular networks are further connected with each other by interlayered CB8 ‘type B’ motifs (Fig. 1E and F) to finally produce a 3D polythreading U3(bcbpy)3(CB8) framework (Fig. 1G). It is notable that, due to the steric hindrance from the bulky CB8 macrocycles, two equivalent bcbpy groups that coordinate in a bidentate manner with uranyl centres are free from supramolecular encapsulation by any CB8 host thus avoiding overcrowding around the hexanuclear uranyl unit. These two bcbpy groups only form weak interactions with spatially adjacent CB8 molecules through a large number of C–H⋯O hydrogen bonds (the yellow stick model in Fig. S10†).
) plane is formed through ‘type A’ host–guest complexation between arrays of hexanuclear units along axis b (Fig. 1C and D). These 2D supramolecular networks are further connected with each other by interlayered CB8 ‘type B’ motifs (Fig. 1E and F) to finally produce a 3D polythreading U3(bcbpy)3(CB8) framework (Fig. 1G). It is notable that, due to the steric hindrance from the bulky CB8 macrocycles, two equivalent bcbpy groups that coordinate in a bidentate manner with uranyl centres are free from supramolecular encapsulation by any CB8 host thus avoiding overcrowding around the hexanuclear uranyl unit. These two bcbpy groups only form weak interactions with spatially adjacent CB8 molecules through a large number of C–H⋯O hydrogen bonds (the yellow stick model in Fig. S10†).
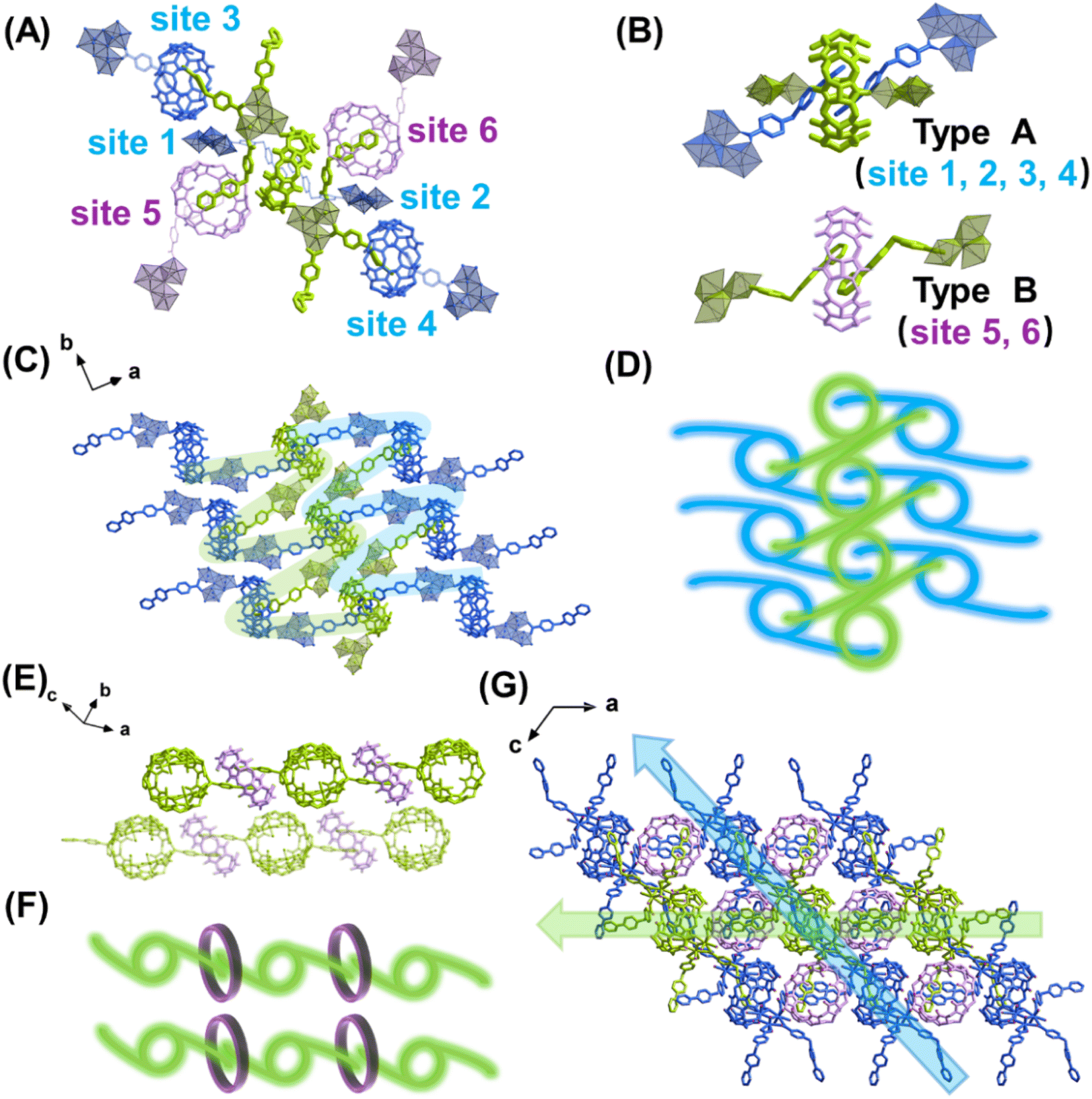 | ||
| Fig. 1 Crystal structure of uranyl-organic polythreading framework U3(bcbpy)3(CB8): (A) CB8-bridged hexanuclear uranyl unit with six connecting sites that can be extended via (bcbpy)2@CB8-typed host–guest complexation; (B) two different kinds of CB8 for formation of (bcbpy)2@CB8 motifs (type A: uranyl-tagged CB8, type B: non-coordinated CB8); (C and D) the interwoven 2D network formed along the (10) plane through ‘type A’ supramolecular motifs ((C): stick model; (D): schematic diagram); (E) ‘type B’ supramolecular motifs for linking interwoven 2D networks along the a-axis ((E): stick model; (F): schematic diagram); and (G) the supramolecular 3D framework of U3(bcbpy)3(CB8) extending along the [101] axis and [100] axes. | ||
A comparison of the molecular structures of the (bcbpy)2@CB8 complexes in [(Hbcbpy)2@CB8]Cl2 and U3(bcbpy)3(CB8) reveals that, different from the relatively rigid macrocyclic CB8 host which undergoes little structural change (Fig. S11 and Table S3†), the bcbpy component encapsulated in the CB8 cavity exhibits significant structural dynamics due to its linkage with the flexible benzyl group (Fig. S12†). The dynamic characteristics of the (bcbpy)2@CB8 motif inspired us to further explore the temperature responsivity of the U3(bcbpy)3(CB8) framework that employs this supramolecular motif as the main building block. As revealed by variable-temperature powder X-ray diffraction (VT-PXRD) analysis within the temperature range from 170 K to 320 K, the PXRD pattern of U3(bcbpy)3(CB8) shows an interesting temperature-dependent change. Specifically, typical diffraction peaks corresponding to the (20![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) and (00) planes first move towards low 2θ, and then reverse that trend around 260 K (Fig. 2A). The shifts of these indices of diffraction to larger 2θ angles indicate an anomalous contraction of the corresponding crystal plane spacing, i.e. negative thermal expansion (NTE) with increasing temperature. Thermal analyses of U3(bcbpy)3(CB8) (Fig. S15 and S16†) reveal that the sample undergoes no phase transition but only a simple endothermic process within a temperature range of 223.15–313.15 K which covers the possible NTE region (from 260 K to near room temperature).
) and (00) planes first move towards low 2θ, and then reverse that trend around 260 K (Fig. 2A). The shifts of these indices of diffraction to larger 2θ angles indicate an anomalous contraction of the corresponding crystal plane spacing, i.e. negative thermal expansion (NTE) with increasing temperature. Thermal analyses of U3(bcbpy)3(CB8) (Fig. S15 and S16†) reveal that the sample undergoes no phase transition but only a simple endothermic process within a temperature range of 223.15–313.15 K which covers the possible NTE region (from 260 K to near room temperature).
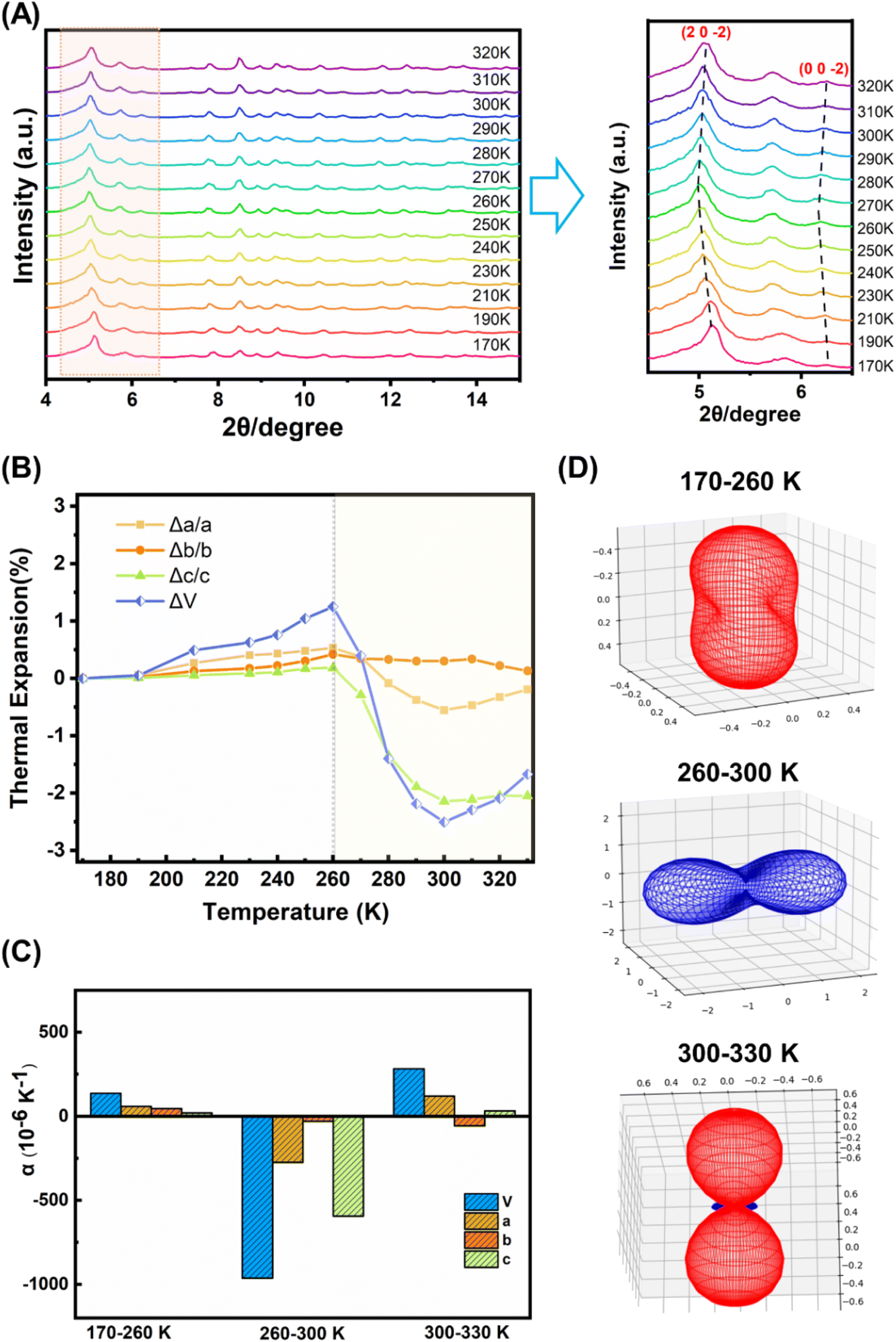 | ||
| Fig. 2 Thermal responsive behaviour of uranyl-organic polythreading U3(bcbpy)3(CB8) framework: (A) variable-temperature powder X-ray diffraction (VT-PXRD) analysis of U3(bcbpy)3(CB8) within the temperature range of 170 K to 320 K (left, full diagram; right, local enlarged view between 2θ of 4.5° to 6.5°); (B) changes in unit cell sizes and total volume with increasing temperature (170 K is set as the starting point); (C) thermal expansion coefficients α of different cell axes and total volume in three different temperature ranges (170–260 K, 260–300 K, 300–330 K), where αL = 1/L(dL/dT) ≈ ΔL/(LΔT) while L refers to a, b, c, and αV = 1/V(dV/dT) ≈ ΔV/(VΔT); and (D) plots of expansivity indicatrices of U3(bcbpy)3(CB8) within different temperature ranges calculated using the PASCal software. | ||
The temperature-related lattice parameters of U3(bcbpy)3(CB8) were extracted from the experimental VT-PXRD data in the range of 170 K to 320 K through profile fitting using the Pawley refinement (Table S4†),54 while more accurate lattice parameters as a function of temperature are given in the following section on single-crystal diffraction analysis. Interestingly, the change of cell parameters is basically the same as that reflected by PXRD, most notably an inflection temperature point around 260 K (Fig. S17†). Before and after this temperature point, the unit cell undergoes two different processes: common expansion and abnormal contraction, respectively. Specifically, the volumetric NTE coefficients (αV) of U3(bcbpy)3(CB8) at 300 K and 310 K reach up to −1043.3 × 10−6 K−1 and −1073.1 × 10−6 K−1, respectively. Moreover, the linear NTE coefficient (αL) along a certain direction, such as the particularly significant c-axis, is as high as −556.0 × 10−6 K−1 within this NTE temperature range.
