DOI:
10.1039/D3SC01382K
(Edge Article)
Chem. Sci., 2023,
14, 7334-7345
Catalyst-free thiazolidine formation chemistry enables the facile construction of peptide/protein–cell conjugates (PCCs) at physiological pH†
Received
15th March 2023
, Accepted 7th June 2023
First published on 8th June 2023
Abstract
Although numerous genetic, chemical, and physical strategies have been developed to remodel the cell surface landscape for basic research and the development of live cell-based therapeutics, new chemical modification strategies capable of decorating cells with various genetically/non-genetically encodable molecules are still urgently needed. Herein, we describe a remarkably simple and robust chemical strategy for cell surface modifications by revisiting the classical thiazolidine formation chemistry. Cell surfaces harbouring aldehydes can be chemoselectively conjugated with molecules containing a 1,2-aminothiol moiety at physiological pH without the need to use any toxic catalysts and complicated chemical synthesis. Through the combined use of thiazolidine formation and the SpyCatcher–SpyTag system, we have further developed a SpyCatcher–SpyTag Chemistry Assisted Cell Surface Engineering (SpyCASE) platform, providing a modular approach for the construction of large protein–cell conjugates (PCCs) in their native state. Thiazolidine-bridged molecules can also be detached from the surface again through a biocompatible Pd-catalyzed bond scission reaction, enabling reversible modification of living cell surfaces. In addition, this approach allows us to modulate specific cell–cell interactions and generate NK cell-based PCCs to selectively target/kill several EGFR-positive cancer cells in vitro. Overall, this study provides an underappreciated but useful chemical tool to decorate cells with tailor-made functionalities.
Introduction
Cell surfaces are highly dynamic interfaces that play important roles in intracellular signaling and cell–cell and cell–environment communications.1 The cell membrane presents a very complex network of distinct types of proteins, lipids, and carbohydrates.2 Methods for engineering and functionalizing the eukaryotic cell surface have been heavily sought after for various applications, including the study of fundamental cell biology, tissue engineering and development of cell-based drug delivery systems and therapeutics.3,4 To date, various genetic, chemical, chemoenzymatic, and physical strategies have been successfully designed to tailor the cell surface with a wide range of functionalities, including DNA, antibodies, enzymes, synthetic polymers, nanoparticles, etc.5–9 Among them, genetic engineering is used to remodel the cell surface landscape by manipulation of the cell genome and in recent years it also has brought about breakthroughs in therapeutic intervention. One of the prominent examples is Kymriah, a chimeric antigen receptor T-cell (CAR-T) therapy that was approved as the first cell-based gene therapy in the United States for the treatment of patients with B-cell precursor acute lymphoblastic leukemia.10 However, this “inside” strategy is subjected to technical complications and safety concerns, such as the inconsistent reproducibility of viral transduction efficiency of primary cells, heterogeneous expression levels, and the potential for endogenous gene disruption.11 Additionally, one limitation to this method is that only genetically encodable molecules can be displayed on the cell surface.
By contrast, the decoration of the cell surface from the “outside” using various chemical biology tools has attracted more interest due to its featuring advantages, such as the ability to tailor the cell surface with a more diverse set of unnatural functionalities. For example, the technique of metabolic oligosaccharide engineering (MOE) developed by the Nobel laureate Bertozzi's group has revolutionized the field of chemical biology and provided great opportunities for selective glycan labeling, super-resolution imaging of glycans, cancer cell-selective immunotherapy, etc.12–14 This two-step approach for remodeling cell surface glycans first incorporates unique reactive chemical handles into surface glycans by feeding cells with synthetic sugars that can hijack endogenous sugar metabolism pathways, followed by a biorthogonal reaction to achieve desired modifications.15 However, the incorporation efficiency of sugar analogs varies greatly between different cell lines.16 To precisely modify the proteins located on the cell membrane, genetic code expansion technology17 and several post-translational modifying (PTM) enzymes, such as sortase, butelase-1, glycosyltransferase, lipoic acid ligase, biotin ligase, and phosphopantetheine transferase, among others,18–22 have emerged as powerful tools. However, these approaches also require genetic manipulation of the target cells to express genes encoding the orthogonal aminoacyl-tRNA synthetase/tRNA pair and/or enzyme substrate, suffering from the aforementioned limitations. Therefore, the development of a novel, simple and general cell surface conjugation approach is still extraordinarily demanded. Ideally, this strategy should have the following features: no catalyst, no complicated chemical synthesis, genetic manipulation-free, low concentration of reactants, fast reaction kinetics, finely tuned product stability, and proceeding under physiological conditions.
The condensation between 1,2-aminothiols and aldehydes to form thiazolidines is an interesting bioorthogonal reaction as the 1,2-aminothiol group naturally exists in the proteins being an N-terminal Cys. This reaction is highly specific to 1,2-aminothiol even in the presence of other internal competing sulfhydryl groups on the biomolecule. Over the years, this chemistry has been utilized to immobilize protein for biosensor analysis,23 prepare ubiquitin dimer and antibody–drug conjugates,24,25 as a very common N-terminal Cys protecting group for peptide/protein chemical synthesis,26 and on-demand control the function of caged protein probes for profiling deubiquitinases.27 Despite these intriguing studies, this chemistry remains an underutilized bioconjugation tool. Thiazolidine ring formation is generally believed to proceed at an acidic pH of 4–5, requires long reaction times (up to several days) and high concentrations of reactants, and is in most cases a reversible process.25,28 However, conflicting results suggest that thiazolidine chemistry is actually a fast click-type reaction that can proceed efficiently under physiological conditions for bioconjugation, and the modified peptide/protein conjugate with a thiazolidine