
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress in the chemistry of molecular actinide-nitride compounds
Megan
Keener
 a,
Leonor
Maria
b and
Marinella
Mazzanti
*a
a,
Leonor
Maria
b and
Marinella
Mazzanti
*a
aGroup of Coordination Chemistry, Institute of Chemical Sciences and Engineering – ISIC, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, 2695-066 Bobadela, Portugal
First published on 31st May 2023
Abstract
The chemistry of actinide-nitrides has witnessed significant advances in the last ten years with a large focus on uranium and a few breakthroughs with thorium. Following the early discovery of the first terminal and bridging nitride complexes, various synthetic routes to uranium nitrides have since been identified, although the range of ligands capable of stabilizing uranium nitrides still remains scarce. In particular, both terminal- and bridging-nitrides possess attractive advantages for potential reactivity, especially in light of the recent development of uranium complexes for dinitrogen reduction and functionalization. The first molecular thorium bridged-nitride complexes have also been recently identified, anticipating the possibility of expanding nitride chemistry not only to low-valent thorium, but also to the transuranic elements.
 Megan Keener | Megan Keener obtained her B.Sc. degree in Chemistry in 2015 at California State University, Chico, where she did undergraduate research with Professor David Ball. In 2020, she received her Ph.D. in Chemistry from the University of California, Santa Barbara, under the supervision of Professor Gabriel Ménard. Her Ph.D. research was focused on synthetic inorganic chemistry, in which her thesis was entitled, “From Transition Metals to Actinides: A Homogenous Approach for Clean Alternative Energy and Storage Applications”. She is a current postdoctoral researcher in the group of coordination chemistry of Professor Marinella Mazzanti at École Polytechnique Fédérale of Lausanne (EPFL). Her current research interests are focused on developing highly reactive f element complexes for the transformation of widely available feedstocks into higher-added value compounds. |
 Leonor Maria | Leonor Maria was born in Caparica (Portugal). She graduated in chemical engineering at Faculdade de Ciências e Tecnologia - Universidade Nova de Lisboa in 1996 and obtained a PhD degree in Chemistry in 2003 from Instituto Superior Técnico – Universidade Técnica de Lisboa (IST). After postdoctoral fellowships at Instituto Tecnológico Nuclear (ITN, Bobadela) and Centro de Química Estrutural – Instituto Superior Técnico (CQE-IST), she was awarded an Assistant Researcher contract under FCT program Ciência-2008, and in 2009, she joined the f-Element Chemistry Group at ITN (later Centro de Ciências e Tecnologias Nucleares of IST). Since January 2020, she is a member of the Inorganic and Organometallic Architectures, Reactivity and Catalysis (IOARC) Group of CQE-IST. Her main research interests are in the synthesis, structure, reactivity and bonding of lanthanide and actinide complexes, with focus on multi-electron transfer reactions and metal-ligand multiple bond formation. |
 Marinella Mazzanti | Marinella Mazzanti is a Professor at the Ecole Polytechnique Fédérale of Lausanne (EPFL) in Switzerland since 2014. She was born in Vinci (Italy). She obtained a master degree in Inorganic Chemistry from the University of Pisa and a PhD in Chemistry from the University of Lausanne with Carlo Floriani. She was post-doc in UC-Berkeley and at UC-Davis working with Alan Balch. From 1996 to 2014 she held a position as research scientist at a National Laboratory in France (CEA-Grenoble) working in f-elements chemistry. She is an associate editor of Chemical Communications. She was awarded the ACS F. Albert Cotton Award in Synthetic Inorganic Chemistry in 2021 and the Le Coq de Boisbaudran award in 2023 for her work on uranium and lanthanide chemistry. Her research interests are centered on synthetic inorganic chemistry and in the development of new molecules with unusual properties and original reactivity with focus on small molecule activation. |
1. Introduction
Metal nitrides are important intermediates in the biological and industrial conversion of dinitrogen (N2) to ammonia (NH3), as well as in stoichiometric and catalytic N-transfer reactions. As a result, the synthesis, characterization, and reactivity of molecular nitride compounds of transition metals have been, and continue to be, extensively studied.1–7 In contrast, the chemistry of molecular actinide-nitrides is significantly less developed than their d-block counterparts, however, inorganic actinide-nitride materials (uranium, thorium, neptunium, and plutonium) have attracted large attention as advanced nuclear fuels.8–14 Notably, uranium nitride (UN) materials were some of the first compounds reported as active catalysts in the industrial Haber–Bosch process for the production of NH3 from N2 and H2.15,16Three solid uranium nitrides are known (UN, UN2, and U2N3).11 The synthesis of phase pure ThN,17–19 PuN,20 and NpN21 are also well established, however, these are less studied than uranium nitrides, with the most common synthetic route for actinide mono-nitrides involving carbothermic reduction of dioxides.22
Molecular actinide-nitrides were first isolated in noble gas matrices at cryogenic temperatures, and characterized by infrared spectroscopy for uranium (UN, N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) UF3, N
UF3, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UN, NUN–H, NUN(NN)x, (UN)(NN)x),23–28 plutonium (PuN, PuN2),29 and thorium (ThN, NThN, NThO),30–32 but are only stable at cryogenic temperatures. Since, molecular actinide-nitrides have harnessed significant attention due to their key role in understanding actinide bonding interactions, covalency, and electronic structure properties, as well as their ability to mediate important small molecule transformations.33–41 Early attempts to prepare molecular uranium-nitride complexes were already covered in a review published in 2014,42 therefore those systems will not be described in detail here.
UN, NUN–H, NUN(NN)x, (UN)(NN)x),23–28 plutonium (PuN, PuN2),29 and thorium (ThN, NThN, NThO),30–32 but are only stable at cryogenic temperatures. Since, molecular actinide-nitrides have harnessed significant attention due to their key role in understanding actinide bonding interactions, covalency, and electronic structure properties, as well as their ability to mediate important small molecule transformations.33–41 Early attempts to prepare molecular uranium-nitride complexes were already covered in a review published in 2014,42 therefore those systems will not be described in detail here.
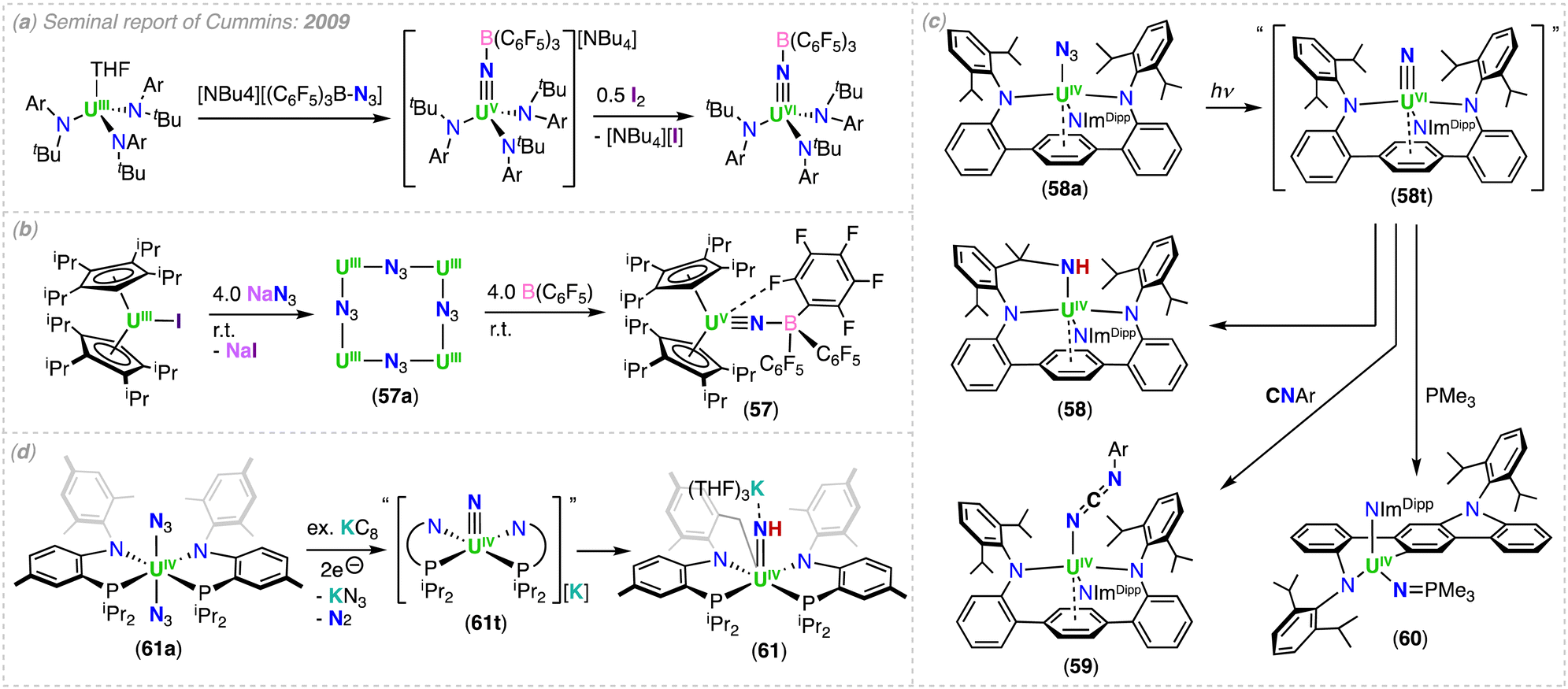
However, we would like to summarize the main milestones in nitride chemistry that occurred before 2014 (Scheme 1).33,34 Seminal attempts from the Cummins group to prepare uranium nitride complexes by N2 cleavage,35 or N-transfer reactions36 with the Hdbabh (2,3,5-dibenzo-7-azabicyclo[2.2.1.]hepta-2,5-diene), led to stable molybdenum/uranium (Mo/U) dinitrogen and uranium hexakis–imido complexes. Remarkably, the first example of a U(IV)/U(V) uranium-nitride was obtained in 2002 by reduction of a U(III) tetrapyrrole complex, [K(DME)][(Et8-calix[4]tetrapyrrole)UIII(DME)] (DME = 1,2-dimethoxyethane), with [K(naphthalenide)] under N2, and presented a diamond core UN2 motif (A, Scheme 1).37 Shortly after, two uranium azide (N3−)/nitride (N3−) clusters of very different structure (one presenting a UNU motif, and the other with an interstitial μ-N3), were obtained from the reaction of U(III) complexes with alkali azides (complexes B and C).38,39
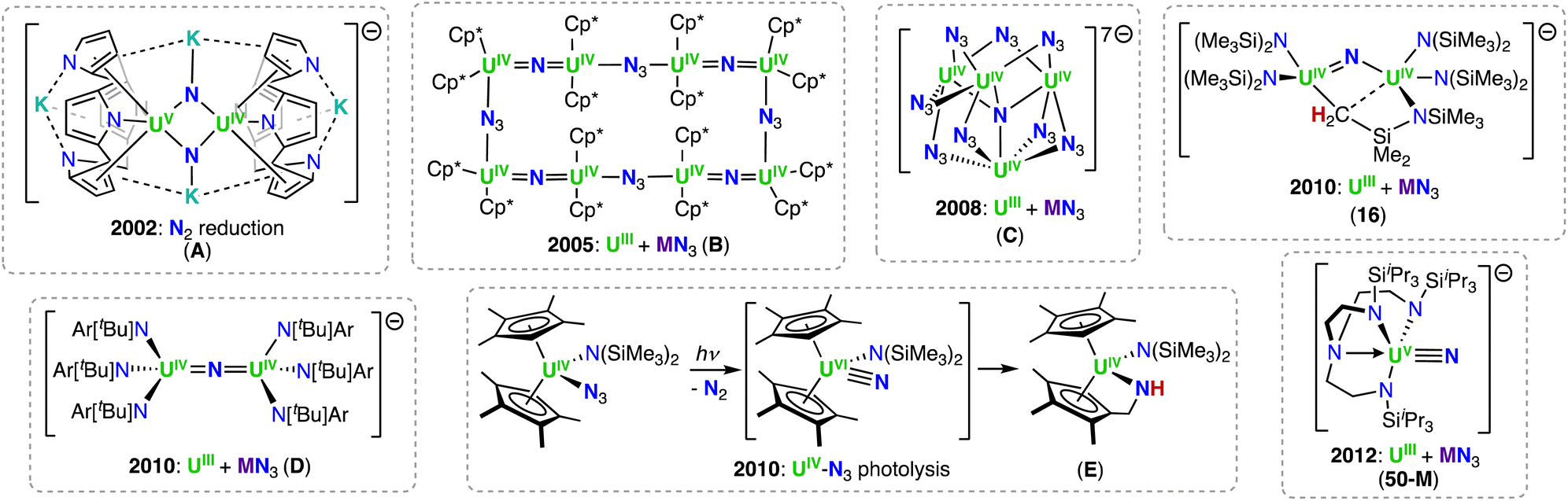 | ||
| Scheme 1 Molecular nitrides of uranium in the +4 and +5 oxidation states reported before 2014. | ||
Utilizing bulkier ligand environments, and the methodology involving the reduction of inorganic azides with U(III) complexes, the groups of Hayton and Cummins were able to effect the controlled synthesis of dinuclear bridged-nitride complexes in varying oxidation states (complexes 16 (Section 3.1.1) and D).40,41 Finally, the group of Liddle demonstrated that utilization of the bulky triamidoamine (TRENTIPS) ligand, and the analogous U(III) azide reduction route, allowed the preparation and isolation of the first uranium(V) terminal nitride (50-M), which was then subsequently oxidized to yield the U(VI) terminal-nitride analogue (Section 4.2).43,44 It should be noted, prior to the isolation of the relatively stable U(V) and U(VI) terminal nitrides by the Liddle group, Kiplinger and co-workers demonstrated that a putative terminal-nitride could be generated by a different synthetic route, involving photolysis of a U(IV) azide precursor.45 The high reactivity of the putative nitride towards C–H activation of one of the methyl groups of the ligand resulted in the isolation of a U(IV) amido derivative (E; Scheme 1).45 These early studies demonstrated that different synthetic routes could be used to access bridging- and terminal-nitrides, such as azide reduction, photolysis, or N2 reduction, but choice of the supporting ligands was crucial in controlling both the nuclearity and reactivity of the nitride products. Since then, careful tuning of the reaction conditions has significantly expanded the chemistry of molecular uranium nitrides, leading to the isolation of nitrides in a broad range of oxidation states (+3 to +6), which have shown high reactivity towards small molecules such as CO, CO2, H2, and N2.46–53 Notably, the use of the monodentate amide ligand, –N(SiMe3)2, in the groups of Hayton and Mazzanti, and the siloxide ligand, –OSi(OtBu)3, in the group of Mazzanti, have led to the expansion of nitride-bridged complexes of uranium and thorium in the last 20 years. The use of the polydentate tripodal amido ligands (TrenR: N(CH2CH2NR)3−) in the Liddle group has led to important advancements in terminal nitride chemistry and An–N bonding analysis. This chemistry, and all other actinide-nitrides reported since 2014, will be reviewed herein according to the supporting ligand environment, synthetic method, and actinide ion utilized.
2. Actinide nitride complexes derived from N2 cleavage
Metal complexes capable of effectively splitting N2 to nitrides remain rare, and are mostly limited to complexes of d-block metals.1,2 The N2 chemistry of actinides is significantly less developed, and typically involves labile N2 binding, the 2e− reduction of N2, or more recent examples demonstrating the 4e− reduction by two U(III) complexes or dinuclear U(III)/U(III) species.35,48,54–61 Based on the structure of the final nitride complex, multimetallic cooperation of transition metals and Lewis acids, potassium (K) in particular, was invoked to be crucial in the 6e− cleavage of N2 to nitride involving alkali ions as reducing agents.62,63 Notably, the first actinide-nitride complex isolated in any significant quantity was synthetized in 2002 by Gambarotta and co-workers via full reductive cleavage of N2, promoted by the reaction of the trivalent uranium complex, [K(DME)][(Et8-calix[4]tetrapyrrole)UIII(DME)], with [K(naphthalenide)] under N2 (A, Scheme 1).37 It is interesting to note that the U(III) precursor was not reported to react with N2 in the absence of reducing agent, or at least in the polar solvent, DME. However, it is likely that reversible binding may occur, based on recent results from the Mazzanti group, where the mechanism of N2 cleavage to nitride involves reduction of the U(III)-bound N2 by excess alkali metal, yielding a U(V)/U(V) bis-nitride.64 However, an alternative mechanism involving the binding and reduction of N2 by a putative “U(II)” species cannot be ruled out without further studies.Falcone, Mazzanti and co-workers first demonstrated that a well-defined, multimetallic oxide-bridged diuranium(III) complex, [K2(UIII(OSi(OtBu)3)3)2(μ-O)] (1-O), supported by siloxide ligands (–OSi(OtBu)3), is capable of effecting the 4e− reduction of N2, forming the bridging hydrazido (N24−) complex [K2(UV(OSi(OtBu)3)3)2(μ-O)(μ-η2:η2-N2)], (1-N2).59 In a later study by Jori, Mazzanti and co-workers, full reduction of the N24− moiety to nitride was accomplished by addition of KC8, with formation of the U(VI)/(U(IV))3 tetranitride cluster, [K6((OSi(OtBu)3)2UIV)3((OSi(OtBu)3)2UVI)(μ4-N)3(μ3-N)(μ3-O)2] (1) (Scheme 2).64 Computational studies showed that the reduction of the N2 complex 1-N2 proceeds via successive 1e− transfer from the alkali ion first to the uranium center, and then to the coordinated hydrazido (N24−) moiety, generating a putative diuranium(V) bis-nitride terminal oxo complex, [K3(UV(OSi(OtBu)3)3)(μ-N)2(UV(OSi(OtBu)3)2(κ-O))][KOSi(OtBu)3] (1-(N)2), that could not be isolated.64 Cooperative K binding to the uranium–N24− ligand facilitates the N–N bond cleavage during electron transfer. Further reduction of the putative 1-(N)2 resulted in the multimetallic U(VI)/U(IV) tetranitride 1. This combined experimental and computational study of the stepwise reduction of N2 clearly shows that N2 splitting proceeds via a N24− intermediate (Scheme 2).
 | ||
| Scheme 2 (a) Synthesis of uranium nitrides supported by siloxide (–OSi(OtBu)3) ligands via N2 cleavage. –OtBu substituents were omitted for clarity. (b) Computed enthalpy profile in kcal mol−1 at room temperature for the formation of 1-(N)2, 2-(N)2, and 3-(N)2 from the reduction of N2 by 1-O, 2-O, and 3-O, respectively. | ||
The molecular structure of the complex 1 shows three U(IV), one U(VI), and six K cations bridged by four nitride and two oxo ligands. Remarkably, the U4N4O2K6 cluster contains a U(VI) coordinated by four bridging-nitride ligands, with two nitride groups bound in a trans mode with an almost linear N–U–N angle (178.7(3)°), providing the second example of a trans-(NUVIN) moiety analogue of UO22+.65,66 The two trans U–Nnitride bond lengths (1.963(8) and 1.902(8) Å) are shorter than the cis-coordinated nitrides (2.256(6) and 2.265(7) Å) in 1; however, they are longer than the trans UVI–Nnitride bonds (1.799(8)–1.870(9) Å) reported by Rudel, Kraus and co-workers for [(NH3)8U(μ-N)(NH3)3(X)2U(μ-N)U(NH3)8]Yn·ZNH3 (where X = NH3, Br−, or Cl−; n = 6–8; Y = Cl− or Br−; Z = 26, 21, or 6) (Section 3.2; Scheme 11).65 The elongated UVI–N distances in 1 are most likely due to the presence of a more electron-rich uranium center bound by four additional anionic siloxide ligands compared to the neutral NH3 ligands in the complexes reported by Rudel, Kraus and co-workers. The UIV–Nnitride bond distances in 1 range from 2.183(7) to 2.319(78) Å and are comparable with those found in other reported nitride-bridged U(IV) clusters and consistent with U–N single bonds.39,67 In a more recent study, Jori, Mazzanti and co-workers reported the reactivity of the analogous rubidium (Rb) and cesium (Cs) complexes, [M2(UIII(OSi(OtBu)3)3)2(μ-O)] (M = Rb (2-O), Cs (3-O)) with N2 (Scheme 2a).61 The three alkali cations (K, Rb, Cs) display differences in the ability to bind N2, which are not correlated to differences found in the redox potentials of the three complexes,61 but instead, a decrease with increasing size and decreasing Lewis acidity of the alkali ion binding to the UIII–O–UIII complex. Complex 1-O irreversibly reduces N2 at ambient conditions, while N2 is reversibly reduced by the Rb2UIII–O–UIII complex 2-O in the same conditions. In contrast, the reaction of the Cs2UIII–O–UIII complex (3-O) with N2, could only be detected by 1H NMR spectroscopy at high pressures (12 to 100 atm) and low temperatures. DFT analysis showed that N2 binding to 3-O is hindered by steric effects, but, the calculated uranium(V)–N24− species presented a similar degree of activation of the bound N2 for the three uranium-alkali ion M2UV-(N2)-UV complexes (M = K, Rb, Cs; Scheme 2b).61 In the case of Rb, it was possible to isolate the intermediate hydrazido (N24−) complex [Rb2(UV(OSi(OtBu)3)3)2(μ-O)(μ-η2:η2-N2)] (2-N2). Analysis by 1H NMR spectroscopy indicated that addition of 2.0 equiv. of RbC8 to 2-N2 (or 2-O) under N2 generates a putative bis-nitride (2-(N)2), that could not be isolated. Even though the formation of the Cs–N24− intermediate complex, “[Cs2(UV(OSi(OtBu)3)3)2(μ-O)(μ-η2:η2-N2)]” (3-N2), was not observed by 1H NMR spectroscopy under 1 atm of N2, the reaction of 3-O with 2.0 equiv. of CsC8 at low temperature under N2 led to full N2 cleavage and isolation of the U(V)/U(V) bis-nitride complex, [Cs3(UV(OSi(OtBu)3)3)(μ-N)2(UV(OSi(OtBu)3)2(κ-O))][CsOSi(OtBu)3] (3-(N)2) (Scheme 2a).61 The molecular structure of 3-(N)2 shows two Cs-bound nitrides linking two U(V) ions in a U2N2 diamond-shaped core with asymmetric U–N bond lengths ranging from 1.85(1) to 2.34(1) Å. The shorter and longer U–Nnitride bond distances are those of the bridging nitride which is trans to the oxo group (U–O 1.856(4) Å, O–U–N 172.6(4)°), suggesting that the nitride ligand binds the oxo-bound U(V) ion with a multiple bond and the other U(V) ion with a single bond. Complex 3-(N)2 represents a rare example of a U(V) molecular compound featuring a OUN moiety analogue of uranyl(V). The only other known example of a uranium complex with such a moiety was reported by Fortier, Hayton and co-workers for a trans oxo-nitride U(VI) complex.40
In 2020, Xin, Zhu and co-workers reported the multimetallic (U(IV))4/(Rh(–I))2 bis-nitride complex, [(U2IV(NNP2)2(Rh−I)(μ-N))2] (4), that was isolated from the reduction of the U2–Rh complex, [(UIV(NNP2)2Cl2)2(μ-Cl)(μ-RhI)] (4a) (NNP2 = (N(CH3)(CH2CH2NPiPr2)2)2−), under N2 with excess KC8 (top, Scheme 3).68 To confirm the nitride ligands in 4 originated from N2 cleavage, the isotopically enriched complex 15N-4 was prepared under 1 atm of 15N2. Acidification of complexes 4 and 15N-4 resulted in 14NH4+ and 15NH4+, respectively.68 The bis-nitride complex 4 was also obtained from reduction with 4.0 equiv. of KC8 under N2 of the U(IV)/Rh(–I) complex, [(UIV(NNP2)2Cl)2(μ-Cl)(μ-Rh−I)], obtained from the reduction of 4a under Argon. Computational studies suggested that the N2 cleavage proceeds through N2 binding by two [(UIV(NNP2)2Cl)2(μ-Cl)(μ-Rh−I)] complexes following addition of 2.0 equiv. of KC8, and is promoted by the cooperative multimetallic binding of N2 by four uranium centers and two Rh ions. However, the intermediates involved in the N2 splitting could not be isolated and the oxidation state of the uranium ions involved in N2 binding remains ambiguous.