Immediately after, we employed variable-temperature single-crystal X-ray diffraction (VT-SCXRD) analysis to further explore the NTE behaviour of U3(bcbpy)3(CB8). Tracking of the temperature-dependent changes of the lattice parameters of U3(bcbpy)3(CB8) clearly illustrates anisotropic thermal expansion behaviour along the three different crystal axes of U3(bcbpy)3(CB8) (Table S5† and Fig. 2B). It can be seen that, with the increase of temperature, both the cell parameters along the c-axis and a-axis experience a drastic contraction at around 260 K after a gradual increase, while along the b-axis only minor changes are seen over the entire temperature range. The inflection point of 260 K observed here for the NTE phenomenon is consistent with the results of the VT-PXRD analysis. The NTE responsivity can be characterized further by the αL coefficients for the a, b and c-axis of −273.6, −29.1 and −594.3 × 10−6 K−1, respectively. Moreover, the total unit cell volume also exhibits a change of NTE in the temperature range of 260–300 K, giving a remarkable volumetric coefficient αV of −962.9 × 10−6 K−1 (Fig. 2C). It is notable that the NTE coefficients for the c-axis and total cell volume agree well with those determined from the VT-PXRD analysis discussed above, demonstrating the consistency of the single-crystal diffraction analysis and powder diffraction analysis. Up to now, although the solid-state thermal responsiveness of metal–organic rotaxane compounds has drawn much attention,41–43,45,55,56 there are still very cases that show a NTE effect, and only one case (namely URCP1) which was reported by our group recently.42 Due to the difference between the interlocking structures of U3(bcbpy)3(CB8) and URCP1, the volumetric coefficient αV of U3(bcbpy)3(CB8) is much larger than that of URCP1 (αV = −46.8 × 10−6 K−1). Moreover, the NTE effect of U3(bcbpy)3(CB8) here is also superior to those of previously reported NTE MOFs, such as MOF-5 (αV = −39.3 × 10−6 K−1), MOF-14 (αV = −26 × 10−6 K−1) or UiO-66 (Hf) (αV = −97 × 10−6 K−1) (Fig. S18 and Table S6†).24–33
U3(bcbpy)3(CB8) crystallizes in the monoclinic space group C2/c and the PASCal program57 was used to determine the principal axes and principal expansivities, so as to get further insight into the huge NTE effect (Fig. 2D and S19†). Within the NTE temperature range of 260–300 K, the three principal axes, x1, x2 and x3, are [103], [100] and [010], respectively, the first two of which contribute the most to the total NTE effect. Since the main directions responsible for the huge NTE lie on planes x1 and x2, perpendicular to the x3 principal axis (i.e. the (010) plane across both crystallographic a-axis and c-axis), the (010) plane should have particular importance to the NTE behaviour. We find that along all the directions of extension the (bcbpy)2@CB8 complexing motifs fall on the (010) plane without exception. Therefore, vital clues about the driving forces for the NTE effect could be gained, and strongly suggest that the NTE behaviour of the material is likely to be related to the weakly bonded nature of the CB8-based pseudorotaxane motif that is attached to the hexanuclear uranyl node through a flexible methylene group in the bcbpy guest.
To understand the underlying molecular mechanism for the huge NTE effect, crystal structures of U3(bcbpy)3(CB8) at different temperatures were determined. Of these, the specific data obtained at different critical temperature points including 170 K, 260 K, 300 K and 330 K are compared. It should be mentioned that, besides the first two expansion-contraction steps, the short range after 300 K, where a minor increase of total cell volume occurs, is also analysed for comparison. Analysis of the stacking of the entire lattice shows that the intertwined supramolecular chain containing an array of hexanuclear uranyl units that extend along different directions (Fig. S20–S22†) undergoes a temperature-triggered expansion and contraction similar to that observed during the lattice parameter analysis. The process begins with stretching from 170 K to 260 K followed by a contracting motion as the temperature increases up to 300 K. The overlaid structures at different temperatures confirm that the theory, derived from the PASCal program above, that the thermal response of U3(bcbpy)3(CB8) should originate from flexible connectivity along the [101] axis and [100] axis, i.e. a weakly bonded (bcbpy)2@CB8 motif composed of a pair of bcbpy side groups joined by one CB8 macrocycle.
Further analysis of the local structures of the (bcbpy)2@CB8 supramolecular motif suggests that the well-defined host–guest inclusion pattern within the cavity of macrocyclic CB8 undergoes no noticeable change, whilst the molecular conformation of the V-shaped bcbpy guest connected by the flexible methylene linkage undergoes thermally induced distortion (Fig. S23†). When viewed in a hexanuclear uranyl unit of U3(bcbpy)3(CB8), the conformational changes of bcbpy are even more significant (Fig. 3). We next conducted a detailed analysis of the temperature-triggered structural changes of the bcbpy moiety during the heating process. A comparison of the overlaid molecular structures of the hexanuclear uranyl units at different temperatures shows that the terminal bipyridinium motif, hinged by a methylene group, first experiences a small stretching movement upon heating, and then undergoes a contraction after 260 K accompanied by remarkable lateral movement (Fig. 3A). Interestingly, the temperature-triggered motion of the terminal bipyridinium motif can be clearly tracked by the variation of the corresponding methylene opening angle Ψ and dihedral angle Θ of the ‘type A’ bcbpy moiety (Fig. 3B and Table S7†). Here, the onset temperature of 260 K for the NET effect is just the inflection point of the curves that give the changes of the Ψ and Θ angles as a function of temperature, suggesting a strong correlation between the flexibility of the CB8-encapsulated bcbpy moiety with the NTE behaviour of the supramolecular framework of U3(bcbpy)3(CB8). Specifically, as heating starts from 170 K, the opening angle Ψ of bcbpy motif increases gradually; after reaching a threshold at ∼260 K (black dashed line in the right diagram of Fig. 3B), it recovers back accompanied by a dramatic perturbation in dihedral angle Θ (after 260 K, the slope of the curve becomes larger, corresponding to a more significant change in Θ).
 | ||
| Fig. 3 Local structural analysis of the bcbpy moieties in a single hexanuclear uranyl unit in the supramolecular framework of U3(bcbpy)3(CB8) at different temperatures: (A) overlaid structures of the hexanuclear uranyl units at 170 K, 260 K, 300 K and 330 K with two enlarged diagrams of the type A’ (top) and ‘type B’ (bottom) bcbpy moieties; and (B) temperature-dependent trends of the opening angle of methylene, Ψ, and dihedral angle, Θ, corresponding to bcbpy moieties of ‘type A’, where Ψ is defined as the opening angle between the C–N bond and the C–C bond at the methylene group, and Θ is defined as the dihedral angle between the plane consisting both the C–N bond and C–C bond around the methylene and phenyl plane. | ||
Generally, it has been established that the NTE effect in framework structure materials like MOFs mainly originates from a combination of local and collective transverse vibrations as low-energy phonon modes.4,5,58,59 On the microscopical scale these vibrations correspond to contracting motions of rigid motifs driven by transverse displacement of flexible bridging ligands and can be described using a well-known hinging model.58 For U3(bcbpy)3(CB8) with a weakly bonded structure, transverse vibrations of the flexible bcbpy groups are also observed as the temperature increases, but seem to be present within the entire temperature range including both the PTE and NTE regions (Fig. S24†). This surprising observation seems to be due to the difference in intrinsic structural flexibility and adaptivity of the weakly-bonded polythreading framework which is different to MOFs that commonly have relatively strong coordination bonds. Explaining framework structure NTE simply through the transverse motion of organic linkers does not apply to such a highly flexible supramolecular framework like U3(bcbpy)3(CB8), and a new mechanism should be proposed. Based on the step-by-step nature of the thermo-responsive behaviour of U3(bcbpy)3(CB8), which includes a quick recovery following a gradual stretching of the structure, it can be speculated that the contraction-related NTE effect corresponding to the recovery stage is a typical relaxation process after a progressive thermal disturbance.
This relaxation-related mechanism is proved by the irreversibility of the NTE effect when a reverse cooling protocol and a second heating protocol are used after the first heating protocol (Fig. S25†). It can be seen that, after undergoing structural contraction, the crystal of U3(bcbpy)3(CB8) is nearly unchanged upon cooling from 320 K to 240 K (red line in Fig. S25†). As the crystal remains intact throughout the experiment, and the single-crystal X-ray diffraction analysis does not show any signs of collapse into a polycrystalline state, the possibility of cracking of the crystal after the thermally induced NTE effect is ruled out. On the other hand, considering the necessity of thermo-triggered cumulative stress for the relaxation-related NTE effect, it is reasonable that no reverse NTE process is observed during the reverse cooling protocol. Moreover, it is observed that, the second heating protocol from 240 K to 320 K only provides a slight increase in cell volume, but no NTE process reappears (blue line in Fig. S25†), which might be attributed to the irreversible change of crystal structure of U3(bcbpy)3(CB8) after a dramatic thermo-triggered transformation.
More interestingly, monitoring the kinetics of this NTE process through fast recording of lattice parameters at different time points covering the NTE temperature range (Fig. 4A and S26†) reveals the time-dependent structural dynamics of U3(bcbpy)3(CB8) on the observation time frame (up to 2 hours for each temperature), and thus serves as another important piece of evidence for the mechanism of the relaxation process. Specifically, before the inflection temperature point, for example at 240 K, all the lattice parameters remain largely unchanged during the period of monitoring, which means that the crystal sample can achieve both temperature equilibrium and undergo a minor structural expansion at the first time point of 10 min. Upon heating over the onset temperature for NTE, the lattice volume shrinks continuously over time, maintaining a constant temperature at each temperature point (most remarkably for the first three temperature points, i.e. 260 K, 270 K and 280 K) and reaching a transient equilibrium after 1–2 hours. Since a relaxation time of over 2 hours is adopted at each temperature point, rather than a short period of 10 minutes used in the VT-PXRD and VT-SCXRD experiments, the relaxation-based NTE effect is accelerated and completed at 280–290 K. Furthermore, the snapshots of the crystal structures of U3(bcbpy)3(CB8) at two different relaxation times provides more detailed molecular information on the changes that occur in this polythreading framework during the time-dependent relaxation process (Fig. 4B). It can be observed that a prolonged relaxation time is conducive to the continuous regulation of the molecular conformation of the flexible bcbpy chain of the (bcbpy)2@CB8 complex during the NTE process. Notably, as far as we know, this is the first report in NTE materials of these unique time-dependent structural dynamics observed during the relaxation process of U3(bcbpy)3(CB8).
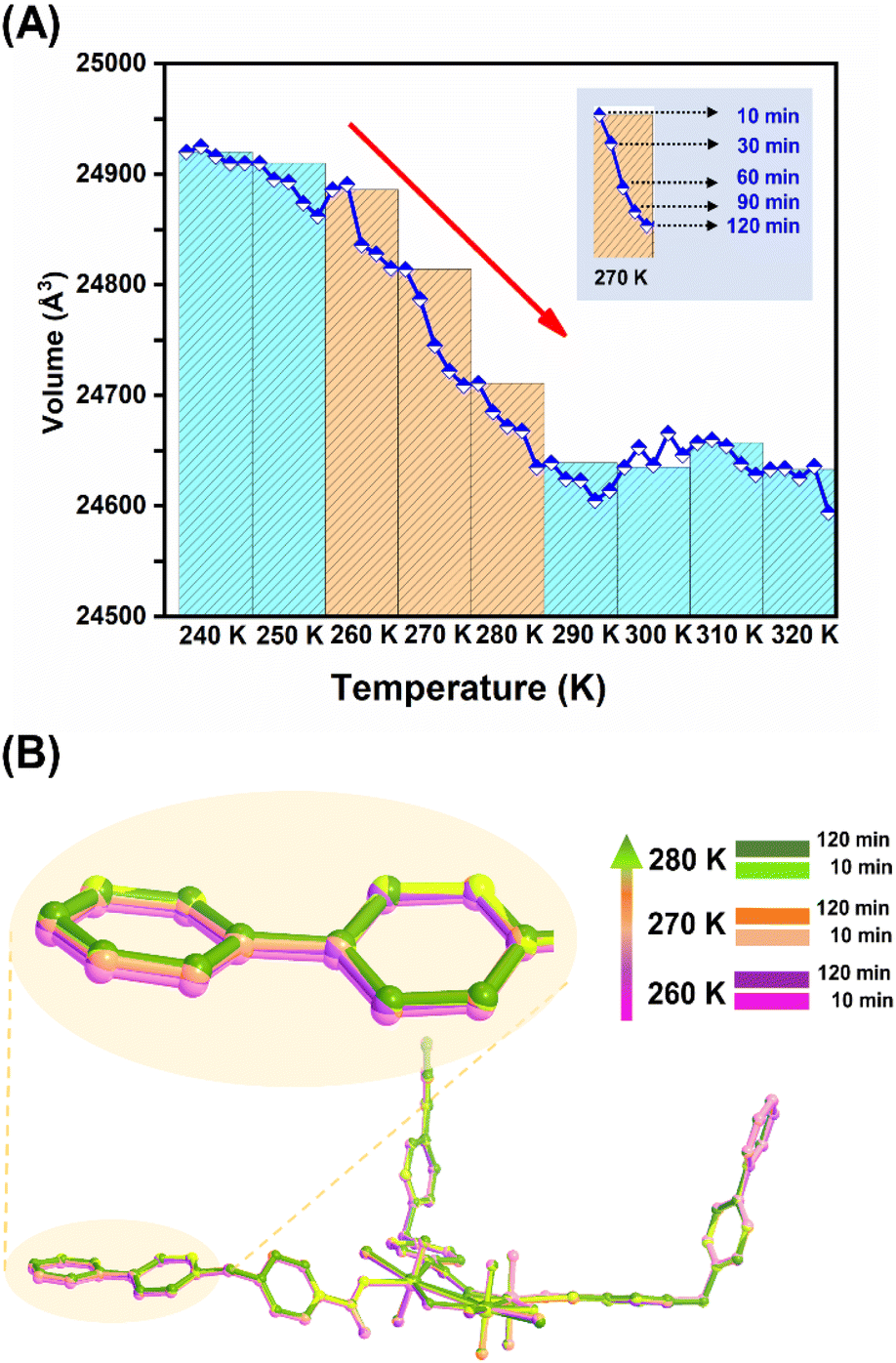 | ||
| Fig. 4 Kinetics of the NTE effect of U3(bcbpy)3(CB8) and crystallographic snapshots after different relaxation periods during NTE: (A) a diagram showing the lattice parameter changes at different time points from 10 to 120 minutes covering the NTE temperature range from 240 K to 320 K; and (B) snapshots of the crystal structures of U3(bcbpy)3(CB8) after different relaxation periods (10 min and 120 min) at three typical temperature points during the relaxation process with time-dependent structural dynamics. | ||
Based on the comprehensive analysis detailed above, a schematic diagram illustrating the proposed mechanism of relaxation-related contraction for the NTE effect is given. Considering that this temperature-driven relaxation process resembles the stretching and contraction process of an ideally functioning spring,60–62 the stepwise thermal response behaviour of the (bcbpy)2@CB8 complex in U3(bcbpy)3(CB8) is simplified as a molecular spring-like model so as to depict the mechanism more clearly (Fig. 5). The CB8-enabled host–guest complexation with high flexibility endows the V-shaped methylene segments of the bcbpy moiety more degrees of freedom under solid conditions. In turn this means that the opening angle of the methylene group can increase gradually through enhanced thermal vibration upon heating, thus promoting the polythreading framework to undergo thermo-triggered expansion (corresponding to the PTE step in Fig. 5). When the internal stress accumulation exceeds the threshold that the lattice can withstand as the expansion progresses, the stretching conformation of the bcbpy moiety will contract drastically to achieve a recovery through a relaxation process, which ultimately results in a huge NTE effect (corresponding to the NTE step in Fig. 5). Moreover, by compared to the hinging model for NTE in MOFs with higher skeleton rigidity, it can be found that the weakly bonded nature of the polythreading framework is crucial to afford this spring-like model with relaxation-induced contraction/NTE effect (Fig. S27†).
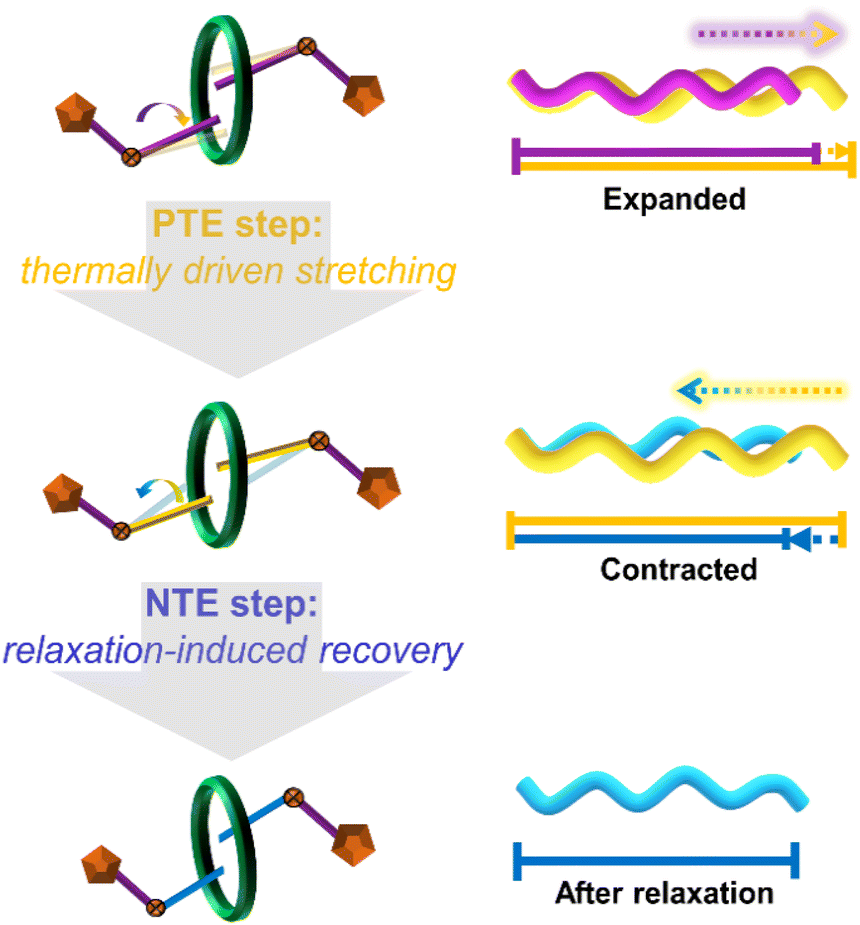 | ||
| Fig. 5 A proposed mechanism of relaxation-induced contraction as a rationale for the NTE effect of the polythreading U3(bcbpy)3(CB8) framework. The mechanism includes two continuous thermo-responsive stages: (1) PTE step with thermally driven stretching; and (2) NTE step with relaxation-induced recovery of flexible bcbpy motifs. | ||
Conclusions
In summary, a host–guest complex (bcbpy)2@CB8 was utilized as a weakly bonded linkage to construct a flexible hexanuclear uranyl-organic polythreading framework. The bcbpy moieties are connected in couples via supramolecular encapsulation in CB8 and, following progressive stretching, undergo thermo-triggered contraction resulting in a huge NTE effect. The volumetric coefficient of −962.9 × 10−6 K−1 outperforms those of NTE metal–organic rotaxane compounds and MOFs that have been reported to date. The NTE proceeds via a relaxation-related mechanism that requires a cumulative thermo-induced expansion, as proved by the time-dependent structural dynamics of this polythreading framework and the irreversibility of the NTE effect. This work proves the feasibility of employing well-defined supramolecular motifs featuring noncovalent interactions to efficiently improve the thermal response of framework-structured materials. It is expected that more NTE materials based on supramolecular motifs will be developed.Experimental
Hydrothermal synthesis of uranyl-organic polythreading framework, U3(bcbpy)3(CB8)
[Hbcbpy]Cl (16.4 mg, 0.05 mmol), CB8 (16.6 mg, 0.0125 mmol), 0.5 M uranyl nitrate mother solution (100 μL, 0.05 mmol), 0.5 M sodium hydroxide solution (80 μL, 0.04 mmol) and 1 mL of deionized water were added into a 10 mL Teflon autoclave. The initial pH of the reaction mixture was 3.07. The autoclave was then sealed with a stainless-steel kettle jacket and heated at 150 °C for 48 h. After naturally cooling to room temperature, the pH of the solution was measured to be 2.99. Large yellow block crystals together with an unknown precipitate were obtained. Pure crystal samples of U3(bcbpy)3(CB8) were picked out before being subjected to subsequent structure determination and analysis.Variable-temperature powder X-ray diffraction (VT-PXRD) measurements
VT-PXRD analysis of U3(bcbpy)3(CB8) was carried out using a Bruker D8 Discover diffractometer with Cu Kα radiation (λ = 1.5406 Å) in the range of 3–50° (step size: 0.02°) with a scanning rate of 1° min−1. The measurement covers the temperature range of 170–320 K including a set of temperature points, 170 K, 190 K, 210 K, 230 K, 240 K, 250 K, 260 K, 270 K, 280 K, 290 K, 300 K, 310 K and 320 K. The experiment started at 170 K, and gradually raised the temperature to the temperature points set in sequence. After heating to the target temperature, the sample was kept at target temperature for 10 minutes before being subjected to measurement. The spacing between certain crystal planes, as well as corresponding linear NTE coefficients (αL) along certain crystal planes, was extracted from the VT-PXRD pattern based on Bragg's law, nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ.
sinθ.
Variable-temperature single crystal X-ray diffraction (VT-SCXRD) analysis
Single crystal X-ray diffraction characterization of U3(bcbpy)3(CB8) at different temperatures were carried out with a Cryostream 700 low temperature device. Liquid nitrogen was used to control the testing temperatures of the cooling or heating atmosphere. Specifically, a series of temperature points within the range of 170–330 K were set according to the experimental requirements (170, 190, 210, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320 and 330 K). The experiment started at 170 K, and gradually raised the temperature to the temperature points set in sequence with a heating rate of 120 K h−1. The crystal sample was kept at target temperature for 10 min before being subjected to measurement. The crystal data of all compounds are given in ESI Tables S1–S4. Crystallographic data in this work have been deposited with Cambridge Crystallographic Data Centre, and the CCDC numbers are 2170887–2170901, including: ([Hbcbpy]Cl)2@CB8 (2170894), U3(bcbpy)3(CB8)_170 K_A (2170893), U3(bcbpy)3(CB8)_190 K_A (2170890), U3(bcbpy)3(CB8)_210 K_A (2170889), U3(bcbpy)3(CB8)_230 K_A (2170892), U3(bcbpy)3(CB8)_240 K_A (2170891), U3(bcbpy)3(CB8)_250 K_A (2170888), U3(bcbpy)3(CB8)_260 K_A (2170887), U3(bcbpy)3(CB8)_270 K_A (2170901), U3(bcbpy)3(CB8)_280 K_A (2170900), U3(bcbpy)3(CB8)_290 K_A (2170899), U3(bcbpy)3(CB8)_300 K_A (2170898), U3(bcbpy)3(CB8)_310 K_A (2170897), U3(bcbpy)3(CB8)_320 K_A (2170896), and U3(bcbpy)3(CB8)_330 K_A (2170895).†Monitoring the kinetics of the NTE process of U3(bcbpy)3(CB8)
The monitoring of the time-dependent dynamic process of U3(bcbpy)3(CB8) was conducted using VT-SCXRD analysis covering the temperature range of 170–330 K with a total of thirteen temperature points examined (170 K, 210 K, 240 K, 250 K, 260 K, 270 K, 280 K, 290 K, 300 K, 310 K, 320 K, 330 K and 340 K). The experiment started at 170 K, and gradually raised the temperature to the temperature points set in sequence. For the temperature range around NTE effect from 240 K to 320 K, the crystal sample was kept at target temperature for an extended period of 120 minutes. During this period, the diffraction data at each duration of 10, 30, 60, 90 and 120 minutes were collected using a fast scanning protocol to understand the corresponding cell parameters at different time duration (the structural change within the short period for fast scanning can be ignored).Based on the analysis of the dynamic process of thermal responsive behaviour, single-crystal X-ray diffraction images at two different relaxation times (10 min and 120 min) were collected and a set of snapshot crystal structures of U3(bcbpy)3(CB8) were obtained. Crystallographic data in this work have been deposited with Cambridge Crystallographic Data Centre, and the CCDC numbers are 2218693–2218710, including U3(bcbpy)3(CB8)_170 K_B (2218693), U3(bcbpy)3(CB8)_210 K_B (2218694), U3(bcbpy)3(CB8)_250 K_B (2218695), U3(bcbpy)3(CB8)_260 K_B1 (2218696), U3(bcbpy)3(CB8)_260 K_B2 (2218697), U3(bcbpy)3(CB8)_270 K_B1 (2218698), U3(bcbpy)3(CB8)_270 K_B2 (2218699), U3(bcbpy)3(CB8)_280 K_B1 (2218700), U3(bcbpy)3(CB8)_280 K_B2 (2218701), U3(bcbpy)3(CB8)_290 K_B1 (2218702), U3(bcbpy)3(CB8)_290 K_B2 (2218703), U3(bcbpy)3(CB8)_300 K_B1 (2218704), U3(bcbpy)3(CB8)_300 K_B2 (2218705), U3(bcbpy)3(CB8)_310 K_B1 (2218706), U3(bcbpy)3(CB8)_310 K_B2 (2218707), U3(bcbpy)3(CB8)_320 K_B1 (2218708), U3(bcbpy)3(CB8)_320 K_B2 (2218709), and U3(bcbpy)3(CB8)_330 K_B (2218710). The suffix of ‘_B’ or ‘_B1’ represents data obtained at 10 min and ‘_B2’ is at 120 min.†
Data availability
Data available in the ESI.†Author contributions
W.-Q. S. and L. M. conceived the idea and wrote the manuscript. Q.-Y. J. and Y.-Y. L. initiated and conducted the single crystal synthesis and variable temperature single crystal X-ray diffraction analysis, with assistance from L.-Y. Y. and J.-P. Y. K.-Q. H. and L. Meng solved the X-ray structures. W.-Q. S., Z.-H. Z. and Z.-F. C. reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support from the National Natural Science Foundation of China (22122609, 22076186 and U20B2019). The National Science Fund for Distinguished Young Scholars (21925603) and the Youth Innovation Promotion Association of CAS (2020014) are also acknowledged.Notes and references
- V. A. Drebushchak, J. Therm. Anal. Calorim., 2020, 142, 1097–1113 CrossRef CAS.
- R. Mittal, M. K. Gupta and S. L. Chaplot, Prog. Mater. Sci., 2018, 92, 360–445 CrossRef CAS.
- Q. Li, K. Lin, Z. N. Liu, L. Hu, Y. L. Cao, J. Chen and X. R. Xing, Chem. Rev., 2022, 122, 8438–8486 CrossRef CAS.
- N. K. Shi, Y. Z. Song, X. R. Xing and J. Chen, Coord. Chem. Rev., 2021, 449, 214204 CrossRef CAS.
- J. D. Evans, J. P. Durholt, S. Kaskel and R. Schmid, J. Mater. Chem. A, 2019, 7, 24019–24026 RSC.
- E. Liang, Q. Sun, H. Yuan, J. Wang, G. Zeng and Q. Gao, Front. Phys., 2021, 16, 53302 CrossRef.
- J. Chen, L. Hu, J. X. Deng and X. R. Xing, Chem. Soc. Rev., 2015, 44, 3522–3567 RSC.
- K. Takenaka, Sci. Technol. Adv. Mater., 2012, 13, 013001 CrossRef PubMed.
- B. A. Marinkovic, P. I. Ponton, C. P. Romao, T. Moreira and M. A. White, Front. Mater., 2021, 8, 741560 CrossRef.
- L. Hu, J. Chen, J. L. Xu, N. Wang, F. Han, Y. Ren, Z. Pan, Y. C. Rong, R. J. Huang, J. X. Deng, L. F. Li and X. R. Xing, J. Am. Chem. Soc., 2016, 138, 14530–14533 CrossRef CAS.
- X. X. Jiang, M. S. Molokeev, P. F. Gong, Y. Yang, W. Wang, S. H. Wang, S. F. Wu, Y. X. Wang, R. J. Huang, L. F. Li, Y. C. Wu, X. R. Xing and Z. S. Lin, Adv. Mater., 2016, 28, 7936–7940 CrossRef CAS PubMed.
- A. E. Phillips and A. D. Fortes, Angew. Chem., Int. Ed., 2017, 56, 15950–15953 CrossRef CAS PubMed.
- H. Zhu, Q. Li, C. Yang, Q. H. Zhang, Y. Ren, Q. L. Gao, N. Wang, K. Lin, J. X. Deng, J. Chen, L. Gu, J. W. Hong and X. R. Xing, J. Am. Chem. Soc., 2018, 140, 7403–7406 CrossRef CAS.
- A. B. Cairns and A. L. Goodwin, Chem. Soc. Rev., 2013, 42, 4881–4893 RSC.
- T. A. Mary, J. S. O. Evans, T. Vogt and A. W. Sleight, Science, 1996, 272, 90–92 CrossRef CAS.
- B. K. Greve, K. L. Martin, P. L. Lee, P. J. Chupas, K. W. Chapman and A. P. Wilkinson, J. Am. Chem. Soc., 2010, 132, 15496–15498 CrossRef CAS PubMed.
- J. C. Hancock, K. W. Chapman, G. J. Halder, C. R. Morelock, B. S. Karlan, L. C. Gallington, A. Bongiorno, C. Han, S. Zhou and A. P. Wilkinson, Chem. Mater., 2015, 27, 3912–3918 CrossRef CAS.
- Q. L. Gao, E. J. Liang, X. R. Xing and J. Chen, Chem. J. Chin. Univ., 2020, 41, 388–400 CAS.
- S. D’Ambrumeni, M. Zbiri, A. M. Chippindale and S. J. Hibble, Phys. Rev. B, 2019, 100, 094312 CrossRef.
- S. D’Ambrumenil, M. Zbiri, A. M. Chippindale, S. J. Hibble, E. Marelli and A. C. Hannon, Phys. Rev. B, 2019, 99, 024309 CrossRef.
- S. J. Hibble, A. M. Chippindale, E. Marelli, S. Kroeker, V. K. Michaelis, B. J. Greer, P. M. Aguiar, E. J. Bilbe, E. R. Barney and A. C. Hannon, J. Am. Chem. Soc., 2013, 135, 16478–16489 CrossRef CAS.
- S. G. Duyker, V. K. Peterson, G. J. Kearley, A. J. Ramirez-Cuesta and C. J. Kepert, Angew. Chem., Int. Ed., 2013, 52, 5266–5270 CrossRef CAS PubMed.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929–8932 CrossRef CAS PubMed.
- Y. Wu, V. K. Peterson, E. Luks, T. A. Darwish and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 5175–5178 CrossRef CAS PubMed.
- Z. N. Liu, Q. Li, H. Zhu, K. Lin, J. X. Deng, J. Chen and X. R. Xing, Chem. Commun., 2018, 54, 5712–5715 RSC.
- Z. N. Liu, R. Ma, J. X. Deng, J. Chen and X. R. Xing, Chem. Mater., 2020, 32, 2893–2898 CrossRef CAS.
- Y. Kim, R. Haldar, H. Kim, J. Koo and K. Kim, Dalton Trans., 2016, 45, 4187–4192 RSC.
- Z. N. Liu, X. X. Jiang, C. M. Wang, C. X. Liu, Z. S. Lin, J. X. Deng, J. Chen and X. R. Xing, Inorg. Chem. Front., 2019, 6, 1675–1679 RSC.
- N. Lock, Y. Wu, M. Christensen, L. J. Cameron, V. K. Peterson, A. J. Bridgeman, C. J. Kepert and B. B. Iversen, J. Phys. Chem. C, 2010, 114, 16181–16186 CrossRef CAS.
- C. Schneider, D. Bodesheim, M. G. Ehrenreich, V. Crocella, J. Mink, R. A. Fischer, K. T. Butler and G. Kieslich, J. Am. Chem. Soc., 2019, 141, 10504–10509 CrossRef CAS.
- M. Asgari, I. Kochetygov, H. Abedini and W. L. Queen, Nano Res., 2021, 14, 404–410 CrossRef CAS.
- M. J. Cliffe, J. A. Hill, C. A. Murray, F. X. Coudert and A. L. Goodwin, Phys. Chem. Chem. Phys., 2015, 17, 11586–11592 RSC.
- J. E. Auckett, A. A. Barkhordarian, S. H. Ogilvie, S. G. Duyker, H. Chevreau, V. K. Peterson and C. J. Kepert, Nat. Commun., 2018, 9, 4873 CrossRef PubMed.
- Z. N. Liu, Q. L. Gao, J. Chen, J. X. Deng, K. Lin and X. R. Xing, Chem. Commun., 2018, 54, 5164–5176 RSC.
- H. Y. Liu, M. J. Gutmann, H. T. Stokes, B. J. Campbell, I. R. Evans and J. S. Evans, Chem. Mater., 2019, 31, 4514–4523 CrossRef CAS.
- X. Y. Shen, C. Viney, E. R. Johnson, C. C. Wang and J. Q. Lu, Nat. Chem., 2013, 5, 1035–1041 CrossRef CAS PubMed.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- J.-s. Geng, L. Mei, Y.-y. Liang, L.-y. Yuan, J.-p. Yu, K.-q. Hu, L.-h. Yuan, W. Feng, Z.-f. Chai and W.-q. Shi, Nat. Commun., 2022, 13, 2030 CrossRef CAS PubMed.
- W. J. Meng, S. Kondo, T. Ito, K. Komatsu, J. Pirillo, Y. Hijikata, Y. Ikuhara, T. Aida and H. Sato, Nature, 2021, 598, 298–303 CrossRef CAS PubMed.
- F. Z. Li, J. S. Geng, K. Q. Hu, L. W. Zeng, J. Y. Wang, X. H. Kong, N. Liu, Z. F. Chai, L. Mei and W. Q. Shi, Chem.–Eur. J., 2021, 27, 8730–8736 CrossRef CAS PubMed.
- K. L. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS.
- V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896–5906 RSC.
- V. N. Vukotic, K. J. Harris, K. L. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed.
- Q. W. Li, W. Y. Zhang, O. S. Miljanic, C. H. Sue, Y. L. Zhao, L. H. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS PubMed.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. Garcia, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- Y. L. Liu, H. Yang, Z. Q. Wang and X. Zhang, Chem.–Asian J., 2013, 8, 1626–1632 CrossRef CAS PubMed.
- Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305–1315, 10.1039/b615103e.
- F. Z. Li, J. S. Geng, K. Q. Hu, J. P. Yu, N. Liu, Z. F. Chai, L. Mei and W. Q. Shi, lnorg. Chem., 2021, 60, 10522–10534 CrossRef CAS PubMed.
- F. Z. Li, L. Mei, K. Q. Hu, S. W. An, S. Wu, N. Liu, Z. F. Chai and W. Q. Shi, lnorg. Chem., 2019, 58, 3271–3282 CrossRef CAS PubMed.
- F. Z. Li, L. Mei, K. Q. Hu, J. P. Yu, S. W. An, K. Liu, Z. F. Chai, N. Liu and W. Q. Shi, lnorg. Chem., 2018, 57, 13513–13523 CrossRef CAS PubMed.
- J. Y. Wang, L. Mei, Z. W. Huang, X. W. Chi, J. S. Geng, K. Q. Hu, J. P. Yu, C. S. Jiao, M. Zhang, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2022, 61, 3058–3071 CrossRef CAS PubMed.
- G. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- N. Zhu, K. Nakazono and T. Takata, Chem. Lett., 2016, 45, 445–447 CrossRef CAS.
- N. Farahani, K. L. Zhu, C. A. O'Keefe, R. W. Schurko and S. J. Loeb, ChemPlusChem, 2016, 81, 836–841 CrossRef CAS PubMed.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- I. E. Collings, M. G. Tucker, D. A. Keen and A. L. Goodwin, CrystEngComm, 2014, 16, 3498–3506 RSC.
- W. Z. Cai and A. Katrusiak, Nat. Commun., 2014, 5, 4337 CrossRef CAS PubMed.
- L. Mei, S. W. An, K. Q. Hu, L. Wang, J. P. Yu, Z. W. Huang, X. H. Kong, C. Q. Xia, Z. F. Chai and W. Q. Shi, Angew. Chem., Int. Ed., 2020, 59, 16061–16068 CrossRef CAS PubMed.
- C. B. Huang, A. Ciesielski and P. Samori, Angew. Chem., Int. Ed., 2020, 59, 7319–7330 CrossRef CAS PubMed.
- Z. N. Liu, C. X. Liu, Q. Li, J. Chen and X. R. Xing, Phys. Chem. Chem. Phys., 2017, 19, 24436–24439 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic details, X-ray crystallographic data, crystal structures, PXRD pattern, TGA curves, DSC results and FTIR spectra. CCDC 2170887–2170901 and 2218693–2218710. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01343j |
| This journal is © The Royal Society of Chemistry 2023 |