linkage is highly stable at both acidic and neutral pH.29 Furthermore, accumulating evidence indicates that the reaction kinetics, conditions and product stability can be controlled by using different aldehydes as the condensation partner. The earliest example is the preparation of aldehydes having a labile ester in the proximity such that the thiazolidine product can undergo ring rearrangement to afford a pseudoproline as the irreversible linker.30,31 Subsequently, it is further revealed that if an ortho-boronic acid modified benzaldehyde was used as the condensation substrate, it could be rapidly conjugated with 1,2-aminothiol to give a stable thiazolidino boronate (TzB) complex at neutral pH.32,33 Recently, this TzB complex formation strategy in combination with an acyl transfer reaction enabled the fast formation of irreversible conjugates under neutral pH, thus addressing the notorious reversibility issue.34 Surprisingly, despite these advances, this classical biorthogonal chemistry has been rarely used for living cell surface engineering. Therefore, we envision that thiazolidine formation may be used as a new alternative chemical biology tool to engineer and functionalize a living cell surface under physiological conditions, while retaining cell viability and function (Scheme 1).
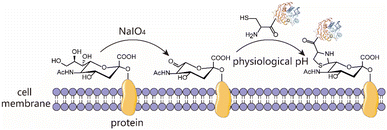 |
| | Scheme 1 Schematic representation of chemical modification of the living cell surface mediated by thiazolidine ring formation chemistry in two steps. Step (1) generation of aldehydes on cell surfaces by periodate oxidation; step (2) decorating the cell surfaces with molecules harbouring a 1,2-aminothiol moiety under physiological conditions. | |
Results and discussion
Conjugation of fluorescent peptides to the cell surfaces
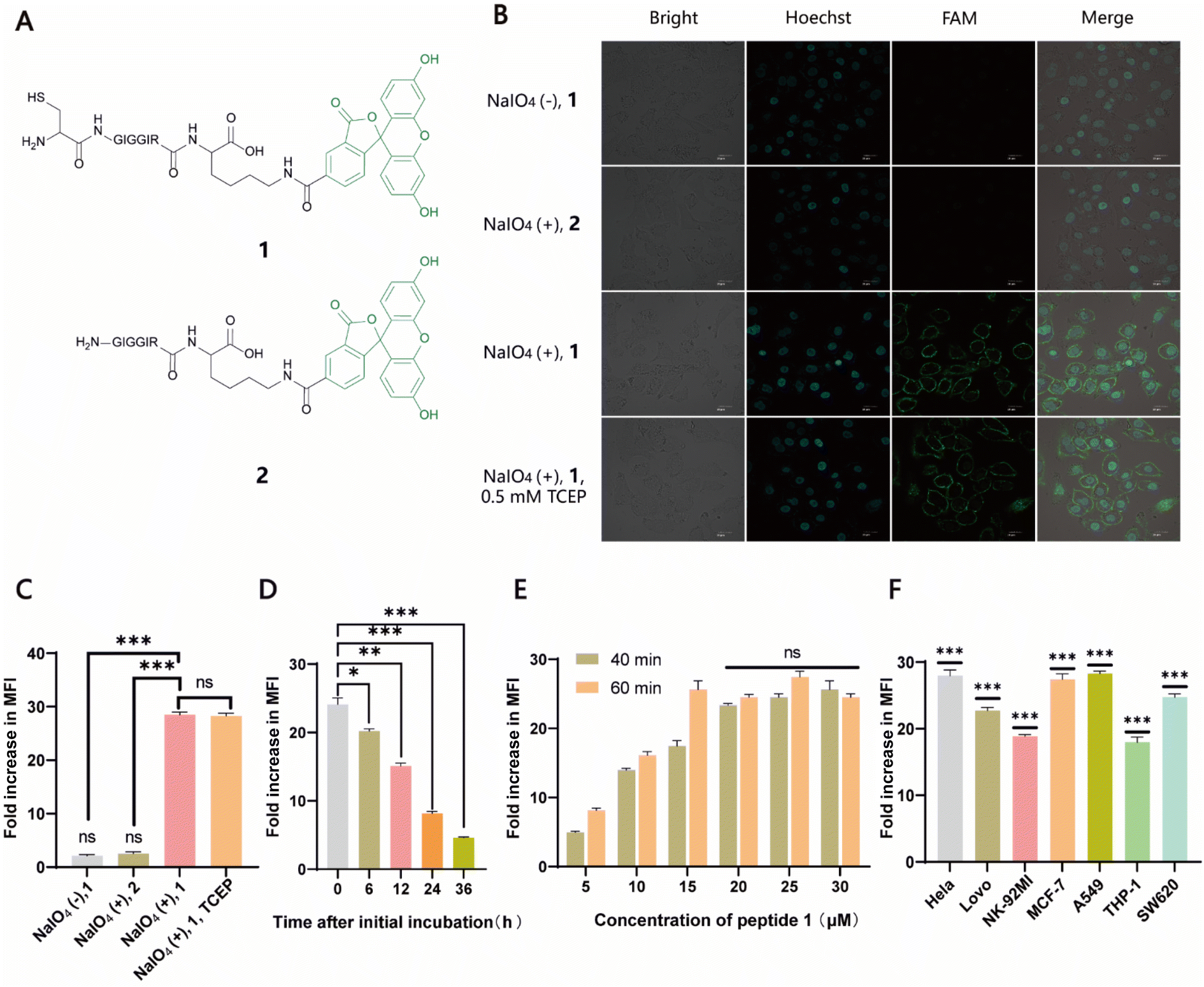
Naturally, there is no aldehyde or 1,2-aminothiol on the living cell surface. In a proof-of-concept study, MCF-7 cells were used as the model and oxidized with 1 mM NaIO4 in PBS (pH 7.2) for 5 min to generate aldehydes by selective oxidation of the adjacent diols of sialic acids at the surface glycans. It has been validated that this treatment is fully compatible with a wide range of cell lines and the exposure of cells to a low concentration of NaIO4 (1–5 mM) for a short period of time (2–5 min) has no effect on cell viability.35 To conveniently monitor thiazolidine formation-mediated cell surface labeling, we designed and synthesized a fluorescent peptide 1, which has a 1,2-aminothiol moiety at its N-terminus and a 5-carboxyfluorescein (5-FAM) dye coupled with the side chain of a C-terminal Lys residue for fluorescence detection. Peptide 2 lacking 1,2-aminothiol was prepared in parallel as the negative control (Fig. 1A and S1†). To test whether cells could be labeled at physiological pH, 1 and 2 were added to periodate-exposed MCF-7 cells and control counterparts, respectively, at a final concentration of 15 μM in PBS (pH 7.4) and incubated for 1 h at room temperature. Then the cells were extensively washed with fresh PBS to remove unreacted 1 and 2, followed by staining the cell nuclei with Hoechst dye for confocal microscopy imaging. To our delight, only in the oxidized cells labeled with 1 could an apparent green rim be detected at the cell surface. In contrast, there was no significant fluorescent signal in non-oxidized cells even in the presence of 1, or in the oxidized cells treated with 2 (Fig. 1B). Since there are many free sulfhydryl groups on the cell surface, this may also lead to the fluorescent labeling of MCF-7 cells due to the oxidative disulfide formation reaction with 1.36 To exclude this possibility, we treated 1-MCF-7 with 0.5 mM TCEP for 30–60 min in PBS (pH 7.4) to reduce any plausible disulfide bonds into free thiols. The data again showed that there was no change in fluorescence intensity in the TCEP-treated cells compared to the non-treated counterparts (Fig. 1B). Consistently, flow cytometry data further confirmed that only the oxidized cells could be efficiently labeled with 1 compared to the other counterpart controls (Fig. 1C). In the following experiments, we also performed the cell labeling assay in the presence of both 1 and TCEP, but there was no difference in the labeling efficiency, regardless of whether TCEP was added or not, indicating that the potential thiol exchange reaction could not cause obvious cell labeling under the conditions we tested. As cell membranes are highly dynamic, involving processes such as internalization of membrane proteins and glycan,37,38 or due to the potential reversibility of thiazolidine linkage under our test conditions, the abundance of thiazolidine tethered-1 on the surface may change along with cell growth. It was shown, as expected, that the intensity did diminish gradually with prolonged incubation time. However, the detected fluorescence can persist at appreciable levels for up to 24 h (i.e., >8-fold compared to unlabeled cells) (Fig. 1D), which is comparable to previous reports.39–41 We then further optimized the labeling reaction by increasing the probe concentration and incubation time. The results showed that maximum cell labeling could be achieved within 1 h using 15 μM of 1, while a slightly higher concentration (20 μM) could shorten the time to 40 min (Fig. 1E). To demonstrate whether our method is generally applicable to other types of cells, the labeling reaction was tested on a panel of cell lines derived from both hematological and solid tumors (Fig. 1F). It was found that all the cells could be successfully labeled, but the labeling levels varied among the cell lines. This may be attributed to different abundances of sialic acids on the cell surface.42,43 Overall, as an expansion to the toolbox for cell surface engineering, our results clearly demonstrate that thiazolidine chemistry is fully compatible with living cells, and it enables the efficient labeling of cell surfaces at physiological pH by only using a low concentration of reagents (μM range).
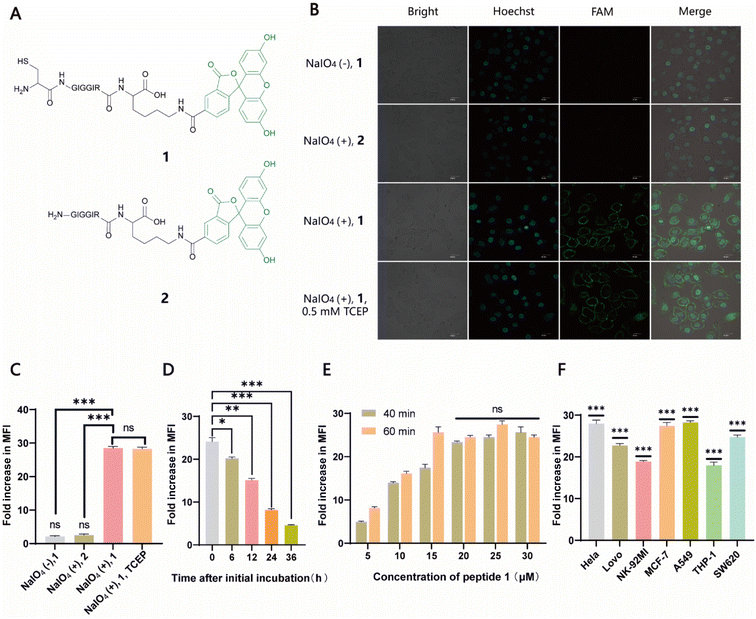 |
| | Fig. 1 Labeling of cell surfaces with fluorescent probes. (A) Chemical structures of two fluorescent peptide-based probes used here, 1 and 2. (B) Confocal microscopy of fluorescein-labeled cells (green) in different treatments. Nuclei were stained with Hoechst (blue). (C) Flow cytometry analysis of fluorescein-labeled MCF-7 cells in different treatments. (D) Detection of fluorescence intensity of cells conjugated with 1 at different incubation time points using flow cytometry. (E) Optimization of labeling reactions in which periodate-treated cells were labeled with different concentrations (5, 10, 15, 20, 25, and 30 μM) of 1 for 40 and 60 min, respectively, followed by flow cytometry analysis. (F) Levels of fluorescence intensity on the surface of a panel of cell lines labeled with 1. Significance is determined relative to the non-labeled control. | |
Conjugation of recombinant proteins to the cell surfaces via SpyCASE
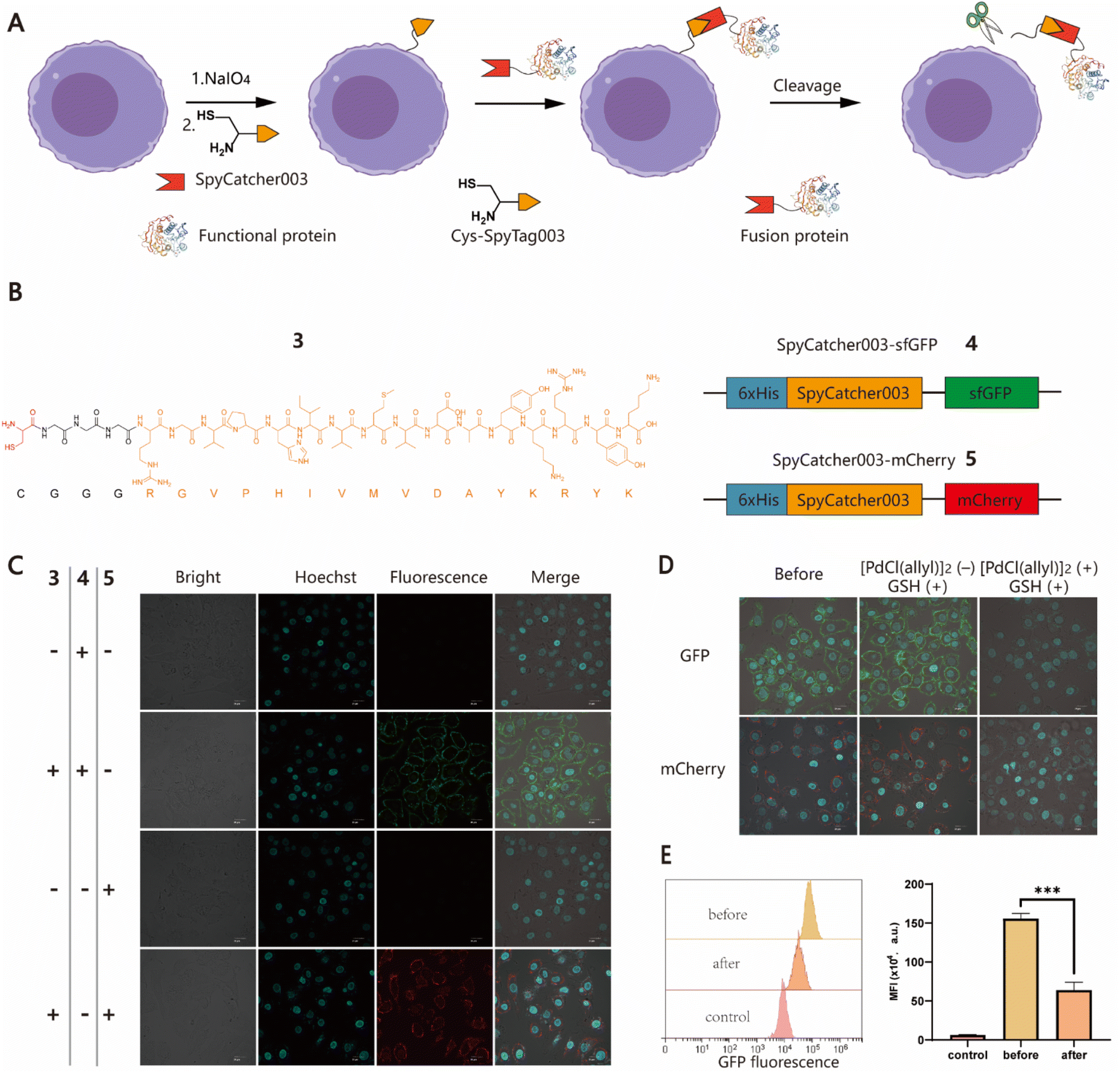
To further expand the potential utility of this strategy, we attempted to test whether recombinant proteins could also be chemically conjugated to cell surfaces for the construction of protein–cell conjugates (PCCs) to develop novel drug modalities for cancer therapy, such as antibody or nanobody–cell conjugates.44,45 Although several approaches have been developed to produce proteins with an N-terminal free Cys, it remains a labor-intensive task involving enzyme-mediated cleavage and subsequent stepwise protein purification procedures.46,47 For ease of manipulation, we decided to develop a SpyCatcher–SpyTag Chemistry-Assisted Cell Surface Engineering (SpyCASE) strategy through the combined use of thiazolidine formation and the SpyCatcher–SpyTag system (Fig. 2A). SpyCatcher and SpyTag are derived from the CnaB2 domain of the FbaB protein from Streptococcus pyogenes via protein engineering, and have the fascinating ability to self-assemble through spontaneous isopeptide bond formation in a wide range of aqueous buffers. They have emerged as a powerful conjugation tool for a plethora of applications, including vaccine development, nanoparticle functionalization, and protein hydrogel engineering.48–50 SpyCatcher003 and SpyTag003, another robust pair with the fastest reaction kinetics to date, were developed with great potential for use in the intracellular environment.51 To demonstrate the concept of SpyCASE, we conveniently prepared a SpyTag003-derived peptide 3 containing a 1,2-aminothiol at its N-terminus (Fig. 2B and S2†), and two fluorescent proteins sfGFP 4 and mCherry 5, and both were fused with SpyCatcher003, via chemical synthesis or recombinant expression (Fig. 2B). Referring to the labeling condition mentioned above, NaIO4-treated MCF-7 cells were first incubated with 20 μM of 3 in PBS (pH 7.4) for 1 h to incorporate SpyTag003 into the cell surface. The cells were then washed and incubated in normal DMEM media instead of PBS containing 10 μM 4 or 5 for another 1 h in an incubator (37 °C and 5% CO2). This change of conditions was made because extended exposure of cells to PBS would cause alterations in the cell morphology and cell detachment from the Petri dish. However, if the cells were immediately transferred to normal cell media after thiazolidine formation in PBS, they could retain optimal viability after the treatment. The results showed that NaIO4-treated cells were successfully labeled with 4 and 5 only in cells treated with 3, whereas in the absence of 3, no distinct fluorescent rims were observed in either oxidized or non-oxidized cells (Fig. 2C and S3†). This suggested that both thiazolidine formation and isopeptide bond formation are required for the cell surface modification. As it is relatively easy to prepare SpyCatcher-containing molecules of interest, SpyCASE has the potential to be a simple, versatile, and modular approach for the construction of various PCCs.
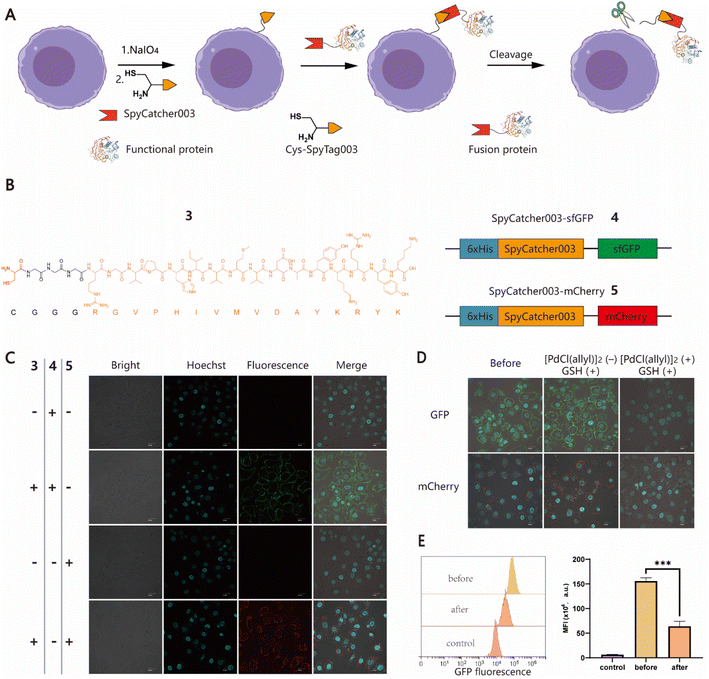 |
| | Fig. 2 Generation of protein–cell conjugates (PCCs) using the SpyCASE platform. (A) Schematic representation for the functionalization of cell surfaces with large recombinant proteins achieved by SpyCASE and palladium-mediated reversible chemical modification via thiazolidine cleavage. (B) Molecular structure of 3 containing a Cys residue marked with red and a G3 linker, followed by the sequence of SpyTag003 (RGVPHIVMVDAYKRYK) highlighted in orange (left panel), and schematic representations of SpyCatcher003-fused sfGFP and mCherry constructs, 4 and 5 (right panel). (C) Confocal microscopy images of MCF-7 cells labeled with fluorescent proteins 4 and 5 using SpyCASE. Scale bars represent 20 μm. (D) Confocal microscopy images of labeled MCF-7 cells conjugated with sfGFP and mCherry before and after palladium treatment. (E) Flow cytometry analysis of the fluorescence intensity change of sfGFP–cell conjugates after palladium treatment. | |
Metal ion-mediated reversible cell surface chemical modifications
To control cell behaviour spatially and temporally, reversible cell surface modification strategies are always highly desired. Currently, most of them rely on internal/external pH, light or enzymes to trigger bond-breaking reactions, leading to reversible chemical modifications. However, metal ion-mediated reversible cell surface engineering is still poorly studied with a few examples.52,53 The reversible thiazolidine linkage has been used to demonstrate traceless release of anticancer drugs from antibody–drug conjugates in vivo and Pd-mediated activation of chemically synthesized probes to inhibit deubiquitinases in living cells.24,27 However, to the best of our knowledge, the application of this chemistry in reversible chemical modifications of the cell surface has not been fully explored yet. Inspired by the work of Brik's group, sfGFP/mCherry–cell conjugates prepared using SpyCASE were treated with 5 μM [PdCl(allyl)]2 and reduced GSH in PBS (pH 7.4) at 4 °C for 15 min, as it has been shown that reduced GSH can protect cells from Pd-induced cytotoxicity.27 As a control, the cells were treated with only 5 μM GSH. The results showed that the fluorescent signal at the cell surface remained unchanged in the cells treated with GSH alone compared to the untreated counterparts. However, for the cells treated with [PdCl(allyl)]2 and GSH together, the intensity of fluorescence at the surface was substantially reduced, as shown by confocal microscopy imaging (Fig. 2D). Flow cytometry analysis further confirmed that the fluorescence intensity of Pd-treated cells was reduced by more than 60% compared to that of cells prior to Pd treatment (Fig. 2E). Subsequently, although we attempted to further increase the decaging efficiency by increasing the concentration of Pd or extending the reaction time, significant cytotoxicity was observed (Fig. S4†). Overall, this proof-of-concept study demonstrates that Pd-mediated deprotection of thiazolidine could be a potentially useful tool for the reversible chemical modification of living cell surfaces. Follow-up studies to screen and select less toxic metal ions to improve overall efficiency and exploration of their biomedical applications in vitro and in vivo are also possible for further investigation.
Induction of specific cell–cell interactions
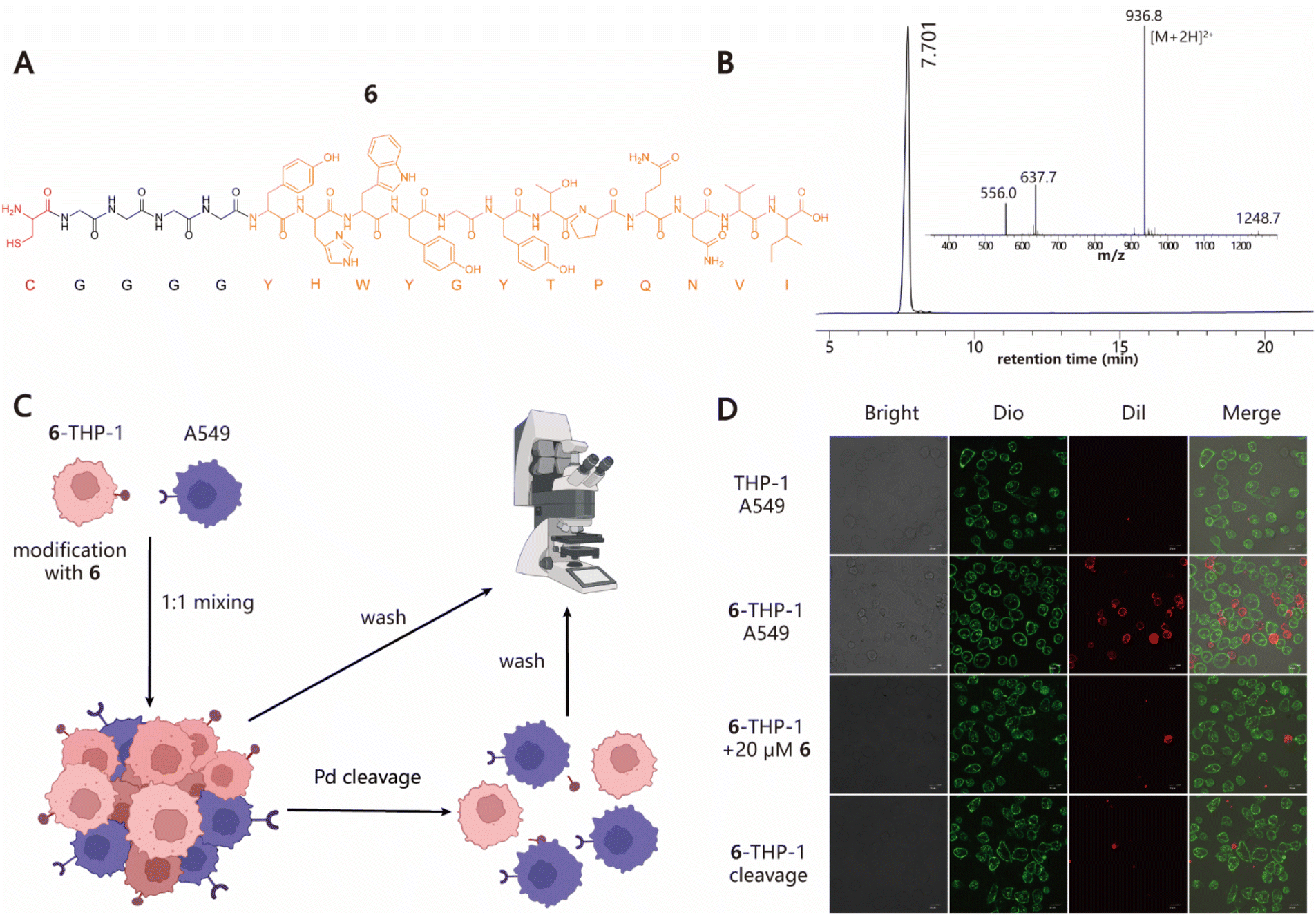
Next, we explored the ability of our approach to induce cell–cell interactions for modulating cell behaviours. To this end, we synthesized peptide 6 with a Cys at its N-terminus followed by a G4 linker and GE11 sequence, which is a previously reported tumor-targeting peptide that can specifically recognize the epidermal growth factor receptor (EGFR) with high affinity (Fig. 3A and B).54 To test whether 6–cell conjugates retain their EGFR-specific targeting ability, cell adhesion experiments using THP-1 and A549 cell lines as the model system were performed (Fig. 3C). 6–THP-1 conjugates were stained with DiI (red) and EGFR-positive A549 cells were labeled with Dio (green). Then two color-labeled cells were mixed at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (6–THP-1:A549) while unmodified THP-1 mixed with A549 under the same conditions served as the control. The cell mixtures were allowed to settle and interact with each other for 1 h in a cell incubator (37 °C and 5% CO2). Finally, the cells were washed to remove unbound 6–THP-1 and imaged using confocal microscopy. The results showed that a greater proportion of red 6–THP-1 was attached to the surface of green A549 cells compared to the unmodified counterparts. However, when 20 μM of free 6 (as the competitive inhibitor) was added to the A549 cell culture prior to cell mixing, the induced interactions between 6–THP-1 and A549 were significantly blocked, indicating that their interactions depend on the target selectivity of 6. In addition, if A549 and 6–THP-1 cell complexes were treated with 5 μM [PdCl(allyl)]2 to hydrolyse the thiazolidino linkage, approximately 85% of the bound 6–THP-1 would be separated from A549 cells again (Fig. 3D and S8†), thereby achieving a reversible control of cell–cell interactions. These data together demonstrate the potential utility of our method in modulating cell–cell interactions.
:1 (6–THP-1:A549) while unmodified THP-1 mixed with A549 under the same conditions served as the control. The cell mixtures were allowed to settle and interact with each other for 1 h in a cell incubator (37 °C and 5% CO2). Finally, the cells were washed to remove unbound 6–THP-1 and imaged using confocal microscopy. The results showed that a greater proportion of red 6–THP-1 was attached to the surface of green A549 cells compared to the unmodified counterparts. However, when 20 μM of free 6 (as the competitive inhibitor) was added to the A549 cell culture prior to cell mixing, the induced interactions between 6–THP-1 and A549 were significantly blocked, indicating that their interactions depend on the target selectivity of 6. In addition, if A549 and 6–THP-1 cell complexes were treated with 5 μM [PdCl(allyl)]2 to hydrolyse the thiazolidino linkage, approximately 85% of the bound 6–THP-1 would be separated from A549 cells again (Fig. 3D and S8†), thereby achieving a reversible control of cell–cell interactions. These data together demonstrate the potential utility of our method in modulating cell–cell interactions.
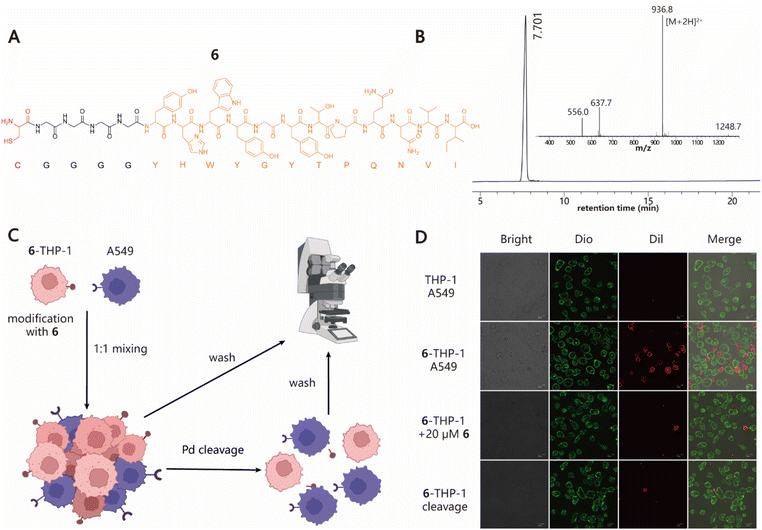 |
| | Fig. 3 Programming cell–cell interactions. (A) Structural information of 6 containing one Cys residue at the N-terminus marked with red and a G4 linker, followed by the sequence of GE11 peptide (YHWYGYTPQNVI) highlighted in orange. (B) Analytical HPLC and MS profile of 6. Calculated MS: 1872.03; observed MS: 1871.6. (C) Diagram for the induction of specific interactions between THP-1 and A549 cells and its reversible regulation. (D) Confocal microscopy images of cell–cell interactions between 6–THP-1 and A549 in different treatments. Scale bars represent 20 μm. | |
Selective targeting/killing of cancer cells by NK cell-based conjugates
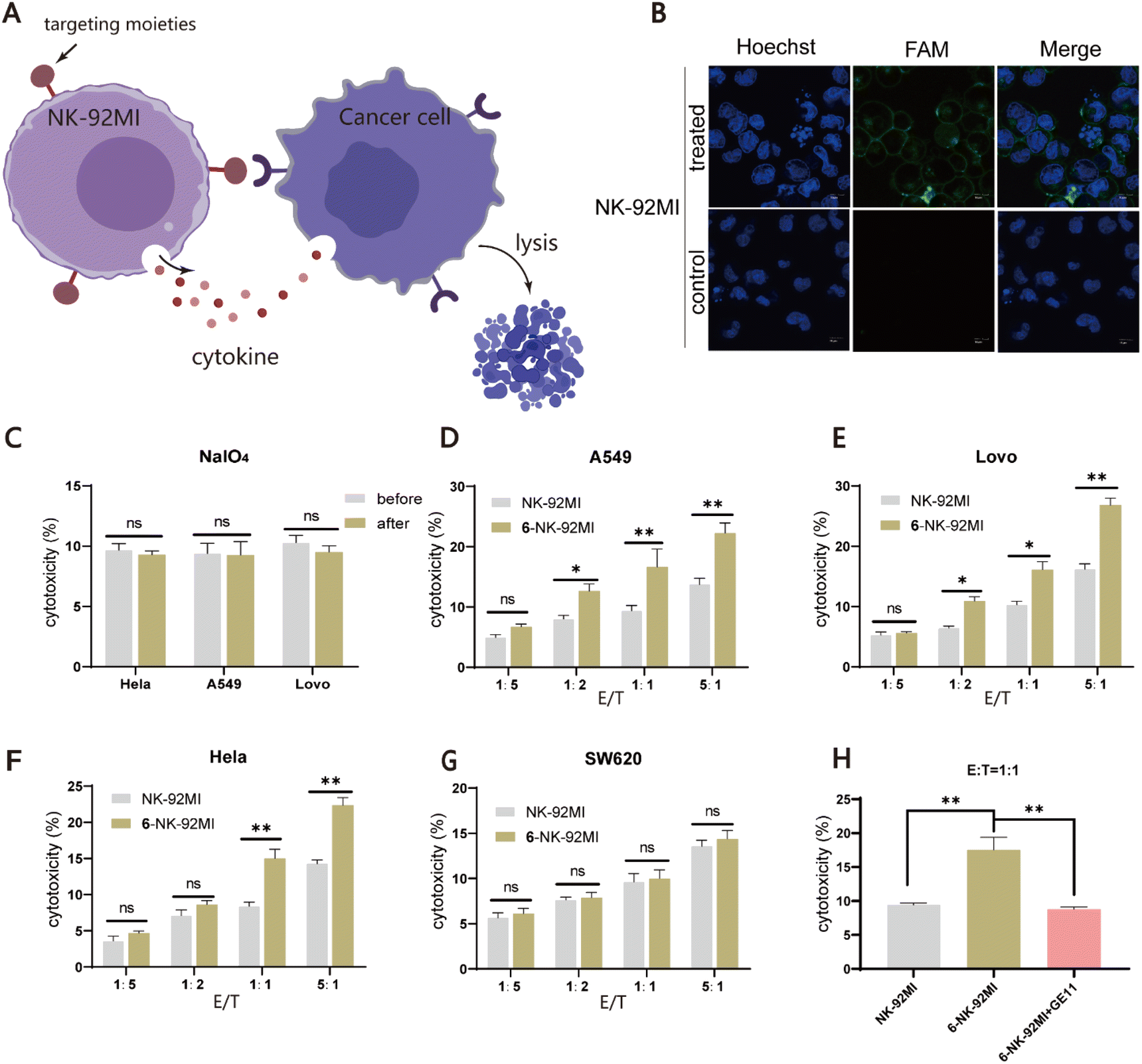
Finally, we expanded our method to generate peptide/protein-natural killer (NK) cell conjugates for the development of novel cancer immunotherapies. NK cells have broad-spectrum tumor-killing abilities and do not cause graft-versus-host disease, making them promising agents for allogeneic cellular immunotherapy.55,56 However, NK cells do not have an intrinsic targeting ability against cancer cells, limiting their overall efficacy. Therefore, we speculated that the installation of a tumor-targeting moiety into NK cells using our method may offer an alternative solution to this issue (Fig. 4A). Towards this goal, NK-92MI cells, an IL-2-independent variant of the NK-92 cell line, were used for the demonstration. NK-92MI cells are constantly active and nonimmunogenic, can be easily and reproducibly expanded in vitro, and have already shown promising value in clinical trials.57 As there are no previous reports on the study of the periodate oxidation of NK-92MI, we first tested whether NK-92MI could be labeled with fluorescent probe 1. Flow cytometry analysis showed that cells treated with 0.5 mM NaIO4 at 4 °C for 3 min, followed by incubation with 15 μM of 1 for 40 min, produced a strong fluorescence signal. Increasing the reaction time (60 or 80 min) helped improve the labeling level but without a significant difference (Fig. S5†). Confocal microscopy imaging also confirmed that only cells treated with NaIO4 and 1 were efficiently labeled (Fig. 4B). Furthermore, assays were performed to validate that periodate exposure had a negligible effect on the viability and functions of NK-92MI (Fig. 4C and S6†). Next, to endow NK-92MI cells with cancer-targeting ability, 6 was conjugated to NK-92MI using the optimized conditions mentioned above. Three EGFR-positive cancer cell lines with different EGFR expression levels, including LoVo (CRC), A549 (lung cancer), and HeLa (cervical cancer), were selected as target cells, while the EGFR-negative CRC cell line SW620 was used as a negative control (Fig. S7†). NK-92MI cells and 6–NK-92MI conjugates were incubated with these cancer cell lines at effector/target (E/T) ratios of 1:5, 1:2, 1:1, and 5:1 for 6 h, respectively. The ability of the NK cells to destroy cancer cells was measured using a lactate dehydrogenase cytotoxicity test kit. The result showed that at an E/T ratio of 1:5, neither unmodified NK-92MI nor 6–NK-92MI cells exhibited cytotoxicity against cancer cells. As the E/T ratio was increased, more cell death could be observed in the group treated with 6–NK-92MI. Fig. 4D–F show that at an E/T ratio of 5:1, about 22% of HeLa and A549 cells and 28% of LoVo cells were killed. The higher expression level of EGFR may account for the higher percentage of dead LoVo cells, as confirmed by western blot data (Fig. S7†). However, under the same conditions, there was no difference in the EGFR-negative SW620 cells treated with 6–NK-92MI or unmodified NK-92MI (Fig. 4G). In addition, if A549 cells were co-cultured with 6–NK-92MI or unmodified NK-92MI in the presence of free GE11 peptide (20 μM) as a competitor, at an E/T ratio of 1:1 for 6 h in the culture, the killing activity of 6–NK-92MI against A549 cells was significantly inhibited (Fig. 4H). This suggested that the interaction between 6 and EGFR is responsible for the improved killing ability of 6–NK-92MI towards target cells.
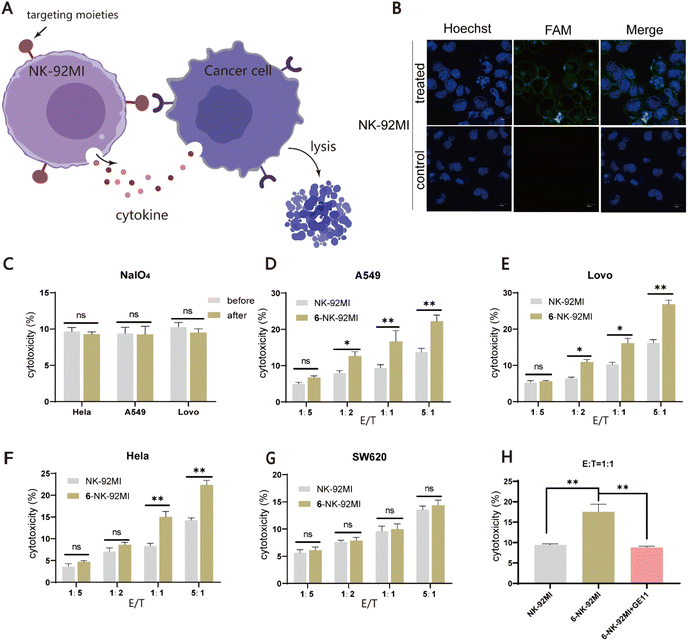 |
| | Fig. 4 Assessment of the killing activity of NK-92MI cell-based conjugates towards EGFR positive cancer cells. (A) Schematic representation of selective targeting and killing of cancer cells by NK-92MI cell-based conjugates. (B) Confocal microscopy images of fluorescently labeled NK-92MI cells by probe 1. Scale bars represent 20 μm. (C) Comparison of the killing activity of periodate-treated and untreated NK-92MI against HeLa, A549 and LoVo cells. (D–G) Evaluation of the cellular cytotoxicity of NK-92MI cells or 6–NK-92MI conjugates towards the EGFR-positive (A549, HeLa, and SW620) and the EGFR-negative (SW620) cancer cells in vitro. Values represent the mean ± SD from three independent experiments. (H) Comparison of the cellular cytotoxicity of NK-92MI or 6–NK-92MI against the A549 cell after adding free 6 (20 μM). | |
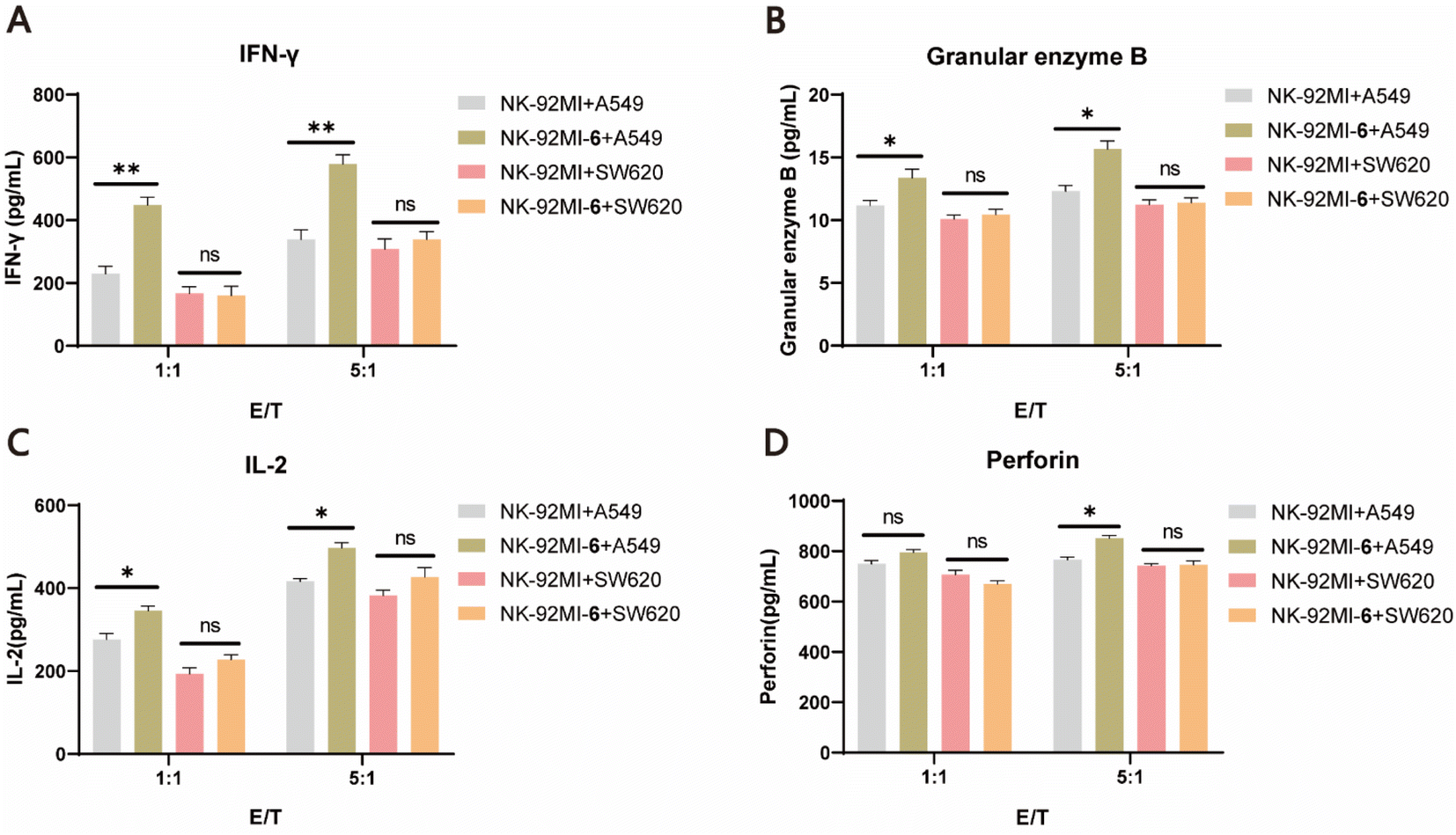
It is well known that once NK cells are activated, they secrete a range of immunomodulatory cytokines, such as granzyme B, perforin, IFN-γ, and IL-2, which serve as markers of NK cell activation.58 To further validate the improved killing capability of 6–NK-92MI, ELISAs were performed to characterize the levels of cytokines after incubating 6–NK-92MI and NK-92MI cells with either A549 cells or SW620 cells at different E/T ratios (1:1 and 5:1) for 6 h. The results showed that granzyme B, IFN-γ, and IL-2 were all significantly increased in supernatants of A549 cell culture treated with 6–NK-92MI compared with those treated with NK-92MI. However, for the EGFR-negative SW620 cells there was no difference in the level of detected cytokines produced by NK cells in the supernatants, regardless of whether they were treated with 6–NK-92MI or NK-92MI (Fig. 5A–D). Altogether, these results convincingly demonstrate that NK-92MI cells decorated with 6 have enhanced targeting ability and cytotoxicity in vitro, unleashing the potential power of our method for cancer treatment.
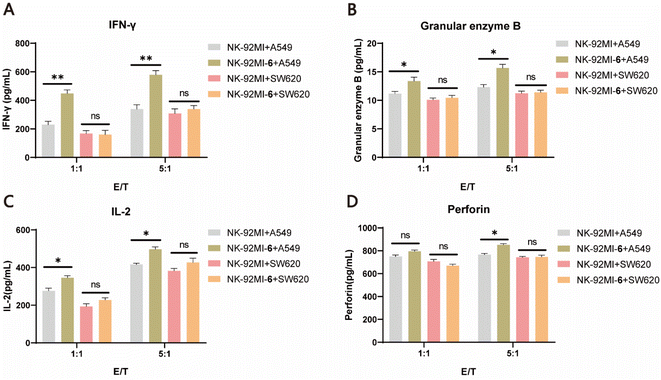 |
| | Fig. 5 Activation of 6–NK-92MI conjugates in the EGFR-positive cancer cell cultures. Detection of the levels of IFN-γ (A), granular enzyme B (B), IL-2 (C), and perforin (D) released by 6–NK-92MI or unmodified NK-92MI cells in the cell-free supernatants of A549 and SW620, respectively, using ELISAs. | |
Conclusions
In this study, we have developed a general chemical strategy to functionalize living cell surfaces with various functional molecules, including synthetic peptides and recombinant proteins, at physiological pH by leveraging the classical thiazolidine ring formation chemistry, which serves a significant addition to the toolbox for live cell surface engineering. Using this method, we successfully induced specific cell–cell interaction, achieved Pd-mediated reversible cell surface chemical modification, and generated NK cell-based PCCs for the selective targeting/killing of several EGFR-positive cancer cells. We further developed an efficient and modular SpyCASE platform by combining thiazolidine formation and the SpyCatcher–SpyTag system. This platform enables the decoration of cells with large functional proteins, such as antibodies or enzymes, in their native states.
Compared to previously developed strategies, our approach has several appealing features, such as its ease of manipulation, high efficiency, physiological conditions, and tunable bond stability. Currently, it is undisputed that the combination of MOE with click chemistry remains a powerful strategy for functionalizing living cell surfaces.13,59 However, certain types of unnatural sugar analogs with click handles are not readily available due to their complicated organic synthesis and high price. In comparison, our method greatly simplifies the experimental procedure for cell surface labeling by using common and inexpensive reagents. Additionally, much lower concentrations of labeling reagents (μM) were used in this study than in previously reported oxime ligation-mediated methods,35,60 and no catalyst or reducing reagents are required. Nevertheless, it should be noted that caution needs to be taken if the decorated molecules are required to reside on the cell surface for longer time, such as a couple of days or even longer. Overall, we believe that this simple method will find broad application in the future, such as the development of a pH-responsive live cell-based drug delivery system.
Experimental
Materials
Allylpalladium(II) chloride dimer ([Pd(allyl)Cl]2) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Bide Pharmatech Ltd (Shanghai, China). Human serum and trypsin–EDTA (0.25%), RPMI 1640 medium, Dulbecco's Modified Eagle's Medium (DMEM), and 1× PBS (pH 7.2–7.4), penicillin–streptomycin solution, and phenol-red-free MEM-α were purchased from Solarbio (Beijing, China). The BCA protein assay kit, CCK-8 kit, anti-EGFR antibody, anti-rabbit IgG antibodies, DiI dye, and Dio dye were purchased from Beyotime Biotechnology (Shanghai, China). Glutathione, IPTG, 50× TAE buffer and oligonucleotides (Table S1, ESI†) were obtained from Sangon (Shanghai, China). Phanta max super-fidelity DNA polymerase was purchased from Vazyme (Nanjing, China). The NK-92MI cell culture medium, one step freezing medium, and NK-92MI cell line were obtained from Cas9X (Suzhou, China). Ni-NTA beads were purchased from Smart-Lifescience (Xian, China). FastDigest restriction enzymes (HindIII, NcoI), DNA gel loading dye, SYBR gold nucleic acid gel stain (1000×), and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific Inc (Waltham, MA, USA). The peptides (1, 2, 3, and 6) were purchased from GenScript (Nanjing, China). The plasmid extraction kit and Gel extraction kit were available from TransGen Biotech (Beijing, China). ELISA kits for the detection of IFN-γ, IL-2, perforin, and granular enzyme B were all from Wuhan Saipei Biotechnology Co., Ltd (Wuhan, China). Commercial sources were used to get other unspecified reagents.
Bacterial strains and culture conditions
Escherichia coli DH5α was used as a sub-cloning host, whereas BL21(DE3) was used as an expression host. Unless stated otherwise, all strains were grown at 37 °C in LB medium (10 g L−1 tryptone, 5 g L−1 yeast extract, and 5 g L−1 NaCl). If required, antibiotics and inducers were added, for instance, chloramphenicol (50 μg mL−1), ampicillin (100 μg mL−1), and IPTG (0.5–1 mM).
Cell culture
The NK-92MI cell line (human) was incubated in MEM-α medium supplemented with 12.5% FBS, 12.5% HS, and 0.2 mM inositol + 0.1 mM 2-mercaptoethanol + 0.02 mM folic acid + 1% penicillin–streptomycin (P/S). LoVo (CRC), HeLa (cervical cancer), MCF-7 (human breast cancer), and A549 (non-small cell lung cancer) cell lines were grown in DMEM containing 10% FBS and 1% P/S. SW620 (CRC) and THP-1 (human monocytic leukemia) cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% P/S. All cells were incubated at 37 °C in a humidified 5% CO2 atmosphere.
Plasmid construction and protein expression
Plasmids of pETDuet–SpyCatcher003–GFP and pETDuet–SpyCatcher003–mCherry were constructed to produce SpyCatcher003–sfGFP 4 and SpyCatcher003–mCherry 5. The SpyCatcher003 gene was amplified using primers SC3-G F1/SC3-G R1 or SC3-M F1/SC3-M R1 from SpyCatcher003 (addgene, plasmid #133447). The sfGFP gene was amplified from pBad–sfGFP (addgene, plasmid #85482) using SC3-G F2/SC3-G R2. The mCherry gene was amplified using primers SC3-M F2/SC3-M R2 from pETDuet-SI–mCherry–SpyCatcher constructed before in our lab. Plasmid pETDuet was linearized with NcoI and HindIII at 37 °C for 30 min. The corresponding PCR products were ligated into pETduet to generate pETDuet–SpyCatcher003–sfGFP and pETDuet–SpyCatcher003–mCherry by using a Hieff Clone Plus Multi One Step Cloning Kit (Shanghai, China). Plasmids were transformed into E. coli DH5α competent cells, and positive colonies were selected by PCR analysis for sequencing. For SpyCatcher003–sfGFP 4 and SpyCatcher003–mCherry 5 expression and purification, BL21(DE3) cells were transformed with the corresponding plasmids described above, and a single colony was picked and cultured in 3 mL LB at 37 °C overnight. The cultures were then sub-cultured to 400 mL LB and induced by 1 mM IPTG for 5 h. The bacterial cultures were harvested and lysed in Tris buffer (20 mM Tris, 50 mM NaCl, and pH 7.5). The cell fragments were removed from the supernatant by centrifugation at 10000 × g for 20 min at 4 °C. After filtering the supernatant with a 0.22 μm filter, the target protein in the supernatant was separated by using Ni-NTA beads. The beads were then washed with different concentrations of imidazole. The solution containing the target protein was then washed with PBS in a 10 KDa ultrafiltration tube to concentrate and remove the imidazole.
Western blot analysis of EGFR expression
Tumor cells in a 1.5 mL tube were lysed in boiling water for 15 min after being suspended in an SDS-PAGE loading buffer. After BCA assay, the samples underwent SDS-PAGE gel separation and then transferred to a PVDF membrane. The PVDF membrane after being blocked by 5% skimmed milk was exposed to the anti-EGFR antibody at 4 °C overnight. Then HRP-conjugated goat anti-rabbit IgG antibodies was added at rt. for 1 h. After washing three times with TBST, the Chemical Scope series (Clinx Science Instruments Co., Ltd, China) was used to capture the membrane which was treated with an ECL kit. The β-actin protein was used as the internal reference.
The effects of periodate oxidation on the killing activity of NK-92MI cells
NK-92MI cells was treated with 0.5 mM NaIO4 at 4 °C for 3 min. After washing with PBS, treated NK-92MI cells and untreated NK-92MI were incubated with the three cancer cell lines (HeLa, LoVo, and A549) at an effector/target (E/T) ratio of 1:1 for 6 h, respectively. A lactate dehydrogenase cytotoxicity test kit was then used to measure the ability of the NK cells to destroy cancer cells.
Pd-induced cytotoxicity
Cells (A549, HeLa, MCF-7; 1 × 105 cells) were treated with 5 μM [PdCl(allyl)]2 in PBS (pH 7.4) supplemented with 5 μM reduced GSH at 4 °C for 10, 15, and 20 min. The treated cells were washed 3 times using PBS buffer, and then resuspended in phenol-red-free MEM-α supplemented with both 10% FBS and 10% CCK-8 and incubated for 1 h at 37 °C. The optical density at 450 nm wavelength was then detected using a microplate reader (BioTek Instrument, Winooski, VT, USA).
Cell viability of NK-92MI cells after periodate oxidation
NK-92MI cells (1 × 105 cells) were treated with different concentrations of NaIO4 with a final concentration (0, 0.25, 0.5, and 1 mM) for different times (1, 3, and 5 min). The treated NK-92MI cells were washed 3 times using PBS buffer, and then resuspended in phenol-red-free MEM-α supplemented with both 10% FBS and 10% CCK-8 and incubated for 1 h at 37 °C. The optical density at 450 nm wavelength was then detected using a microplate reader (BioTek Instrument, Winooski, VT, USA).
Quantification of cytokines secreted by NK-92MI cells
A549 cells and SW620 cells (1 × 104 cells per well) were seeded into 96-well plates. After attachment, the wells were washed with PBS and then co-incubated with the engineered 6–NK-92MI and unmodified cells at an effector/target (E/T) ratio of 1:1 or 5:1. After 6 h, the levels of IFN-γ, IL-2, perforin, and granular enzyme B in cell-free supernatants were measured by ELISA assays according to the manufacturer's instructions (Wuhan Saipei Biotechnology, Wuhan, China).
Statistical analysis
The data presented were performed at least three times. The data were presented as mean ± SD. Statistical analyses and image processing were performed by using ImageJ, Adobe Illustrator 2022, Cytexpert, and GraphPad Prism 8.0.1. The comparison of two groups was performed by a two-tailed unpaired t-test. The significant difference was arranged as follows: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, and ns, not significant.
Data availability
Information supporting this article has been uploaded as part of the ESI.† Additional data are available from the authors on reasonable request.
Author contributions
X. B. conceived and designed the project. X. L. performed the experiments and collected data. Y. W. helped to purify the proteins. X. L. and X. B. analyzed the data, and wrote and proofread the manuscript. X. B. and B. Y. were responsible for the acquisition of funding and supervised the project. All authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by a grant from the National Key Research and Development Program of China (2018YFA0900404), the National Natural Science Foundation of China (22134003) and Zhejiang University of Technology High-Level Talents Startup Funds (X. B. Bi).
Notes and references
- S. Abbina, E. M. J. Siren, H. Moon and J. N. Kizhakkedathu, Surface Engineering for Cell-Based Therapies: Techniques for Manipulating Mammalian Cell Surfaces, ACS Biomater. Sci. Eng., 2018, 4, 3658–3677 CrossRef CAS PubMed.
- M. D. Mager, V. LaPointe and M. M. Stevens, Exploring and exploiting chemistry at the cell surface, Nat. Chem., 2011, 3, 582–589 CrossRef CAS PubMed.
- Y. H. Lai, C. Y. Su, H. W. Cheng, C. Y. Chu, L. B. Jeng, C. S. Chiang, W. C. Shyu and S. Y. Chen, Stem cell-nanomedicine system as a theranostic bio-gadolinium agent for targeted neutron capture cancer therapy, Nat. Commun., 2023, 14, 285 CrossRef CAS PubMed.
- L. Yang, Y. Yang, Y. Chen, Y. Xu and J. Peng, Cell-based drug delivery systems and their in vivo fate, Adv. Drug Delivery Rev., 2022, 187, 114394 CrossRef CAS PubMed.
- T. Thomsen and H. A. Klok, Chemical Cell Surface Modification and Analysis of Nanoparticle-Modified Living Cells, ACS Appl. Bio Mater., 2021, 4, 2293–2306 CrossRef CAS PubMed.
- R. Zhou, Y. Wu, K. Chen, D. Zhang, Q. Chen, D. Zhang, Y. She, W. Zhang, L. Liu, Y. Zhu, C. Gao and R. Liu, A Polymeric Strategy Empowering Vascular Cell Selectivity and Potential Application Superior to Extracellular Matrix Peptides, Adv. Mater., 2022, 34, e2200464 CrossRef PubMed.
- S. C. Hsiao, B. J. Shum, H. Onoe, E. S. Douglas, Z. J. Gartner, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns, Langmuir, 2009, 25, 6985–6991 CrossRef CAS PubMed.
- T. J. Gardner, J. P. Lee, C. M. Bourne, D. Wijewarnasuriya, N. Kinarivala, K. G. Kurtz, B. C. Corless, M. M. Dacek, A. Y. Chang, G. Mo, K. M. Nguyen, R. J. Brentjens, D. S. Tan and D. A. Scheinberg, Engineering CAR-T cells to activate small-molecule drugs in situ, Nat. Chem. Biol., 2022, 18, 216–225 CrossRef CAS PubMed.
- J. H. Collier and M. Mrksich, Engineering a biospecific communication pathway between cells and electrodes, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2021–2025 CrossRef CAS PubMed.
- V. Prasad, Immunotherapy: tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers, Nat. Rev. Clin. Oncol., 2018, 15, 11–12 CrossRef PubMed.
- C. M. Csizmar, J. R. Petersburg and C. R. Wagner, Programming Cell-Cell Interactions through Non-genetic Membrane Engineering, Cell Chem. Biol., 2018, 25, 931–940 CrossRef CAS PubMed.
- S. Letschert, A. Göhler, C. Franke, N. Bertleff-Zieschang, E. Memmel, S. Doose, J. Seibel and M. Sauer, Super-resolution imaging of plasma membrane glycans, Angew. Chem., Int. Ed. Engl., 2014, 53, 10921–10924 CrossRef CAS PubMed.
- X. Wang, S. Lang, Y. Tian, J. Zhang, X. Yan, Z. Fang, J. Weng, N. Lu, X. Wu, T. Li, H. Cao, Z. Li and X. Huang, Glycoengineering of Natural Killer Cells with CD22 Ligands for Enhanced Anticancer Immunotherapy, ACS Cent. Sci., 2020, 6, 382–389 CrossRef CAS PubMed.
- M. F. Debets, O. Y. Tastan, S. P. Wisnovsky, S. A. Malaker, N. Angelis, L. K. R. Moeckl, J. Choi, H. Flynn, L. J. S. Wagner, G. Bineva-Todd, A. Antonopoulos, A. Cioce, W. M. Browne, Z. Li, D. C. Briggs, H. L. Douglas, G. T. Hess, A. J. Agbay, C. Roustan, S. Kjaer, S. M. Haslam, A. P. Snijders, M. C. Bassik, W. E. Moerner, V. S. W. Li, C. R. Bertozzi and B. Schumann, Metabolic precision labeling enables selective probing of O-linked N-acetylgalactosamine glycosylation, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25293–25301 CrossRef CAS PubMed.
- P. Shi, E. Ju, Z. Yan, N. Gao, J. Wang, J. Hou, Y. Zhang, J. Ren and X. Qu, Spatiotemporal control of cell-cell reversible interactions using molecular engineering, Nat. Commun., 2016, 7, 13088 CrossRef CAS PubMed.
- C. Agatemor, M. J. Buettner, R. Ariss, K. Muthiah, C. T. Saeui and K. J. Yarema, Exploiting metabolic glycoengineering to advance healthcare, Nat. Rev. Chem., 2019, 3, 605–620 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, Genetic Encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- N. Pishesha, A. M. Bilate, M. C. Wibowo, N. J. Huang, Z. Li, R. Deshycka, D. Bousbaine, H. Li, H. C. Patterson, S. K. Dougan, T. Maruyama, H. F. Lodish and H. L. Ploegh, Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3157–3162 CrossRef CAS PubMed.
- X. Bi, J. Yin, D. Zhang, X. Zhang, S. Balamkundu, J. Lescar, P. C. Dedon, J. P. Tam and C. F. Liu, Tagging Transferrin Receptor with a Disulfide FRET Probe To Gauge the Redox State in Endosomal Compartments, Anal. Chem., 2020, 92, 12460–12466 CrossRef CAS PubMed.
- S. Hong, Y. Shi, N. C. Wu, G. Grande, L. Douthit, H. Wang, W. Zhou, K. B. Sharpless, I. A. Wilson, J. Xie and P. Wu, Bacterial glycosyltransferase-mediated cell-surface chemoenzymatic glycan modification, Nat. Commun., 2019, 10, 1799 CrossRef PubMed.
- M. Fernández-Suárez, H. Baruah, L. Martínez-Hernández, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef PubMed.
- N. George, H. Pick, H. Vogel, N. Johnsson and K. Johnsson, Specific labeling of cell surface proteins with chemically diverse compounds, J. Am. Chem. Soc., 2004, 126, 8896–8897 CrossRef CAS PubMed.
- J. D. Wade, T. Domagala, J. Rothacker, B. Catimel and E. Nice, Use of thiazolidine-mediated ligation for site specific biotinylation of mouse EGF for biosensor immobilisation, Lett. Pept. Sci., 2001, 8, 211–220 CrossRef CAS.
- G. Casi, N. Huguenin-Dezot, K. Zuberbuhler, J. Scheuermann and D. Neri, Site-specific traceless coupling of potent cytotoxic drugs to recombinant antibodies for pharmacodelivery, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS PubMed.
- X. Bi, K. K. Pasunooti, A. H. Tareq, J. Takyi-Williams and C. F. Liu, Genetic incorporation of 1,2-aminothiol functionality for site-specific protein modification via thiazolidine formation, Org. Biomol. Chem., 2016, 14, 5282–5285 RSC.
- K. Nakatsu, A. Okamoto, G. Hayashi and H. Murakami, Repetitive Thiazolidine Deprotection Using a Thioester-Compatible Aldehyde Scavenger for One-Pot Multiple Peptide Ligation, Angew. Chem., Int. Ed. Engl., 2022, 61, e202206240 CrossRef CAS PubMed.
- G. Mann, G. Satish, R. Meledin, G. B. Vamisetti and A. Brik, Palladium-Mediated Cleavage of Proteins with Thiazolidine-Modified Backbone in Live Cells, Angew. Chem., Int. Ed. Engl., 2019, 58, 13540–13549 CrossRef CAS PubMed.
- N. Asiimwe, M. F. Al Mazid, D. P. Murale, Y. K. Kim and J.-S. Lee, Recent advances in protein modifications techniques for the targeting N-terminal cysteine, Pept. Sci., 2022, 114, e24235 CAS.
- D. Bermejo-Velasco, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Thiazolidine chemistry revisited: a fast, efficient and stable click-type reaction at physiological pH, Chem. Commun., 2018, 54, 12507–12510 RSC.
- M. Wathier, C. S. Johnson, T. Kim and M. W. Grinstaff, Hydrogels formed by multiple peptide ligation reactions to fasten corneal transplants, Bioconjugate Chem., 2006, 17, 873–876 CrossRef CAS PubMed.
- C. F. Liu and J. P. Tam, Peptide segment ligation strategy without use of protecting groups, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6584–6588 CrossRef CAS PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation, Chem. Sci., 2016, 7, 4589–4593 RSC.
- H. Faustino, M. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalisation, Chem. Sci., 2016, 7, 5052–5058 RSC.
- K. Li, W. Wang and J. Gao, Fast and Stable N-Terminal Cysteine Modification through Thiazolidino Boronate Mediated Acyl Transfer, Angew. Chem., Int. Ed. Engl., 2020, 59, 14246–14250 CrossRef CAS PubMed.
- Y. Zeng, T. N. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, High-efficiency labeling of sialylated glycoproteins on living cells, Nat. Methods, 2009, 6, 207–209 CrossRef CAS PubMed.
- S. Aubry, F. Burlina, E. Dupont, D. Delaroche, A. Joliot, S. Lavielle, G. Chassaing and S. Sagan, Cell-surface thiols affect cell entry of disulfide-conjugated peptides, FASEB J., 2009, 23, 2956–2967 CrossRef CAS PubMed.
- S.-Y. Zhang, Z.-R. Zhou and R.-C. Qian, Recent Progress and Perspectives on Cell Surface Modification, Chem.–Asian J., 2021, 16, 3250–3258 CrossRef CAS PubMed.
- M. A. McNiven and A. J. Ridley, Focus on membrane dynamics, Trends Cell Biol., 2006, 16, 485–486 CrossRef PubMed.
- J. C. Maza, D. M. García-Almedina, L. E. Boike, N. X. Hamlish, D. K. Nomura and M. B. Francis, Tyrosinase-Mediated Synthesis of Nanobody–Cell Conjugates, ACS Cent. Sci., 2022, 8(7), 955–962 CrossRef CAS PubMed.
- M. J. Frank, N. Olsson, A. Huang, S. W. Tang, R. S. Negrin, J. E. Elias and E. H. Meyer, A novel antibody-cell conjugation method to enhance and characterize cytokine-induced killer cells, Cytotherapy, 2020, 22, 135–143 CrossRef CAS PubMed.
- J. Li, M. Chen, Z. Liu, L. Zhang, B. H. Felding, K. W. Moremen, G. Lauvau, M. Abadier, K. Ley and P. Wu, A Single-Step Chemoenzymatic Reaction for the Construction of Antibody-Cell Conjugates, ACS Cent. Sci., 2018, 4, 1633–1641 CrossRef CAS PubMed.
- X.-N. He, Y.-N. Wang, Y. Wang and Z.-R. Xu, Accurate quantitative detection of cell surface sialic acids with a background-free SERS probe, Talanta, 2020, 209, 120579 CrossRef CAS PubMed.
- A. Matsumoto, H. Cabral, N. Sato, K. Kataoka and Y. Miyahara, Assessment of Tumor Metastasis by the Direct Determination of Cell-Membrane Sialic Acid Expression, Angew. Chem., Int. Ed., 2010, 49, 5494–5497 CrossRef CAS PubMed.
- Y. Zhao, Y. Dong, S. Yang, Y. Tu, C. Wang, J. Li, Y. Yuan and Z. Lian, Bioorthogonal Equipping CAR-T Cells with Hyaluronidase and Checkpoint Blocking Antibody for Enhanced Solid Tumor Immunotherapy, ACS Cent. Sci., 2022, 8, 603–614 CrossRef CAS PubMed.
- L. Gong, Y. Li, K. Cui, Y. Chen, H. Hong, J. Li, D. Li, Y. Yin, Z. Wu and Z. Huang, Nanobody-Engineered Natural Killer Cell Conjugates for Solid Tumor Adoptive Immunotherapy, Small, 2021, 17, e2103463 CrossRef PubMed.
- I. E. Gentle, D. P. De Souza and M. Baca, Direct production of proteins with N-terminal cysteine for site-specific conjugation, Bioconjugate Chem., 2004, 15, 658–663 CrossRef CAS PubMed.
- J. P. Hempfling, E. R. Sekera, A. Sarkar, A. B. Hummon and D. Pei, Generation of Proteins with Free N-Terminal Cysteine by Aminopeptidases, J. Am. Chem. Soc., 2022, 144, 21763–21771 CrossRef CAS PubMed.
- F. Sun, W. B. Zhang, A. Mahdavi, F. H. Arnold and D. A. Tirrell, Synthesis of bioactive protein hydrogels by genetically encoded SpyTag-SpyCatcher chemistry, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11269–11274 CrossRef CAS PubMed.
- Y. F. Kang, C. Sun, Z. Zhuang, R. Y. Yuan, Q. Zheng, J. P. Li, P. P. Zhou, X. C. Chen, Z. Liu, X. Zhang, X. H. Yu, X. W. Kong, Q. Y. Zhu, Q. Zhong, M. Xu, N. S. Zhong, Y. X. Zeng, G. K. Feng, C. Ke, J. C. Zhao and M. S. Zeng, Rapid Development of SARS-CoV-2 Spike Protein Receptor-Binding Domain Self-Assembled Nanoparticle Vaccine Candidates, ACS Nano, 2021, 15, 2738–2752 CrossRef CAS PubMed.
- C. C. S. Pedroso, V. R. Mann, K. Zuberbühler, M. F. Bohn, J. Yu, V. Altoe, C. S. Craik and B. E. Cohen, Immunotargeting of Nanocrystals by SpyCatcher Conjugation of Engineered Antibodies, ACS Nano, 2021, 15, 18374–18384 CrossRef CAS PubMed.
- A. H. Keeble, P. Turkki, S. Stokes, I. N. A. Khairil Anuar, R. Rahikainen, V. P. Hytonen and M. Howarth, Approaching infinite affinity through engineering of peptide-protein interaction, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26523–26533 CrossRef CAS PubMed.
- R. C. Qian, Z. R. Zhou, W. Guo, Y. Wu, Z. Yang and Y. Lu, Cell Surface Engineering Using DNAzymes: Metal Ion Mediated Control of Cell-Cell Interactions, J. Am. Chem. Soc., 2021, 143, 5737–5744 CrossRef CAS PubMed.
- X. Wang, Y. Liu, X. Fan, J. Wang, W. S. C. Ngai, H. Zhang, J. Li, G. Zhang, J. Lin and P. R. Chen, Copper-Triggered Bioorthogonal Cleavage Reactions for Reversible Protein and Cell Surface Modifications, J. Am. Chem. Soc., 2019, 141, 17133–17141 CrossRef CAS PubMed.
- T. Hailing, P. Yonghong, Z. Yufeng and T. Haitao, Challenges for the application of EGFR-targeting peptide GE11 in tumor diagnosis and treatment, J. Controlled Release, 2022, 349, 592–605 CrossRef PubMed.
- M. G. Morvan and L. L. Lanier, NK cells and cancer: you can teach innate cells new tricks, Nat. Rev. Cancer, 2016, 16, 7–19 CrossRef CAS PubMed.
- T. Bald, M. F. Krummel, M. J. Smyth and K. C. Barry, The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies, Nat. Immunol., 2020, 21, 835–847 CrossRef CAS PubMed.
- G. Suck, M. Odendahl, P. Nowakowska, C. Seidl, W. S. Wels, H. G. Klingemann and T. Tonn, NK-92: an 'off-the-shelf therapeutic' for adoptive natural killer cell-based cancer immunotherapy, Cancer Immunol., Immunother., 2016, 65, 485–492 CrossRef CAS PubMed.
- N. K. Wolf, D. U. Kissiov and D. H. Raulet, Roles of natural killer cells in immunity to cancer, and applications to immunotherapy, Nat. Rev. Immunol., 2023, 23, 90–105 CrossRef CAS PubMed.
- H. Y. Yoon, H. Koo, K. Kim and I. C. Kwon, Molecular imaging based on metabolic glycoengineering and bioorthogonal click chemistry, Biomaterials, 2017, 132, 28–36 CrossRef CAS PubMed.
- D. Zhang, Z. Wang, S. Hu, J. Lescar, J. P. Tam and C. F. Liu, Vypal2: A Versatile Peptide Ligase for Precision Tailoring of Proteins, Int. J. Mol. Sci., 2021, 23, 458 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Xiaobao
Bi
*ab and
Xiaobao
Bi