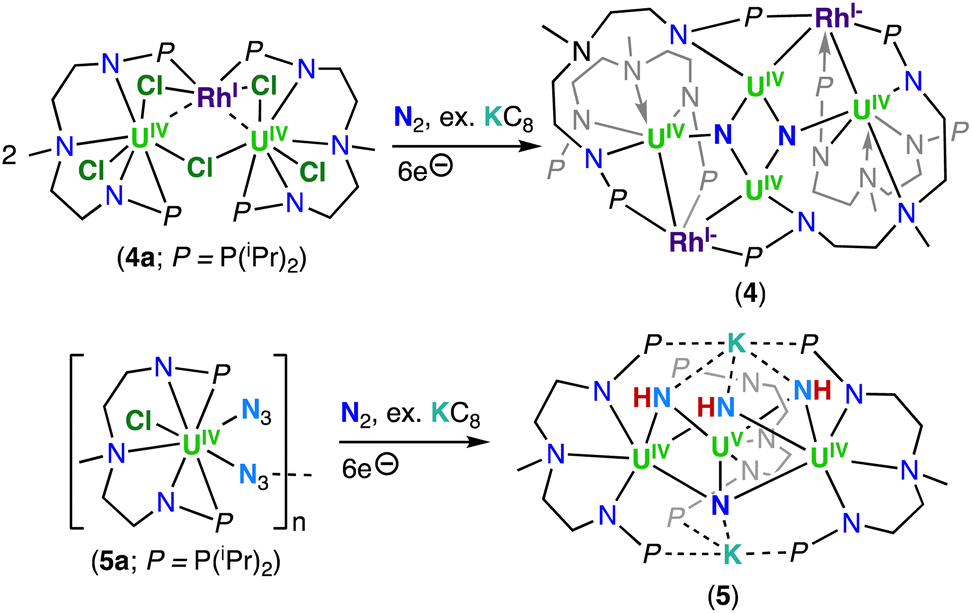 | ||
| Scheme 3 Synthesis of uranium nitrides supported by the diamidoamine (NNP2) ligand via N2 cleavage. | ||
The importance of cooperativity between the U(III) and inner sphere alkali ions had already been identified in the reduction of N2 by well-defined multimetallic uranium complexes.48,59,64 The solid-state structure of 4 revealed a centrosymmetric structure with two bridging nitride ligands in a U4Rh2N2 core, with U and Rh in oxidation states +4 and −1, respectively. The oxidation state of uranium was confirmed by magnetic studies. Each nitride binds three U(IV) ions, with one U–Nnitride bond (2.302(6) Å) being significantly longer than the other two, which are similar (2.158(7) and 2.154(7) Å). The three U–Nnitride bond lengths are comparable to those found in a hydrazide-bridged uranium complex (2.163(13)–2.311(13) Å)48 and are consistent with U–N single bonds.
More recently, Xin, Zhu and co-workers also reported that the reduction of a U(IV) azide complex, supported by the (NNP2)2− ligand (5a), with excess KC8 under N2 generates the trinuclear imido–nitride complex, [K2((U(NNP2)2(μ-NH)3(μ-N))] (5) (bottom, Scheme 3).69 The molecular structure of 5 consists of three uranium centers that are bridged by three imido (NH) ligands, and one nitride (N3−) ligand with U–Nnitride bond lengths which range from 2.204 to 2.218 Å. These distances are comparable to those reported for the U(IV)/U(VI) tetranitride cluster 1, and slightly longer than those found in the nitride bridged cluster, [(UIVCp*(μ-I)2)3(μ-N)], reported by Evans and co-workers (2.138(3)–2.157(3) Å).67 The U–NH bond lengths range from 2.188 to 2.31 Å and are comparable to other bridged U–Nimido distances reported previously (2.10–2.55 Å).59,60,70
Variable temperature magnetic data, UV-visible-NIR absorption, and X-ray photoelectron spectroscopy (XPS) were consistent with the presence of two U(IV) ions and one U(V) ion in the cluster complex 5, which was corroborated by computational studies. The authors proposed that the nitride ligand in 5 arises from N2 reduction, while the imido ligands arise from reduction of the azide ligand which yields a nitride that is subsequently protonated. The origins of the protons remain ambiguous. To support that the nitride originated from N2 reduction, an experiment was conducted in the presence of 15N-labelled N2. Acidification of the isotopically enriched complex 15N-5 resulted in 15NH4Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14NH4Cl in a ratio of 0.5:3, demonstrating that the nitride moiety was a result of N2 cleavage, and not from azide reduction. The lower-than-expected ratio (expected ratio was 1:3) was proposed by the authors to result from the 14N2 generated in situ by the reduction of three U–14N3 moieties. DFT calculations demonstrated that the K cations are involved in the activation of N2, therefore, once again demonstrating that N2 cleavage is the result of a multimetallic synergetic processes.
:14NH4Cl in a ratio of 0.5:3, demonstrating that the nitride moiety was a result of N2 cleavage, and not from azide reduction. The lower-than-expected ratio (expected ratio was 1:3) was proposed by the authors to result from the 14N2 generated in situ by the reduction of three U–14N3 moieties. DFT calculations demonstrated that the K cations are involved in the activation of N2, therefore, once again demonstrating that N2 cleavage is the result of a multimetallic synergetic processes.
3. Mono- and bis-nitride bridged actinide complexes
3.1. Chemical reduction of azide precursors
As discussed in a previous review of the subject,42 the rational synthesis of actinide nitride complexes remained elusive, despite the isolation in 2002 of the bis-nitride complex from N2 reduction by Gambarotta.37 The most explored synthetic routes to actinide nitrides involves: (i) reduction of alkali metal-azides with U(III) complexes and (ii) the reaction of an actinide-azide complex with a reducing agent. Azides can act both as a 2e− oxidant and a source of nitride (i.e., (N3)− + 2e− → N3− + N2). These synthetic strategies led first to the isolation of multimetallic uranium nitride/azide clusters,38,39,67 and later the isolation of the first dinuclear nitride bridged complexes presenting either a bent UN–U40 or a linear UNU core in three different oxidation states,41 which were discussed in detail in the previous review by Liddle.42 The only reactivity of bridging nitride complexes reported up to 2014 was the reaction of the linear UVNUV with cyanide (CN−), where the complex behaved as an electrophilic nitride to yield the bimetallic U(IV)/U(IV) bridging cyanoimide complex, [UIV(N[tBu](3,5-Me2C6H3)3)2(μ-NCN)].41
This work incited other researchers to pursue the synthesis of uranium bridged-nitride complexes utilizing other supporting ligand frameworks. However, the isolation of actinide bridged-nitride complexes remains not trivial, and requires careful tuning of the supporting ligands steric and electronic properties, as well as the reaction conditions. Successful production of uranium bridged-nitride complexes in different oxidation states is reviewed in the following sections.
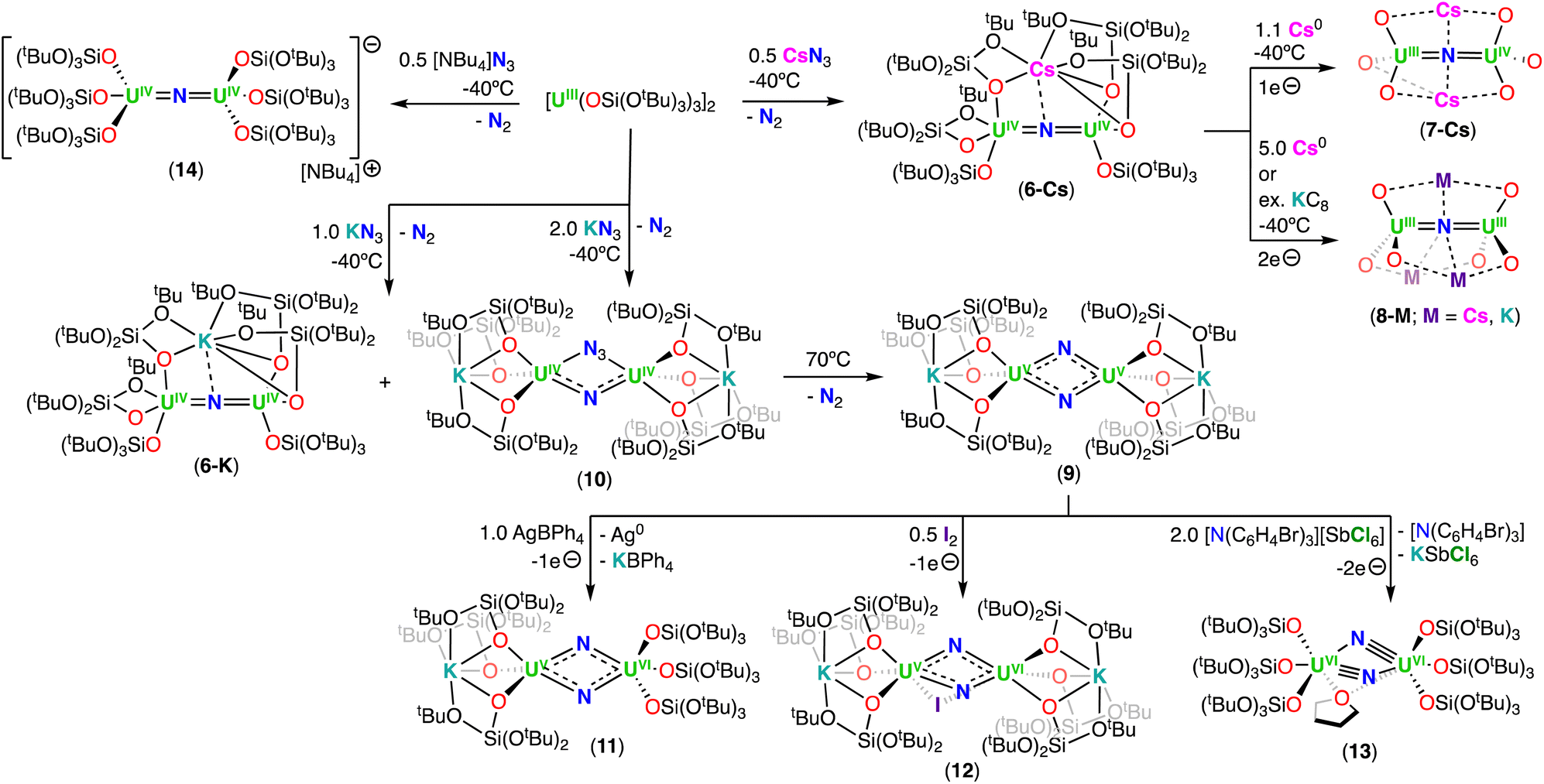 | ||
| Scheme 4 Synthesis of uranium nitrides supported by the siloxide (–OSi(OtBu)3) ligand. The ligands on complexes 7-Cs and 8-M were omitted for clarity. | ||
The X-ray crystallographic structures of 6-Cs and 6-K are analogous and consist of a heterometallic configuration, U2M (M = K, Cs), in which the two U(IV) cations are bridged by an azide (N3−) ligand in an almost linear fashion (U–N–U: 170.2(3)° 6-Cs; 170.1(5)° 6-K), with short U–N bond distances (2.058(5)–2.089(9) Å), consistent with U–N multiple bond character. The linear UIVNUIV core shows metrical parameters similar to those found in the dinuclear U(IV) nitride complex, Na[(UIV(N[tBu](3,5-Me2C6H3))3)2(μ-N)], reported by Cummins and co-workers.41 The main difference is the presence of the inner sphere alkali cation in the neutral complexes, 6-K and 6-Cs. In these, either five or six oxygen atoms of the –OSi(OtBu)3 ligands tightly bind the alkali cation, which also binds the nitride ligand at the exact apical position, with M–N distances of 3.393(4) Å and 3.245(8) Å for 6-Cs and 6-K, respectively. Bond valence sum calculations and magnetization data are consistent with the U(IV) oxidation state for both uranium ions.
The reduction of the CsUIVNUIV nitride complex (6-Cs) with 1.0 equiv. or excess of metallic cesium at low temperature, resulted in the formation of the U(III)/U(IV) and U(III)/U(III) bridging-nitride complexes, [Cs2(UIII/IV(OSi(OtBu)3)3)2(μ-N)] (7-Cs) and [Cs3(UIII(OSi(OtBu)3)3)2(μ-N)] (8-Cs), respectively, which were the first isolated uranium nitride complexes incorporating U(III) ions (Scheme 4).72 As observed for 6-Cs, the uranium nitrides, 7-Cs and 8-Cs, are also stabilized by two and three Cs cations, respectively, which are bound in the pocket formed by the –OSi(OtBu)3 ligands, and have a bonding interaction with the bridging nitride moiety. In complex 7-Cs, the two OSi(OtBu)3-ligated Cs cations lie at the apical position of the UIII–N–UIV core, while in 8-Cs, the three OSi(OtBu)3-ligated Cs cations bind the nitride ligand and form a distorted triangular array that is perpendicular to the UIII–N–UIII fragment. The U–N–U moiety is almost linear in both compounds and the U–Nnitride bond lengths (2.081(1)–2.150(1) Å) are consistent with a non-delocalized UNU bonding mode. The U–Nnitride bond distances increase by about 0.08 Å from CsUIVNUIV to the fully reduced Cs3UIIINUIII species, which can be related to the differences in ionic radii of U(III) and U(IV). Complexes 7-Cs and 8-Cs are very reactive and can only be handled in solution at low temperatures. The cyclic voltammogram of 6-Cs, measured in THF with [NBu4][BAr4F] as the supporting electrolyte, showed two irreversible redox events at −2.34 and −0.92 V (vs. [Cp2Fe]0/+). The irreversibility of these redox events was attributed to the rearrangement of the coordination sphere during the redox processes, but supports access to the low-valent U(III) species 7-Cs and 8-Cs.
The diuranium(III) potassium analogue of 8-Cs, [K3(UIII(OSi(OtBu)3)3)2(μ-N)] (8-K), was prepared by reduction of the CsUIVNUIV complex 6-Cs with excess of KC8 in THF.48 The presence of the inner sphere K cations in 8-K resulted in a higher stability when compared to the Cs3UIIINUIII complex (8-Cs), both in solid- and solution-state, making 8-K more suitable for reactivity studies with small molecules (Section 6.2.3). It should be noted that the alkali ion is so strongly bound in 8-K, that addition of 2.2.2-cryptand does not allow its removal.
With the aim of generating terminal-nitrides, rather than bridging moieties, Camp, Mazzanti and co-workers found that the anionic U(III) tetrakis complex, [K(18C6)][UIII(OSi(OtBu)3)4], which is sterically saturated by the coordination of four –OSi(OtBu)3 ligands, reacts with 1.0 equiv. of CsN3 at low temperature. However, in this case, a mixture of compounds was obtained, most likely resulting from the decomposition of a putative terminal nitride intermediate. Among the products, the second example of a diuranium(V) bis-nitride complex was identified, [K2({UV(OSi(OtBu)3)3}2(μ-N)2)] (9), comprising of a diamond-shaped U2N2 core.71
In 2019, the same group reported an improved and reproducible route for complex 9. The reaction of [UIII(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2 with 2.0 equiv. of KN3 at low temperature afforded the U(IV)/U(IV) bridged nitride-azide complex, [K({UIV(OSi(OtBu)3)3}2(μ-N)(μ-N3)] (10), which is unstable at room temperature and can be fully converted at 70 °C into the diuranium(V) bridged bis-nitride complex 9 (Scheme 4).52 Complex 9 has a crystallographically imposed symmetry center in between the two uranium cations with a U⋯U distance of 3.2960(6) Å, and two distinct short U–N bond lengths of 2.101(6) and 2.023(5) Å,71 which are similar to those reported by Gambarotta for the bis-nitride complex, [K(DME)4][(K(DME)((calix[4]tetrapyrrole)U)2(μ-NK)2].37 Bond valence sum calculations agree with a U(V) oxidation state, in which no EPR signal was observed for 9,52 indicative of a 3/2 magnetic ground state.73 The magnetic data measured for the UV–(μ-N)2–UV complex showed an unusually strong antiferromagnetic coupling between the two f1 ions with a TN (Néel temperature) of approximately 77 K,52 slightly higher than that found in the UV–(μ-O)2–UV complex [(UV((nP,MeArO)3tacn))2(μ-O)2] (tacn = triazacyclononane, nP = neopentyl) (∼70 K).74
More recently, Barluzzi, Mazzanti and co-workers reported that oxidation of complex 9 afforded the first bis-nitride uranium complexes containing U(VI) ions.75 Addition of AgBPh4 or I2 afforded the mixed-valent, U(V)/U(VI) bis-nitride complexes, [K(UV/VI(OSi(OtBu)3)3)2(μ-N)2] (11) and [K2(UV/VI(OSi(OtBu)3)3)2(μ-N)2(μ-I)] (12), respectively, whereas addition of the strong oxidant, [N(C6H4Br)3][SbCl5] (“magic blue”), afforded the U(VI)/U(VI) bis-nitride complex, [(UVI(OSi(OtBu)3)3)2(μ-N)2(μ-THF)] (13) (Scheme 4).75 All the U–N bond distances in 11 (avg. 2.06(3) Å) and in 12 (avg. 2.13(5) Å) are nearly equivalent for the two uranium centers, suggesting the presence of a charge delocalized species. In the U(VI)/U(VI) complex 13, short U–N bond distances (1.966(16) and 1.850(12) Å) are observed with much longer distances (2.292(13) and 2.252(16) Å), indicating one nitride ligand binds the U(VI) ion with a triple bond, whereas the other nitride with a single dative bond. An equivalent asymmetric bonding was calculated for the matrix isolated U(VI) bis-nitride, [UN(μ-N)]2, (U–Nbridging: 1.859 and 2.281 Å).30 Computational studies were consistent with a delocalized bond across the U–N–U bonding for the U(V)/U(V) bis-nitride 9 and for the mixed valent U(V)/U(VI) bis-nitride 11, but an asymmetric bonding mode was established for the U(VI)/U(VI) bis-nitride complex, 13, which showed a U–N σ orbital well localized over the UN, and π orbitals which are partially delocalized to form a U–N single bond with the other U(VI) ion.75
Using a similar strategy previously applied by Cummins,41 in 2019, Palumbo, Mazzanti and co-workers verified that by using the non-metallic azide, [NBu4][N3], in the oxidation of [UIII(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2, the U(IV)/U(IV) bridged nitride complex, [NBu4][(UIV(OSi(OtBu)3)3)2(μ-N)] (14) (Scheme 4) could be obtained.76 In the molecular structure of 14, the –OSi(OtBu)3 ligands coordinate to the uranium centers in a monodentate mode, κ1-OSi(OtBu)3, in a staggered orientation with respect to those of the adjacent [UIV(OSi(OtBu)3)3]+. Complex 14 shows average U–Nnitride bond lengths of 2.05(2) Å, comparable to those observed for the Cs-capped nitride, CsUIVNUIV (6-Cs) (avg. 2.09(1) Å),71 while the U–N–U angle is slightly more linear (avg. 174(3)°).
Using the same approach, the authors demonstrated that reduction of the azide precursor, [NBu4][N3], could be carried out with the analogous monometallic U(III) amide (–N(SiMe3)2) complex, [UIII(N(SiMe3)2)3], at low temperature (−40 °C) (Scheme 5),76 affording the U(IV)/U(IV) nitride complex, [NBu4][(UIV((N(SiMe3)2)3)2(μ-N)] (15). Performing the reaction at low temperature prevented cyclometallation, which had been previously described by Fortier, Hayton and co-workers for the U(IV)/U(IV) bridging nitride complex, [Na(DME)2(TMEDA)][((N(SiMe3)2)2UIV)(μ-N)(μ-κ2:C,N–CH2SiMe2NSiMe3)(UIV(N(SiMe3)2)] (16).40 The molecular structure of complex 15 shows two staggered [UIVN(SiMe3)3]+ units bridged by a nitride ligand. The U–N–U bond angle is close to linearity (avg. 179.3°), and the U–Nnitride bond lengths are equivalent, consistent with a delocalized UNU bonding mode, similar to that observed for the U(IV)/U(IV) amido complex, [NBu4][(UIV(N[tBu](3,5-Me2C6H3))3)2(μ-N)], reported by the Cummins group.41 The 1e− oxidation of 15 with AgBPh4, afforded the neutral nitride-bridged U(IV)/U(V) complex, [(UIV/V(N(SiMe3)2)3)2(μ-N)] (17) (Scheme 5).76 The molecular structure shows two different U–Nnitride bond lengths (2.080(5) and 2.150(5) Å), consistent with the presence of a localized valence.
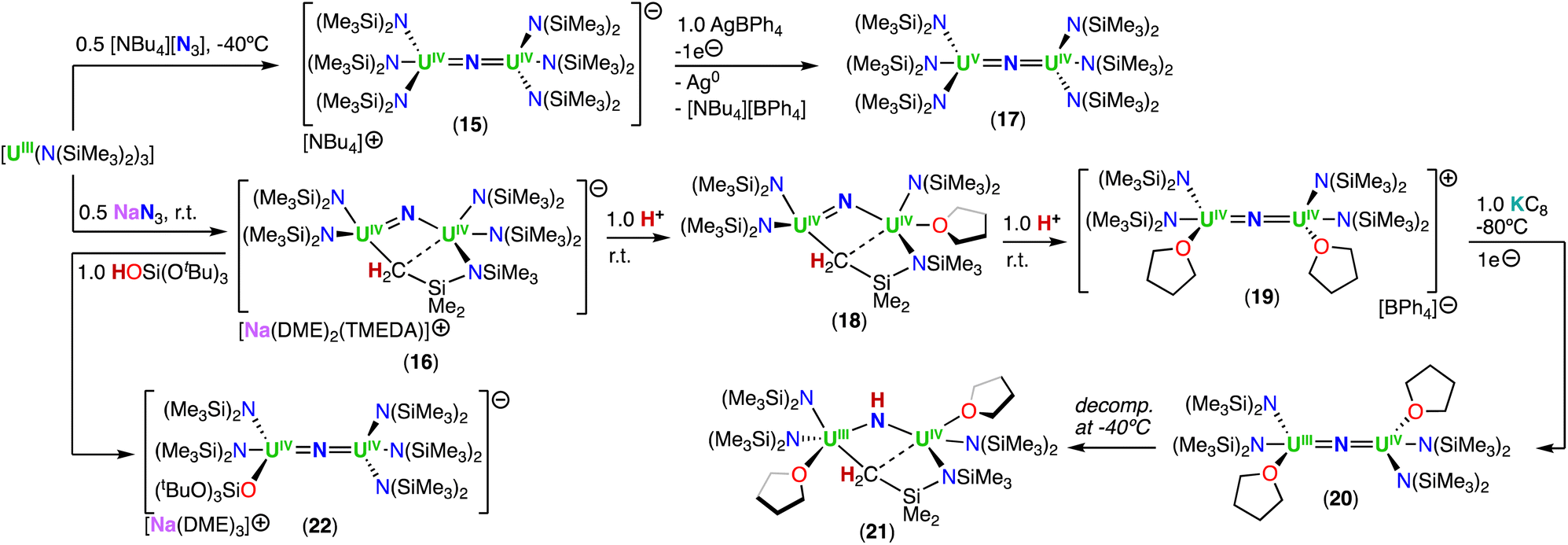 | ||
| Scheme 5 Synthesis of uranium nitrides supported by the amide (–N(SiMe3)2) ligand. | ||
In a later study, the authors found that reacting complex 16 (ref. 40) with 1.0 and 2.0 equiv. of HNEt3BPh3, resulted in the neutral [(N(SiMe3)2)2UIV(μ-N)(μ-κ2:C,N–CH2SiMe2NSiMe3)U(N(SiMe3)2)(THF)] (18) and cationic [(UIV((N(SiMe3)2)2(THF))2(μ-N)][BPh4] (19), U(IV)/U(IV) nitride complexes by successive protonolysis of one –N(SiMe3)2 ligand and of the uranium–CH2 bond, respectively (Scheme 5).70 Notably, the authors found that reducing complex 19 with 1.0 equiv. KC8 at −80 °C led to the formation of a rare U(III)/U(IV) bridging-nitride complex, [(UIII/IV(N(SiMe3)2)2(THF)2(μ-N)] (20).70 The solid-state molecular structure of the U(III)/U(IV) bridged nitride 20 is similar to the parent U(IV)/U(IV) complex 19, but with U–N(SiMe3)2 and U–THF bond distances consistent with a reduction to U(III). The U–Nnitride bond lengths are comparable (19: 2.0634(3) Å; 20: 2.055(3) and 2.064(3) Å), however, the U–N–U bond angle is completely linear in complex 19. The U(III)/U(IV) complex 20 is highly reactive and above −40 °C, undergoes 1,2-addition of the C–H bond of a N(SiMe3)2 ligand across the UIIINUIV moiety, generating a U(III)/U(IV) imido–cyclometalate complex, [(N(SiMe3)2)2(THF)UIII(μ-NH)(μ-κ2:C,N–CH2SiMe2NSiMe3)UIV(N(SiMe3)2)(THF)] (21).
These results demonstrated that a U(III)/U(IV) nitride could be isolated with strong sigma donor supporting ligands (–N(SiMe3)2) upon careful choice of the reaction conditions. However, further reduction of the U(IV) ion to yield a U(III)/U(III) was not accessible in this N(SiMe3)2-nitride complex. This incited the same group to pursue heteroleptic complexes to maximize the electron-rich character without hampering access to a U(III)/U(III) nitride (vide infra).
Treatment of complex 16 (ref. 40) with 1.0 equiv. of HOSi(OtBu)3 generated the heteroleptic U(IV)/U(IV) nitride complex [Na(DME)3][(N(SiMe3)2)3UIV(μ-N)UIV(N(SiMe3)2)2(OSi(OtBu)3)] (22) (Scheme 5).76 The presence of one –OSi(OtBu)3 ligand resulted in a significant deviation of the U–N–U bond angle from linearity (168.4(3)°).
The magnetic behavior of complexes 14, 15, and 22 were studied. The χ versus T plots for the all-N(SiMe3)2 and heteroleptic complexes, 15 and 22, respectively, are consistent with an antiferromagnetic coupling of the U(IV)/U(IV) centers, with maximums at 90 and 55 K, respectively.76 The highest χ maximum (at 110 K) ever measured for an actinide complex was reported by Vlaisavljevich, Cummins and co-workers for a dimeric arene-bridged U(III)/U(III).77 Additionally, Liddle and co-workers reported a strong antiferromagnetic coupling (∼60 K maximum) for a triamidoamine (TrenTIPS) bridged nitride complex with a linear UIVNUIV core.78
Shorter U⋯U bond distances were found for the all-OSi(OtBu)3 nitride complexes, [Cs(UIV(OSi(OtBu)3)3)2(μ-N)] (6-Cs) and [NBu4][(UIV(OSi(OtBu)3)3)2(μ-N)] (14) (4.121(1) and 4.107(2) Å, respectively), compared to the all-N(SiMe3)2 complex, [NBu4][(UIV(N(SiMe3)2)3)2(μ-N)] (15) (4.15 Å), but magnetic coupling was only detected in 15. The heteroleptic complex 22 exhibits antiferromagnetic coupled U(IV)/U(IV) centers, but the structure displays a U⋯U bond distance of 4.101(1) Å, and the U–N–U bond angle is similar to the all-OSi(OtBu)3 complex 6-Cs, suggesting there is no correlation between the geometric parameters and magnetic properties in these complexes.76 Computational studies showed that the all-N(SiMe3)2 complex 15 has a singlet antiferromagnetic ground-state, while the quintet state is the most stable for the all-OSi(OtBu)3 complex 14. The difference in magnetic properties were rationalized in terms of the different nature and strength of the U–L bonding interaction for the –N(SiMe3)2 and –OSi(OtBu)3 ligands.
The synthetic strategy used in the preparation of [Cs(UIV(OSi(OtBu)3)3)2(μ-N)] (6-Cs)71 was also successfully applied by Keener, Mazzanti and co-workers for the isolation of other heteroleptic U(IV)/U(IV) bridged nitrides.79 First, the heteroleptic complex, [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (23), containing a combination of –OSi(OtBu) and –N(SiMe3)2 ligands (4:2 ratio), was obtained from the reaction of CsN3 with the in situ generated heteroleptic U(III) precursor, “[UIII(OSi(OtBu)3)2(N(SiMe3)2)]” (Scheme 6).79 Complex 23 is quite stable at low temperatures in toluene, but slowly decomposes at −40 °C in THF, and undergoes full decomposition at room temperature forming the U(IV)/U(IV) bridged imido–cyclometallate complex, [Cs((OSi(OtBu)3)3UIV)(μ-NH)(μ-η2:C,N–CH2SiMe2NSiMe3)UIV(N(SiMe3)2)(OSi(OtBu)3)] (23b), involving a 1,2-addition of a C–H bond of N(SiMe3)2 across the uranium–nitride bond, similar to what was observed for the U(III)/U(IV) nitride complex 20. From the same study, a second heteroleptic U(IV)/U(IV) nitride complex, [K(UIV(OSi(OtBu)3)(N(SiMe3)2)2)2(μ-N)] (24), containing a different combination of –OSi(OtBu)3 and –N(SiMe3)2 ligands (2:4 ratio), was prepared by the addition of 2.0 equiv. KOSi(OtBu)3 to the cationic nitride complex 19 (ref. 70) (Scheme 6), and was found to be stable in various solvents.79 The increased stability of 24 towards C–H bond activation of a N(SiMe3)2 ligand, in comparison to complex 23, was attributed to the number of –OSi(OtBu)3 ligands (two vs. four). The authors found that a higher number of –OSi(OtBu)3 ligands resulted in an increased nucleophilicity of the nitride in 23, leading to 1,2-addition of a C–H bond of a N(SiMe3)2 ligand. This is consistent with the lower nucleophilicity of bridging nitrides in U(IV) complexes supported by amide compared to those supported by siloxides (see reactivity studies). The X-ray diffraction analysis of the bridging nitride complexes, 23 and 24, revealed similar U–Nnitride bond distances to those of 6-Cs71 and 6-K.52 As expected, the U–Nimido bond distances in 23b (2.167(8) and 2.198(8) Å) are longer than those in the parent U(IV)/U(IV) nitride precursor, 23.
 | ||
| Scheme 6 Synthesis of heteroleptic uranium nitrides supported by varying combinations of siloxide (–OSi(OtBu)3) and amide (–N(SiMe3)2) ligands. | ||
The heteroleptic CsUIVNUIV complex 23, which contains more –OSi(OtBu)3 ligands, could be reduced at −40 °C with excess KC8 (Et2O) or CsC8 (THF), generating the U(III)/U(IV) analogues, [K2(UIII/IV(OSi(OtBu)3)2(N(SiMe3)2))2(μ-N)] (25-K) and [Cs2(UIII/IV(OSi(OtBu)3)2(N(SiMe3)2))2(μ-N)] (25-Cs), respectively. However, by changing the solvent from Et2O to THF in the reduction of CsUIVNUIV complex 23 with KC8, afforded a putative heteroleptic U(III)/U(III) bridging nitride, “[K3(UIII(OSi(OtBu)3)2(N(SiMe3)2))2(μ-N)]” (26). Alternatively, reduction of the heteroleptic KUIVNUIV complex 24, containing more –N(SiMe3)2 ligands, with excess KC8 at −80 °C yielded the U(III)/U(IV) nitride complex, [K(THF)6][K(UIII/IV(OSi(OtBu)3)(N(SiMe3)2)2)2(μ-N)] (27) (Scheme 6).79 Interestingly, the two additional –OSi(OtBu)3 ligands in the U(III)/U(IV) complex 27, compared to the cationic U(III)/U(IV) complex 20, results in an increased stability towards the 1,2-addition of the C–H bond of a –N(SiMe3)2 ligand across the uranium–nitride, most likely due to the OSi(OtBu)3-bound K cation capping the nitride. Removal of the K cation by addition of 2.2.2-cryptand, increases nitride reactivity towards C–H activation, affording the imido cyclometallated complex, [((Me3Si)2N)2UIV(OSi(OtBu)3)(μ-NH)(μ-κ2:C,N–CH2SiMe2NSiMe3)UIII(N(SiMe3)2)(OSi(OtBu)3)][(K(2.2.2-cryptand))2] 28. Reduction of the complexes, Cs2UIIINUIV (25-Cs) and [K(THF)6][KUIIINUIV] (27), with excess CsC8 and KC8, respectively, to afford their M3UIIINUIII (M = K, Cs) analogues revealed impossible due to the very electron-rich character of uranium by influence of the different ancillary ligands or alkali-metal (Cs versus K) (Scheme 6). This contrasts with what was observed for the all-OSi(OtBu)3 complex, 6-Cs, which could be reduced to the analogous M3UIIINUIII (M = K (8-K), Cs (8-Cs)) complexes.
3.1.2. Complexes with triamidoamine ligands
The triamidooamine ligand (TrenR: N(CH2CH2NR)3−) was introduced in actinide coordination chemistry by Scott and co-workers in 1994.80 Since 2012,43 the Liddle group has used this tripodal-framework to promote the formation of terminal uranium nitrides and to investigate their chemistry.43 In 2019, Du, Liddle and co-workers reported the use of TrenDMBS (DMBS = N(CH2CH2NSiMe2tBu)3−) in the synthesis of mono nitride-bridged uranium complexes.78 The chemical reduction of the uranium(IV) azide complex, [(UIV(TrenDMBS)(μ-N3))4] (29), with KC8 afforded the bridged nitride complexes, [K(THF)6][(UIV(TrenDMBS))2(μ-N)] (30) and [(UV/IV(TrenDMBS))2(μ-N)] (31),78 while photolysis of the U(IV) azide precursor, 29, led only to formation of the mixed-valent U(IV)/U(V) complex 31 (Scheme 7). It should be noted that this work provided the first example of nitride complexes isolated from the reduction or photolysis of a U(IV)-azide precursor, rather than reaction of a U(III) complex with a metal azide. Seminal attempts to form nitrides using these routes had failed to result in N2 elimination (as for the reduction of [U(N(SiMe3)2)3(N3)2] reported by Fortier, Hayton and co-workers)81 or generated highly reactive nitride intermediates that underwent insertion into the ligand framework.45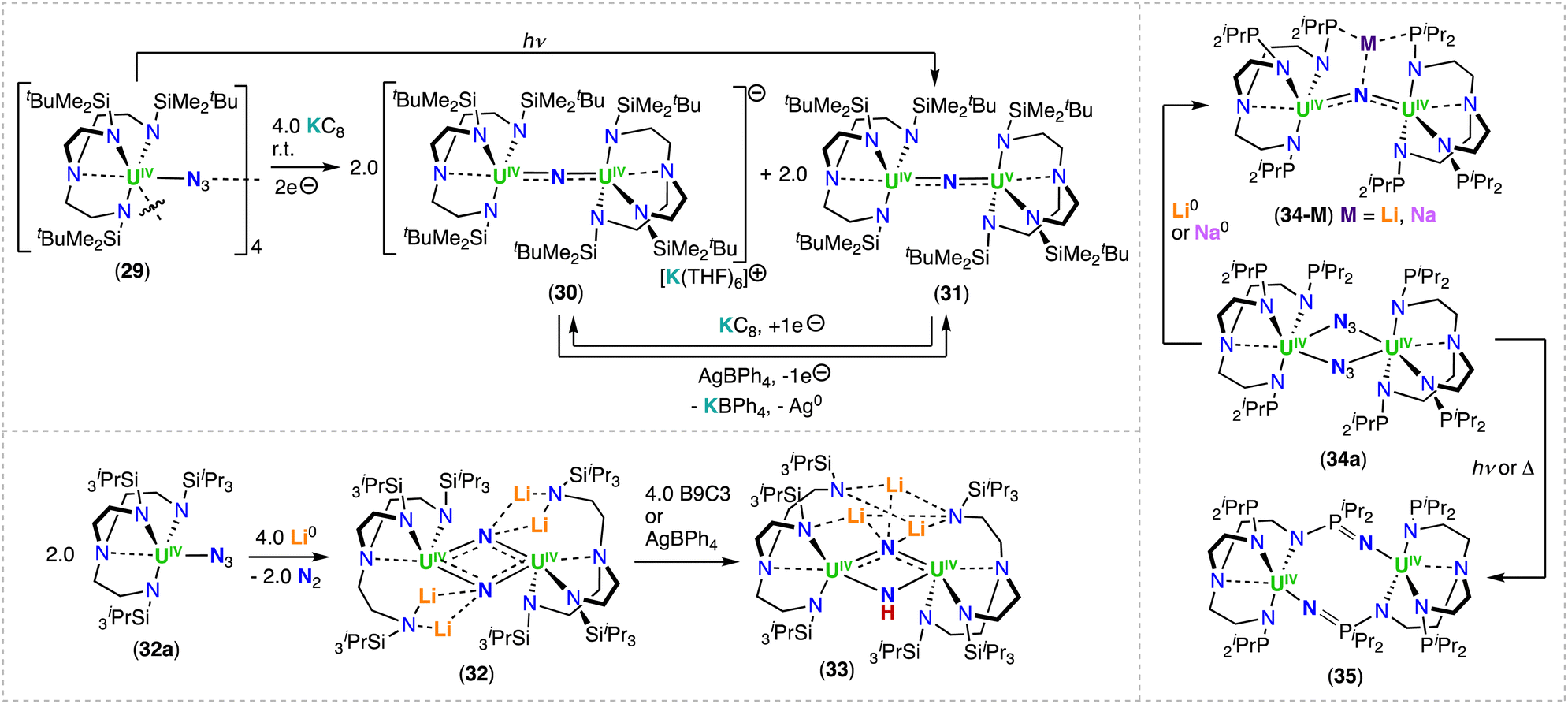 | ||
| Scheme 7 Synthesis of uranium-nitrides supported by the triamidoamine (TRENDMBS and TRENTIPS) or phosphorus-TREN (TRENNP) ligands. | ||
In the solid-state molecular structure of 30, the anion lies on a crystallographic 3-fold rotation axis and therefore the UIV–N–UIV angle is 180°, and the U–Nnitride bonds are equivalent (2.0648(2) Å). In the mixed-valent U(IV)/U(V) complex, 31, the U–Nnitride bond lengths are inequivalent (2.081(5) and 2.136(5) Å) and the UV–N–UIV bond angle is bent (161.2(2)°). The magnetic data of complexes 30 and 31 are consistent with the U(IV)/U(IV) and U(IV)/U(V) combinations, respectively. The presence of U(V) in complex 31 was unambiguously confirmed by S- and X-band EPR spectroscopies.
Moreover, antiferromagnetic U(IV)/U(IV) coupling was suggested by a maximum at 60 K in the χ vs. T plot for complex 30.
The uranium bridged nitrides 30 and 31 are chemically robust, and are reversibly interconverted by oxidation and reduction, respectively, with no evidence of C–H bond activation, which is instead observed with the analogous Th(IV) system (Section 3.1.3) indicating a lower reactivity of the uranium nitride.78
More recently, the sterically bulkier TrenTIPS (N(CH2CH2NSiiPr3)3−) ligand, was utilized to stabilize the bridged U(IV)/U(IV) bis-nitride complex, [Li4(UIV(TrenTIPS))2(μ-N)2] (32),82 which was obtained by reduction of the U(IV) azide precursor, [UIV(TrenTIPS)(N3)] (32a) (Scheme 7), with excess metallic lithium (Li), and concomitant formation of the U(III) complex, [UIII(TrenTIPS)]. The solid-state structure of complex 32 comprises of a U2N2 diamond-shaped core, with a U⋯U distance of 3.404(1) Å). Each nitride (N3−) ligand is bridging two U(IV) centers, and both nitrides are bound to two Li cations, which seemingly facilitate formation of the UIV–(μ-N)2–UIV bonds. Attempts to prepare high-valent species by addition of benzo-9-crown-3 ether (B9C3) or AgBPh4, resulted in exchange of the Li cation for a proton (H+), converting complex 32 to the U(IV)/U(IV) nitride-imido complex, [Li3(UIV(TrenTIPS))2(μ-NH)(μ-N)] (33).82 The U–Nnitride bond lengths of 32 (2.153(2) Å) and 33 (2.171(4) Å) are longer than in the U(IV)/U(IV) bridging mono-nitride complexes (1.95(1)–2.12(1) Å), but are more consistent with the U(V)/U(V) bis-nitride complex, [K2([UV(OSi(OtBu)3)3]2(μ-N)2)] (9) (2.101(6)–2.022(5) Å).71
Variable-temperature magnetic data for the U(IV)/U(IV) bridging nitride complexes, 32 and 33, revealed high, low-temperature magnetic moments, consistent with doubly degenerate ground states, where the effective symmetry of the strong crystal field of the nitride dominates over the spin–orbit coupled nature of the ground state multiplet of U(IV). Spin Hamiltonian modelling of the magnetic data for complexes, 32 and 33, suggest U⋯U anti-ferromagnetic coupling of −4.1 and −3.4 cm−1, respectively. Computational studies show a borderline case where the prospect of direct U–U bonding was raised, but high-level ab initio calculations revealed that if any U–U bonding is present, it is weak, and instead the bridging nitride ligands facilitate the U⋯U electronic communication.82 This is consistent with the strong antiferromagnetic coupling measured for the U(V)/U(V) bis-nitride complex, [K2([UV(OSi(OtBu)3)3]2(μ-N)2)] (9).52
Recently, Zhu and co-workers investigated the reactivity of U(IV) azide complexes supported by a phosphorus-substituted Tren ligand, (N(CH2CH2NPiPr2)3)3− = TrenNP), containing three rigid N–P units.83 Reaction of the U(IV) complex, [UIV(TrenNP)Cl], with NaN3 led to the U(IV)/U(IV) azide complex, [(UIV(TrenNP))2(μ-N3)2] (34a). Subsequent addition of metallic Li or sodium (Na) afforded the U(IV)/U(IV) bridging nitride complexes, [Li(UIV(TrenNP))2(μ-N)] (34-Li) and [Na(THF)(UIV(TrenNP))2(μ-N)] (34-Na), respectively (Scheme 7).84 In contrast, the reduction with metallic K generated the U(IV)/U(IV) complex, [K2(UIV(TrenNP))(μ-NH)2], with two bridging imido (μ-NH2−) ligands linking the two uranium centers, while coordinated to the K cations. The X-ray diffraction structures of 34-Li and 34-Na are similar and both complexes exhibit an alkali metal-capped uranium nitride unit, MUIV–N–UIV (M = Li, Na).84 The U–N–U bond angle in complex 34-Na (127.8(2)°) is bent when compared to 34-Li (146.6(3)°), which could arise from the bound THF molecule coordinated to the Na cation in 34-Na. Photolysis and thermolysis of [(UIV(TrenNP))2(μ-N3)2] (34a), led to a U(IV)/U(IV) ligand-insertion product, [UIV(N(CH2CH2NPiPr2)2(CH2CH2NPiPr2N)]2 (35) (Scheme 7), in which a uranium-nitride intermediate was proposed by the authors for its formation.
3.1.3. Thorium nitrides from azide reduction
Thorium nitrides are less common than uranium nitrides, and until 2019, examples of thorium nitrides had only been isolated under cryogenic matrix conditions, such as ThN, NThN and NThO molecular species.30–32 Du, Liddle and co-workers reported the reduction of Th(IV)/Th(IV) azide precursors supported by two different triamidoamine ligands, [(ThIV(TrenDMBS)(μ-N3))3] (36a) and [ThIV(TrenTIPS)(N3)] (37a) (TrenDMBS: N(CH2CH2NSiMe2tBu)33−; TrenTIPS: N(CH2CH2NSiiPr3)33−), which generate highly nucleophilic and transient thorium-nitrides. These complexes were found to activate C–H bonds from various solvents, yielding parent Th(IV)/Th(IV) bridging imido complexes, [(ThIV(TrenDMBS))2(μ-NH)] (36) and [M2(ThIV(TrenTIPS)(μ-NH))2] (M = Li, Na, K, Rb, Cs; 37-M), respectively (Scheme 8a).78,85 These results provided the first evidence of transient ThIVNThIV species that could be generated by chemical reduction of thorium azides; however, the transient thorium–nitride complex can easily activate C–H bonds of the solvent, or of the supporting TrenDMBS ligand, demonstrating that thorium-nitride bonds are highly reactive, and are more so than their uranium analogues. Indeed, DFT calculations suggested that the thorium complex contains more polar and ionic metal-nitride linkages, and importantly the frontier molecular orbitals that comprise the ThNTh bonding interactions are destabilised by ∼1.1 (σ) and ∼0.4 (π) eV compared to the corresponding UNU orbitals. Instead, the first example of a dinuclear thorium nitride isolated in any significant quantity was prepared by Staun, Hayton and co-workers, via a synthetic approach that comprises of the reaction with a Th–NH2 precursor (Section 3.2).86
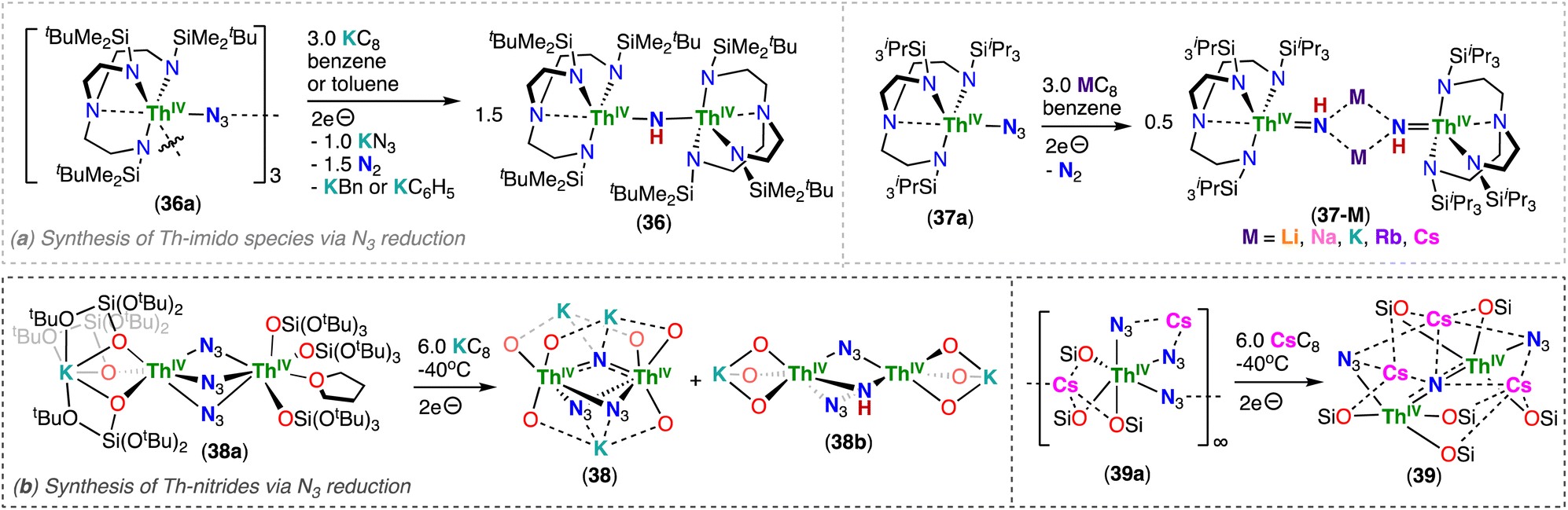 | ||
| Scheme 8 Synthesis of thorium (a) imidos and (b) nitrides supported by triamidoamine (TRENDMBS and TRENTIPS) and siloxide (–OSi(OtBu)3) ligands, respectively. | ||
In 2022, Hsueh, Mazzanti and co-workers demonstrated that stable multimetallic thorium nitride complexes supported by –OSi(OtBu)3 ligands could be generated from the reduction of actinide azide precursors.87 In fact, the reaction of the Th(IV) bridging azide complexes, [K(OSi(OtBu)3)3ThIV(μ-N3)3ThIV(OSi(OtBu)3)3(THF)] (38a) and [Cs(OSi(OtBu)3)3ThIV(N3)(μ-N3)2Cs]∞ (39a), with excess KC8 or CsC8, respectively, at low temperatures generated the respective Th(IV)/Th(IV) bridged-nitride complexes, [K3(ThIV(OSi(OtBu)3)3)2(μ-N)(μ-N3)2] (38) and [Cs3(ThIV(OSi(OtBu)3)3)2(μ-N)(μ-N3)2] (39) (Scheme 8b). A Th(IV) bridging-imido complex, [K(ThIV(OSi(OtBu)3)3)2(μ-NH)(μ-N3)] (38b), was also formed during the synthesis of 38, showing that the nature of the alkali ion plays an important role in generating the nitride complex. The presence of three alkali ions bound to the nucleophilic nitride moiety provides high stability for the azide/nitride clusters, 38 and 39, but the nature of the cation led to vast structural differences. The molecular structure of 38 exhibits a bent Th–N–Th angle (114.4(3)°) with a short K–Nnitride distance (2.760(3) Å), while in complex 39, the ThIVNThIV fragment adopts a linear geometry (178.1(15)°) with a Cs–Nnitride distance of (avg. 3.49(6) Å). The Th–Nnitride bond lengths (38: 2.185(4) Å; 39: avg. 2.13(5) Å) are shorter than those found in reported thorium bridging imido complexes,78 but consistent with those found in the linear ThIVNThIV complex reported by Staun, Hayton and co-workers.86
Computational studies of Th(IV)/Th(IV) bridged nitrides demonstrated that the An–N–An bonding involves three-center-two-electron interactions of σ and π-type, with two Th–N σ bonds strongly polarized toward N (90%). However, some differences were also revealed from the theoretical calculations: in the bent nitride structure 38, the π orbitals are lower in energy than the σ orbital, whereas in the linear nitride structure 39 the σ is the lowest one. The σ > π phenomenon is known for terminal uranium nitrides43,44 and uranyl,88,89 and was recently reported for Th–nitrogen multiple bonds complexes and assigned to the “pushing from below effect”.85
3.1.4. Other ligands
In 2015, Maria and co-workers reported the solid-state structure of a U(IV)/U(IV) nitride-bridged complex supported by a bis-aryloxide cyclam ligand, ((tBu2ArO)2Me2-cyclam)2−.90 Few crystals suitable for XRD analysis of the complex, [(UIV(κ4-((tBu2ArO)2Me2-cyclam)(N3))(μ-N))(UIV(κ5-((tBu2ArO)2Me2-cyclam))] (40), were obtained from a NMR scale reaction of the U(III) precursor [UIII(κ6-((tBu2ArO)2Me2-cyclam))I] (40a) with CsN3 in pyridine at room temperature (Scheme 9).90 The asymmetric U(IV)/U(IV) nitride complex 40 exhibits a U–N–U bond angle of 173.2(2)°, and U–Nnitrido bond distances of 2.086(3) and 2.035(3) Å, that are within the values reported for other mono-nitride bridged U(IV)/U(IV) complexes (1.95(1)–2.09(1) Å).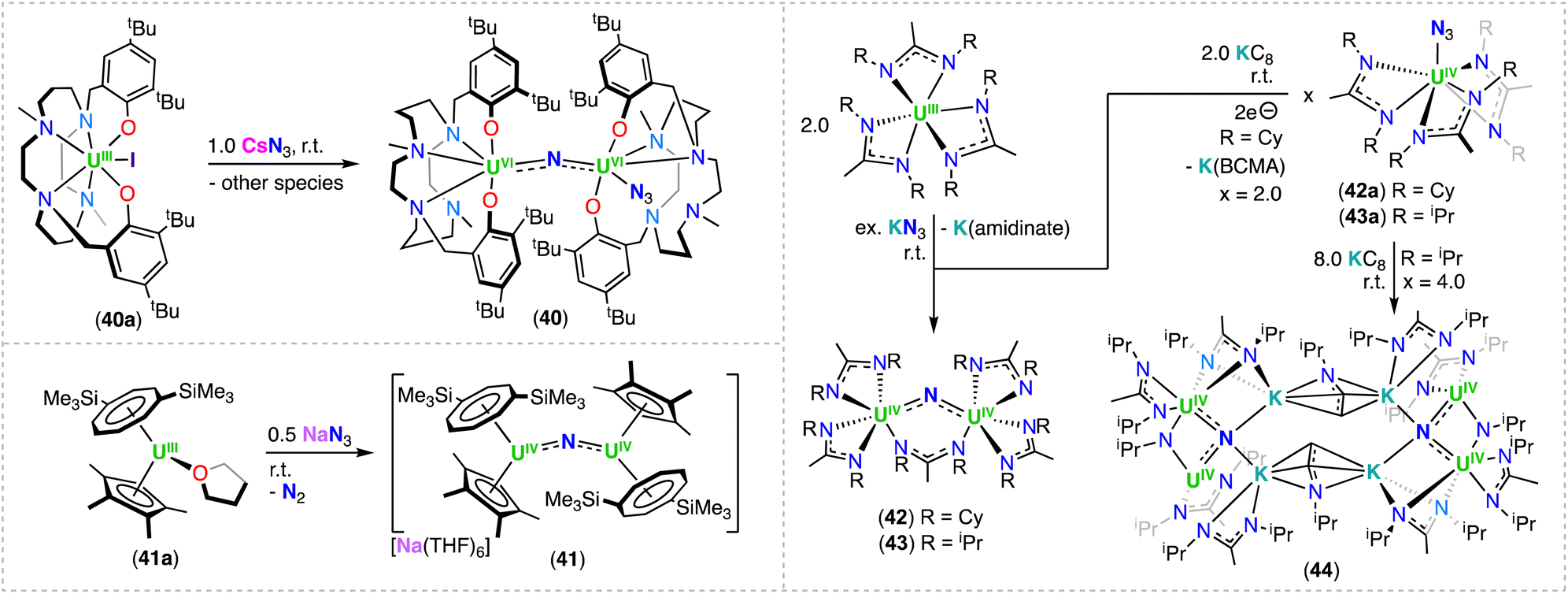 | ||
| Scheme 9 Synthesis of uranium-nitrides supported by other ligand systems. | ||
In 2016, Tsoureas, Cloke and co-workers reported an organometallic U(IV)/U(IV) bridging nitride complex, [Na(THF)6][(UIV(η8-C8H6(1,4-SiMe3)2(Cp*))2(μ-N)] (41), by reacting the monometallic heteroleptic U(III) complex, [(UIII(η8-C8H6(1,4-SiMe3)2)(Cp*)] (41a), with NaN3 at room temperature, leading to a delocalized UIVNUIV bonding interaction (Scheme 9).91 Alternatively, with use of bulkier substituents, the same group was able to isolate a capped terminal nitride, [Na(UV(η8-C8H6(1,4-SiiPr3)2)(η5-Cp*))(μ-N)] 66 (see Section 4.2; Scheme 13).
More recently, Straub, John Arnold and co-workers explored the ability of amidinate ligands to stabilize uranium nitride complexes. The homoleptic U(III) amidinate complexes, [UIII(BCMA)3] (BCMA = N,N-bis(Cy)methylamidinate) and [UIII(BIMA)3] (BIMA = N,N-bis(iPr)methylamidinate), reduce KN3 to give the diuranium complexes, [(UIV(BCMA)2)2(μ-N)(μ-κ1:κ1-BCMA)] (42) and [(UIV(BIMA)2)2(μ-N)(μ-κ1:κ1-BIMA) (43), respectively (Scheme 9).92 The molecular structures of complexes 42 and 43 are similar, with the two U(IV) centers bridged by a nitride and a μ-κ1:κ1 bound amidinato ligand, where the U–Nnitrido bond distances range from 2.023(3) to 2.057(3) Å. Complex 42 can also be obtained by reduction of the U(IV) azide complex, [UIV (BCMA)3(N3)] (42a), with 2.0 equiv. KC8. Alternatively, addition of excess KC8 to [UIV (BCMA)3(N3)] (43a) led to the formation of a tetra-U(IV) cluster incorporating two UIV–N–UIV fragments, [K(UIV(BIMA)2(μ-NiPr))2(μ-N)(μ-η3:η3-CH2CHNiPr)]2 (44). Interestingly, the solid-state structure of 44 also revealed the presence of imido ((NiPr)2−) and vinylamido ((CH2CHNiPr)−) ligands formed by amidinate cleavage. In the structure of 44 all four uranium atoms are N-bound to ligands having formal mono-, di-, and tri-anionic character. Overall, 44 is stabilized by additional interactions of K cations with the nitride and amidinato ligands.92 Preparation of the isotopically enriched complexes, 15N-42 and 15N-43, and acidification with acid (H+) to 15NH4+, and IR spectroscopic analysis, allowed confirmation of the presence the bridging nitride ligand.92 This study provides a rare example in which uranium nitride complexes could be isolated by reduction of a U(IV) precursor with alkali ions, where this methodology was previously unsuccessful in generating uranium nitride complexes.
3.2. Reactions involving NH3 and derivatives
In 2019, Staun, Hayton and co-workers reported the first molecular thorium nitride isolated outside low temperature matrices.86 The Th(IV)/Th(IV) bridged nitride complex, [Na(18C6)(Et2O)][(ThIV(N(SiMe3)2)3)2(μ-N)] (45-Na), was obtained from addition of 1.0 equiv. NaNH2 to the metallacycle, [ThIV(N(SiMe3)(SiMe2CH2))(N(SiMe3)2)2] (46),93 in the presence of 18-crown-6 (18C6) (Scheme 10).86 Based on mechanistic studies, the authors suggested that the first step is the deprotonation of 46 with NaNH2, which results in the thorium bis-metallacycle complex, [Na(THF)x][Th(N(SiMe3)(SiMe2CH2))2(N(SiMe3)2)] (47-Na), and NH3. Then, NH3 reacts with another molecule of 46 to create the Th(IV)–NH2 complex, [ThIV(N(SiMe3)2)3(NH2)] (48), which protonates complex 47-Na to produce the thorium nitride complex 45-Na (mechanistic scheme not shown here). This hypothesis was supported by the reaction of 48 with the bis–metallacycle complex 47-K94 in the presence of 18-crown-6, which led to the formation of [K(18c6)(THF)2][(Th(N(SiMe3)2)3)2(μ-N)] (45-K).86 | ||
| Scheme 10 Synthesis of thorium- and uranium–thorium nitrides supported by the amide (–N(SiMe3)2) ligands via amine derivatives. | ||
The solid-state structure of complex 45-M (M = Na, K) exhibits a linear Th–N–Th bond angle (179(1)°), and the Th–Nnitride bond lengths (2.14(2) and 2.11(2) Å) are much shorter than the Th–Nsilylamido bond lengths (avg. 2.41 Å), consistent with Th–N multiple bond character. NBO/NLMO analysis revealed two orthogonal π bonds and two predominant σ bonds in Th–N–Th bonding, with σ-bonding having three-center character, consistent with ThN double bonds. Covalency in the Th–Nnitride bonds in 45-M was found to be larger than that reported for the Th–Nimido in [ThIV(NAriPr2)(N(SiMe3)2)3]− (AriPr2 = 2,6-iPr2C6H3)94 and for the Th–Ooxo in [Th(O)(N(SiMe3)2)3]−,95 with a greater participation of the 5f orbital in bonding and higher Wiberg bond index.
15N NMR spectroscopy was also used for the first time to evaluate Th–Nnitride bond covalency. To achieve that, the isotopically enriched analogues of complexes 48 and 45-K were prepared. The thorium nitride 15N-45-K was obtained in low yield by addition of 15NH4Cl to complex 47-K. Whereas, [ThIV(N(SiMe3)2)3(15NH2)] (15N-48) was isolated from the reaction of 46 with 1.0 equiv. of 15NH3 gas. The solution-state 15N NMR spectroscopy chemical shifts of the bridging nitride in 15N-45-K appeared at 298.8 ppm (referenced to CH3NO2), whereas the 15N resonance of the (–NH2−) in 15N-48 appeared at −198.4 ppm. The magnitude of the downfield shift correlates with the degree of 5f character in the An–N bond, which is lower in 48.86 Similar conclusions about covalency were achieved when the 15N NMR solid-state spectra of 15N-48 and 15N-45-K were measured.96
Using a similar synthetic approach to the one used for the synthesis of 45-K, Staun, Hayton and co-workers also prepared the first mixed-actinide nitrides.97 The Th(IV)/U(IV) complex, [K(18C6)(THF)2][(N(SiMe3)2)3UIV(μ-N)ThIV(N(SiMe3)2)3] (49), was isolated in moderate yield from the reaction of complex 47-K94 with the U(IV)–NH2 complex, [UIV(N(SiMe3)2)3(NH2)] (50), previously prepared by reaction of the uranium metallacycle [UN(SiMe3{(SiMe2CH2)}(N(SIMe3)2)2] with NH3, in the presence of 18-crown-6. The cyclometallate mixed-actinide nitride complex, [K(18c6)][K(18c6)(Et2O)2][(N(SiMe3)2)2UIV(μ-N)(μ-κ2-C,N–CH2SiMe2NSiMe3)ThIV(N(SiMe3)2)2]2 (51), was also isolated from the reaction mixture, and was thought to proceed through a similar pathway as the analogous cyclometallate U(IV)/U(IV) nitride complex 16.40 Oxidation of 49 with 0.5 equiv. of iodine afforded the U(V)/Th(IV) bridged nitride [(N(SIMe3)2)3UV(μ-N)ThIV(N(SiMe3)2)3] (52) (Scheme 10).97
The solid-state molecular structure of the mixed-actinide Th(IV)/U(IV) bridging nitride complex 49 showed that the uranium and thorium atoms are mutually disordered over both actinide sites in a 50:50 ratio. The An–N–An moiety is linear, while the An–Nnitride bond length is 2.1037(9) Å.97 The Th(IV)/U(IV) cyclometallate complex 51 displays a bent U–N–Th angle (122.2(2)°), a U–Nnitride bond length of 2.002(4) Å, and a Th–Nnitride bond length of 2.160(5) Å.97 Comparable crystallographic data were observed for the isostructural U(IV)/U(IV) analogue, complex 16,40 which is indicative of a localized Th–NU bonding motif. In the solid-state structure of 52, the uranium and thorium atoms were refined over both metal sites in a 50:50 ratio, resulting in An–Nnitride bond lengths of 2.10(1) Å and 2.17(1) Å, and a U–N–Th bond angle of (177.9(6)°), which can be compared with those observed for the U(IV)/U(V) nitride complex, [(UIV/V(N(SiMe3)2)3)2(μ-N)] (17), with bond metrics of 2.080(5), 2.150(5) Å and 179.4(3)°, respectively.76
The disorder found in the structure of 49 did not definitively confirm the presence of Th and U in the molecule, however, preparation of the Th(IV)/U(V) bridged nitride 52 from complex 49, coupled with spectroscopic analysis, reinforced the heterobimetallic formulation of 52. A comparative study with the U(V) Li-capped nitride complex, [Li(12C4)(UV(TrenTIPS)(μ-N))] (64-Li),98 based on EPR spectroscopy, SQUID magnetometry, NIR-visible spectroscopy, and crystal field analysis supported the presence of a U(V) ion in 52. Moreover, the study demonstrated that the energies of the 5f orbitals of the U(V)/Th(IV) bridged nitride complex 52 are essentially determined by the strong ligand field of the nitride ligand.
In 2020, Rudel, Kraus and co-workers described the first crystallographically authenticated complexes incorporating the linear NUN moiety, isoelectronic to the uranyl cation, prepared outside matrix conditions.65 Three closely related molecular uranium nitrides were prepared by reaction of uranium pentahalides UCl5 or UBr5 with anhydrous liquid NH3 (Scheme 11), instead of an azide (N3−) precursor, which is the most commonly utilized pathway to produce actinide nitrides. In the structures of the trinuclear complexes, [U3(μ-N)2(NH3)21]8+ (53), [U3Br(μ-N)2(NH3)20]7+ (54), and [U3Cl2(μ-N)2(NH3)19]6+ (55), the N–Ucenter–N angle is nearly linear and the U(VI)center is coordinated by five additional ligands (NH3, Cl− or Br−) in a pentagonal bipyramidal geometry, which is regularly found in complexes of uranyl (UO22+). Additionally, each nitride ligand binds a [U(NH3)8]4+ fragment, forming almost linear trinuclear complex cations.
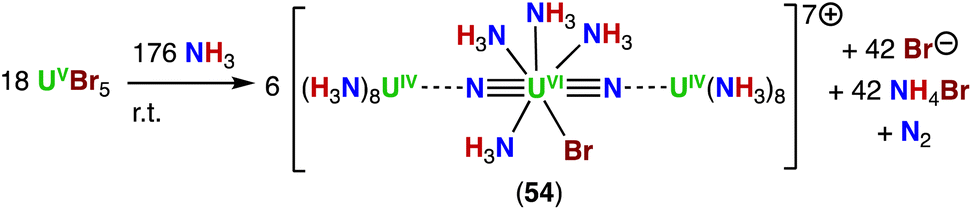 | ||
| Scheme 11 Synthesis of uranium-nitrides via ammonia (NH3) addition. | ||
The Ucenter–Nnitrido bond distances in the three compounds are similar, with an average value of 1.84(4) Å that is longer than those calculated at the CASPT2 level for [NUN] (1.73 Å),24 but close to the terminal UVIN triple bond of the uranium complex with the TrenTIPS supporting ligand reported by Liddle and co-workers (Section 4.2).44 The UIV–Nnitrido bond distances in complexes 53–55 (2.235(5) to 2.392(8) Å) are longer than those found in the single-bonded U(IV) nitride complex, [(UIVCp*(μ-I)2)3(μ-N)] (2.138(3) to 2.157(3) Å),67 consistent with less donation of the nitride ligand to the terminal U(IV) ions. Quantum chemical calculations, as well as Raman and infrared spectroscopy studies of the 14N/15N-complexes of [U3Br(μ-N)2(NH3)20]7+ (54), support the presence of two U–N triple bonds in the trans-(NUVIN) core.
The formation of the three U(VI) bis-nitride complexes was proposed to proceed via disproportionation of the U(V) precursor in NH3 to U(IV) and U(VI) species. The authors suggest that some NH3 is oxidized by such a U(VI) species to N2, while formation of the trinuclear U complexes 53–55 proceeds via deprotonation of ammine ligands. The resulting nitrido ligands stabilize the central U(VI) species, while the nitrido complexes are stabilized by the coordination of [U(NH3)8]4+ complexes.
3.3. Other methods
Li, Chen and co-workers reported the first actinide-nitride fullerene cluster, U2N@Ih(7)-C80 (56),99 which was produced by a modified Krätschmer–Huffman DC arc-discharge method.100 Graphite rods, packed with U3O8 and graphite powders, were vaporized in the arcing chamber under helium atmosphere with 1 torr NH3, and after work-up, the cluster was isolated and fully characterized by single X-ray crystallography and multiple other spectroscopic methods. The stabilization of this uranium-nitride cluster demonstrated that the combination of the unique properties of actinides and the special chemical environment inside the fullerene cage allows stabilization of a bimetallic uranium nitride moiety.The UNU encaged moiety exhibits an angle of 150.0(2)°, which is significantly bent when compared with other diuranium bridged mono-nitride compounds supported by chelating ligands, in which the U–N–U units adopt close to linear configurations. The bent angle was rationalized to have resulted from the steric effect of encapsulation in the C80 cage. The two U–N bond distances are inequivalent (2.058(3) Å and 1.943(3) Å), creating an unsymmetrical bonding. The shortest U-cage distances range from 2.448(4) to 2.543(5) Å. Although the U–Nnitride bond distances are shorter, the UNU bonding resemble that of the U(IV)/U(V) nitride complex 17, reported by the Mazzanti group, where the two different U–Nnitride bond lengths are the result of localized valences.76 Computational studies combined with X-ray absorption spectroscopy suggest two uranium ions with different oxidation states of U(IV) and U(V). Quantum-chemical investigation further revealed that f1/f2 population dominantly induces a distortion of the UNU moiety, which leads to the unsymmetrical structure. A comparative study of U2X@C80 (X = C, N and O) revealed that the U–X interaction in the UXU clusters can hardly be considered as classical multiple bonds, but is more like an anionic central ion Xn− with biased overlaps with the two metal ions, which decrease as the electronegativity of the ligand X increases.99
4. Terminal and Lewis acid-capped nitride actinide complexes
4.1. Seminal studies and putative terminal nitrides
The synthesis of terminal nitrides has been a long sought-after target in actinide chemistry because of the potential reactivity and fundamental relevance in the study of An–N multiple bonds. The most typical route to generate terminal nitride species involves the reaction of azide (N3−) anions with reducing metal complexes containing bulky ligands, in order to prevent the formation of bridging nitrides (Section 3), however, an alternative route involves the photolysis of terminally bound azides ligands.Seminal studies (2009) from the Cummins group led to the isolation of the first uranium nitridoborate complexes, [NBu4][UV((μ-N)B(C6F5)3)(N[tBu])(3,5-Me2C6H3))3] and the U(VI) analogue, that could also be viewed as borane-capped uranium nitrides.101 Related to this work, in 2020, Boreen, John Arnold and co-workers reported the reaction of [UIII(CpiPr4)2I] with NaN3, yielding an unusual U(III) azide-bridged tetrameric complex, [(CpiPr4)2UIII(μ-η1:η1-N3)]4 (57a), but the subsequent addition of a strong Lewis acid, B(C6F5)3, results in loss of N2 and trapping of a nitride fragment, forming the U(V) nitridoborate complex, [UV(CpiPr4)2(μ-N)B(C6F5)3] (57) (Scheme 12).102 The metrical parameters of 57 are comparable to the borane-capped complex, [NBu4][UV((μ-N)B(C6F5)3)(N[tBu])(3,5-Me2C6H3))3], reported by Cummins. This result highlights that not all U(III) complexes necessarily reduce the coordinated azide ligand, but reduction of the azide can be promoted by the presence of a strong Lewis acid.
 | ||
| Scheme 12 Synthesis of putative uranium-nitrides (a)–(d). | ||
The isolation of uncapped terminal nitrides became more attainable with the first report by Kiplinger and co-workers, in which they proposed a transient terminal nitride generated by photolysis of a U(IV) azide complex.45 This study demonstrated that nitrides are photochemically accessible, but suggested an inherent instability and highlighted the very high reactivity of terminal nitrides towards C–H activation of supporting ligands. This work inspired more recent studies, in which the aim has been to utilize more robust, bulky ligand frameworks, and properly tune reaction conditions. However, this task remains far from trivial, in which isolated terminal nitrides are still limited to primarily three ligand systems (Section 4.2).
Recently, Yadav, Fortier and co-workers reported the photolytic production of a putative terminal nitride that can access both intra- and intermolecular chemistry.47 Photolysis of the U(IV) azide complex, [UIV(LAr)(NImDipp)(N3)] (58a) ((LAr)2− = 2,2′-bis(2,6-diisopropylanilide)-p-terphenyl and (NImDipp)− = 1,3-bis(Dipp) imidazolin-2-iminato), generated the U(IV) amido complex, [UIV(N–LAr)(NImDipp)] (58). The formation of complex 58, was rationalized in terms of the intramolecular insertion of the terminal nitride group of the transient U(VI)–nitride, “[U(LAr)(N)(NImDipp)]” (58t), into a C–H bond of a pendant isopropyl group of the bis-anilide terphenyl ligand (Scheme 12). Furthermore, formation of the putative terminal nitride is supported by its intermolecular capture during the photolysis of complex 58a in the presence of an isocyanide (CNAr) or PMe3, to give the U(IV)-imine products, [UIV(LAr)(NCN(C6H3–Me2))(NImDipp)] (59) and [UIV(N,C–LAr*)(NPMe3)(NImDipp)] (60), respectively (Scheme 12).47 The formation of complex 60 is associated with a major structural rearrangement, which was suggested to arise from the steric congestion resulting from formation of the phosphinimide, which subsequently resulted in various unidentified chemical events that gives the complicated reaction mixture containing 60.
An alternative mechanism for the intramolecular C–H activation via a transient terminal nitride was reported by Mullane, Schelter and co-workers.103 The transient uranium terminal nitride moiety, generated from the reduction of the U(IV) bis-azide precursor, [UIV(PN)2(N3)2] (61a), with KC8 led to the parent imido complex, [UIV(PN)(iPr2P(C6H3Me)N–C6H2Me2CH2)(μ-NH)K(THF)3] (61); (PN)− = (N-(2-(diisopropylphosphino)-4-methylphenyl)-2,4,6-trimethylanilide), with a very short UNH bond length (1.997(2) Å). Calculated reaction energy profiles strongly suggested that the transient terminal U(IV) nitride, “[K][(PN)2UIV(N)]” (61t), engages in an unprecedented 1,2-addition of a C–H bond, instead of a reductive C–H insertion, in a distinct reaction pathway than previously observed for other uranium nitride complexes.44,45
4.2. Isolated terminal nitrides
In 2012, King, Liddle and co-workers described the first isolated molecular terminal uranium nitride complex synthetized under conventional experimental techniques.43 The 2e− oxidation of the monometallic U(III) complex, [U(TrenTIPS)], supported by the sterically demanding ligand TrenTIPS, with NaN3 allowed the isolation of the dimeric U(V)/U(V) complex, [Na2(UV(TrenTIPS)(μ-N))2] (62-Na), where both N3− ligands are stabilized by bridging Na cation interactions (Scheme 13a). Sequestration and encapsulation of the Na cations with 4.0 equiv. of 12-crown-4 (12C4) generated the first terminal U(V) nitride complex, [Na(12C4)2][UV(TrenTIPS)(N)] (63-Na), with a U–Nnitride bond length of 1.825(15) Å (Scheme 13a). In contrast, treatment of 62-Na with 15-crown ether (15C5), resulted in the U(V) nitride-capped complex, [Na(15C5)(UV(TrenTIPS)(μ-N)] (64-Na), where the nitride ligand is capped by the Na cation.44 Oxidation of 63-Na with I2 resulted in the clean formation of the first terminal U(VI) nitride complex, [UVI(TrenTIPS)(N)] (65) (Scheme 13a).44 Attempts to prepare the analogous U(IV) terminal nitride afforded the uranium terminal parent imido complex, [K(15C5)2][UIV(TrenTIPS)(NH)], which can also be viewed as a masked U(IV) terminal nitride. The high nucleophilic character of the nitride moiety prevented its isolation in the used reaction conditions.43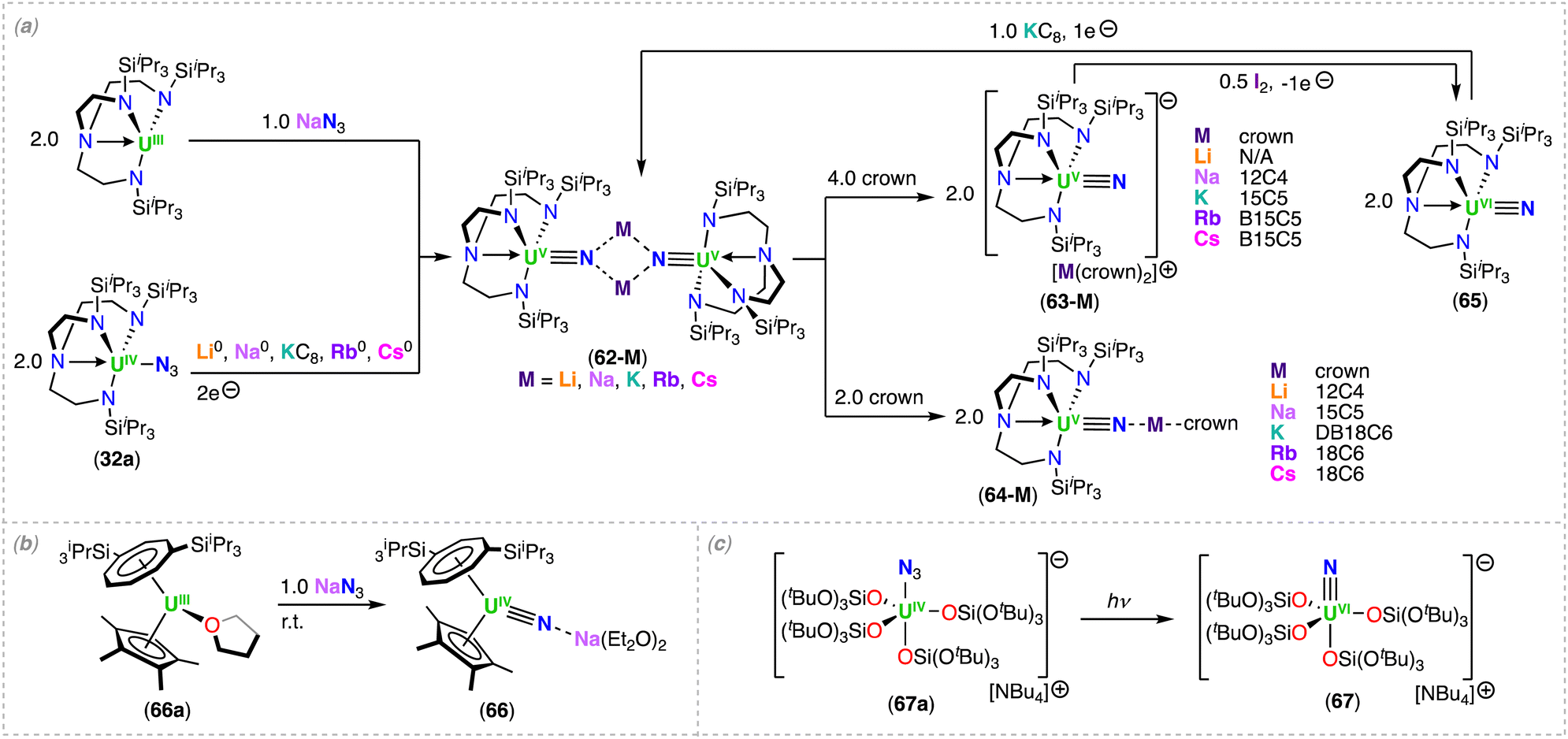 | ||
| Scheme 13 Synthesis of (a) alkali-capped and terminal uranium-nitrides supported by the triamidoamine (TRENTIPS) ligands; (b) organometallic alkali-capped uranium(V)-nitride; (c) terminal uranium(VI)-nitride supported by siloxide (–OSi(OtBu)3) ligands. | ||
In a later study, the authors prepared a library of alkali metal-capped dimeric U(V) nitride complexes, [M2(U(TrenTIPS)(μ-N))2] (62-M) (M = Li, K, Rb and Cs), by reduction of the terminal U(VI) nitride (65) with KC8, or by reduction of the U(IV) azide (32a) with Li, Na, KC8, Rb, or Cs (Scheme 13a).46,98 Carefully tuning the addition of a variety of crown ethers to 62-M, affording either the U(V) terminal- or capped-nitride complexes, [M(crown)2][UV(TrenTIPS)(N)] (63-M) or [M(crown)(UV(TrenTIPS)(μ-N))] (64-M) (M = Li, Na, K, Rb, Cs), respectively.98
The nitride complexes, 62-M (M = K, Rb, Cs), were also reported to be generated together with a U(III)-amide complex, [M][UIII(TrenTIPS)(NH2)] (M = K, Rb, Cs), by disproportionation of the U(IV)-imido complex, [(UIV(TrenTIPS)(μ-NH)(μ-M))2], in moderate heating.104 This reaction was facilitated by benzene, but not toluene, as benzene was found to engage in further redox chemistry with the [M][UIII(TrenTIPS)(NH2)] complex (M = K, Rb, Cs), generating [UIV(TrenTIPS)(NH2)], and concomitant reduced arene, [M][C6H6] (M = K, Rb, Cs). This process occurred with the K, Rb, and Cs cation complexes, but not with Li or Na, reflecting the stability of the corresponding alkali metal-arene byproducts.
The U–Nnitride bond distances in the terminal U(V) nitride complexes, 63-M, range from 1.801(3) to 1.825(15) Å, while the U–Nnitride bond length of the terminal U(VI) nitride complex 65 (1.799(7) Å) is statistically equivalent with these distances, and can be explained by the fact that a non-bonding 5f-electron is removed upon oxidation from U(V) to U(VI).
The structural assignment of the uranium nitrides 62-M to 65 was supported by electronic absorption spectroscopy, EPR, magnetometry, electronic structure calculations, and 15N-isotopic labelling.43,44,46,98 The uranium(V) nitrides behave as single-molecule magnets (SMMs); the first uranium SMM was reported in 2009,105 involving a U(III) complex, whereas two examples involving U(V) ions were reported in a later study106,107 The observed 14N/15N isotopomer shift for the U–Nnitride IR stretching frequencies range from 955 to 930 cm−1, 936 to 900 cm−1, and 914 to 883 cm−1, respectively for complexes, 62-Na, 64-Na, and 65, where the corresponding 15N-labelled complexes are consistent with the nitride terminal formulation.43,44 DFT calculations for the terminal uranium nitride complexes reveal similar composition for the UN bond in the two uranium oxidation states (+5 and +6). One σ and two π bonds are present in the U–N interaction, in agreement with a formal UN triple bond. Like in the uranyl (UO22+) case,107 the σ component is higher in energy than the π, whereas uranium–imido complexes follow the normal energetic ordering of π > σ. This energetic ordering is suggested to occur due to the terminal U–Nnitride bond manifesting an antibonding interaction between a σ-orientated nitrogen p-orbital overlapping with the toroidal 5f and 6d lobes on uranium. QTAIM analysis of the U(V) and U(VI) TrenTIPS terminal-nitrides and isovalent group 6 nitrides suggested that the level of covalency in UN bonds are comparable with those in group 6 transition-metal nitride complexes, or even greater.44 Moreover, the authors evaluated the covalency of the terminal U(VI)-nitride 15N-65 by 15N nuclear magnetic resonance spectroscopy and found an exceptional nitride chemical shift (δ 968.9 ppm referenced to CH3NO2) and chemical shift anisotropy.108 Recently, two U(VI) imido complexes were also characterized by 15N NMR spectroscopy and revealed lower downfield shifts (δ 302.4 and 256.7 ppm referenced to CH3NO2) as expected for UNR bonding.109 Computational benchmarking of the 15N NMR spectroscopy analysis of 15N-65 showed that the large chemical shift results from a large contribution of the σ paramagnetic component (almost negligible contribution of the spin orbital coupling component σSO) resulted from the UN triple bond σ and π components. In addition, this work enabled to build a predictive M-ligand isotropic 15N chemical shift-bond order correlations to guide future studies on understanding of An–N bonding.108
In 2016, Tsoureas, Cloke and co-workers reported a new example of the Na-capped U(V) terminal-nitride stabilized by (COT)2− and (Cp*)1− ligands. This work confirmed the importance of steric factors in determining the formation of terminal versus bridging nitride complexes. The sodium-capped U(V) nitride complex, [Na(UV(η8-C8H6(1,4-SiiPr3)2)(η5-Cp*))(μ-N)] (66), was obtained by treating the U(III) mixed sandwich complex, [UIII(η8-C8H6(1,4-SiiPr3)2)(η5-Cp*)] (66a), with NaN3 (Scheme 13b).91 The steric protection afforded by the bulky –SiiPr3 substituents on the COT ligand help in facilitating a 2e− oxidation of a single U(III) center. However, when the steric hindrance around the uranium center is reduced, for example, by the use of less bulky substituents, the U(IV)/U(IV) bridged nitride complex, [Na(THF)6][(UIV(η8-C8H6(1,4-SiMe3)2)(Cp*))2(μ-N)] (41), could be isolated instead (Scheme 9).91
Photolysis of alkali-metal azide (MN3) complexes has been an effective route for the synthesis and isolation of terminal metal nitrides with transition metals.110–114 However, this strategy, until recently, had not been successful to generate stable terminal actinide nitride complexes. Photolysis of U(IV) azides with bulky amide supporting ligands such as, TrenTIPS and bis(anilide) terphenyl (LAr)2− (2,2′-bis(2,6-diisopropylanilide)-p-terphenyl), also failed in producing terminal uranium-nitrides, and in its place, intramolecular insertion of the nitride in the ligand framework was observed.44,47
Recently, Barluzzi, Mazzanti and co-workers, by carefully tuning the reaction conditions and by using the sterically crowded monometallic U(IV) azide precursor, [NBu4][UIV(OSi(OtBu)3)4(N3)] (67a), were able to generate the first stable U(VI) terminal-nitride by photochemical synthesis (Scheme 13c).115 Complex [NBu4][UVI(OSi(OtBu)3)4(N)] (67) exhibits a U–Nnitride bond length of 1.769(2) Å, that is slightly shorter than the only other isolated U(VI) terminal-nitride complex, [UVI(TrenTPS)(N)] (65) (1.799(7) Å),44 and compares with the calculated U–N distance for the matrix-isolated nitride [UNF3] (1.759 Å).26
This work provided the second example of a terminal U(VI) nitride, and showed that these complexes can be prepared by photolysis, however, the steric contributions of the uranium-azide precursors, and the type of anionic azide utilized, should be carefully considered in order to maximize stability. Despite the stability of 67 under ambient conditions, the terminal nitride displays high reactivity towards electrophiles (Section 6.1).
4.3. Other methods
Charged UN diatomic species captured in fullerene cages, UN@Cs(6)-C82 (68) and UN@C2(5)-C82 (69), were synthesized by a modified Krätschmer–Huffman DC arc-discharge method100 using a mixture of U3O8 and graphite powders deposited into hollow graphite rods, and reacting in a arcing chamber under He and N2 atmosphere.116 X-ray crystallographic analysis of the isolated crystals showed short U–N bond lengths of 1.760(7) and 1.760(20) Å for complexes 68 and 69, respectively, which are very close to those calculated for gas phase molecules and clusters obtained only in low temperature matrix-isolation conditions, such as UN, NUF3 and NUNH.23,26,31 Quantum-chemical calculations were consistent with a f1 electronic structure for uranium and the presence of a UVN triple bond in the cluster fullerenes. This work establishes an approach to study fundamentally important actinide multiply bonded species.
5. Heterometallic actinide/transition metal nitride complexes
In 2022, Mazzanti, Agapie, and co-workers reported the first example of a heterometallic uranium-transition metal nitride complex, [Na(U(OSi(OtBu)3(μ-N)(MoP2))] (70) (P2 = terphenyl diphosphine ligand), generated from partial N-transfer to the U(III) precursor, [UIII(OSi(OtBu)3)3(THF)2], from the Na-capped molybdenum(II) nitride, [Na(N)MoIIP2] (70a) (Scheme 14a).117 The solid-state molecular structure of 70 displays a U–Mo heterometallic complex bridged by a N3− ligand, where the U and Mo centers are in close proximity with a U–Mo distance of 3.1032(2), which was slightly shorter than the distances found in other heterometallic U–Mo complexes bridged by phosphinoamide ligands (3.1682(4), 3.159(2) Å).118 The short U–Mo bond distance highlights the ability of the bridging nitride and bridging arene of the P2 ligand in 70 to promote metal–metal interactions.119,120 The U–Nnitride bond distance is very short (1.856(2) Å), indicative of a multiple UN bond, and is shorter than previously reported U(V) nitride complexes (range: 2.022(5)-2.2101(6) Å),48,52,71 and longer than U(VI) nitrides (1.769(2) and 1.799(7) Å),44,115 however, the distances are most consistent with Na-capped U(V) terminal nitrides (1.883(4) and 1.835(5) Å).43,91 Structural, EPR, and computational studies indicated that N-transfer is accompanied by a 2e− transfer from the uranium to molybdenum, resulting in a UVN triple bond and a Mo0–N single bonding profile.
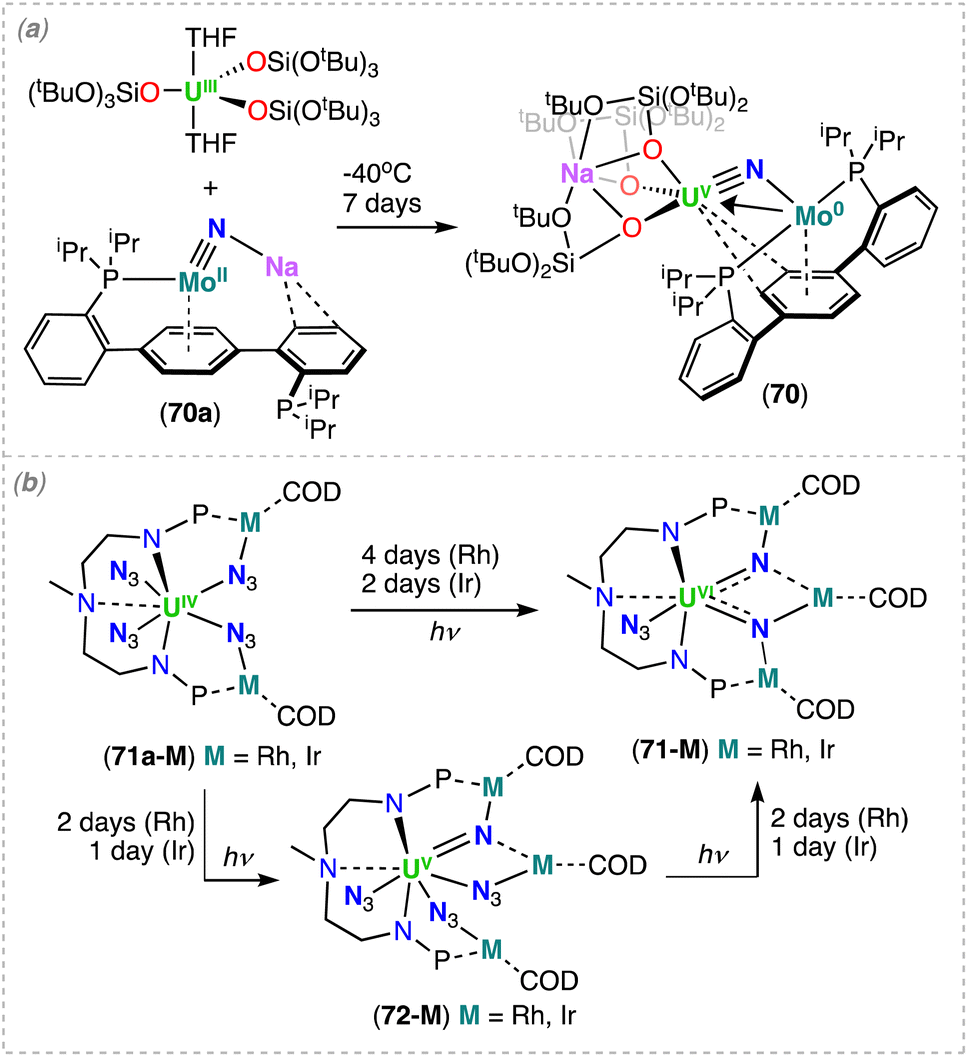 | ||
| Scheme 14 Synthesis of heterometallic uranium-nitrides with d-block metals (a) molybdenum (Mo) and (b) rhodium (Rh) and iridium (Ir). | ||
In the same year, Xin, Zhu and co-workers presented the isolation of two heterometallic uranium-transition metal nitrides. The transition metal-stabilized U(VI) nitrides, [{UVI{N(CH3)(CH2CH2NPiPr2)2}(N3){[(μ-N)M(COD)]2[M(COD)]}}] (71-M; M = Rh, Ir), were generated by photolysis of the U(IV)-azide precursors, [{UIV{N(CH3)(CH2CH2NPiPr2)2}(N3)2[(μ-N3)M(COD)]2}] (71a-M; M = Rh, Ir). The U(V) nitride intermediates, [{UV{N(CH3)(CH2CH2NPiPr2)2}(N3)[(μ-N3)M(COD)]2[(μ-N)M(COD)]}] (72-M; M = Rh, Ir), could also be successfully isolated by carefully controlling the irradiation timeframe (Scheme 14b).121
The solid-state molecular structures were determined for complexes 72-M and 71-M. The uranium(V) centers in 72-M are coordinated by three azides and a nitride ligand, where the nitride moiety is bound to one U and two transition-metal atoms (M = Rh, Ir). The U–Nnitride distances are 1.963(5) Å and 2.011(4) Å in 72-Rh and 72-Ir, respectively, which are shorter than the bond distances of the U(V)/U(V) bridged nitride complex, [(UV(N[tBu](3,5-Me2C6H3)3)2)(μ-N)][B(ArF)4] (2.0470(3), 2.0511(3) Å),41 but similar to the U(V) Na- or borane-capped nitride complexes, 62-Na (1.883(4) Å) and [NBu4][UV{(μ-N)B(C6F5)3}(N(tBu)(3,5-Me2C6H3))3] (1.916(4) Å), respectively.43,101 Complexes 71-M display two bridged nitride moieties, which are capped by three Rh or Ir ions. The bond lengths of the U(VI)–Nnitride in 71-M (1.975(5) (Rh) and 2.005(6) Å (Ir)) are comparable with the U(V)–Nnitride bond lengths found in 72-M, however, they are longer than those in the U(VI)–Nimido bond in a trans-imido uranium complex (1.840(4)–1.866(2) Å)122 or in the U(VI)–Nnitride bond in a uranyl analog [OUN]+ (1.818(9) Å),40 altogether indicating UN multiple bonds. The U(V) and U(VI) nitride complexes showed excellent stabilities, which was attributed to the nitrides being stabilized by capped-transition metals (Rh or Ir).
6. Reactivity of actinide nitride complexes
Reactivity studies of terminal- and bridging-nitrides provide important information on the nature of the An–N bond in these complexes and in related actinide nitride materials. Moreover, promoting the reactivity of uranium nitrides towards CO, CO2, or H2 allows the possibility of establishing stoichiometric and catalytic N–C and N–H bond formation from U–N, with the long-term objective of using N2 as a nitride source, thus allowing N2 functionalization by H2, CO, or CO2 in ambient conditions.6.1. Alkali- and transition-metal capped, and terminal actinide-nitride complexes
In 2012, the group of Liddle reported the first isolable U(V) terminal nitride (Section 4.2), isolated by addition of 12-crown-4 (12C4) to the Na-capped U(V)-nitride complex, [Na2(UV(TrenTIPS)(μ-N))2] (62-Na), and were able to demonstrate the reactive nature of these complexes, with electrophiles such as acid (H+) and Me3SiCl. Addition of Me3SiCl to complexes 63-Na43 and 62-Na,44 yielded the U(V) imide complex, [UV(NSiMe3)(TrenTIPS)] (73). They were able to further confirm the imide formation via an alternative synthetic route, by reacting the U(III) complex, [UIII(TrenTIPS)], with Me3SiN3.44 They were also able to further confirm the presence of a nitride moiety in 63-Na by addition of excess water (15 equiv.) in the presence of cobaltocene (Cp2Co) to afford NH3 in 41% yield (Scheme 15).43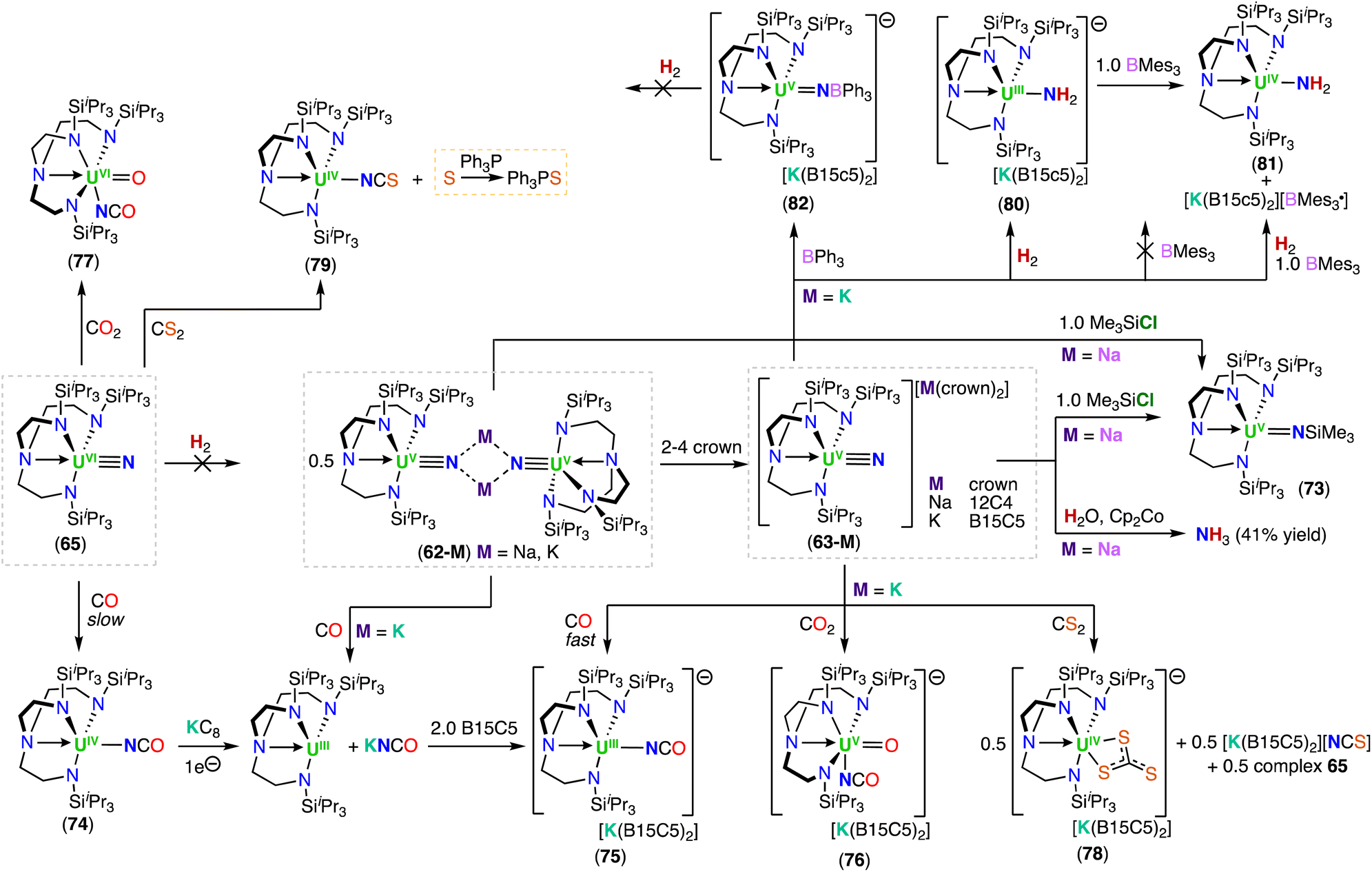 | ||
| Scheme 15 Reactivity of alkali-capped and terminal uranium-nitrides supported by the triamidoamine (TRENTIPS) ligands. | ||
In 2014, the same group reported the reductive carbonylation to U–NCO isocyanates upon addition of CO to a series of U(VI)- and U(V)-nitride complexes (Scheme 15).46 They found the U(VI)-nitride complex 65 reacts with CO and undergoes a two-electron reduction, yielding the U(IV) cyanate complex, [UIV(TrenTIPS)(NCO)] (74). Reduction of 74 with KC8 resulted in cyanate dissociation to give the U(III) complex, [UIII(TrenTIPS)], and KNCO, or cyanate association when the K cation is sequestered with B15C5 forming complex, [K(B15C5)2][UIII(TrenTIPS)(NCO)] (75). Complexes 75, and [UIII(TrenTIPS)]/KNCO, were also prepared from CO and the U(V)-nitrides, [(UV(TrenTIPS)(N)K)2] (62-K) and [K(B15C5)2][UV(TrenTIPS)(N)] (63-K), respectively. Notably, 63-K reacts with CO much faster than 65. This unprecedented f-block reactivity was modelled theoretically, revealing nucleophilic attack of the π* orbital of CO by the nitride, with activation energy barriers of 24.7 and 11.3 kcal mol−1 for U(VI) and U(V), respectively. They were also able to devise a two-step synthetic cycle for the conversion of azide to U(V)-nitride to cyanate using the U(III) complex, [UIII(TrenTIPS)], NaN3 and CO.46
In a later study, Cleaves, Liddle and co-workers found that the U(V)- and U(VI)-terminal nitride complexes, 63-K and 65, respectively, were also reactive towards heteroallenes, such as CO2 and CS2, resulting in well-defined CE (E = O, S) cleavage followed by zero-, one-, and two-electron redox events.123 The U(V)-nitride complex 63-K reacts with CO2 to give [UV(TrenTIPS)(O)(NCO)][K(B15C5)2] (76), whereas the U(VI)-nitride complex 65 reacts with CO2 to give isolable, [UVI(TrenTIPS)(O)(NCO)] (77), which rapidly decomposes to yield the previously reported terminal U(V) oxo, [U(TrenTIPS)(O)],124 with the proposed concomitant formation of N2 and CO. In contrast, 63-K reacts with CS2 to give [UIV(TrenTIPS)(κ2-CS3)][K(B15C5)2] (78), 65, and [K(B15C5)2][NCS], whereas complex 65 reacts with CS2 yielding, [UIV(TrenTIPS)(NCS)] (79) and “S”, with the latter trapped as Ph3PS. Calculated reaction profiles reveal outer-sphere reactivity for U(V) and inner sphere mechanisms for U(VI).123
Despite the wide divergence of products, the initial activation of CE2 (E = O, S) follows mechanistically related pathways, while uranium oxidation state, type of chalcogen, and NCE products govern the subsequent divergent redox reactions that include common one-electron reactions and less-common two-electron redox events.123 In 2020, Chatelain, Liddle and co-workers expanded upon this work and reported the hydrogenolysis of the terminal U(V)-nitride complex 63-K under mild conditions (Scheme 15).53 Notably, this is only the fifth reported example of H2 splitting via a uranium nitride, with the four other examples involving H2 splitting by the U(IV)/U(IV) mono-nitride complexes, 6-Cs49 and 14,76 and the U(V)/U(V) and U(V)/(VI) bis-nitride complexes, 9 (ref. 52) and 11 (ref. 75) (Sections 6.2.1 and 6.2.2). The hydrogenation of metal-nitrides is highly relevant towards understanding the mechanism for ammonia production through the industrial Haber–Bosch process, however, studies are limited by the paucity of metal-nitrides capable of effecting H2 splitting in ambient conditions.
In the hydrogenolysis of 63-K, two divergent mechanisms were found; (i) direct 1,2-dihydrogen addition of H2 across the U(V)-nitride bond in 63-K, coupled with H-atom migration to give a U(III)-amide complex, [K(B15C5)2][UIII(TrenTIPS)(NH2)] (80), or (ii) a Frustrated Lewis Pair (FLP) pathway when hydrogenation is performed in the presence of trimesitylborane (BMes3), producing a U(IV)-amide complex, [UIV(TrenTIPS)(NH2)] (81), and a BMes3 radical anion, [K(B15C5)2][BMes3] (Scheme 15).53 The isostructural U(VI)-nitride complex 65 is inert to hydrogenolysis, suggesting the 5f1 electron of the U(V)-nitride in 63-K is not purely non-bonding. Moreover, the authors found that the nature of the Lewis acid plays an important role in the hydrogenolysis mediated by a FLP. Notably, addition of BPh3 to complex 63-K, results in the formation of the uranium(V)-imido-borate complex, [K(B15C5)2][UV(TrenTIPS)(NBPh3)] (82), which does not react with H2. In contrast, 63-K does not form an adduct with BMes3, however, BMes3 promotes the reactivity of 63-K with H2. The computed mechanism indicated that when 63-K is reacted with H2 in the presence of BMes3, the [UV(TrenTIPS)(N)]− and BMes3 components constitute the FLP complex, which reacts with H2 and facilitates its cleavage. In the absence of BMes3, the formation of 81 is slower and proceeds via the direct hydrogenation pathway previously reported for the hydrogenation of the U(IV)/U(IV) bridging nitride, [Cs(UIV(OSi(OtBu)3)3)2(μ-N)] (6-Cs).49 Thus, this work establishes a unique example of actinide and FLP chemistries combined. Furthermore, a synthetic cycle for ammonia (NH3) synthesis was demonstrated, where treatment of the U(IV) complex, [UIV(TrenTIPS)Cl], with NaN3, KC8, and 2.0 equiv. of B15C5 produces the terminal U(V)-nitride 63-K, which in turn reacts with H2 and BMes3 to give 81. Treatment of 81 with Me3SiCl, followed by work-up and acidification steps, produces ammonia.53
In the same year, Barluzzi, Mazzanti and co-workers reported the synthesis of the first stable terminal U(VI)-nitride complex, [NBu4][UVI(OSi(OtBu)3)4(N)] (67), generated by azide photolysis.115 They found complex 67 is stable, but readily reacts with electrophiles such as acid (H+) sources and carbon monoxide (CO). They found that the addition of HCl or H2O/decamethylcobaltocene (Cp2*Co) to complex 67 yields NH3 in 100% and 56% yields, respectively. Additionally, reaction of 67 with 1 atm of CO yields the reductive carbonylation product, [NBu4][UIV(OSi(OtBu)3)4(NCO)] (83) (Scheme 16a), which is an analogous reactivity to that previously observed for the terminal U(VI) nitride complex, 65. Additionally, reaction of 1.0 equiv. of Me3SiI to complex 83 led to the formation of [U(OSi(OtBu)3)4], as the main uranium-containing species, and Me3SiNCO. Based on the formation of [U(OSi(OtBu)3)4], the authors were able to develop a synthetic cycle for the formation of Me3SiNCO from NBu4N3 and CO.115 In 2022, Mazzanti, Agapie and co-workers found that preliminary reactivity studies of the heterometallic U(V)–Mo(0)-bridged nitride complex 70 with 13CO (1–10 equiv.) resulted in the reductive carbonylation of the nitride, leading to the formation of NaN13CO (10–50% yield) and the previously reported Mo(0) carbonyl complexes, [P2Mo0(13CO)] or [P2Mo0(13CO)3] (in low yield), as well as multiple unidentified diamagnetic species (Scheme 16b).117 The uranium species formed in the reaction could not be identified, but the formation of NaN13CO was unambiguously demonstrated by 13C NMR spectroscopy of the reaction mixture in D2O. The reductive carbonylation of the U(V) nitride most likely proceeds with release of the Mo(0) complex, but reactivity with complex 70 was not ruled out.
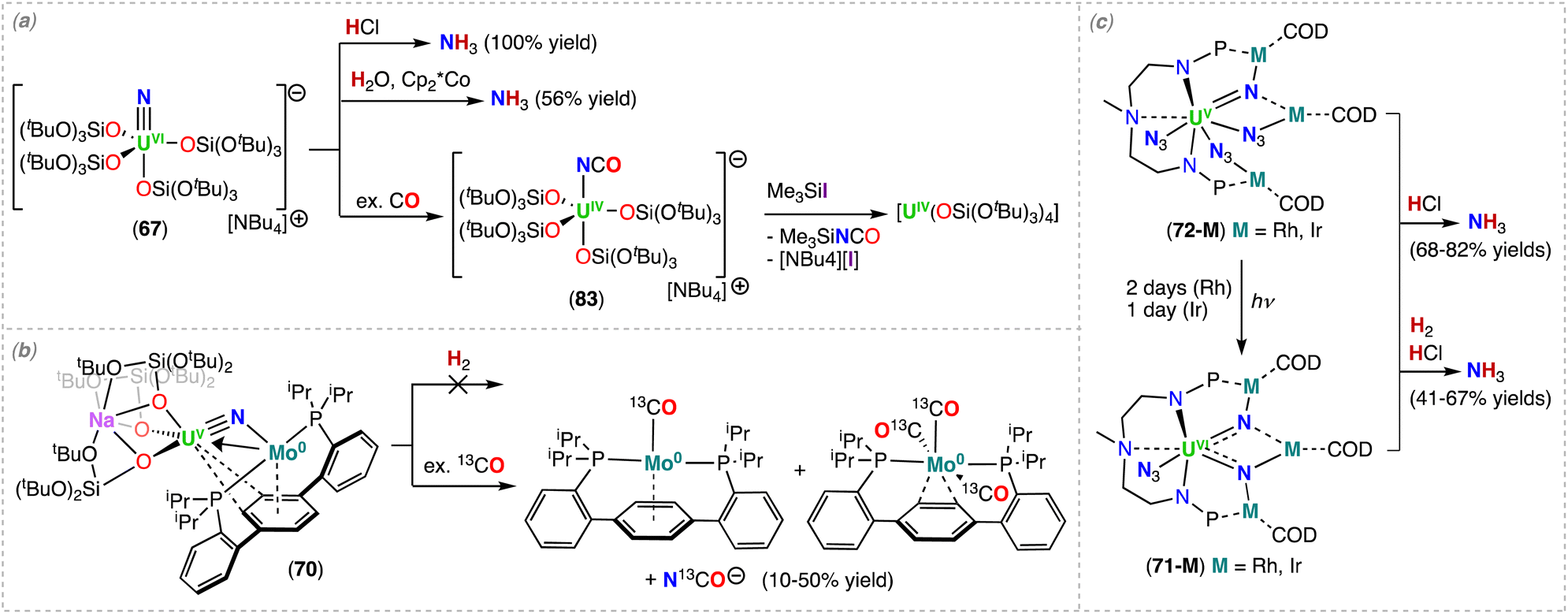 | ||
| Scheme 16 Reactivity of terminal and transition metal-capped uranium-nitrides supported by various ligands. | ||
In the same year, Xin, Zhu and co-workers reported the reactivity of the transition metal-capped U(V) and U(VI) nitride complexes (72-M), and (71-M) (M = Rh, Ir), towards acid (H+) and H2.121 They found that addition of excess PyHCl to the heterometallic uranium nitride complexes, 71-M and 72-M, resulted in NH4Cl in 68–82% yields, respectively (Scheme 16c). Moreover, addition of 1 atm H2 to complexes, 71-M and 72-M, led to a color change in the reaction mixture, however, attempts to isolate single crystals of the products were unsuccessful. The subsequent addition of excess PyHCl to the reaction mixtures led to the formation of NH4Cl in 41–67% yields. In comparison, no NH4Cl was observed from the reactions of U(IV) azide complexes (71a-M) with excess PyHCl. These results suggested that the nitride moieties in complexes 71-M and 72-M are the sources of the NH4Cl.121
6.2. Actinide bridged-nitride complexes
The study of the reactivity of dinuclear bridging uranium nitrides have so far been limited to three ligand systems, namely, complexes of –NtBuAr (Ar = 3,5-Me2C6H3), –OSi(OtBu)3, and –N(SiMe3)2 ancillary ligands.6.2.1.1. CO reactivity. In 2010, Cummins and co-workers first demonstrated the reactivity of the U(V)/U(V) nitride complex, [(UV(N[tBu](3,5-Me2C6H3)3)2)(μ-N)][B(ArF)4], with NaCN, in which cyanide acts as a 2e− reducing agent with insertion into the electrophilic nitride moiety to yield a [UIV–N
CN–UIV] species. The same complex was found not to react with carbon monoxide (CO).41
Conversely, in a later study, Barluzzi, Mazzanti and co-workers found that the nitrides in the U(V)/U(V) bis-nitride complex, [K2([UV(OSi(OtBu)3)3]2(μ-N)2)] (9), bearing –OSi(OtBu)3 ligands, are highly nucleophilic and react with a variety of small molecules (top, Scheme 17).52
 | ||
| Scheme 17 Reactivity of U(V) and U(VI)-bridged nitrides supported by siloxide (–OSi(OtBu)3) ligands. | ||
Addition of 3.0 equiv. CO to 9 afforded the bridging cyanide–cyanate–oxo complex, [K2([UIV(OSi(OtBu)3)3]2(μ-NC)(μ-O)(μ-NCO))] (84), resulting from different reactivities of the two bridging nitride moieties. Notably, one nitride promotes the cleavage of CO affording a CN− and an oxo ligand, similar to the C–O cleavage observed by the bridging U(IV) mono-nitride complex (6-Cs; Section 6.2.2). Alternatively, the second nitride undergoes the reductive carbonylation to cyanate (NCO−), altogether with the concomitant 2e− reduction of the two U(V) centers to U(IV).52 A similar 2e− reductive carbonylation had been observed for the monometallic terminal U(V) (63-K) and U(VI) (65) nitrides, to yield U(III)–NCO (75) and U(IV)–NCO (74) complexes, respectively.
In a later study, the reactivity of the analogous U(VI)/U(VI) and U(VI)/U(V) bis-nitride derivatives (13), and (11), were studied and resulted in differences in reactivity when compared to 9.75 For example, when the U(VI)/U(VI) complex (13) was reacted with 1 atm of CO, there was no evidence of reactivity, even when left for long reaction times. Alternatively, the U(VI)/U(V) bis-nitride complex (11) promoted the reductive carbonylation of two CO molecules, forming N13CO− as the only 13C-containing species when hydrolyzed and analyzed by 13C NMR spectroscopy (top, Scheme 17). The difference in reactivity for the U(V)/U(V) versus the U(VI)/U(V) bis-nitride complexes was suggested to stem from a decreased nucleophilicity of the second nitride moiety in 11, due to an increase in oxidation state of one uranium, favoring reductive carbonylation opposed to nucleophilic C–O cleavage in 9.75 Similar to the reactivity of 9, the U(VI)/U(IV)3 tetranitride-cluster (1) reported by Jori, Mazzanti and co-workers, also performs the cleavage and reductive carbonylation of CO to yield CN− and NCO−, respectively.64 From the reaction with stoichiometric quantites of CO, two additional U(IV)/U(V) clusters were obtained, namely, [K4((OSi(OtBu)3)2UIV)2((OSi(OtBu)3)2UV)2(μ4-N)2(μ-O)2(μ3-O)2] (85), and [K3((OSi(OtBu)3)2UIV)2((OSi(OtBu)3)2UV)2(μ3-N)(μ-O)(μ3-O)4] (86), demonstrating that the bridging nitride moieties in 1 could be replaced by oxo-ligands without disrupting the primary structure, and leads only to valence redistribution (bottom, Scheme 17). In the same study, characterization of the putative U(V) oxo bis-nitride, (1-(N)2; Section 2), an intermediate formed in the synthesis of the tetranitride cluster 1, was carried out by reaction with CO.
Addition of 3.0 equiv. CO to 1-(N)2 resulted in an analogous CO cleavage and reductive carbonylation pathway when compared to complex 9, however, due to the additional oxo-ligand in 1-(N)2, this resulted in the release of KCN and KNCO upon formation of the bis-oxo complex, [K2([UIV(OSi(OtBu)3)3]2(μ-O)2)] (87-K).64 Reactivity and structural confirmation of the putative oxo bis-nitride (1-(N)2) was later supported by isolation of the bridging bis-nitride, terminal oxo complex, [Cs3(UV(OSi(OtBu)3)3)(μ-N)2(UV(OSi(OtBu)3)2(κ-O))][CsOSi(OtBu)3] (3-(N)2; Section 2), where reaction of 3-(N)2 with 3.0 equiv. CO led to isolation of the bis-oxo U(IV) complex, [Cs2([UIV(OSi(OtBu)3)3]2(μ-O)2)] (87-Cs), and release of CN− and NCO−, analogous to 1-(N)2.64
6.2.1.2. CO2 and CS2 reactivity. Complex 9 also reacts with CO2 or CS2 leading to the redox-neutral, C–O or C–S bond cleavage with concomitant N–C bond formation of one nitride ligand. Addition of 1.0 equiv. CO2 or CS2 resulted in isolation of the nitride-oxo-cyanate complex, [K2([UV(OSi(OtBu)3)3]2(μ-N)(μ-O)(μ-NCO))] (88), and the nitride–thiocyanate–sulfide complex, [K2([UV(OSi(OtBu)3)3]2(μ-N)(μ-S)(μ-NCS))] (89), respectively (top, Scheme 17).52 Formation of the oxo-NCO ligands from CO2, and NCS− from CS2 cleavage, has also been observed for the respective U(V) and U(VI) terminal nitrides, 63-K and 65 (Section 6.1), as well as the U(IV) bridging mono-nitride, CsUIV
NUIV (6-Cs) (Section 6.2.2). In these previous examples with CS2, however, formation of a bridging sulfide, similar to 89, had never been observed. Instead, the terminal U(V) nitride (63-K) and the U(IV)/U(IV) bridging mono-nitride (6-Cs) complexes form a putative uranium-sulfide which then reacts with CS2 to yield a trithiocarbonate ligand. The formation of complex 89 was rationalized by the increased sterics in the core of the U(V)/U(V) bis-nitride complex 9, preventing formation of the trithiocarbonate ligand.
It is important to note that the analogous U(VI)/U(VI) bis-nitride (13) also reacted immediately with CO2, but the reaction products could not be identified.75 The formation of thiocyanate (N13CS−), cyanate (N13CO−), and cyanide (13CN−) were all confirmed by structural and 13C NMR spectroscopy analysis of the products after reactions with 13CS2, 13CO2, and 13CO.
6.2.1.3. H2 reactivity. Complex 9 also effects the oxidative cleavage of H2 to yield the U(IV) bis-imido complex, [K2[UIV(OSi(OtBu)3)3(μ-NH)]2] (90).52 Similarly, the analogous U(IV)/U(V) complex (11) also promotes H2 cleavage, but instead yields an equimolar amount of the U(IV) bis-imido and bis–amido complexes, 90 and [K2([UIV(OSi(OtBu)3)3]2(μ-NH)2(μ-THF)] (91), respectively (top, Scheme 17).75 The U(IV)/U(IV) complex (90) does not react further with H2; therefore, it was proposed that the reaction proceeds by first an oxidative cleavage of 1.0 equiv. of H2, affording transient high-valent and ligand-scrambling bis-μ-NH products, which further react with an additional 1.0 equiv. of H2, affording a mixture of complexes 90 and 91. Complex 91 could be independently synthesized by reacting 2.0 equiv. of HNEt3BPh4 to complex 90. Alternatively, the U(VI)/U(VI) bis-nitride (13) does not effect H2 cleavage.75 Similar oxidative cleavage of H2 was observed for the monometallic U(V) terminal nitride (63-K), whereas the U(VI) derivative (65) did not react, similar to 13.
6.2.1.4. Acid (H+) reactivity. All nitride complexes described in this section, 9, 11, 13, 1, 1-(N)2, and 3-(N)2, were found to react with strong acid (HCl) to afford NH4Cl typically in high or quantitative yields.
6.2.2.1. CO reactivity. The –OSi(OtBu)3 complexes, CsUIV
NUIV (6-Cs) and [NBu4][UIVNUIV] (14),76 show immediate reactivity with CO resulting in C–O cleavage. Complex 6-Cs cleanly reacts with CO in toluene to afford the bridging oxo/cyanide complex [Cs(UIV(OSi(OtBu)3)3)2(μ-CN)(μ-O)] (92) (Scheme 18a), in which the resultant cyanide ligand can be alkylated by Me3SiI or MeOTf to afford Me3SiCN or MeCN, respectively.50 Initial computational studies indicated the importance of cooperative binding of the CO by the multimetallic U–N–Cs moiety. However, the reaction of CO with complex 14 in both toluene and THF solutions also led to the formation of CN− (Scheme 18b), but in toluene, resulted in more products compared to 6-Cs, indicating ligand scrambling.76 The differences in reactivity of 6-Cs and 14 with CO in toluene could be explained by an increased stability the inner-sphere Cs cation provides to 92, by binding four –OSi(OtBu)3 ligands and the bridging oxo. For the CO reaction with 14, crystals suitable for XRD studies could not be isolated. However, when the reaction residue was hydrolyzed with D2O (pD = 12), CN− was observed as the only product, similar to 6-Cs. Likewise, the Th(IV)/Th(IV) nitride-azide complexes, M3ThNTh (M = K (38), Cs (39)), containing –OSi(OtBu)3 ligands, were also found to cleave CO to CN−, however, in lower yields (30 and 22% yield, respectively). The reactivity of the nitride moiety in 38 toward CO was increased by removal of two azide ligands and two K cations, in the form of [K(2.2.2-cryptand)][N3]. Formation of the putative mono-nitride complex, “[(KTh2(OSi(OtBu)3)6)(μ-N))]” (38t) and reaction with CO led to an increase in reactivity, forming CN− in 70% yield (Scheme 18c).87
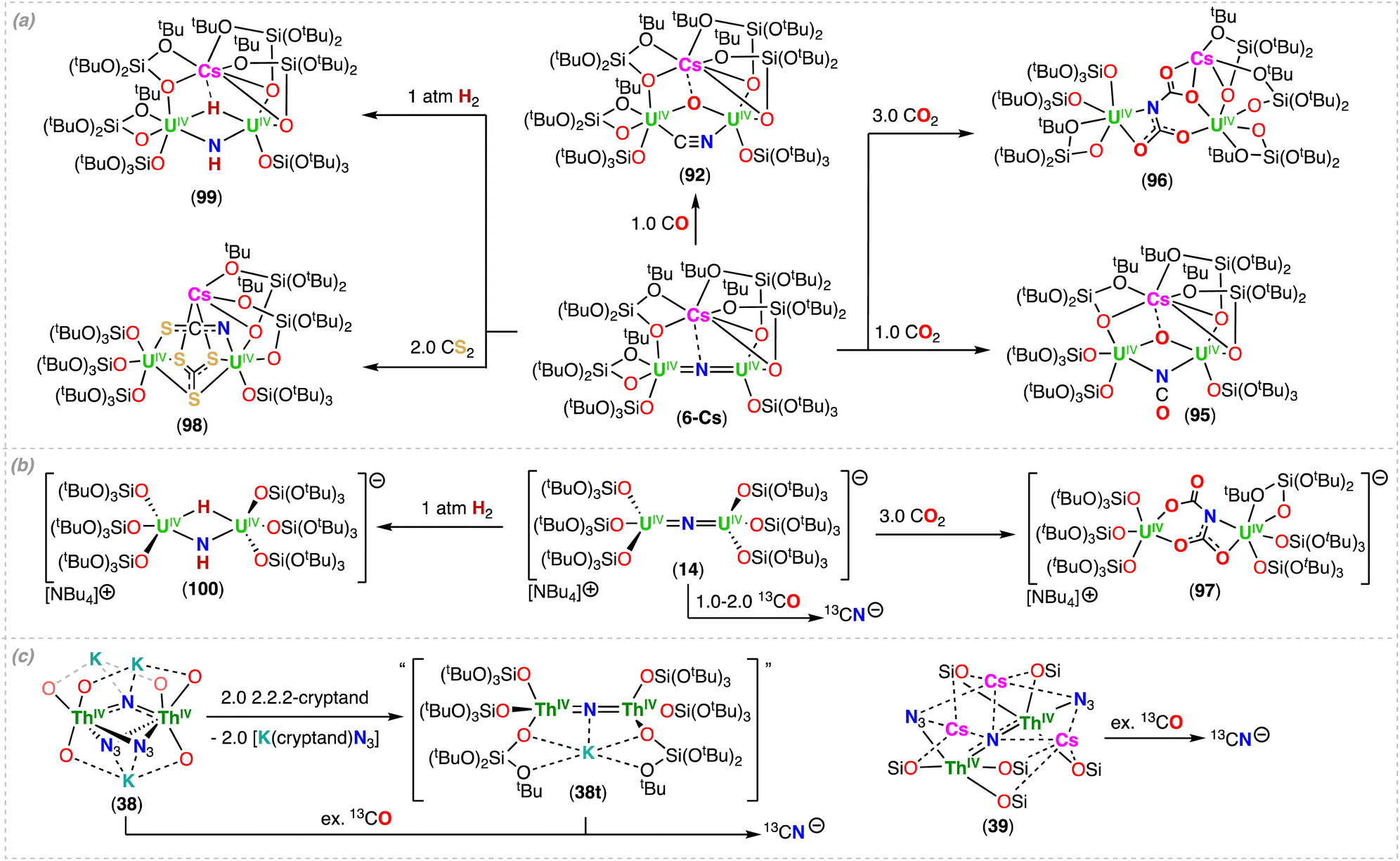 | ||
| Scheme 18 Reactivity of U(IV) and Th(IV)-bridged nitrides supported by siloxide (–OSi(OtBu)3) ligands. | ||
The analogous U(IV)/U(IV) nitride bridged complex, [NBu4][UIVNUIV] (15), supported by –N(SiMe3)2 ligands, was reacted with 1.0 atm of CO, resulting in the formation of decomposition products not derived from C–O bond cleavage. Additionally, the analogous U(IV)/U(V) all-N(SiMe3)2 complex (17) showed no reactivity upon addition of 1.0 atm of CO.76 However, when one –N(SiMe3)2 was replaced with one –OSi(OtBu)3 ligand to generate the mixed-ligand complex, [Na(DME)3][UIVNUIV] (22), the reactivity with 1.0–2.0 equiv. of CO was observed, but was significantly slower than the all –OSi(OtBu)3 complexes, 6-Cs and 14. Single crystals could not be isolated from the reaction mixture, however, CN− was identified as the only product after hydrolysis and analysis by 13C NMR spectroscopy (Scheme 19).76 To further investigate if the U(IV)/U(IV) nitride–N(SiMe3)2 complexes could react with CO, specifically in different oxidation states or in the presence of other ligands, the cyclometallate nitride, first reported by Fortier, Hayton and co-workers, [Na(DME)2(TMEDA)][UIVNUIV] (16),40 and the cationic nitride complex, [UIVNUIV][BPh4] (19),70 were also reacted with CO.76,125 Addition of 1.0–2.0 equiv. of CO to 16 and 19 resulted in their complete consumption, and upon hydrolysis of the reaction residues, similar to 22, resulted in CN−. From the reaction mixture obtained from reacting 16 with CO, several species were identified and characterized by XRD studies, namely the trimeric bis-N(SiMe3)2 bridging oxo complex, [UIV(N(SiMe3)2(μ-O))]3 (93), and a species in which CO inserted into the uranium–methylene bond, [((N(SiMe3)2)2UIV(μ-O))(UIV(μ-κ2-O,N–OC(C)SiMe2NSiMe3)(N(SiMe3)2)2)] (94) (Scheme 19).76 This type of CO insertion had been observed previously in mononuclear U(IV) metallacycles, but this demonstrated an extension to uranium-nitride complexes.126–128
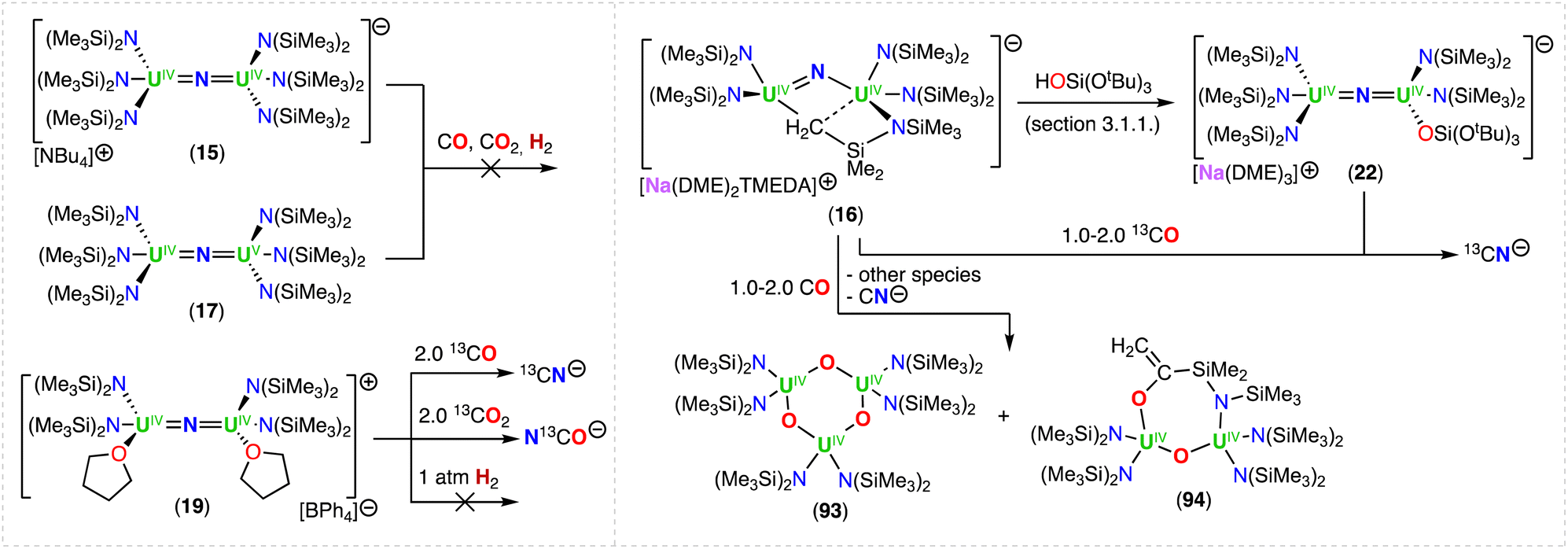 | ||
| Scheme 19 Reactivity of U(IV)-bridged nitrides supported by amide (–N(SiMe3)2) and heteroleptic analogues. | ||
6.2.2.2. CO2 and CS2 reactivity. Addition of 1.0 or 3.0 equiv. of CO2 to 6-Cs leads to differences in reactivity, with the formation of the bridging oxo/cyanate complex, [Cs(U(OSi(OtBu)3)3)2(μ-NCO)(μ-O)] (95), and the unprecedented electrophilic addition of 2 CO2 molecules affording the bis-carbamate complex, [Cs(U(OSi(OtBu)3)3)2(μ-NC2O4)] (96), respectively (Scheme 18a).129 For complex 14, addition of 3.0 equiv. of CO2 under similar reaction conditions led to the formation of the analogous bis-carbamate complex, [NBu4][(UIV(OSi(OtBu)3)3)2(μ-NC2O4)] (97) (Scheme 18b).76 Complexes 96 and 97 display resonances for the bis-carbamate (–N13C2O4) moiety at −134.1 and 141.5 ppm in the 13C NMR spectra, respectively. Hydrolysis of the reaction residue was not carried out for 96, instead, addition of D2O (pD = 12) to 97 and analysis by 13C NMR spectroscopy displayed only D13CO3−.
CS2 reactivity was also investigated, but only for complex 6-Cs, in which C–S bond cleavage was observed upon 2.0 equiv. of CS2, resulting in the formation of the thiocyanate/trithiocarbonate complex, [Cs(UIV(OSi(OtBu)3)3)2(μ-NCS)(μ-CS3)] (98) (Scheme 18a).129
The CO2 reactivity of the all-N(SiMe3)2 complexes (15), and (17) were also investigated, but it was found that exposure to 1 atm of CO2 led to no changes in the 1H NMR spectra, indicating no reactivity. In contrast, the mixed-ligand complexes, 22 and 19, and the cyclometallate, 16,40,76 all react with CO2, but produce complicated mixtures of species. Attempts to obtain single crystals of the products were unsuccessful, however after hydrolysis of the reaction residues with pD = 12 D2O after addition of 13CO2, the formation of N13CO− as the only product generated by the nitride moiety could be detected by 13C NMR spectroscopy (Scheme 19), where evidence of a dicarbamate was not apparent for any system. The formation of cyanate indicates that the reactions of the –N(SiMe3)2 complexes and excess CO2 proceed only via a N–C bond formation and deoxygenation process. Similar reactivity had been observed in 6-Cs129 with stoichiometric quantities of CO2 (Scheme 18a). CS2 reactivity was not investigated for the –N(SiMe3)2 derivatives.
6.2.2.3. H2 reactivity. Addition of H2 to the all –OSi(OtBu)3 complexes, 6-Cs and 14, resulted in the reversible and irreversible formation of the dinuclear U(IV) imide hydride complexes, [Cs(U(OSi(OtBu)3)3)2(μ-NH)(μ-H)] (99)49 and [NBu4][(U(OSi(OtBu)3)3)2(μ-NH)(μ-H)] (100),59 respectively (Scheme 18a and 18b). Complex 99 was found to transfer the hydride to MeCN and CO2 to afford the substrate reduction imido–ketimide and formate insertion products, respectively.49 Contrary to the reversible binding of H2 to complex 99, 100 is stable under dynamic vacuum and shows irreversible binding of H2. The differences in the binding of H2 between the two complexes was rationalized by the solid-state molecular structures. In 99, the Cs cation lies at the apical position of the hydride (–H) ligand, most likely rendering the U–H interaction more labile and facilitating the elimination of H2.
In contrast to the all-OSi(OtBu)3 complexes, H2 cleavage was not observed for the all –N(SiMe3)2 (15), and (17), mixed-ligand, 22 (ref. 76) and 19,125 or cyclometallate (16)76 complexes. This was attributed to the reduced nucleophilicity of the nitride moiety when –N(SiMe3)2 ancillary ligands are incorporated in the system. Altogether, these studies show how inner-sphere cations and supporting ligand can affect the reactivity of the uranium-nitride moiety.
6.2.2.4. Acid (H+), NH3, and alkylation reactivity. Based on the small molecule reactivity described above, complex 6-Cs was suggested to be more reactive towards electrophiles than in the analogous –N(SiMe3)2 complexes, therefore the reactivity with acids (H+) and NH3, was investigated for diuranium(IV) complexes differing in type and number of supporting ligands.125
The nitride in complex 6-Cs,71 was found to be easily protonated by addition of 1.0 equiv. of a weak acid, HNEt3BPh4, resulting in a mono-imido bridged complex, [(UIV(OSi(OtBu)3)3)2(μ-NH)] (101) (Scheme 20). Formation of the stable mono-imido in complex 101 contrasts the disproportionation observed during the alkylation of 6-Cs with MeOTf, resulting in isolation of a mixture of species, namely, the U(V) bis-imido, [(UV(OSi(OtBu)3)3)2(μ-NMe)2] (102), and the U(III) dimeric Cs-OTf, [Cs2((UIII(OSi(OtBu)3)3)(μ-OTf))2] (103), complexes (Scheme 20).50 This reactivity with MeOTf was highly unusual as U(IV) complexes tend to be more stable than their U(V) and U(III) counterparts. Only one additional example of disproportionation of a U(IV)-imido complex, [(UIV(TrenTIPS)(μ-NH)(μ-M))2], has recently been reported (Section 4.2).104 These examples contrasts the formation of the U(IV) mono–imido complex 101, where protonation does not lead to similar disproportionation reactions.
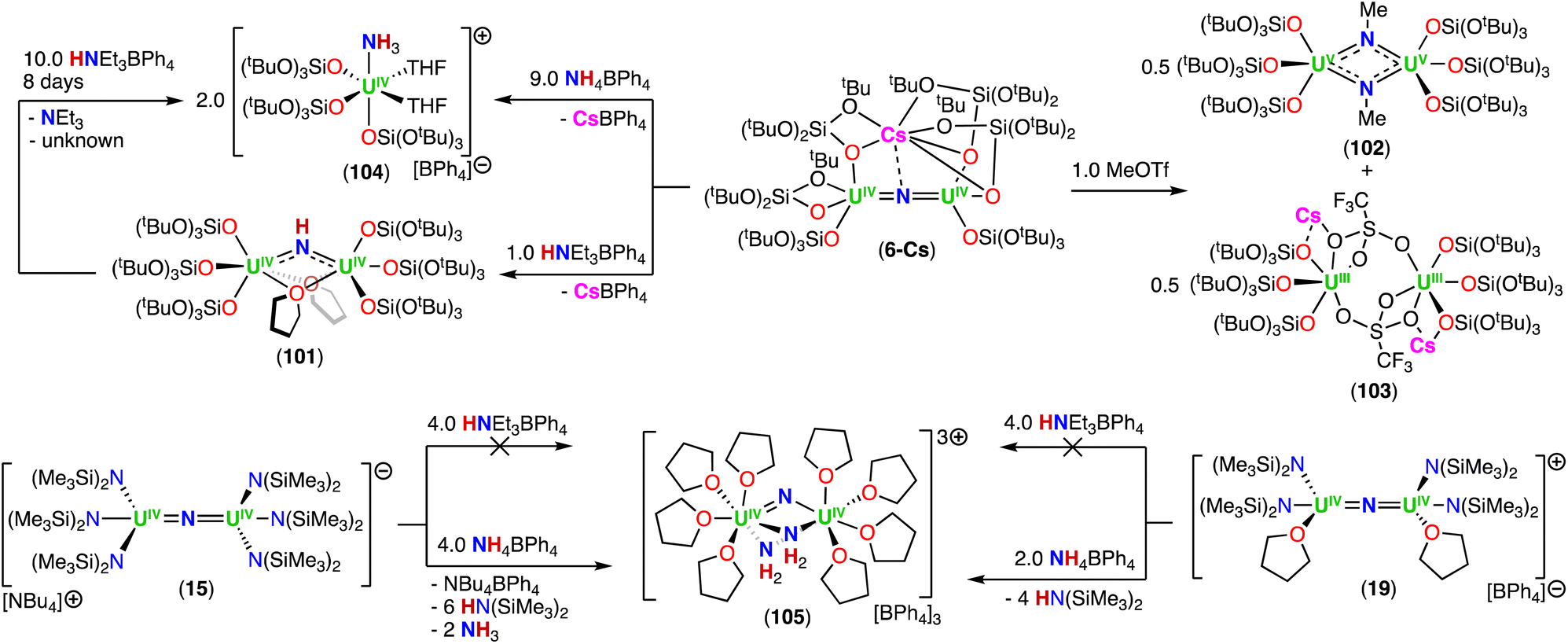 | ||
| Scheme 20 Reactivity of U(IV)-bridged nitrides supported by amide (–N(SiMe3)2) and siloxide (–OSi(OtBu)3) ligands towards acid and alkylating sources. | ||
Further protonation of the imido moiety in 101 to a terminal NH3 complex, [UIV(OSi(OtBu)3)3(THF)2(NH3)][BPh4] (104), could be achieved by using a large excess of a weak acid, HNEt3BPh4, coupled with a longer reaction time, or by means of a stronger acid, NH4BPh4 (Scheme 20). The NH3 binds the U(IV) ion strongly in complex 104, but can be displaced by addition of strong acid (HCl). Additionally, the U–OSi(OtBu)3 bonds were found to be stable, even in the presence of stronger acids, such as NH4BPh4, therefore indicating that –OSi(OtBu)3 supporting ligands are well suited for use when acidic conditions are required, such as in the H+/e− mediated catalytic conversion of N2 to NH3.125
In contrast to the all –OSi(OtBu)3 complexes, different reactivity was observed for both the all –N(SiMe3)2 (15)76 and mixed-ligand (19)70 nitride-bridged complexes. Opposed to complex 6-Cs, the nitride in both 15 and 19 are more resistant toward protonation by acids. For example, when a weak acid, such as HNEt3BPh4, is employed, the nitride in these complexes is unreactive. This is consistent with the lesser nucleophilic character of the bridging nitride in the N(SiMe3)2-containing complexes compared to the analogous –OSi(OtBu)3 systems, which was also supported by their reactivity toward small molecules (CO, CO2 or H2). Alternatively, addition of the stronger acid, NH4BPh4, resulted in the complete loss of –N(SiMe3)2 supporting ligands, while the bridging nitride remains intact. Additionally, when a strong acid is used, such as NH4BPh4, the basic –N(SiMe3)2 ligands promote the N–H heterolytic cleavage of NH3. Addition of NH4BPh4 (2.0–4.0 equiv.) to complexes 15 and 19, yielded a stable U(IV)/U(IV) bis-NH2, mono-nitride bridged complex, [(UIV(THF)4)2(μ-N)(μ-NH2)2][BPh4]3 (105), where only ancillary solvent molecules support the metal center (Scheme 20).125
Overall, this demonstrated that basic supporting ligands, such as –N(SiMe3)2, present several disadvantages compared to the –OSi(OtBu)3 ligands for usage in the development of catalysts for N2 conversion to NH3. Utilizing OSi(OtBu)3-containing complexes is not only advantageous for their resistance toward acids, but also for the high reactivity of bound nitrides to yield NH3. In contrast, N(SiMe3)2-supported complexes may be of interest for studies pertaining to the heterolytic bond activation of NH3.125
6.2.3.1. OSi(OtBu)3 complexes. Addition of 1.0 equiv. of CS2 to the Cs2UIII
NUIV (7-Cs) and Cs3UIIINUIII (8-Cs) complexes, led to the transfer of the nitride moiety with contaminant CS bond cleavage, forming the bis–sulfide complex, [(Cs(THF))2([UIV(OSi(OtBu)3)3]2(μ-S)2)] (106), and NCS−. Formation of U(IV) from the U(III) complexes suggests alternative metal-based reactivity in which unidentifiable species were also formed (Scheme 21).72
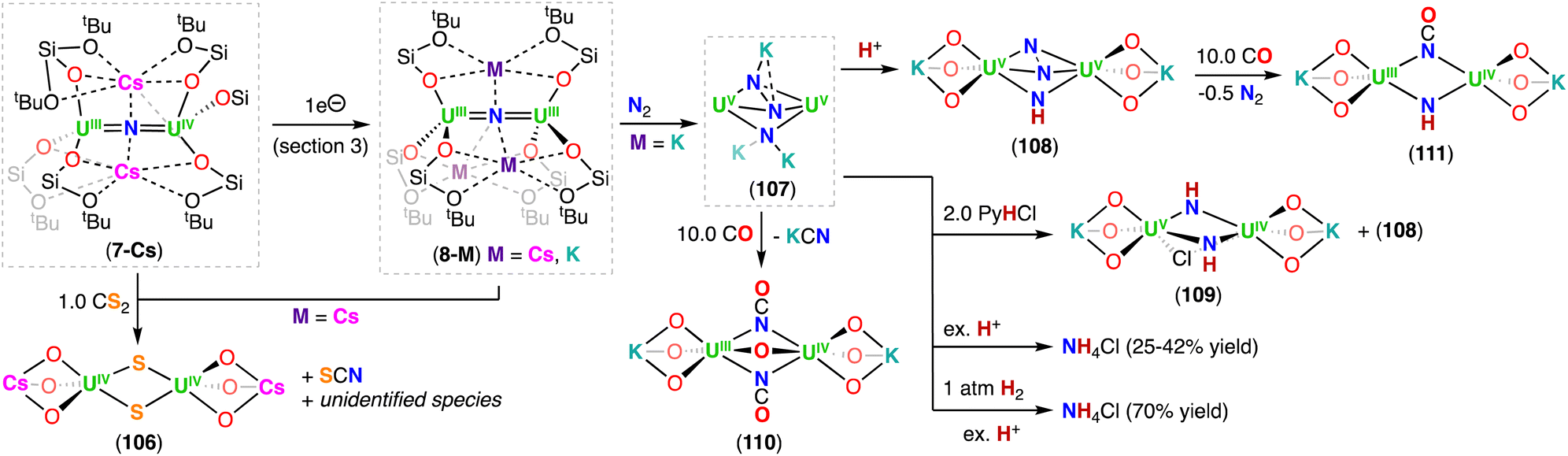 | ||
| Scheme 21 Reactivity of U(III)-bridged nitrides supported by siloxide (–OSi(OtBu)3) ligands. The –Si(OtBu)3 moieties of the ligand for most complexes have been omitted for clarity. | ||
The reaction of 8-Cs with N2 was also investigated, however, the formation of decomposition products led to difficulties in isolation of the newly formed species. Instead, the K-analogue, K3UIIINUIII (8-K) was found to have an increased stability in the solution and solid-state, allowing the study of the reaction with N2 and characterization of its products. Falcone, Mazzanti and co-workers found that addition of N2 to complex 8-K in the solid- or solution-state led to the formation of the U(V)/U(V)–N24− complex, [K3([UV(OSi(OtBu)3)3]2(μ-N)(μ-η2:η2-N2))] (107) (Scheme 21).48 The dinitrogen molecule undergoes a 4e− reduction to the side-on hydrazido moiety (N24−) with subsequent oxidation of the U(III) centers to U(V). The isolation of the hydrazido complex 107 provided the first example of a 4e− reduction of N2 by a molecular complex of uranium, and led to the first example of uranium promoted N2 functionalization. The observed reactivity is the result of the high flexibility of macrocyclic K-siloxide framework that allows bending of the UNU fragment to bind and reduce the N2 moiety. Isolation of the N24− complex allowed further study of the parameters controlling N2 binding and reduction. In particular, the effect of alkali ions and type of ancillary ligands on the reactivity of the U(III)/U(III) nitride was explored (see below).48 Additionally, complex 107 could be further functionalized by addition of acids (H+), CO, and H2, differently from what was previously observed for N2 complexes of the f-elements where addition of these molecules had led only to N2 release. First, addition of 1.0 equiv. of 2,4,6-tri-tertbutylphenol led to nitride protonation forming the mono-imido-hydrazido U(V) complex, [K2[UV(OSi(OtBu)3)3]2(μ-NH)(μ-η2:η2-N2)] (108). In contrast, when a strong acid (PyHCl or HCl) was employed, protonation of the nitride and –N24− ligand occurred, resulting in a mixture of 108 and the U(V)/U(IV) bis-imido complex, [K2((UV/IV(OSi(OtBu)3)3)2(μ-NH)2(μ-Cl))] (109) (Scheme 21). Addition of excess (20.0 equiv.) PyHCl to 107 resulted in the formation of 14NH4Cl, as confirmed by 1H NMR spectroscopy, where a mixture of 15/14NH4Cl was observed when the reaction is carried out with the isotopically enriched analog, [K2([UV(OSi(OtBu)3)3]2(μ-NH)(μ-η2:η2-15N2)] (15N-107). The yields of NH4Cl ranged between 25–42% depending on the acid used, and could be increased by first carrying out a reduction with H2 followed by addition of HCl (77% yield). Although isolable species from H2 reactivity with complex 107 could not be identified, the increase in NH4Cl suggested that H2 cleaved the N–N bond of the –N24− ligand, providing an extremely rare case of N2 reduction by H2 in molecular complexes, and the first in uranium chemistry.48
The reactivity of complexes 107 and 108 were also investigated with CO. Addition of excess CO to 107 afforded the bridging oxo bis–cyanate complex, [K2((UIV(OSi(OtBu)3)3)2(μ-O)(μ-NCO)2)] (110), with concomitant formation of KCN, resulting from the reductive carbonylation of the N24− moiety and CO cleavage by the bridging nitride ligand. The imido-N24− complex, 108, displayed a decreased reactivity in comparison to the nitrido-N24− complex 107, resulting a mixed valent U(III)/U(IV) complex, [K2((UIII/IV(OSi(OtBu)3)3)2(μ-NH)(μ-NCO))] (111), and displacement of N2. Here, the addition of CO results in disproportionation of the –N24− ligand (Scheme 21).48
6.2.3.2. N(SiMe3)2 and OSi(OtBu)3 heteroleptic complexes. To effect the overall 6e− reduction of N2 to nitrides, it was postulated that replacing the ancillary –OSi(OtBu)3 ligands with the more electron-donating –N(SiMe3)2, could possibly increase the reducing potential of the uranium and promote six electron transfer followed by N2 cleavage. Reduction of the previously reported anionic U(IV) complexes, 15 and 16, failed to produce the desired U(III) species. The inability to reduce the uranium center in these complexes was correlated to the more electron-rich character of the –N(SiMe3)2 ligands compared to –OSi(OtBu)3.70 Therefore, reduction of the mixed-ligand U(IV) nitride system, 19, containing four –N(SiMe3)2 ligands and two THF molecules, was pursued, resulting in the 1e− reduction to yield the UIII
NUIV complex (20) (Section 3.1.1).76 This complex was found to be unreactive toward N2. However, it was highly reactive towards C–H bonds, resulting in (1), the 1,2-addition of the C–H bond from the –N(SiMe3)2 ligand across the uranium–nitride bond resulting in complex 21 (Scheme 22); (2) the C–H activation of toluene; and (3) the heterolytic C–H cleavage of phenylacetylene by the nucleophilic nitride. Additionally, complex 20 effects the heterolytic cleavage of H2 yielding a mixture of the U(III)/U(IV) hydride–imide complex, [(UIV/III((N(SiMe3)2)2(THF)(μ-NH)(μ-H)] (112) and complex 21,76 whereas the analogous U(IV)/U(IV) complex was found to be unreactive toward H2 (Scheme 22).125 Similar reactivity was observed for the U(IV)/U(IV) all-OSi(OtBu)3 complex, 6-Cs,49 demonstrating the importance of the oxidation state and ancillary ligands in promoting the reactivity of the nitride.
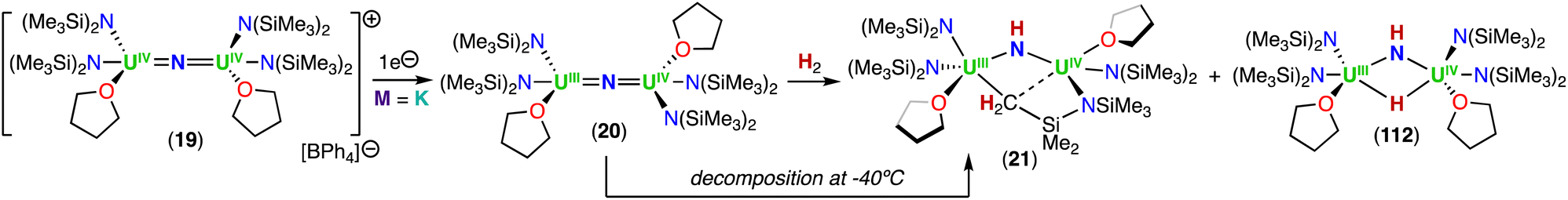 | ||
| Scheme 22 Reactivity of U(III)-bridged nitrides supported by amide (–N(SiMe3)2) ligands. | ||
In a recent study, Keener, Mazzanti and co-workers reported the U(III) mixed-ligand systems containing a varying combination of –OSi(OtBu)3 and –N(SiMe3)2 ligands in order to promote the multielectron transformation of N2. The differences in the electronic and steric properties of these ligands allowed for the tuning of the redox properties and reactivity of the nitride complexes.79 Addition of N2 to the mixed-ligand M2UIIINUIV (M = K or Cs) complexes, 25-K and 25-Cs, resulted in unreacted starting materials. However, addition of N2 to the putative “[K3UIIINUIII]” complex, 26, resulted in the analogous reactivity observed for 8-K,48 in which 26 promotes the 4e− reduction to yield the mixed-ligand U(V)/U(V)–N24− complex, [K3(UV(OSi(OtBu)3)2(N(SiMe3)2)2(μ-N)(μ-η2:η2-N2)] (113) (Scheme 23).79 Converse to the reaction with 8-K, additional N2 cleavage products were isolated from the reaction mixture, namely, [K3(UVI(OSi(OtBu)3)2(N(SiMe3)2)(N))(μ-N)2(UV(OSi(OtBu)3)2(N(SiMe3)2))]2 (114), and [K4((OSi(OtBu)3)2 UV)(N))(μ-NH)(μ-κ2:C,N–CH2SiMe2NSiMe3)(UV(OSi(OtBu)3)2][K(N(SiMe3)2]2, (115) (Scheme 23).
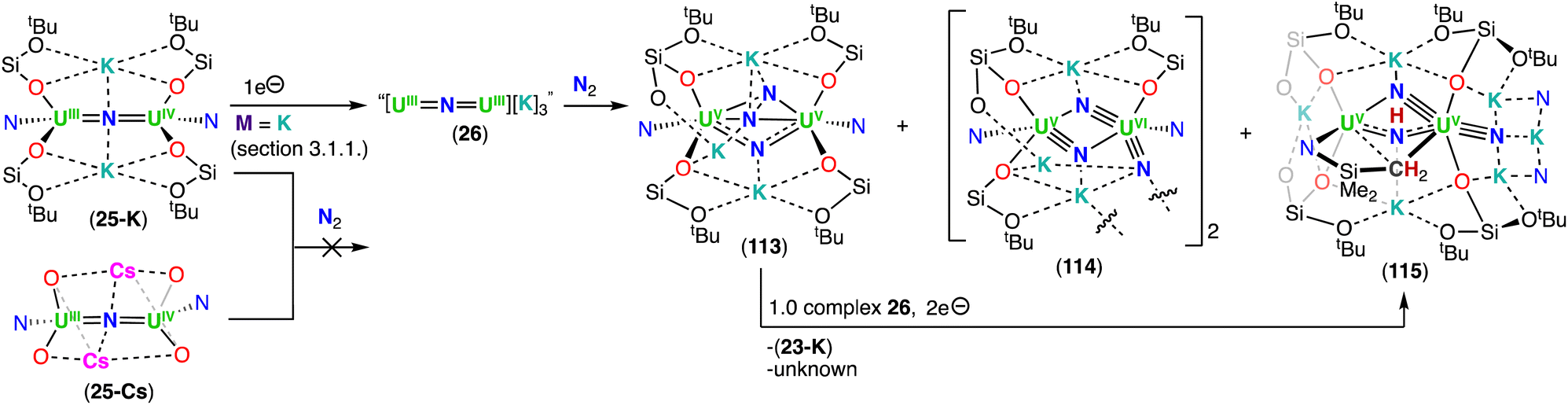 | ||
| Scheme 23 Reactivity of U(III)-bridged nitrides supported by amide (–N(SiMe3)2) and siloxide (–OSi(OtBu)3) ligands. Most of the –OtBu moieties of the ligand have been omitted for clarity. | ||
The tris-nitride complex, 114, was proposed to form from the overall 6e− cleavage of the N2 moiety. It was proposed that 5e− are transferred by the two U(III) centers to yield a U(V)/U(VI) intermediate, where an additional 1e− arises from an external reducing source, most likely the “[K3UIIINUIII]” (26) precursor. In complex 115, N2 cleavage also occurred, but with 2e− arising from an external source, and undergoing a 1,2-addition of a C–H bond of –N(SiMe3)2 across the nitride moiety. Complex 115 could be independently synthesized by addition of 1.0 equiv. of complex 26 (2e−) to the U(V)/U(V)–N24− complex (113), resulting in concomitant formation of the K-analog of the CsUIVNUIV precursor, KUIVNUIV, overall demonstrating that the U(III)/U(III) complex 26 provides 1 or 2e− to further reduce the bound N24− moiety suggesting that the formation of complexes 114 and 115 involves two dinuclear U(III) complexes.79
7. Conclusions
In summary, the past ten years have witnessed an amazing expansion of actinide-nitride chemistry with, most notably, the isolation of several examples of molecular uranium terminal- and bridging-nitrides in a broad range of oxidation states. Although this chemistry remains currently limited to a handful of supporting ligands, several routes have been identified for the synthesis of uranium nitrides. The nature of the supporting ligand is key, in both conferring stability for the targeted nitride complexes, but also in determining the reactivity and electronic structure of the nitride moiety. Notably, bridging nitrides provide an attractive route to promote magnetic coupling between uranium centers, where the bonding interaction with the supporting ligand appears to be critical for the communication to occur. Strong antiferromagnetic coupling between uranium centers in various oxidation states was observed for supporting –OSi(OtBu)3 ligands, but not for –N(SiMe3)2. It could be anticipated that a suitable choice of the supporting ligand may be used in order to implement the electronic and geometric parameters leading to ferromagnetic interactions. Moreover, the short U⋯U distances measured in some bis-nitride complexes suggests that these are well poised to implement U–U bonding interactions, if further reduction of the complex is within reach.Uranium-nitrides formed from dinitrogen cleavage present a great interest for studying the mechanism and key parameters of dinitrogen reduction and functionalization. Moreover, the recent progress in uranium nitride chemistry has shown that uranium nitrides can effect a broad range of reactivity, and that supporting ligands are key in tuning the reactivity towards the desired products.
The first examples of thorium bridged-nitride complexes have also been recently isolated, in which thorium terminal nitrides should be within reach. These preliminary results anticipate that thorium nitride chemistry should also undergo an important development in the near future. By carefully tuning the choice of supporting ligand and reaction conditions, it may also be possible to access homo- and heterometallic nitride bridged complexes containing Th, or Th and U, in low oxidation states, which may result in very interesting bonding interactions.
While no examples of molecular nitride complexes of heavier actinides such as neptunium and plutonium remain unknown, the recent isolation of a terminal Np(V) oxo complex130 suggests that a Np nitride may be in reach.
Nitride bridged An(III) complexes, analogous to 8-M (M = K, Cs), were also computationally identified for Np and Pu by Panthi, Odoh and co-workers,131 suggesting that the possibility of expanding the molecular nitride chemistry beyond uranium exists, and despite the experimental difficulties, is certainly an attractive perspective in transuranic chemistry.
Author contributions
Dr Maria and Dr Keener contributed equally to writing the original draft and to reviewing and editing the manuscript. Prof. Mazzanti conceived the outline, supervised the writing and contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Leonor Maria acknowledges support from Fundação para a Ciência e a Tecnologia (FCT) through projects UIDB/00100/2020 (CQE) and LA/P/0056/2020 (IMS) and contract IST-ID/091/2018. Marinella Mazzanti acknowledges support from Swiss National Science Foundation grant number 200020, 212723 and the Ecole Polytechnique Fédérale de Lausanne (EPFL).References
- S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- J. J. Scepaniak, R. P. Bontchev, D. L. Johnson and J. M. Smith, Angew. Chem., Int. Ed. Engl., 2011, 50, 6630–6633 CrossRef CAS PubMed.
- C. R. Clough, J. B. Greco, J. S. Figueroa, P. L. Diaconescu, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 2004, 126, 7742–7743 CrossRef CAS PubMed.
- J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–470 CAS.
- M. Streit and F. Ingold, J. Eur. Ceram. Soc., 2005, 25, 2687–2692 CrossRef CAS.
- A. V. Lizunov, A. A. Semenov, A. S. Anikin, A. V. Moiseev, A. P. Zhirnov and E. O. Soldatov, At. Energ., 2022, 132, 1–7 CrossRef CAS.
- G. W. C. Silva, C. B. Yeamans, L. Z. Ma, G. S. Cerefice, K. R. Czerwinski and A. P. Sattelberger, Chem. Mater., 2008, 20, 3076–3084 CrossRef CAS.
- G. W. C. Silva, C. B. Yeamans, A. P. Sattelberger, T. Hartmann, G. S. Cerefice and K. R. Czerwinski, Inorg. Chem., 2009, 48, 10635–10642 CrossRef CAS PubMed.
- G. W. C. Silva, C. B. Yeamans, P. F. Weck, J. D. Hunn, G. S. Cerefice, A. P. Sattelberger and K. R. Czerwinski, Inorg. Chem., 2012, 51, 3332–3340 CrossRef CAS PubMed.
- B. J. Jaques, B. M. Marx, A. S. Hamdy and D. P. Butt, J. Nucl. Mater., 2008, 381, 309–311 CrossRef CAS.
- Y. Arai and K. Minato, J. Nucl. Mater., 2005, 344, 180–185 CrossRef CAS.
- F. Haber, Angew. Chem., 1914, 27, 473 CrossRef CAS.
- F. Haber, Ammonia German Patent, DE Pat., 229126, 1909 Search PubMed.
- S. S. Parker, J. T. White, P. Hosemann and A. T. Nelson, J. Nucl. Mater., 2019, 526, 151760 CrossRef CAS.
- S. S. Parker, S. Newman, A. J. Fallgren and J. T. White, JOM, 2021, 73, 3564–3575 CrossRef CAS.
- A. J. Parkison, S. S. Parker and A. T. Nelson, J. Am. Ceram. Soc., 2016, 99, 3909–3914 CrossRef CAS.
- M. Takano, A. Itoh, M. Akabori, T. Ogawa, M. Numata and H. Okamoto, J. Nucl. Mater., 2001, 294, 24–27 CrossRef CAS.
- Y. Suzuki, Y. Arai, Y. Okamoto and T. Ohmichi, J. Nucl. Sci. Technol., 1994, 31, 677–680 CrossRef CAS.
- K. M. Peruski, Front. Nucl. Eng., 2022, 1, 1044657 CrossRef.
- X. Wang, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Inorg. Chem., 2011, 50, 3826–3831 CrossRef CAS PubMed.
- L. Andrews, X. F. Wang, Y. Gong and G. P. Kushto, J. Phys. Chem. A, 2014, 118, 5289–5303 CrossRef CAS PubMed.
- L. Andrews, X. F. Wang, Y. Gong, B. Vlaisavljevich and L. Gagliardi, Inorg. Chem., 2013, 52, 9989–9993 CrossRef CAS PubMed.
- L. Andrews, X. Wang, R. Lindh, B. O. Roos and C. J. Marsden, Angew. Chem., Int. Ed., 2008, 47, 5366–5370 CrossRef CAS PubMed.
- R. D. Hunt, J. T. Yustein and L. Andrews, J. Chem. Phys., 1993, 98, 6070–6074 CrossRef CAS.
- K. Sankaran, K. Sundararajan and K. S. Viswanathan, J. Phys. Chem. A, 2001, 105, 3995–4001 CrossRef CAS.
- D. W. Green and G. T. Reedy, J. Chem. Phys., 1978, 69, 552–555 CrossRef CAS.
- B. Vlaisavljevich, L. Andrews, X. Wang, Y. Gong, G. P. Kushto and B. E. Bursten, J. Am. Chem. Soc., 2016, 138, 893–905 CrossRef CAS PubMed.
- G. P. Kushto, P. F. Souter and L. Andrews, J. Chem. Phys., 1998, 108, 7121–7130 CrossRef CAS.
- D. W. Green and G. T. Reedy, J. Mol. Spectrosc., 1979, 74, 423–434 CrossRef CAS.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- P. L. Diaconescu, Nat. Chem., 2010, 2, 705–706 CrossRef CAS PubMed.
- A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS.
- K. Meyer, D. J. Mindiola, T. A. Baker, W. M. Davis and C. C. Cummins, Angew. Chem., Int. Ed. Engl., 2000, 39, 3063–3066 CrossRef CAS PubMed.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433–3436 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed.
- G. Nocton, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040–3042 CrossRef CAS PubMed.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed.
- M. Yadav, A. Metta-Magaña and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- M. Falcone, L. N. Poon, F. Fadaei Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed.
- M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS PubMed.
- M. Falcone, L. Chatelain, R. Scopelliti and M. Mazzanti, Chimia, 2017, 71, 209–212 CrossRef CAS PubMed.
- L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC.
- L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS PubMed.
- P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS.
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, N. Kaltsoyannis and S. T. Liddle, Nat. Chem., 2019, 11, 806–811 CrossRef CAS PubMed.
- M. Falcone, L. Barluzzi, J. Andrez, F. Fadaei Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS PubMed.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. ivković, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232–9242 RSC.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed.
- K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807–16816 CrossRef CAS PubMed.
- N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivković and M. Mazzanti, J. Am. Chem. Soc., 2021, 143, 11225–11234 CrossRef CAS PubMed.
- S. S. Rudel, H. L. Deubner, M. Müller, A. J. Karttunen and F. Kraus, Nat. Chem., 2020, 12, 962–967 CrossRef CAS PubMed.
- L. Maria and J. Marçalo, Inorganics, 2022, 10, 121 CrossRef CAS.
- W. J. Evans, K. A. Miller, J. W. Ziller and J. Greaves, Inorg. Chem., 2007, 46, 8008–8018 CrossRef CAS PubMed.
- X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, Natl. Sci. Rev., 2022, nwac144, DOI:10.1093/nsr/nwac144.
- C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed.
- C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed.
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens Jr. and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- L. Barluzzi, F.-C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC.
- C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- B. Vlaisavljevich, P. L. Diaconescu, W. L. Lukens Jr, L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef CAS.
- J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- M. Keener, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic and M. Mazzanti, Chem. Sci., 2022, 13, 8025–8035 RSC.
- P. Scott and P. B. Hitchcock, Polyhedron, 1994, 13, 1651–1653 CrossRef CAS.
- S. Fortier, G. Wu and T. W. Hayton, Dalton Trans., 2010, 39, 352–354 RSC.
- D. M. King, B. E. Atkinson, L. Chatelain, M. Gregson, J. A. Seed, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Dalton Trans., 2022, 51, 8855–8864 RSC.
- Q. Zhu, W. Fang, L. Maron and C. Zhu, Acc. Chem. Res., 2022, 55, 1718–1730 CrossRef CAS PubMed.
- P. Wang, Y. Zhao and C. Zhu, Organometallics, 2022, 41, 2448–2454 CrossRef CAS.
- J. Du, C. Alvarez-Lamsfus, E. P. Wildman, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2019, 10, 4203 CrossRef PubMed.
- S. L. Staun, D.-C. Sergentu, G. Wu, J. Autschbach and T. W. Hayton, Chem. Sci., 2019, 10, 6431–6436 RSC.
- F.-C. Hsueh, L. Barluzzi, M. Keener, T. Rajeshkumar, L. Maron, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2022, 144, 3222–3232 CrossRef CAS PubMed.
- N. Kaltsoyannis, Inorg. Chem., 2000, 39, 6009–6017 CrossRef CAS PubMed.
- N. Kaltsoyannis, Chem. Soc. Rev., 2003, 32, 9–16 RSC.
- L. Maria, I. C. Santos, V. R. Sousa and J. Marçalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed.
- N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- M. D. Straub, L. M. Moreau, Y. S. Qiao, E. T. Ouellette, M. A. Boreen, T. D. Lohrey, N. S. Settineri, S. Hohloch, C. H. Booth, S. G. Minasian and J. Arnold, Inorg. Chem., 2021, 60, 6672–6679 CrossRef CAS PubMed.
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919–921 RSC.
- N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492–10495 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu, N. Kaltsoyannis and T. W. Hayton, Chem. Sci., 2015, 6, 3891–3899 RSC.
- D.-C. Sergentu, G. T. Kent, S. L. Staun, X. Yu, H. Cho, J. Autschbach and T. W. Hayton, Inorg. Chem., 2020, 59, 10138–10145 CrossRef CAS PubMed.
- S. L. Staun, G. Wu, W. W. Lukens and T. W. Hayton, Chem. Sci., 2021, 12, 15519–15527 RSC.
- D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed.
- X. Li, Y. Roselló, Y.-R. Yao, J. Zhuang, X. Zhang, A. Rodríguez-Fortea, C. de Graaf, L. Echegoyen, J. M. Poblet and N. Chen, Chem. Sci., 2021, 12, 282–292 RSC.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed.
- M. A. Boreen, G. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC.
- K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M.-H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed.
- J. Du, I. Douair, E. Lu, J. A. Seed, F. Tuna, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2021, 12, 4832 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS PubMed.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- M. Pepper and B. E. Bursten, Chem. Rev., 1991, 91, 719–741 CrossRef CAS.
- J. Du, J. A. Seed, V. E. J. Berryman, N. Kaltsoyannis, R. W. Adams, D. Lee and S. T. Liddle, Nat. Commun., 2021, 12, 5649 CrossRef CAS PubMed.
- L. Maria, N. A. G. Bandeira, J. Marçalo, I. C. Santos, A. S. D. Ferreira and J. R. Ascenso, Inorg. Chem., 2022, 61, 346–356 CrossRef CAS PubMed.
- J. F. Berry, E. Bill, E. Bothe, S. D. George, B. Mienert, F. Neese and K. Wieghardt, Science, 2006, 312, 1937–1941 CrossRef CAS PubMed.
- C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47, 2681–2684 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- E. M. Zolnhofer, M. Käβ, M. M. Khusniyarov, F. W. Heinemann, L. Maron, M. van Gastel, E. Bill and K. Meyer, J. Am. Chem. Soc., 2014, 136, 15072–15078 CrossRef CAS PubMed.
- J. Sun, J. Abbenseth, H. Verplancke, M. Diefenbach, B. de Bruin, D. Hunger, C. Würtele, J. van Slageren, M. C. Holthausen and S. Schneider, Nat. Chem., 2020, 12, 1054–1059 CrossRef CAS PubMed.
- L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed.
- Q. Meng, L. Abella, Y.-R. Yao, D.-C. Sergentu, W. Yang, X. Liu, J. Zhuang, L. Echegoyen, J. Autschbach and N. Chen, Nat. Commun., 2022, 13, 7192 CrossRef CAS PubMed.
- L. Barluzzi, N. Jori, T. He, T. Rajeshkumar, R. Scopelliti, L. Maron, P. Oyala, T. Agapie and M. Mazzanti, Chem. Commun., 2022, 58, 4655–4658 RSC.
- A. J. Ayres, M. Zegke, J. P. A. Ostrowski, F. Tuna, E. J. L. McInnes, A. J. Wooles and S. T. Liddle, Chem. Commun., 2018, 54, 13515–13518 RSC.
- K. T. Horak, A. Velian, M. W. Day and T. Agapie, Chem. Commun., 2014, 50, 4427–4429 RSC.
- S. Lin, D. E. Herbert, A. Velian, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2013, 135, 15830–15840 CrossRef CAS PubMed.
- X. Xin, I. Douair, T. Rajeshkumar, Y. Zhao, S. Wang, L. Maron and C. Zhu, Nat. Commun., 2022, 13, 3809 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941 CrossRef CAS PubMed.
- P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem. - Eur. J., 2017, 23, 2950–2959 CrossRef CAS PubMed.
- D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed.
- M. Keener, R. Scopelliti and M. Mazzanti, Chem. Sci., 2021, 12, 12610–12618 RSC.
- O. Bénaud, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117–8130 CrossRef PubMed.
- S. J. Simpson and R. A. Andersen, J. Am. Chem. Soc., 1981, 103, 4063–4066 CrossRef CAS.
- P. L. Arnold, Z. R. Turner, A. I. Germeroth, I. J. Casely, G. S. Nichol, R. Bellabarba and R. P. Tooze, Dalton Trans., 2013, 42, 1333–1337 RSC.
- M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 4074–4078 CrossRef CAS PubMed.
- M. S. Dutkiewicz, C. A. P. Goodwin, M. Perfetti, A. J. Gaunt, J.-C. Griveau, E. Colineau, A. Kovács, A. J. Wooles, R. Caciuffo, O. Walter and S. T. Liddle, Nat. Chem., 2022, 14, 342–349 CrossRef CAS PubMed.
- D. Panthi, O. Adeyiga, N. K. Dandu and S. O. Odoh, Inorg. Chem., 2019, 58, 6731–6741 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |