
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputer vision as a new paradigm for monitoring of solution and solid phase peptide synthesis†
Chunhui
Yan
 ,
Calum
Fyfe‡
,
Laura
Minty‡
,
Henry
Barrington
,
Craig
Jamieson
* and
Marc
Reid
*
,
Calum
Fyfe‡
,
Laura
Minty‡
,
Henry
Barrington
,
Craig
Jamieson
* and
Marc
Reid
*
WestCHEM Department of Pure & Applied Chemistry, University of Strathclyde, Glasgow, UK. E-mail: craig.jamieson@strath.ac.uk; marc.reid.100@strath.ac.uk
First published on 10th October 2023
Abstract
We report a strategy for the camera-enabled non-contact colourimetric reaction monitoring and optimisation of amide bond formation, mediated by coupling reagents. For amide bond formation in solution phase, investigation of reactions mediated by HATU, PyAOP, and DIC/Oxyma evidenced correlations between colour parameters extracted from video data and conversion to amide product measured by off-line HPLC analysis of concentration. These correlations, supported by mutual information analysis, were further investigated using video recordings of solid phase peptide synthesis (SPPS), co-analysed by off-line HPLC to track remaining unreacted substrate in solution. An optimisation method of coupling time in SPPS was derived from ΔE (a measurement of colour contrast), giving comparable isolated peptide yield and purity at 65–95% reduced overall reaction time. The same colour data enabled data-rich monitoring of reaction rate attenuation, consisted with computationally-derived measures of amino acid steric bulk. These findings provide a foundation for exploring the use of camera technology and computer vision towards automated and online mechanistic profiling of SPPS.
1 Introduction
1.1 Amide formation strategies
Amides are one of the most frequently occurring functional groups in pharmaceutically-relevant chemistry, requiring reliable synthetic transformations to make them.1–3 Their preparation generally requires activation of the carboxylic acid to promote a condensation reaction (Fig. 1).4 The acid is typically activated in the form of an acyl chloride (Fig. 1A), acyl azide (Fig. 1B), anhydride (Fig. 1C and D), or active ester (Fig. 1E). Despite presenting one of the simplest methods of activation,1 the acyl chloride approach comes with a major drawback in the release of HCl, limiting applications involving acid-labile substrates.4 At elevated temperatures, using acyl azides can result in peptide urea byproducts (Curtius rearrangement, Fig. 1B), which are difficult to separate from the desired product.5 When using symmetric anhydrides (Fig. 1C), only half of the parent acid is coupled, wasting the other half, while regioselectivity between the two electrophilic positions of a mixed anhydride (Fig. 1D) can lead to inefficiencies. While these methods still see use in industry, these drawbacks have directed our focus towards amidations mediated by coupling reagents. Specifically, we explored new amide reaction monitoring methods using 1-hydroxy-7-azabenzotriazole (HOAt)-based coupling reagents, as these are arguably the most important class of available amide bond forming methods (Fig. 1E). | ||
| Fig. 1 A representative overview of amide bond-forming strategies. Boxed example: our investigation focused on developing new ways to monitor HOAt coupling agent-mediated amidations. | ||
1.2 HOAt-based coupling reagents
Amidation reactions involving coupling reagents6 often produce distinct colour changes as a result of the anionic by-product.7,8 In our case, coupling reagents based on HOAt (Fig. 2)9 change from colourless to yellow, as the reaction progresses. A range of aminium/uronium or phosphonium reagents have since been developed and become widely available, to improve stability and ease of handling.5,6,10Fig. 2 presents the proposed mechanism for amide coupling reactions mediated by HOAt-based reagents, using HATU as an example. The acid substrate is activated as the OAt ester which then reacts with the amine nucleophile. The HOAt anion released during the reaction is yellow, providing a colorimetric indicator of reaction progress. To the best of our knowledge, related acylation phenomena have only been monitored by Sheppard, whose team fashioned a photometer to monitor resin yellowing in a column reactor.11 More broadly, UV-vis spectroscopic methods have been applied to solid phase peptide synthesis.12–15 However, to date, these colour changes have not been widely used for online quantitative monitoring of amide and peptide synthesis using readily-accessible camera technology. | ||
| Fig. 2 (Top) Structure of HOAt and HOAt-based coupling reagents. (Bottom) General reaction scheme of HATU (HOAt-based) amide coupling reaction. The acid is first converted to active OAt ester by coupling reagent HATU, under basic conditions. The amine nucleophile then reactions with the activated OAt ester to give the amide product and yellow anionic OAt by-product. | ||
1.3 Monitoring methods for solid phase peptide synthesis (SPPS)
The incentive to investigate new approaches to monitoring amide formation was driven by the need for more direct, adaptable, digital-ready, non-contact methods of monitoring SPPS. There are two main reactions in the SPPS cycle – Fmoc deprotection and amide coupling. Both can be monitored by halting the reaction and taking samples for off-line analysis. The quality of the intermediate peptide (anchored to the solid phase resin) can be checked, either by HPLC after acidic cleavage of the peptide from the resin, or directly on resin by IR.16 The Kaiser test,17 and modern variations thereon,18 are often used to provide a colorimetric test for the present of unreacted amine after an SPPS step has concluded. These monitoring methods require the user to pause the synthesis process, making turnover time slow and difficult to automate, during reaction optimisation. Thus, several methods have been reported to provide real-time monitoring based on the electrochemical property changes,19 or physical properties, such as pressure,20 UV absorbance,21 and refractive index.22 During the revision of this manuscript, Wang and co-workers released a study demonstrating the value of NMR kinetics in elucidating mechanistic aspects of amide coupling.23 For monitoring of liquid phase peptide synthesis, Livingston's team have reported a powerful advance in UHPLC-MS.24 Gómez-Bombarelli and Pentelute have demonstrated the value of UV-vis analysis of in-flow Fmoc deprotection reactions to generate data-rich input to build predictive deep learning insights for Fmoc deprotection efficiency.15,25 Otake and co-workers have demonstrated the value of in-line near infrared (NIR) flow cells as a means of tracking liquid phase peptide synthesis in flow.26We used our recently-developed imaging kinetics software, Kineticolor,27,28 to provide camera-enabled ex situ (non-contact) reaction monitoring (Fig. 3). Looking towards complementing (not replacing) known reaction monitoring methods used in amide and peptide synthesis, we sought to demonstrate the ability to derive useful kinetic information from the visible bulk of the reactor, and not the molecular specifics typically captured by established in situ or offline techniques.29 In this study, we focus on the development of kinetic computer vision methods to support reaction monitoring in both liquid and solid phase peptide synthesis.
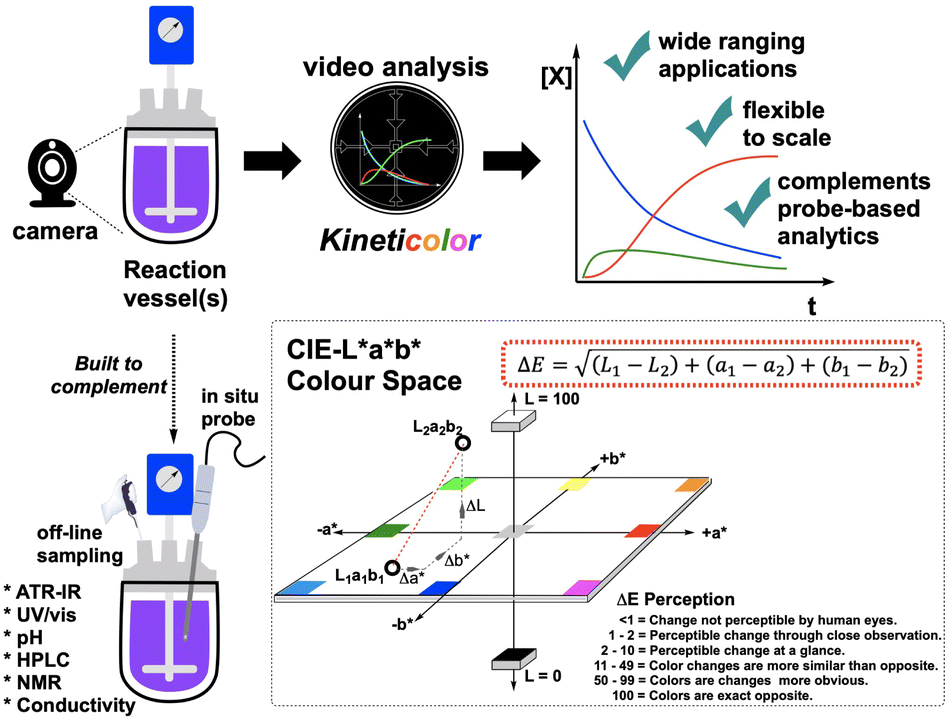 | ||
| Fig. 3 (Top) Conceptual overview of Kineticolor. From video recordings of chemical reactions, the pixel data are converted into colour models such as CIE-L*a*b* (dotted box, bottom right) to generate kinetic profiles from colour. (Bottom left) The non-contact computer vision approach to reaction kinetics is built to complement molecularly specific, often probe-based in situ reaction monitoring methods. | ||
1.4 Computer vision for analytical chemistry
Computer vision for analytical chemistry (CVAC)30,31 is frequently used through single image analysis techniques, providing a promising means of non-invasive analysis using digital camera technology. Comparatively, the extraction of time-dependent colourimetric information from videos of chemical reactions is less mature.32–39 In a previous study, our computer vison-enabled reaction monitoring method helped develop the understanding of palladium catalyzed borylation and catalyst degradation phenomena from colourimetric time profiles of the reaction bulk.28 The same methods have since been extended to tracking the impact of reaction scale-up on mixing times for process chemistry applications.27 Considering the colour change evident in amide coupling reactions, we aimed to expand the application domain of non-contact video analysis to the widely-used amidations described above, for both solution and solid phase reaction environments.2 Results and discussion
2.1 Amide coupling reactions in the solution phase
The first amide coupling reactions to be monitored by video and analysed with the Kineticolor software focused on HATU, one of the most commonly used amide coupling reagents. These reactions formed our initial proof-of-concept.4,6,40 In order to enable off-line HPLC analysis, we employed a sterically encumbered amine to slow the rate of amide bond formation, and thus the release of coloured OAt anion. Under the conditions shown in Fig. 4, HPLC analysis captured a gradual increase in the amide product over 3 hours. A colourless to translucent yellow colour change was observed. As described in our earlier reports, the video recording was analysed using a range of colour models27,28 Delta E (or ΔE), a measure of colour change, was derived from the CIE-L*a*b* colour space (Fig. 3), and calculated over time. In this calculation, each video frame was referenced against the first. Therefore the calculated ΔE value compares the analysed frame's colour to the colour of the first frame. As hypothesised, this approach enabled ex situ capture of the colourless to yellow colour change, registering a ΔE of approximately 60 (a relatively high, visible contrast change) over the 3 hour recording period.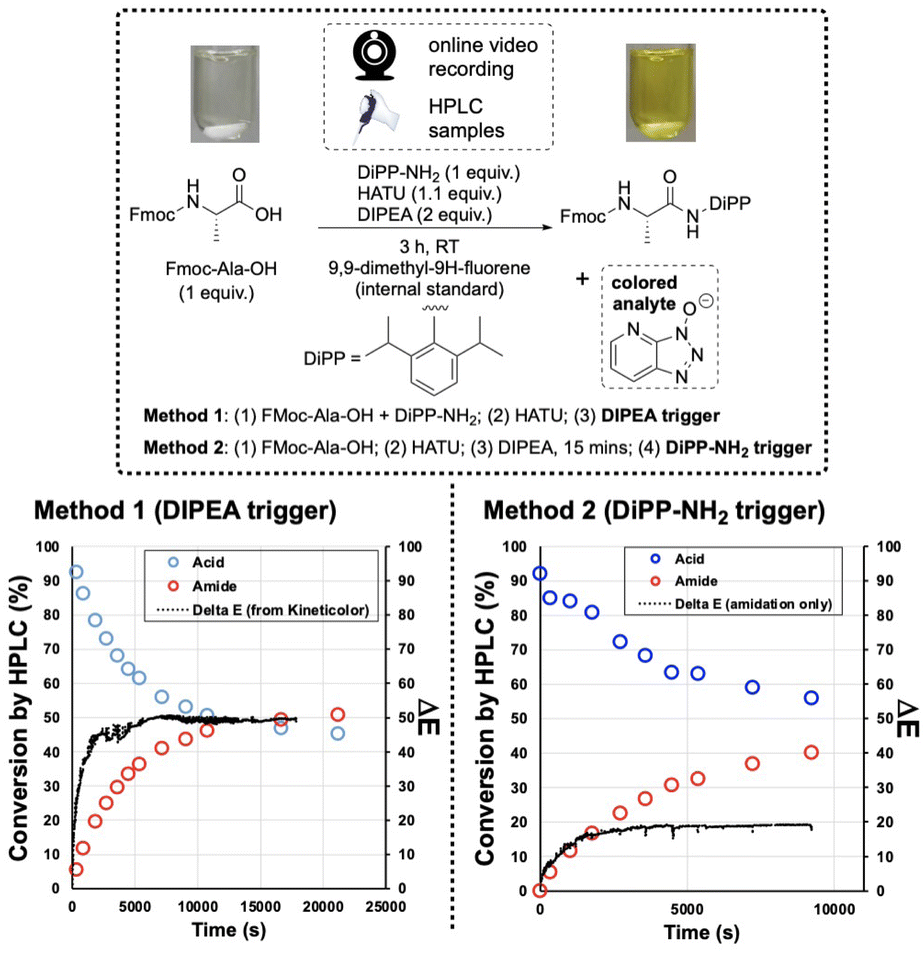 | ||
| Fig. 4 HPLC and ΔE plots of HATU mediated reactions. (Left) Results when initiating the reaction with DIPEA addition. (Right) Results using the amine coupling partner as trigger. Conversion is shown as percentage, calculated by HPLC using 9,9-dimethlyfluorene as internal standard. | ||
The OAt anion is produced by formation of the active ester, prior to the addition of the amine and also during the amide bond forming step itself (Fig. 2, bottom). As a result, activation times were used to examine the impact this has on the colour changes captured (Fig. 4). Triggering the reaction with the amine, rather than the base, produced distinct HPLC concentration and ΔE profiles, by visual inspection. With either trigger, the visual agreement of the HPLC and camera-derived trends remained the same – as conversion by HPLC slowed down, contrast changes by ΔE also plateaued. Interestingly, the overall contrast change is higher for the base-than for the amine-activated coupling (Fig. 4, left versus right, respectively). The lower overall ΔE value of 20 observed over the amine-triggered reaction indicated that the colour at the start of the video recording was closer to the colour at the end, while the base-triggered reaction (ΔE = 60) produced a larger colour change. The more perceptually distinct the reaction start and end colours, the higher the ΔE.
While analysis in Fig. 4, and indeed much of our discussion, focuses on the CIE-L*a*b* colour space, Kineticolor analysis captures 12 parameters spanning the CIE-L*a*b*, RGB, HSV, and XYZ colour spaces, as well as several derived parameters, including ΔE. To explore possible correlations with HPLC analysis, we performed Mutual Information (M.I.) analysis to rank the likelihood of their being any relationship between the independently collected video and HPLC data, linear or otherwise. We did this for three commonly applied coupling reagents. As depicted in Fig. 5, HATU-, PyAOP-, and DIC/Oxyma-mediated couplings evidenced high-ranking, non-zero M.I. scores for saturation (HSV), Lightness (CIE-L*a*b*), and a* (CIE-L*a*b*), respectively. These colour parameters produced correlations with HPLC, inferring that the non-contact camera data may be used as a useful proxy of offline concentration measurements of reaction progress (Fig. 5, bottom). These further experiments, using alternative coupling reagents to circumvent the problematic health issues of using HATU,41 form a broader proof-of-concept, which has revealed an encouraging generality in the video based approach to monitoring amide coupling.
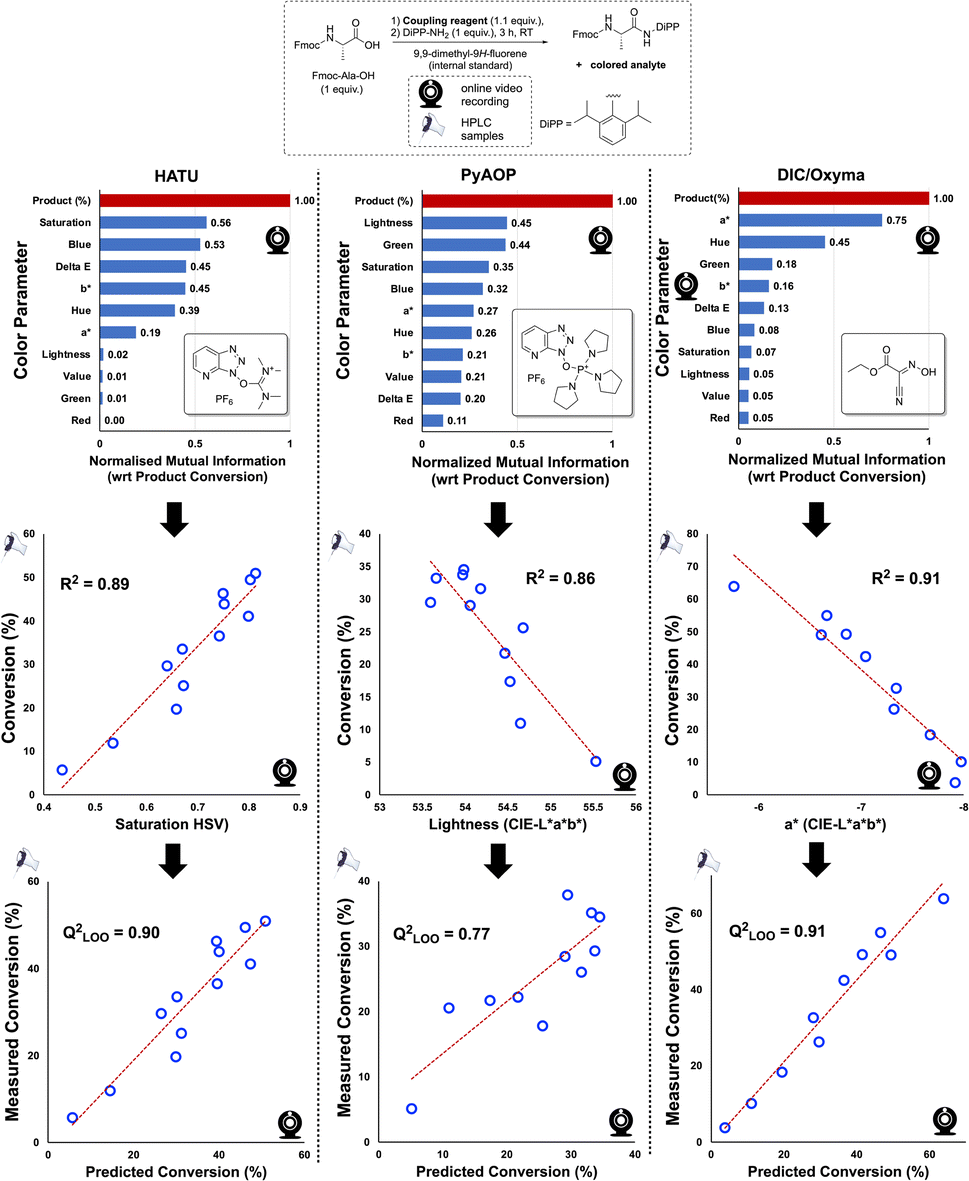 | ||
| Fig. 5 (Top) Generalised scheme of the amide coupling used to investigate correlations between colour measurements and offline HPLC analyses when employing various coupling reagents. (Middle) Ranked and normalised Mutual Information scores, comparing colour parameters with HPLC data, making no assumption of a linear relationship. A score of zero (0) infers that the colour parameter and HPLC data are completely independent of one another and not correlated. The comparison of product (%, by HPLC) against itself denotes the maximum M.I. score of 1 and is shown in red for reference. (Bottom) Inverse single component linear regression and leave-one-out (LOO) cross-validated models using the top-ranking (by M.I.) colour parameter and HPLC data, for each employed coupling reagent, HATU, PyAOP, and Oxyma. | ||
2.2 Amide coupling reactions in the solid phase
We subsequently investigated the applicability of this imaging-based reaction monitoring method to solid phase peptide synthesis (SPPS). Owing to the use of resin-bound reactants, SPPS presents unique mixing challenges and in situ monitoring constraints, further incentivising exploration of the non-invasive reaction monitoring methods.In keeping with the solution-phase processes above, the resin was loaded with Fmoc-protected, sterically-encumbered amino acids. Given that the product synthesised through SPPS is linked to the resin, the formation of product cannot be directly monitored by HPLC analysis of samples taken from solution. Usually, the product is cleaved from the resin under acidic conditions. To avoid errors arising from work-up, it was assumed that conversion to product was proportional to unreacted substrate remaining in solution (based on measured correlations; see ESI†). HPLC was used to analyse unreacted solution-phase substrate for comparison with video-derived analysis of the SPPS reaction medium.
The hindered α,α-disubstituted substrates 1 and 2 (Fig. 6), were each loaded onto Rink amide resin and reacted with Fmoc-Ala-OH using PyAOP as the coupling reagent. In both cases, qualitative visual inspection of the co-plotted HPLC and ΔE profiles indicated that the amidation event was completed within 10 min, suggesting the approx. 2 h reaction time could be appreciably optimised. These findings would later be applied in the optimisation of the solid-supported synthesis of three peptide sequences (see Section 2.3 onward).
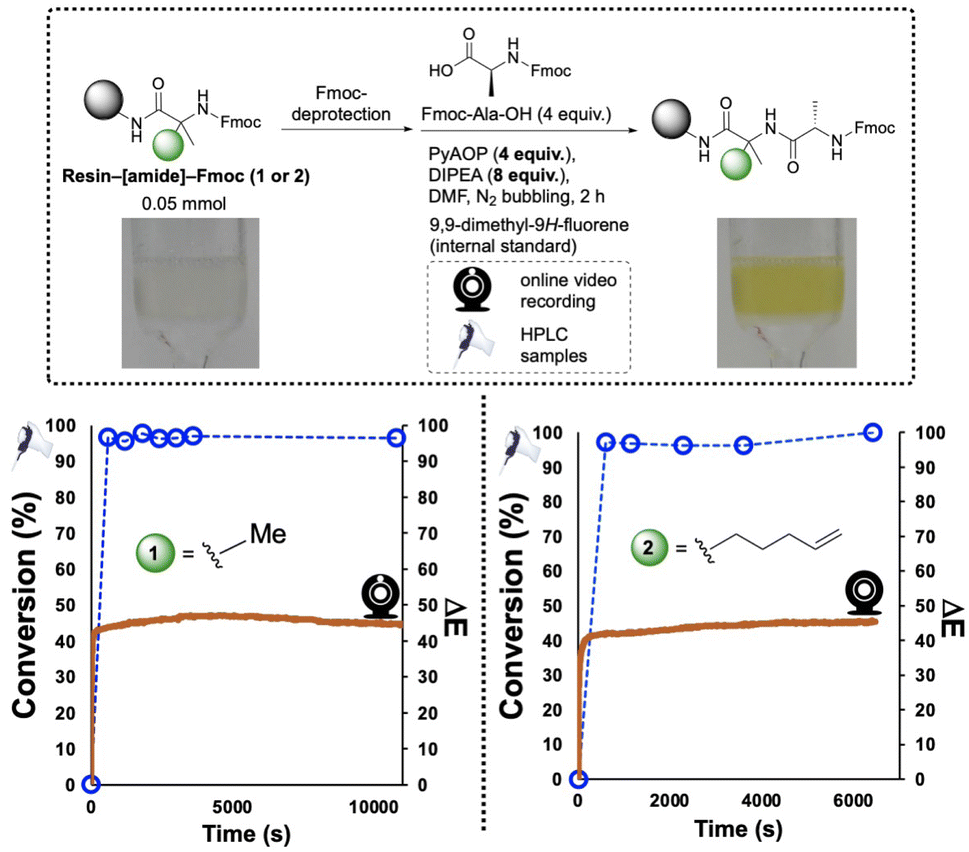 | ||
| Fig. 6 SPPS reaction of using Rink amide resin. In both examples, the amidation step is monitored by camera and HPLC off-line analysis. The HPLC data are plotted as open blue circles. The video-derived ΔE results are co-plotted as the orange lines. | ||
We next investigated the qualitative use of video-based reaction monitoring to assess structure–activity relationships in SPPS, looking specifically at a small series of amine nucleophiles of increasing size, 1–4 (Fig. 7). Guided by M.I. analysis (see ESI†), colour saturation (HSV colour space) was one of several colour parameters evidencing high mutual information with HPLC data. The normalised saturation data increased more rapidly with time across the nucleophile series, in the order 1 > 2 > 3 > 4, from smallest nucleophile to largest (Fig. 7, left). These colourimetric rate data were also qualitatively consistent with the computationally-derived B1 Sterimol parameter,42 capturing proximal steric bulk43–45 perpendicular to the C–N bond of the primary amine nitrogen, revealed after Fmoc deprotection. As the B1 Sterimol parameter increased from substrate 1 to 4, the observed rate of change of colour decreased across the series. To the best of our knowledge, this is the first demonstration of video recordings of the macroscopic (bulk) reaction progress being shown to correlate, even qualitatively, to cheminformatic parameterisation of reactants.
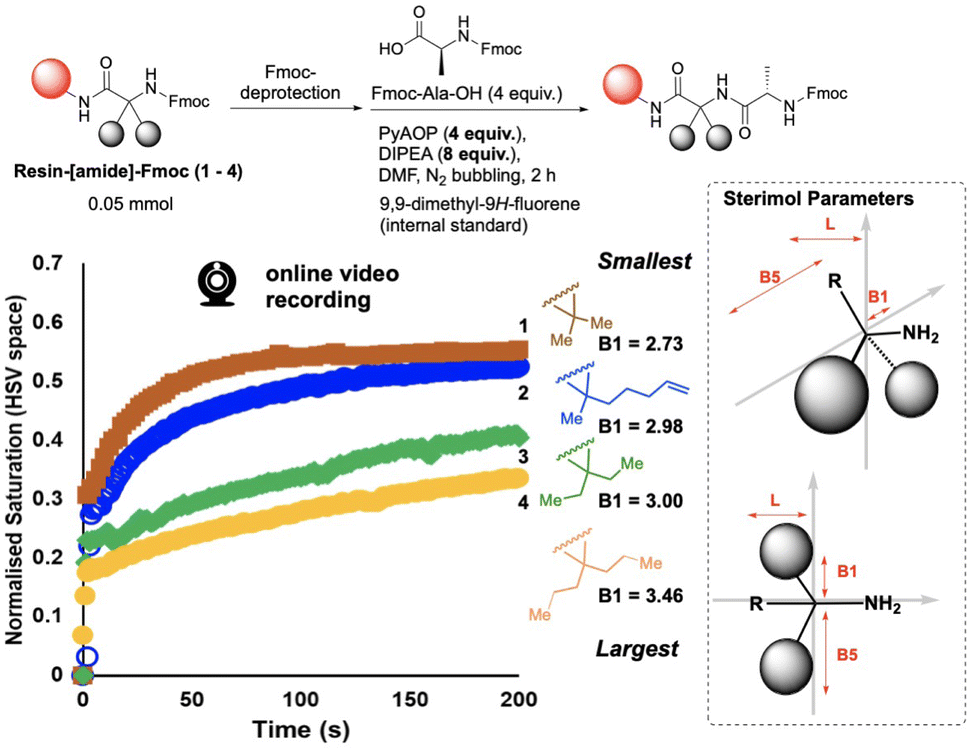 | ||
| Fig. 7 (Left) Normalised co-plot of the saturation parameter tracked for reactions with amino acid nucleophiles 1–4, with increasing steric encumbrance, as measured by the Sterimol B1 parameter. The rate of change of saturation is consistent with the interpretation that coupling reaction rate decreases with increasing substrate size. (Right) Schematic depiction of Sterimol B1 as the shortest distance perpendicular to the primary C–N bond axis defined by the nucleophilic amine center. B1 is used as the measure of sterics least affected by conformational influences, as is known for alternative Sterimol parameters B5 and L. | ||
It should also be noted that these rate differences captured on video (Fig. 7) occurred within the first 200 s (3 min) of each reaction. The HPLC concentration data did not capture these rate differences with the same data density due to the lower temporal resolution of samples taken for HPLC analysis. This difference in rate of data capture between video and offline measurements of the same reaction highlights the complementary value added by online reaction monitoring via video recording of reaction bulk. While sampling captures molecularly-specific concentration data (HPLC), the video captures macroscopic details with several orders of magnitude more data captured per second.
To further investigate the value of our video-derived reaction monitoring method, SPPS reactions with resin-bound diethyl glycine substrate, 3, were repeated using different excesses of the Fmoc-Ala-OH coupling partner, from 1 up to 4 equivalents relative to 3 (Fig. 8). Comparing reactions over 1 hour, HPLC and ΔE profiles followed similar trends, each increasing with the number of equivalents of Fmoc-Ala-OH added.
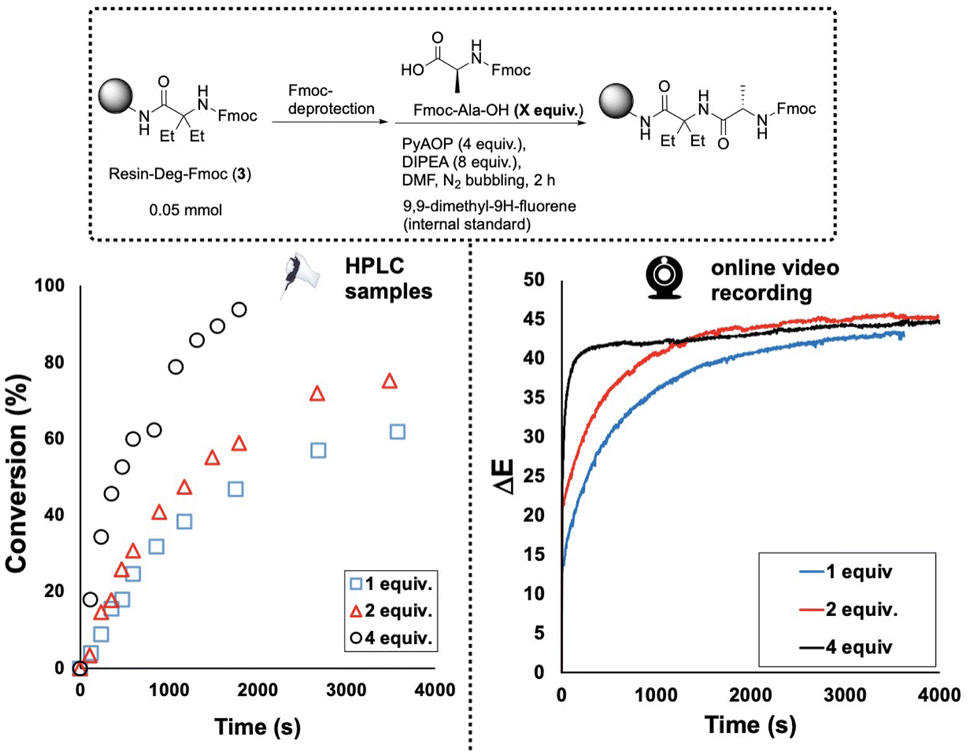 | ||
| Fig. 8 Effects of Fmoc-Ala-OH concentration on coupling reaction rate with 3. As Fmoc-Ala-OH concentration was increased, the rate of its conversion increased. The video-derived ΔE versus time profiles of the same reactions show a trend consistent with that captured by HPLC. | ||
2.3 Optimising the synthesis of Enkephalin
To investigate the application of video-derived colour profiling to the optimisation of amide coupling time, Enkephalin was synthesised via SPPS, following a standard procedure, employing a 5-fold excess of substrate and 2 to 3 hours of coupling reaction time per step (Fig. 9, top). An exemplar ΔE profile from the unoptimised coupling step 4 is shown in Fig. 9 (bottom left). From visual inspection, as was first made apparent in Fig. 6, coupling reactions often exhibited a plateau of colour change sooner than the several hours originally allotted to each unoptimised coupling step. It was hypothesised that the reaction times for many of the steps in peptide synthesis could be reduced without detriment to the yield of product peptide recovered after cleavage from the resin.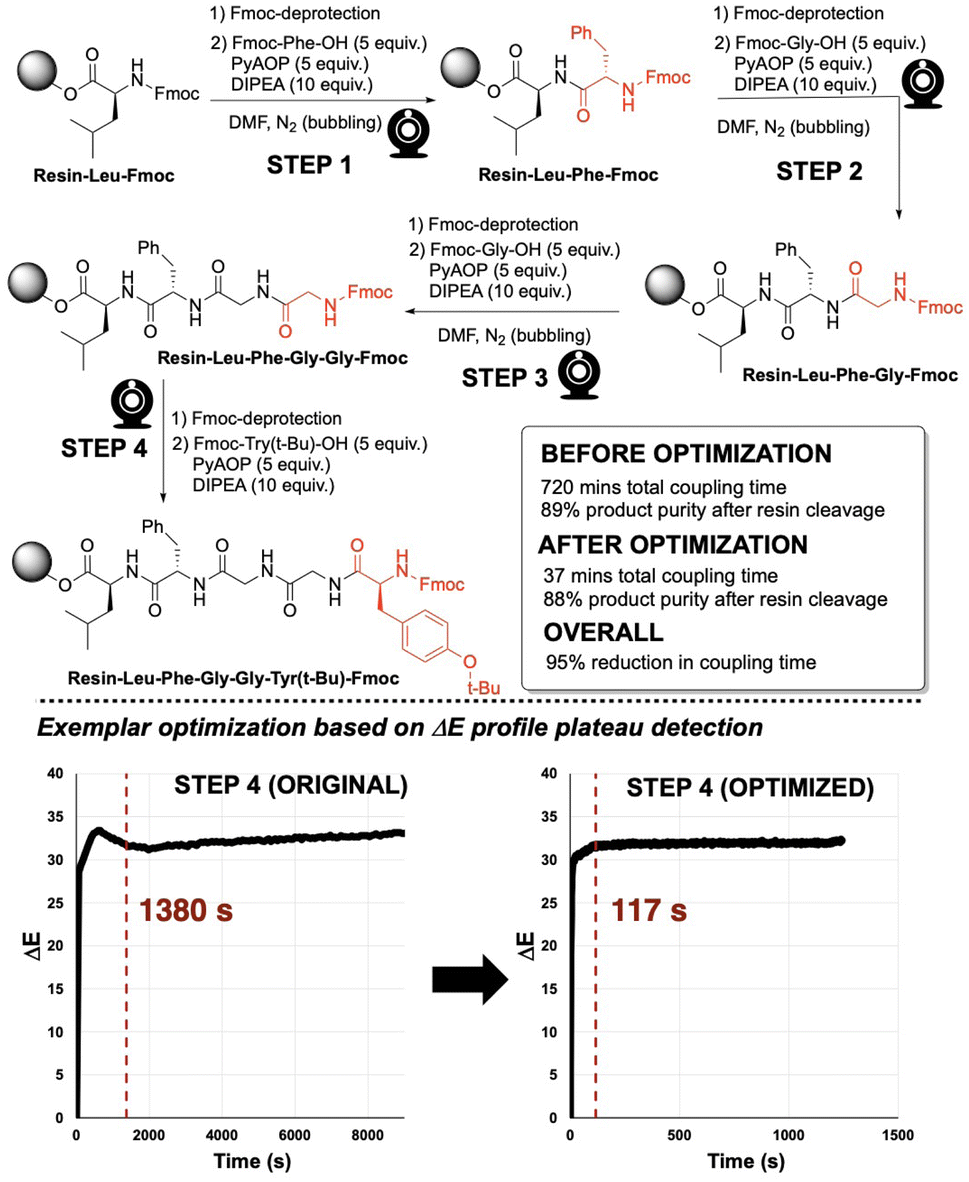 | ||
| Fig. 9 (Top) General reaction scheme of synthesising Enkephalin (bonded to resin; sequence: Y-G-G-F-L via solid phase peptide synthesis. (Bottom) Plateau analysis red dashed line) on ΔE profiles for step 4 in the synthesis. Plateau calculations for all steps are available in the ESI.† | ||
To determine an approximate colourimetric end point or threshold minimum rate of colour change of amide coupling from the computer vision data, indicating the amide coupling end point, plateau detection algorithms were explored. A plateau or threshold minimum rate of change was defined at the point when the rolling average of the gradient was below a target threshold (0.0005–0.004 ΔE/s) for a target duration (3 min). The estimated plateau start times were then used to guide the maximum possible reduction in reaction time.
Enkephalin was re-synthesised employing the pseudo-optimised coupling times for each one of the amide coupling steps 1–4, as derived from the plateau threshold times extracted from ΔE profiles in the original synthesis. In Fig. 9 (bottom), plateau detection for unoptimised and optimised ΔE profiles are exemplified for step 4 in the synthesis of Enkephalin. The original reaction time of 9720 s (162 min) evidenced a plateau at 1380 s (23 min). Re-running step 4 with a pseudo-optimised reaction time of 1246 s (20 min) revealed a plateau of 117 s, suggesting that the reaction time could potentially be optimised further. For the purposes of this study, ΔE plateau estimates were employed for iteration of reaction time optimisation only. The ΔE profiles and plateau calculations for all steps are available in the ESI.†
The qualities of the synthesised sequences were assessed by acidolysis from the solid support and downstream HPLC analysis (Fig. 10). The HPLC profiles of both cases (standard and video-optimised Enkephalin syntheses) evidenced near-identical chromatograms, indicating the parity of the optimised and unoptimised conditions. Overall, the optimised process represented an approximate 95% reduction in overall synthesis time, with no appreciable loss of yield of peptide upon acidolysis. These results highlighted the potential manufacturing application for peptide synthesis kinetics captured via non-contact video recording of the reaction bulk.
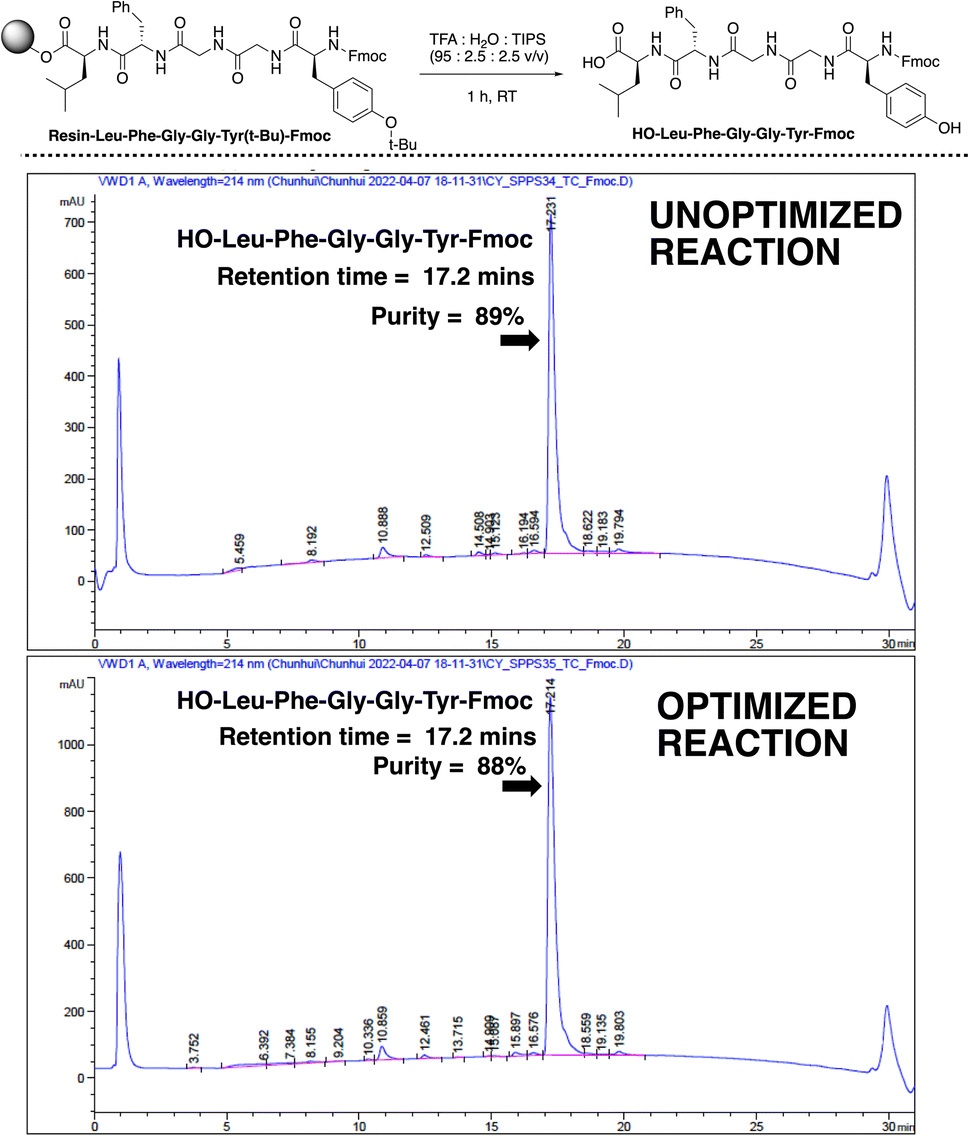 | ||
| Fig. 10 (Top) Acid-mediated cleavage of the target peptide sequence from resin. (Bottom) HPLC profiles for the unoptimised coupling and optimised coupling times. The HPLC suggest similar product quality is achieved. | ||
To track slower kinetics, the synthesis of Enkephalin was next repeated at 0.05 mmol (50%) scale under standard conditions using a 2–3 hour coupling reaction time for each coupling step. The ΔE profiles of each of four unoptimised steps are presented in Fig. 11. Consistent with the 50% reduction of reaction concentration, the observed rate of change of ΔE was lower versus earlier reactions on 0.1 mmol scale. The optimised coupling times, following the aforementioned video-informed plateau estimation method, are shown as red dotted lines in Fig. 11. Similar to the 0.1 mmol scale, optimisation of coupling times in Enkephalin synthesis on the 0.05 mmol scale, using the video-derived plateau estimation, reduced the overall synthesis time by 65%, from approximately 10 hours down to 3.5 hours, with only 4% difference in crude peptide yield (92% versus 88%, Fig. 11, bottom).
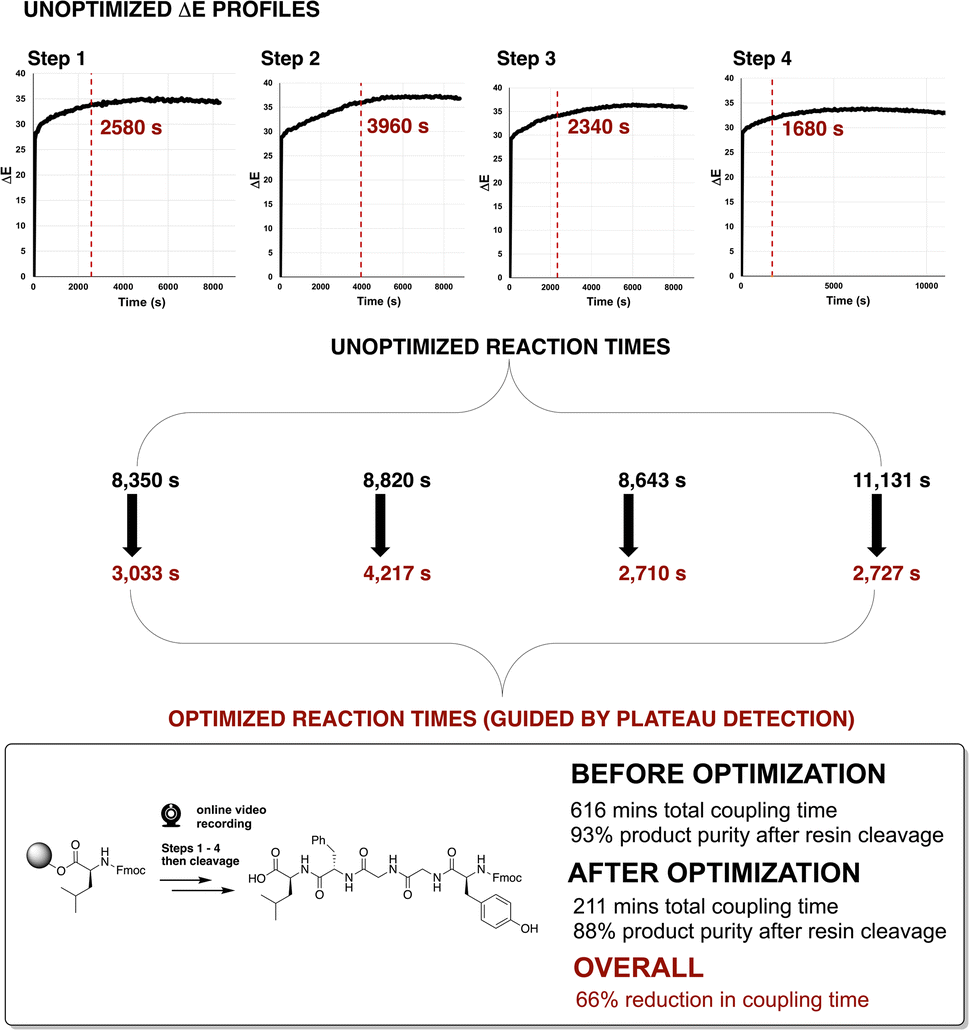 | ||
| Fig. 11 (Top) ΔE profiles for each unoptimised step in the synthesis of Enkephalin at 0.05 mmol scale (cf. 0.1 mmol in Fig. 9). Red dashed lines indicate plateau estimations used to guide reduction in reaction times at each step. HPLC data of product purities are available in the ESI.† | ||
We next applied our video-based reaction monitoring and optimisation methods to synthesise a more highly substituted variant of Enkephalin. Here, the methylene groups from the glycine residues were replaced by geminal dimethyl groups, derived from use of 2-aminoisobutyric acid (Aib) in place of glycine. As illustrated clearly by step 3 of the synthesis (Fig. 12, left), the ΔE profile of the more highly substituted amino acid resulted in a slowed rate of colour change compared to the same reaction employing Fmoc-protected glycine. This observation is consistent with the steric argument presented in Fig. 7. This third case provided a third SPPS example in which the ΔE plateau approach was able to guide a reduction in overall coupling time (66%, Fig. 12, right).
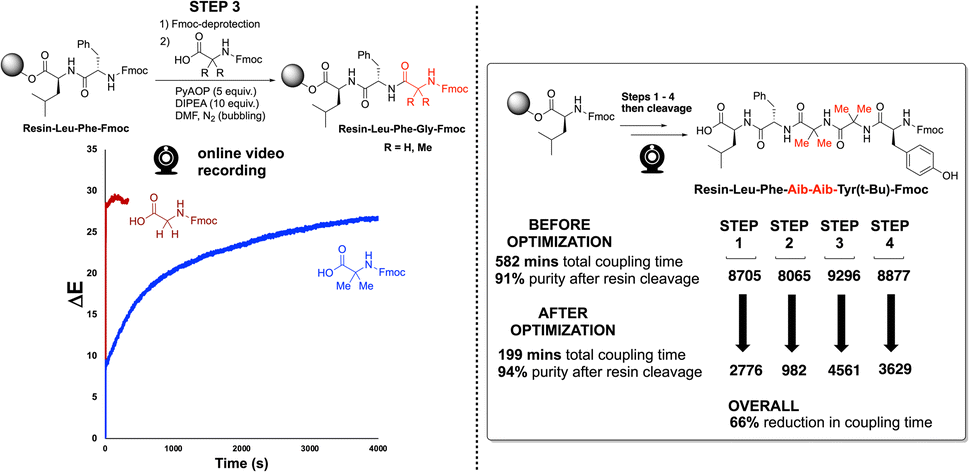 | ||
| Fig. 12 (Left) Reaction scheme and unoptimised ΔE profiles for step 3 in the Enkephalin and Aib-Enkephalin SPPS solid phase peptide synthesis. (Right) Summarised optimisation of overall coupling time for the 4 steps of Aib-Enkephalin synthesis. | ||
2.4 Optimising SPPS a longer sequence
Our video-based reaction monitoring approach was most challengingly applied to the case of the synthesis of a longer peptide sequence.46 A 9 amino acid sequence, KLLQDILDA,47 was selected due to the presence of several sterically encumbered residues and protecting groups (Fig. 13). The results of final analysis from cleaved products of both approaches are shown in Fig. 13. Encouragingly, our camera-enabled reaction monitoring approach supported the colour-based optimisation of all eight steps in the KLLQDILDA. Overall, this resulted in the peptide sequence being made in 51% less time compared to the standard conditions. | ||
| Fig. 13 Summarised optimisation of the reaction times for all eight coupling steps in the synthesis of KLLQDILDA, as enabled by ΔE profile plateau estimation. Output video analysis and HPLC product analysis, after resin cleavage, are all available in the ESI.† | ||
2.5 Investigating isolated yields and automated SPPS
In the final part of our study, we sought to validate the computer vision strategy for monitoring and optimizing SPPS. To this end, we ran end-to-end syntheses of the three peptide sequences described herein, from resin loading to final cleavage, and calculated isolated yields achieved before and after optimization. Encouragingly, for all three peptides, yields obtained when using the computer vision-guided conditions were comparable to the unoptimized yields. Additionally, for the most complex sequence (KLLQDILDA), we demonstrated the optimized conditions produced attractive isolated yields of the peptide in either manual or automated SPPS. All results are summarised in Table 1, and detailed further in the ESI.†3 Conclusions
We have demonstrated the analytical potential of computer vision-based kinetic profiling of reaction bulk to follow amide coupling reaction progress, both in solution and solid phase. Mutual information (M.I.) and single-component linear regression were used in combination to identify useful colour-concentration correlations. Thus, colour data, derived from non-contact video recordings, contains valuable complementary information to that obtained by more traditional sampling for off-line analysis. The method was also demonstrated the optimisation of the coupling time in synthesising peptide sequences via SPPS.Using video-derived reaction time estimates, reductions of 65–95% of the overall coupling time was achieved for several peptide sequences. These time savings were achieved while maintaining comparable isolated yield and qualities of the final peptides to those produced from the unoptimised conditions. Additionally, for the first time, we demonstrate the possibility of comparing DFT-derived steric parameters to bulk reaction kinetics derived from video footage, with increasing B1 Sterimol parameters found to reflect the observed attenuation in ΔE/dt.
These results support the possibility of using the camera-monitoring methods to enable the design a self-optimising automated SPPS instrument. These results also demonstrate the utility of the camera-based method, which could ultimately enable real-time monitoring of processes which might otherwise prove too difficult, or even impossible, to study quantitatively in a non-invasive manner.
Data availability
All machine readable outputs are available in a zipped folder as part of the ESI.†Author contributions
Chunhui Yan: methodology; software; validation; formal analysis; investigation; data curation; writing (original draft). Calum Fyfe: software; validation; formal analysis; data curation. Laura Minty: validation; formal analysis; data curation. Henry Barrington: review & editing. Craig Jamieson: conceptualisation; supervision; resources; project administration; validation. Marc Reid: conceptualisation; methodology; software; formal analysis; resources; data curation; writing (original draft); writing (review & editing); visualisation; supervision; project administration; funding acquisition.Conflicts of interest
M. R. is leading the commercialisation of the Kineticolor software platform.Acknowledgements
M. R. thanks UK Research & Innovation for Future Leaders Fellowship funding (MR/T043458/1). M. R. and C. F. thank the Centre for Process Analytics and Control Technology (CPACT) for funding support. C. J. and L. M. thank the Carnegie Trust for the Universities of Scotland for funding support.References
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS
.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC
- E. Atherton, L. Cameron, M. Meldal and R. C. Sheppard, J. Chem. Soc., Chem. Commun., 1986, 1763–1765 RSC
- A. El-Faham, R. S. Funosas, R. Prohens and F. Albericio, Chem.–Eur. J., 2009, 15, 9404–9416 CrossRef CAS PubMed
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS
- O. Marder and F. Albericio, ChemInform, 2004, 35 DOI:10.1002/chin.200424286
- L. R. Cameron, J. L. Holder, M. Meldal and R. C. Sheppard, J. Chem. Soc., Perkin Trans. I, 1988, 2895–2901 RSC
- E. Atherton, E. Brown, R. C. Sheppard and A. Rosevear, J. Chem. Soc., Chem. Commun., 1981, 1151–1152 RSC
- T. J. Lukas, M. B. Prystowsky and B. W. Erickson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2791–2795 CrossRef CAS PubMed
- A. Dryland and R. C. Sheppard, J. Chem. Soc., Perkin Trans. I, 1986, 125–137 RSC
- S. Mohapatra, N. Hartrampf, M. Poskus, A. Loas, R. Gómez-Bombarelli and B. L. Pentelute, ACS Cent. Sci., 2020, 6, 2277–2286 CrossRef CAS PubMed
- B. Yan, Acc. Chem. Res., 1998, 31, 621–630 CrossRef CAS
- S. Ruhemann, J. Chem. Soc., Trans., 1910, 97, 2025–2031 RSC
- R. Suzuki and H. Konno, Org. Lett., 2020, 22, 3309–3312 CrossRef CAS PubMed
- C. S. Nielsen, P. H. Hansen, A. Lihme and P. M. H. Heegaard, J. Biochem. Biophys. Methods, 1989, 20, 69–79 CrossRef CAS PubMed
- M. Baru, L. Mustaeva, I. Vagenina, E. Y. Gorbunova and V. Cherskii, J. Pept. Res., 2001, 57, 193–202 CrossRef CAS PubMed
- E. T. Sletten, M. Nuño, D. Guthrie and P. H. Seeberger, Chem. Commun., 2019, 55, 14598–14601 RSC
- B. G. de la Torre, S. Ramkisson, F. Albericio and J. Lopez, Org. Process Res. Dev., 2021, 25, 1047–1053 CrossRef
- J. Wang, P. Agrawal, J. Y. Buser, M. C. Embry, A. D. McFarland, M. R. Berglund, J. McClary Groh and S. K. Viswanath, Ind. Eng. Chem. Res., 2023, 62, 14986–14996 CrossRef CAS
- J. Yeo, L. Peeva, S. Chung, P. Gaffney, D. Kim, C. Luciani, S. Tsukanov, K. Seibert, M. Kopach, F. Albericio and A. Livingston, Angew. Chem., Int. Ed., 2021, 60, 7786–7795 CrossRef CAS PubMed
- C. Li, G. Zhang, S. Mohapatra, A. J. Callahan, A. Loas, R. Gómez-Bombarelli and B. L. Pentelute, Adv. Sci., 2022, 9, 2201988 CrossRef CAS PubMed
- Y. Otake, K. Adachi, Y. Yamashita, N. Iwanaga, H. Sunakawa, T. Shamoto, J.-i. Ogawa, A. Ito, Y. Kobayashi, K. Masuya, S. Fuse, D. Kubo and H. Itoh, React. Chem. Eng., 2023, 8, 863–870 RSC
- H. Barrington, A. Dickinson, J. McGuire, C. Yan and M. Reid, Org. Process Res. Dev., 2022, 26, 3073–3088 CrossRef CAS PubMed
- C. Yan, M. Cowie, C. Howcutt, K. M. P. Wheelhouse, N. S. Hodnett, M. Kollie, M. Gildea, M. H. Goodfellow and M. Reid, Chem. Sci., 2023, 14, 5323–5331 RSC
- C. Yan, C. Fyfe, C. Jamieson and M. Reid, Computer Vision as a New Paradigm for Monitoring of Solution and Solid Phase Peptide Synthesis, 2023, https://chemrxiv.org/engage/chemrxiv/article-details/64070568cc600523a3cb7904 Search PubMed.
- L. F. Capitán-Vallvey, N. López-Ruiz, A. Martínez-Olmos, M. M. Erenas and A. J. Palma, Anal. Chim. Acta, 2015, 899, 23–56 CrossRef PubMed
- S. V. Ley, R. J. Ingham, M. O'Brien and D. L. Browne, Beilstein J. Org. Chem., 2013, 9, 1051–1072 CrossRef CAS PubMed
- P. Shiri, V. Lai, T. Zepel, D. Griffin, J. Reifman, S. Clark, S. Grunert, L. P. E. Yunker, S. Steiner, H. Situ, F. Yang, P. L. Prieto and J. E. Hein, iScience, 2021, 24(3), 102176 CrossRef CAS PubMed
- J. Fitschen, S. Hofmann, J. Wutz, A. v. Kameke, M. Hoffmann, T. Wucherpfennig and M. Schlüter, Chem. Eng. Sci., 2021, 10, 100098 CAS
- M. A. Khalid, A. Ray, S. Cohen, M. Tassieri, A. Demčenko, D. Tseng, J. Reboud, A. Ozcan and J. M. Cooper, ACS Nano, 2019, 13, 11062–11069 CrossRef CAS PubMed
- D. Caramelli, D. Salley, A. Henson, G. A. Camarasa, S. Sharabi, G. Keenan and L. Cronin, Nat. Commun., 2018, 9, 3406 CrossRef PubMed
- T. R. Knutson, C. M. Knutson, A. R. Mozzetti, A. R. Campos, C. L. Haynes and R. L. Penn, J. Chem. Educ., 2015, 92, 1692–1695 CrossRef CAS
- M. P. Luna and J. M. Aguilera, Food Biophys., 2014, 9, 61–68 CrossRef
- V. Briones and J. M. Aguilera, Food Res. Int., 2005, 38, 87–94 CrossRef
- M. Shahabi, S. Rafiee, S. S. Mohtasebi and S. Hosseinpour, Food Sci. Technol., 2014, 20, 465–476 CrossRef PubMed
- L. A. Carpino, H. Imazumi, A. El-Faham, F. J. Ferrer, C. Zhang, Y. Lee, B. M. Foxman, P. Henklein, C. Hanay, C. Mügge, H. Wenschuh, J. Klose, M. Beyermann and M. Bienert, Angew. Chem., Int. Ed., 2002, 41, 441–445 CrossRef CAS PubMed
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS PubMed
-
A. Verloop, W. Hoogenstraaten and J. Tipker, Development and Application of New Steric Substituent Parameters in Drug Design, Drug Design, Elsevier, 1976, pp. 165–207 Search PubMed
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, ACS Catal., 2019, 9, 2313–2323 CrossRef
- C.-Y. Lin, S. Lim and E. V. Anslyn, J. Am. Chem. Soc., 2016, 138, 8045–8047 CrossRef CAS PubMed
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366–374 CrossRef CAS PubMed
-
W. Chan and P. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, OUP Oxford, 1999 Search PubMed
- J. N. Naoum, I. Alshanski, G. Mayer, P. Strauss and M. Hurevich, Org. Process Res. Dev., 2022, 26, 129–136 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01383a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |