
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation and redox reactivity of cerium complexes in four redox states†
Fang-Che
Hsueh
 a,
Thayalan
Rajeshkumar
b,
Laurent
Maron
*b,
Rosario
Scopelliti
a,
Andrzej
Sienkiewicz
cd and
Marinella
Mazzanti
*a
a,
Thayalan
Rajeshkumar
b,
Laurent
Maron
*b,
Rosario
Scopelliti
a,
Andrzej
Sienkiewicz
cd and
Marinella
Mazzanti
*a
aGroup of Coordination Chemistry, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, 31077 Toulouse, Cedex 4, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
cLaboratory for Quantum Magnetism, Institute of Physics, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland
dADSresonances Sàrl, 1920 Martigny, Switzerland
First published on 5th May 2023
Abstract
The chemistry of lanthanides is limited to one electron transfer reactions due to the difficulty of accessing multiple oxidation states. Here we report that a redox-active ligand combining three siloxides with an arene ring in a tripodal ligand can stabilize cerium complexes in four different redox states and can promote multielectron redox reactivity in cerium complexes. Ce(III) and Ce(IV) complexes [(LO3)Ce(THF)] (1) and [(LO3)CeCl] (2) (LO3 = 1,3,5-(2-OSi(OtBu)2C6H4)3C6H3) were synthesized and fully characterized. Remarkably the one-electron reduction and the unprecedented two-electron reduction of the tripodal Ce(III) complex are easily achieved to yield reduced complexes [K(2.2.2-cryptand)][(LO3)Ce(THF)] (3) and [K2{(LO3)Ce(Et2O)3}] (5) that are formally “Ce(II)” and “Ce(I)” analogues. Structural analysis, UV and EPR spectroscopy and computational studies indicate that in 3 the cerium oxidation state is in between +II and +III with a partially reduced arene. In 5 the arene is doubly reduced, but the removal of potassium results in a redistribution of electrons on the metal. The electrons in both 3 and 5 are stored onto δ-bonds allowing the reduced complexes to be described as masked “Ce(II)” and “Ce(I)”. Preliminary reactivity studies show that these complexes act as masked Ce(II) and Ce(I) in redox reactions with oxidizing substrates such as Ag+, CO2, I2 and S8 effecting both one- and two-electron transfers that are not accessible in classical cerium chemistry.
Introduction
The chemistry of lanthanides in molecular complexes was for a longtime limited to the +3 oxidation state and to one-electron transfer reactions for the few accessible redox couples (Ce3+/4+, Sm2+/3+, Eu2+/3+, and Yb2+/3+).1 Only in 2013 all lanthanide ions (except the radioactive promethium) have been isolated in the +II oxidation state in molecular complexes expanding significantly the knowledge of this oxidation state.1,2 Molecular species containing rare earths in the +I oxidation state remain elusive with only an example of formal Sc(I) reported 25 years ago,3 and so far the +I oxidation state in f element chemistry has been limited to a single U(I) complex4 or gas phase and solid-state species.5,6The redox chemistry of the cerium ion in particular has received large attention,7 which mostly focused on Ce(III)/Ce(IV) oxidation chemistry because of its application in organic synthesis, catalysis and materials science. In contrast, mononuclear Ce(II) complexes remain limited to five examples shown in Fig. 1.2a,c,f,8,9 including four cyclopentadienide derivatives (complex A in Fig. 1) and the aryloxide complex (complex B in Fig. 1). The redox reactivity of these complexes has not been so far investigated.
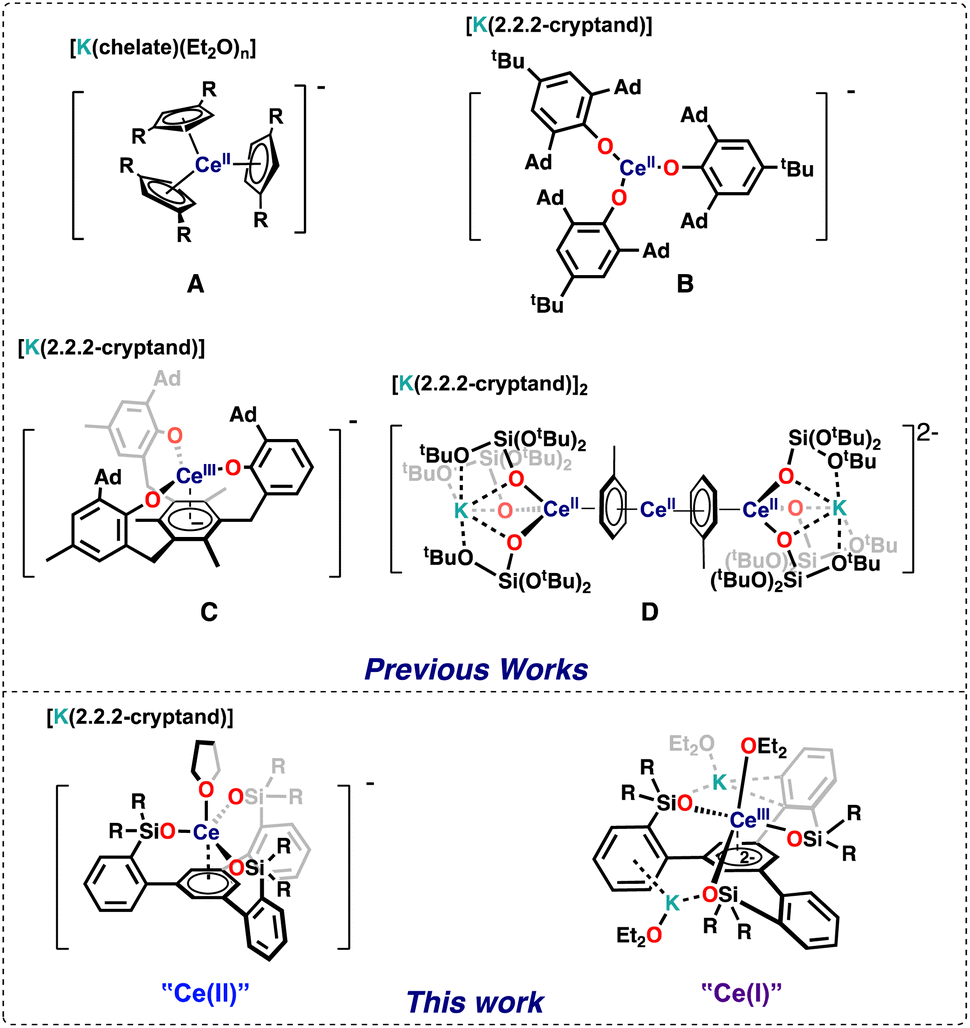 | ||
| Fig. 1 Ce(II) Complexes (chelate = 2.2.2-cryptand or 18-crown-6). | ||
Three additional examples of Ce in the +2 oxidation state have been identified in multimetallic arene sandwich compounds.10 In particular, inverse sandwich complexes of cerium(II) were isolated by reduction of Ce(III) cyclopentadienyl complexes.10a,b Remarkably, the Evans group showed that the four electrons stored in the 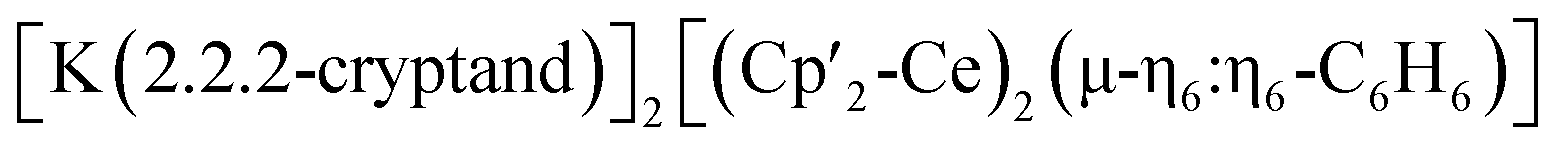 complex, best described as a Ce(II)–arene2−–Ce(II) species,10b become available for the reduction of substrates suggesting that the association of cerium and redox-active ligands may provide an entry into multielectron redox chemistry usually non-accessible for lanthanides. Besides these results, the use of redox-active ligands in low valent lanthanide chemistry remains poorly developed especially in comparison to d-block metals.11
complex, best described as a Ce(II)–arene2−–Ce(II) species,10b become available for the reduction of substrates suggesting that the association of cerium and redox-active ligands may provide an entry into multielectron redox chemistry usually non-accessible for lanthanides. Besides these results, the use of redox-active ligands in low valent lanthanide chemistry remains poorly developed especially in comparison to d-block metals.11
Meyer and coworkers reported tripodal ligands (((ArO)3mes)3−) that incorporate an arene ring as part of a potentially redox-active chelating ligand and showed that these ligand were able to promote unusual reactivity and stabilize the +2 oxidation state in f element chemistry.12 More recently, Evans and Meyer showed that reduced lanthanide complexes of the tripodal (((ArO)3mes)3−) ligand could be isolated for all lanthanide ions2h,13 and they act as electrocatalysts for water reduction.13a Interestingly, the ligand was found to stabilize the low valent oxidation state even for highly reducing Ln2+ ions such as Nd2+,2h but could also act as a redox-active ligand for some lanthanide ions. In particular the reduced Ce complex of the ((Ad,MeArO)3mes)3− ligand was identified as Ce(III) with a negative charge localized on the arene anchor of the tripodal ligand (complex C in Fig. 1).13b
Ligand-based C–H bond activation and loss of hydrogen can also occur under reducing conditions for the aryloxide-arene tripodal ligand ((Ad,MeArO)3mes)3−, to form a tetraanionic ligand (((Ad,MeArO)3mes)4−) leading to the formation of Ln(III) hydride complexes.13b Such reactivity may limit the possibility of accessing higher degrees of Ln reduction.
In our group we showed that the tert-butoxy siloxide ligand can stabilize the cerium cation in different oxidation states (+IV to +II), but the reduction of Ce(IV) with excess KC8 resulted in the partial loss of the supporting ligand leading to the isolation of the tetradecker sandwich complex [K(2.2.2-crypt)]2[{(KL3Ce)(μ-η6:η6-C7H8)}2Ce] (complex D in Fig. 1). Complex D contains Ce(II) in a 4f2 configuration and doubly reduced toluene molecules involved in delta bonds with the 4f orbitals of cerium. Inspired by the molecular and electronic structures of complexes C and D we reasoned that combining three siloxides with an arene ring in a tripodal ligand should lead to the isolation of a series of cerium in different oxidation states and enable a novel route to controlled multi-electron redox chemistry in cerium compounds.
Here we report a new ligand 1,3,5-(2-HOSi(OtBu)2C6H4)3C6H3, and its cerium complexes and show that four different redox states are accessible for this system enabling one- and two-electron transfer reactions with various substrates.
Results and discussion
Tripodal ligand
The tripodal tris(di-tert-butoxylsilanol) arene ligand, 1,3,5-(2-HOSi(OtBu)2C6H4)3C6H3, L(OH)3, was prepared from commercial products on a gram scale in a three step synthesis (Schemes S1† and 1) with a procedure analogous to that reported for the tri-diphenylsiloxide arene ligand.14 Colorless crystals of the tripodal ligand (L(OH)3) were isolated from hexane at room temperature by slow evaporation. The solid-state molecular structure of L(OH)3 as determined by an X-ray diffraction study shows that the three silanol groups are connected by hydrogen bonds that hold the three silanols in the same plane (Fig. S79†).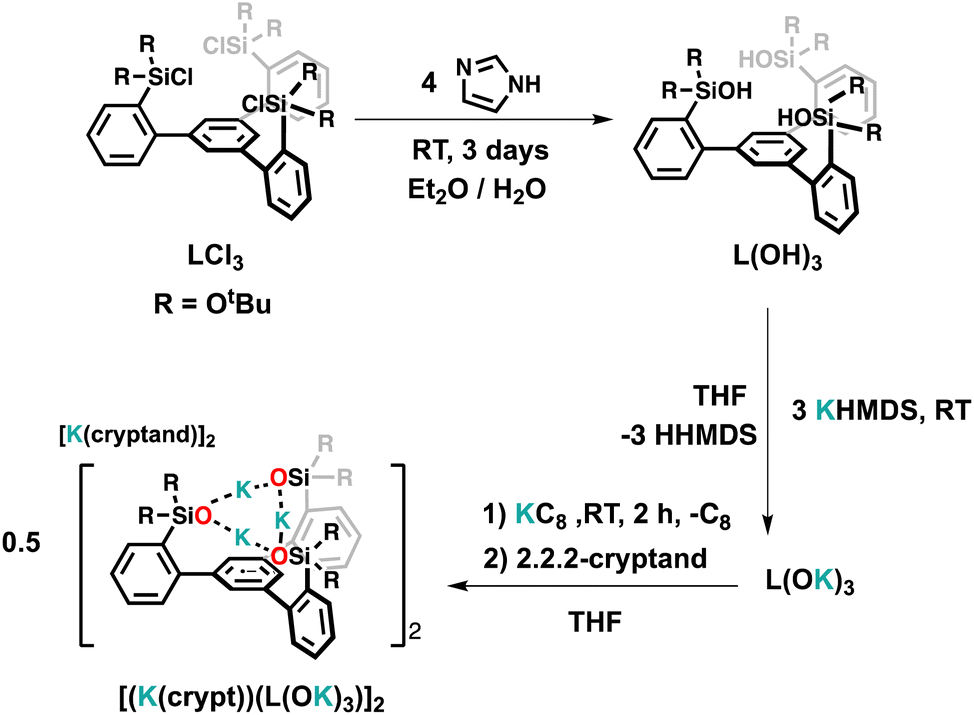 | ||
| Scheme 1 Synthesis of the tripodal ligand L(OH)3 and of the ligand radical salt [(K(crypt))(L(OK)3)]2. | ||
Reduction with 1 equiv. KC8 of the ligand salt L(OK)3 obtained by deprotonation of L(OH)3 with KHMDS afforded the dark blue-green potassium salt of the ligand radical [(K(THF)3)(L(OK)3)]2 which was isolated in 66% yield and crystallographically characterized (see the ESI†). The reduction of [(K(THF)3)(L(OK)3)]2 with additional KC8 only led to decomposition products (Fig. S17†). The molecular structure of [(K(THF)3)(L(OK)3)]2 shows the presence of a dimeric compound where two tetra-anionic radical tripodal moieties are held together by six potassium cations binding siloxide groups from the two molecules (Fig. S80†). The structure is completed by two potassium cations bound to the central arene in a η6-fashion. The C–C bond distances are slightly longer compared to the non-reduced ligand (L(OH)3). We found that two potassium ions can be removed by addition of 2 equiv. of 2.2.2-cryptand to [(K(THF)3)(L(OK)3)]2, yielding [(K(crypt))(L(OK)3)]2. The solid-state structure of [(K(crypt))(L(OK)3)]2 was determined by X-ray diffraction studies and it shows the same dimeric structure as [(K(THF)3)(L(OK)3)]2, but the two arene-η6-bound K+ ions have been removed from the structure and are found as outer sphere [K(2.2.2-cryptand)] cations (Fig. 2).
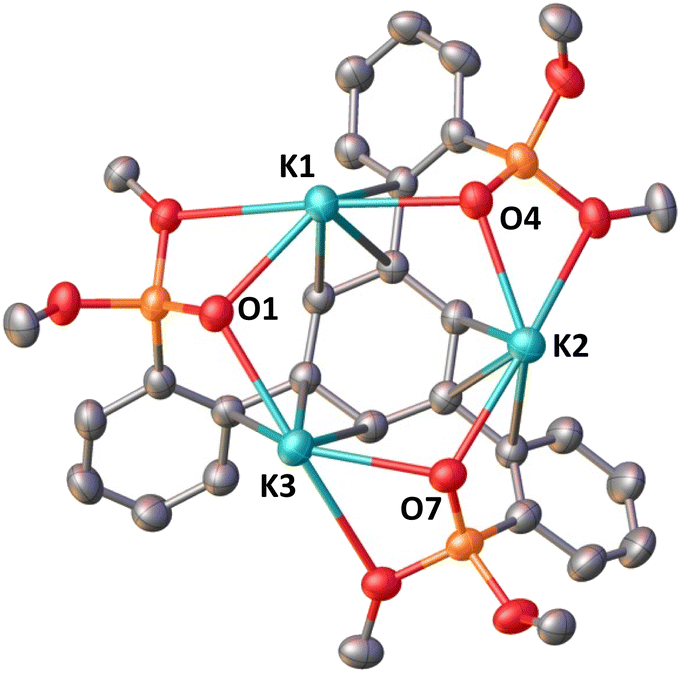 | ||
| Fig. 2 Molecular structure of the monomeric unit L(OK)3 in [(K(crypt))(L(OK)3)]2, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms, methyl groups on the –OSi(OtBu)2 ligands and [K(2.2.2-cryptand)] have been omitted for clarity. | ||
Tripodal Ce(III) and Ce(IV) complexes
At first, we pursued the synthesis of the cerium(III) complex of the ligand L(OH)3 by protonolysis of the [Ce(N(SiMe3)2)3] precursor (Scheme 2). The addition of a THF solution of [Ce(N(SiMe3)2)3] (1 equiv.) to a stirring THF solution of the free ligand (L(OH)3) (1 equiv.) at −40 °C led to the formation of a new species and full consumption of the starting materials as indicated by 1H NMR spectroscopy after 16 hours (Fig. S18†). Light-yellow crystals of the complex, [(LO3)Ce(THF)] (1), were isolated from a concentrated toluene solution at −40 °C. The 1H NMR spectrum of 1 in THF-d8 displayed five paramagnetic shifted resonances (Fig. S19†).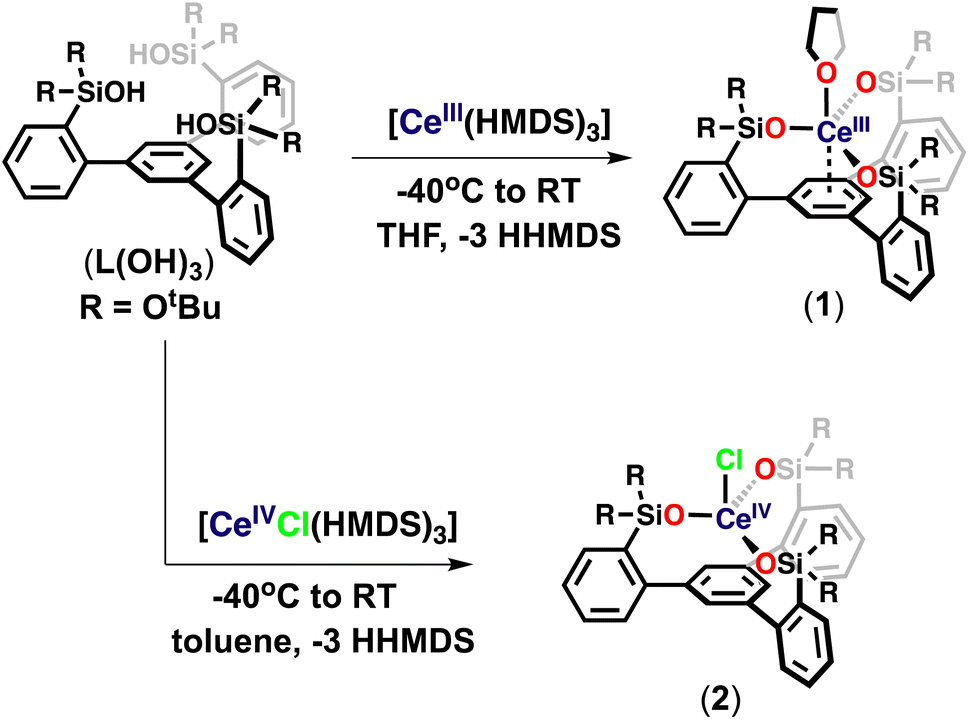 | ||
| Scheme 2 Synthesis of complexes 1 and 2. | ||
We then investigated the possibility of stabilizing Ce in the +4 oxidation state using the tripodal siloxide framework, L(OH)3. Despite the numerous applications of tetravalent cerium compounds in organic synthesis and catalysis,15,7j the coordination and organometallic chemistry of cerium(IV) complexes remain still underdeveloped due to the high oxidizing power of Ce(IV).7a,h,16
Complex 1 did not react with one equivalent of AgCl or AgBPh4 at room temperature or 70 °C after 2 days, as shown by the 1H NMR spectroscopy data (Fig. S44 and S45†). The high stability of 1 toward silver reagents compared to the tetrasiloxide complex [KCe(OSi(OtBu)3)4]10c which can be easily converted into the Ce(IV) analogue is probably explained by the less electron-rich character of complex 1. Therefore, we pursued the synthesis of the Ce(IV) complex by an alternative route using a Ce(IV) precursor. The protonolysis reaction of the heteroleptic [CeCl(N(SiMe3)2)3]17 complex with free ligand L(OH)3 in toluene solution at room temperature for 3 hours (Scheme 2) resulted in an orange reaction mixture and full consumption of the starting material as indicated by 1H NMR spectroscopy (Fig. S20†). Light-yellow crystals of complex [(LO3)CeCl] (2) were isolated from a concentrated toluene solution at −40 °C and characterized by X-ray crystallography. Complex 2 begins immediately to decompose back to 1 in THF at room temperature but is stable in toluene solution at room temperature for up to 2 weeks (Fig. S22 and S23†). These results show that the tripodal LO33− ligand allows the access of robust heteroleptic Ce(IV) complexes that provide attractive precursors for oxidative reactivity.
Low-valent tripodal Ce complexes
With the tripodal Ce(III) complex in hand, we set out to investigate if complex 1 could be further reduced and if lower oxidation states of cerium could be in reach in the environment provided by the tripodal siloxide ligand.The reaction of 1 with 1 equiv. of KC8 in THF at −40 °C resulted in a dark-blue reaction mixture, which displayed only one broad resonance at 1.59 ppm in the 1H NMR spectrum at room temperature (Fig. S25†). Attempts to isolated X-ray suitable single crystals of this new species proved unsuccessful. However, the addition of one equivalent of 2.2.2-cryptand and one equivalent of KC8 at −40 °C to 1 (Scheme 3) resulted in a new set of eight resonances in the 1H NMR spectrum recorded at room temperature (Fig. S29†). Dark-blue crystals of the complex [K(2.2.2-cryptand)][(LO3)Ce(THF)] (3) were isolated from a concentrated Et2O/THF mixture at −40 °C and characterized by X-ray crystallography. Complex 3 is stable in THF at room temperature for at least for 1 week (Fig. S32†).
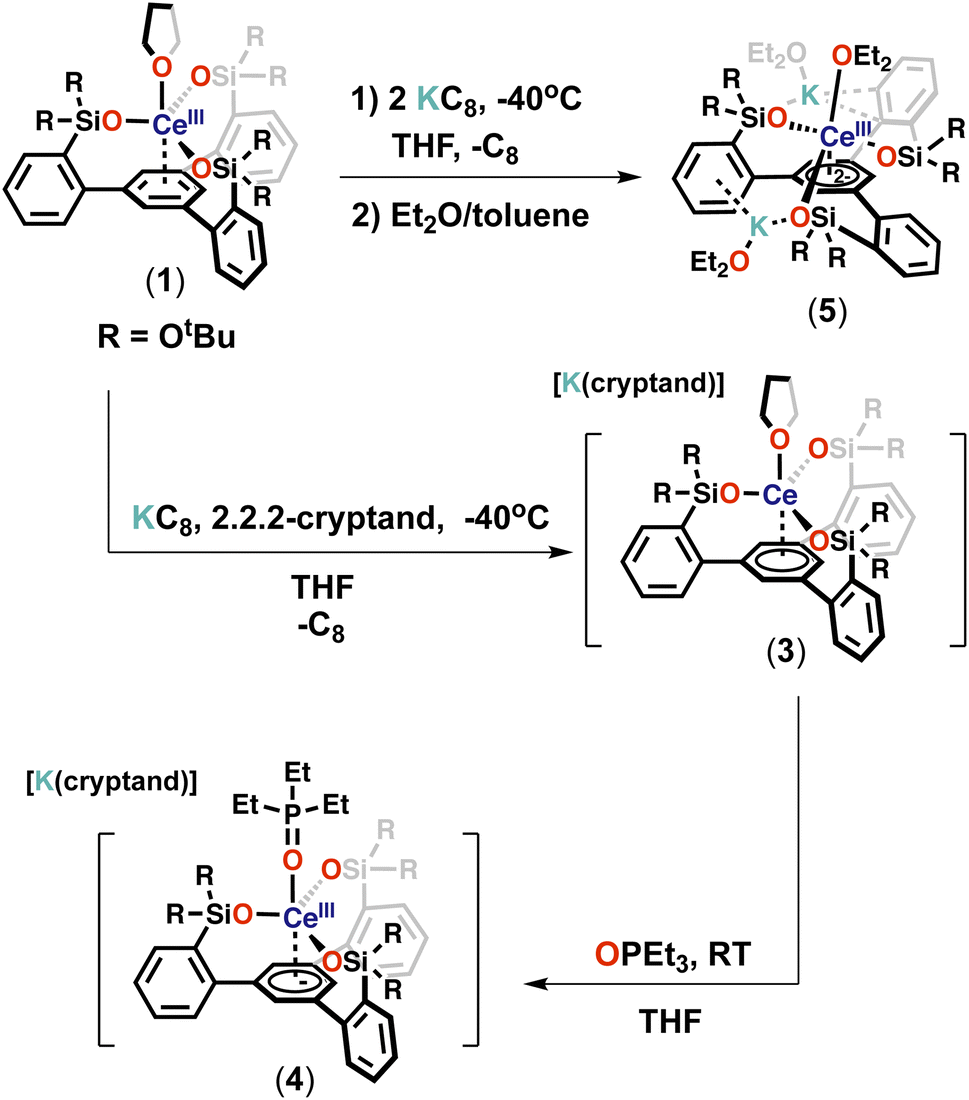 | ||
| Scheme 3 Synthesis of complex 3–5. | ||
In complex 3 the apical THF ligand can be easily replaced by more coordinating ligands. Notably, the reaction of 1 equiv. of Et3PO with a THF solution of 3 at room temperature (Scheme 3) resulted immediately in the replacement of the Ce-bound THF ligand and led to the isolation of dark-blue crystals of the complex [K(2.2.2-cryptand)][(LO3)Ce(Et3PO)] (4).
The 1H NMR spectrum of isolated complex 4 recorded at room temperature displays two additional broad resonances at δ 1.55 and δ 1.06 ppm compared to 3 (Fig. S34†), which were assigned to the ethyl group in the bound Et3PO. The 31P NMR spectrum of 4 exhibits a singlet resonance at δ 45.06 ppm (Fig. S35†), which is in the range typically observed for phosphineoxide complexes.18
A one-electron reduced cerium tripodal complex was isolated previously by Evans and co-workers from the reduction of the tripodal Ce(III) complex [((Ad,MeArO)3mes)Ce] with potassium in the presence of 2.2.2-cryptand.13b Computational and spectroscopic studies led to the assignment of the complex as a 4f1 Ce(III) complex of the reduced tripodal ligand with a population of 1.3 electrons on Ce and 0.7 on the mesitylene ring. Further reduction or reactivity studies with small molecules of this complex were not reported.
In contrast further reduction of complex 3 was found to proceed in a controlled manner leading to a main reduction product.
The addition of two equivalents of KC8 to a THF solution of 1 at −40 °C led to a dark-purple reaction mixture (Scheme 3). The 1H NMR spectrum showed that a new species was formed (Fig. S37†). Dark-purple crystals were isolated from a concentrated Et2O/toluene mixture at −40 °C, and X-ray crystallography revealed a doubly reduced complex [K2{(LO3)Ce(Et2O)3}] (5). The 1H NMR spectrum of isolated complex 5 recorded at −40 °C shows 6 sharp and three broad resonances paramagnetically shifted, between −59.63 and 28.51 ppm (Fig. S39†). Complex 5 is stable in THF at room temperature for at least for 3 days (Fig. S40†). The addition of 2 equiv. of 2.2.2-cryptand resulted in a change in the 1H NMR spectrum at −40 °C with only three broad resonances observed between 0.85 and 3.18 ppm (Fig. S43†) suggesting that the potassium cations are bound in solution and can be removed by cryptand. Attempts to isolate X-ray suitable single crystals of this species proved unsuccessful.
Complexes 3 and 5 can be formally identified as Ce(II) and Ce(I) species, but reduction could occur on the arene of the tripodal ligand as previously observed for some of the mono-reduced [((Ad,MeArO)3mes)Ln] complexes.13b
EPR and UV-vis spectroscopy
To investigate the electronic structure of complexes 1, 3, 4, and 5 X-band EPR spectra were recorded for all complexes and for the ligand radical [(K(crypt))(L(OK)3)]2 (Fig. 3 and S61†). Complex 1 exhibits a strong and distinct EPR signal with a major feature at g ∼ 1.78 in the frozen-solution state at 6 K (Fig. 3) in agreement with the presence of an f1 Ce(III) species.19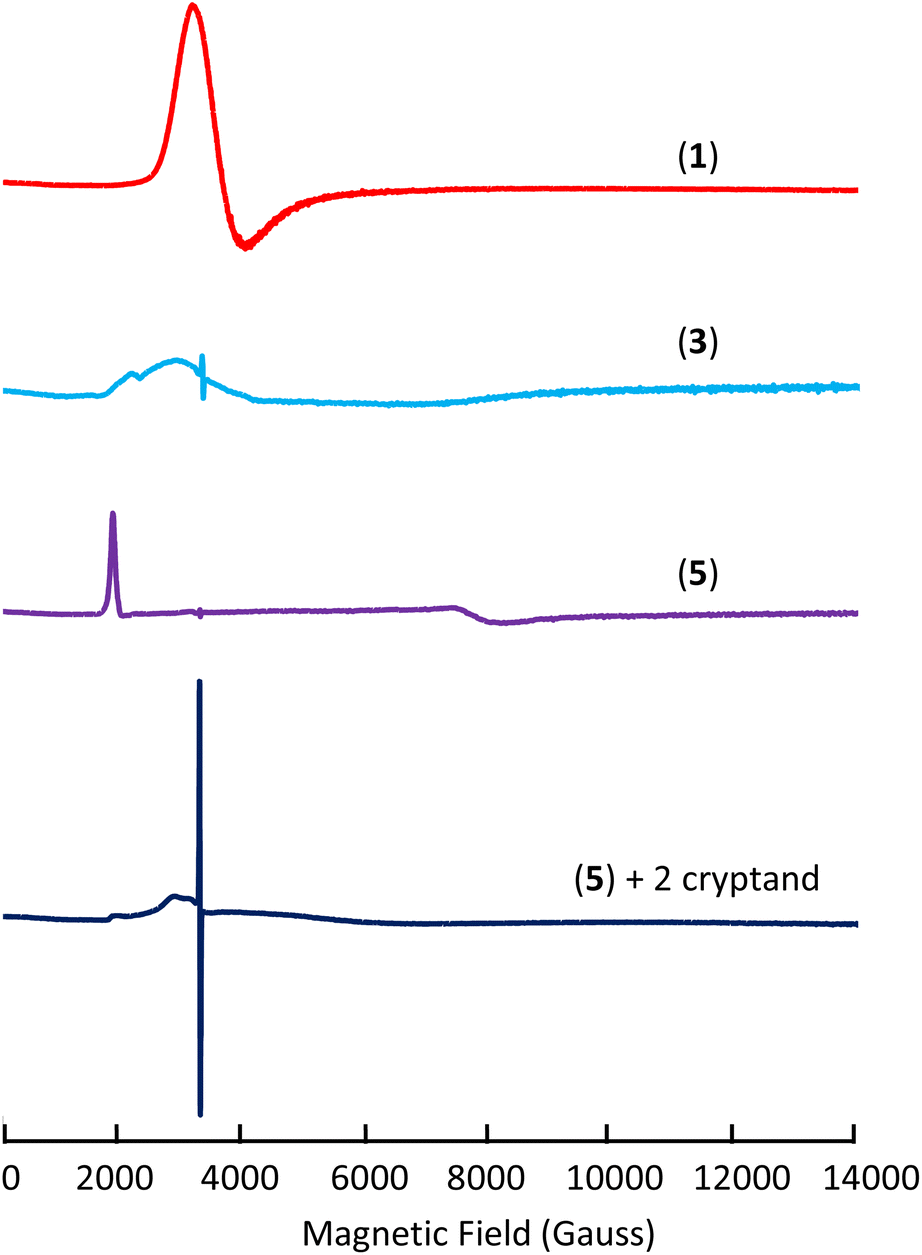 | ||
| Fig. 3 X-band EPR spectra of complexes 1 (frozen-solution), 3 (solid-state), 5 (solid-state), and 5 + cryptand (frozen-solution) at 6 K. | ||
On the other hand, complex 3 exhibits a EPR signal weaker and broader than 1 in the solid-state at 6 K (g1 ∼ 3.02 and g2 ∼ 2.31) (Fig. 3, S62 and S64†) which disappears at higher temperatures (50 K). Since f2 Ce(II) would be EPR silent, the broad and weak nature of the signal for complex 3 could be due to an f1d1 configuration or to the presence of a strong interaction of f1 Ce(III) with the ligand radical. Furthermore, the EPR spectrum of 3 also shows an isotropic signal with a giso of 2.0054 which is consistent with the presence of a ligand radical. A similar signal was also observed in the EPR spectrum recorded at 96 K reported for the tripodal complex [K(2.2.2-cryptand)][((Ad,MeArO)3mes)Ce]13b and assigned to the presence of ligand-radical character. The EPR spectrum recorded before the addition of cryptand is very similar to the EPR spectrum of 3 but more intense (Fig. S65†) suggesting that the electronic configuration is slightly affected by the presence of bound potassium. Moreover, the replacement of the axial THF ligand by Et3OP in complex 4 also results in a EPR spectrum similar to the one recorded for 3 (Fig. S66†). Finally, complex 5 shows a strong signal in the X band solid-state EPR at 6 K which was best fitted as a rhombic signal with gy = 0.870, gz = 3.360 and gx < 0.5 (gx could not be observed because it occurs at magnetic fields outside our accessible magnetic range) (Fig. 3 and S68†). The observed spectrum is in agreement with the assignment of complex 5 as Ce(III)(ligand)2−. Similar rhombic EPR spectra were previously reported for Ce(III) complexes.20 Overall the EPR spectrum of 5 seems to point toward the presence of a Ce(III) component. However, the EPR spectrum is very different after the addition of cryptands showing only a very weak and broad signal and a strong radical signal at g = 2.0054 (Scheme S2,†Fig. 3 and S71†). The spectrum suggests that, in the case of the doubly reduced complex, the removal of potassium results in a significant change in the electron configuration of the cerium cation and indicates the presence of Ce(II) and a mono-reduced arene.
In previous studies of cyclopentadienyl complexes of Ln2+ ions, UV-vis spectroscopy has been crucial for evaluating electronic configurations.2a,f We therefore decided to record UV-vis spectra for complexes 1, 3 and 5.
Complex 1 only shows two intense absorption bands at 210 and 250 nm (Fig. S56†) assigned to the ligand (Fig. S55†). The mono-reduced complex 3 shows the ligand absorption at 210 and 245 nm and three strong absorbances at 375, 545 and 770 nm (Fig. S58†) with calculated molar extinction coefficients, ε, of 7077, 3294 and 4045 M−1 cm−1, respectively. These λmax and ε values are similar to those reported for Ln2+ complexes which have a 4fn5d1 configuration (ε values range from 3000–7000 M−1 cm−1). However, the UV-vis spectrum of the tripodal ligand radical anion, [(K(crypt))(L(OK)3)]2 (Fig. S57†), exhibits similar strong absorbances at 405, 520, and 625 nm with molar extinction coefficients, ε, of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 377, 4462 and 6727 M−1 cm−1. The doubly reduced complex 5 shows different features compared to complex 3 with two strong absorbances at 405 and 520 nm (Fig. S60†) with molar extinction coefficients, ε, of 5913 and 8681 M−1 cm−1, which again could be assigned either to a different degree of ligand reduction or to a 4fn5d1 metal configuration.
377, 4462 and 6727 M−1 cm−1. The doubly reduced complex 5 shows different features compared to complex 3 with two strong absorbances at 405 and 520 nm (Fig. S60†) with molar extinction coefficients, ε, of 5913 and 8681 M−1 cm−1, which again could be assigned either to a different degree of ligand reduction or to a 4fn5d1 metal configuration.
Cyclic voltammetry
Cyclic voltammetry experiments were conducted at ambient temperature in 3 mM THF solutions (0.1 M [NBu4][B(C6H5)4] as the supporting electrolyte) of the ligand salt L(OK)3 and on complexes 1, 3, and 5 (Fig. 4). Only one reversible redox event could be clearly identified for the three complexes. The voltammogram of complex 1 shows one chemically reversible reduction wave at E1/2 = −2.90 V, corresponding to the reduction to complexes 3. The voltammogram of complex 3 confirms this assignment and has very similar E1/2 (−2.95 V). The measured half potential is very similar to that reported for the ligand-based one-electron reduction of the [((Ad,MeArO)3mes)Ce] complex.13a The voltammogram of 5 shows the same redox event but at slightly shifted (E1/2 = −2.81 V) potential probably due to the presence of coordinating potassium cations in the complex, which was assigned to the one-electron reduction. Indeed, the voltammogram of 5 after the addition of cryptands shows a comparable E1/2 value at −2.91 V (Fig. 4). The redox event corresponding to the reduction of 3 to 5 is not observed under the used conditions.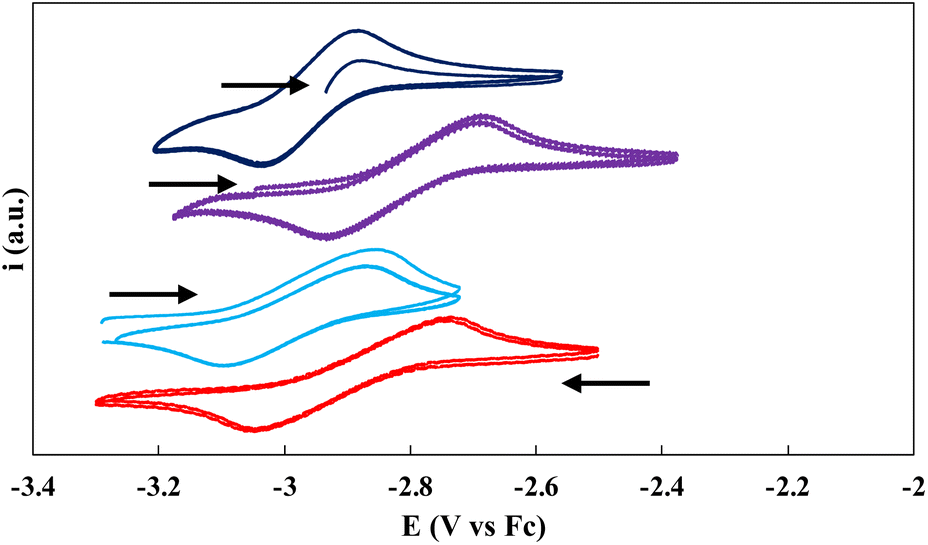 | ||
| Fig. 4 Cyclic voltammograms of complexes 1 (red), 3 (blue), 5 (purple) and 5 + cryptand (dark-blue) in THF (0.1 M [NBu4][BPh4]) at a 250 mV s−1 scan rate versus Fc/Fc+ (arrows indicate the scan direction). | ||
No reduction waves were observed within the window permitted by THF in the voltammogram of the ligand salt L(OK)3 alone or in the presence of 3 equiv. of cryptand (Fig. S76 and S77†), suggesting that the redox events observed in the voltammograms of 1, 3 and 5 may be assigned to a metal centered redox reduction (Ce(III)/Ce(II)).
Structural characterization of tripodal Ce complexes
The solid-state structures of complexes 1–5 were determined by X-ray diffraction studies. The molecular structures of complexes 1–5 are presented in Fig. 5 and the selected structural parameters are in Table 1.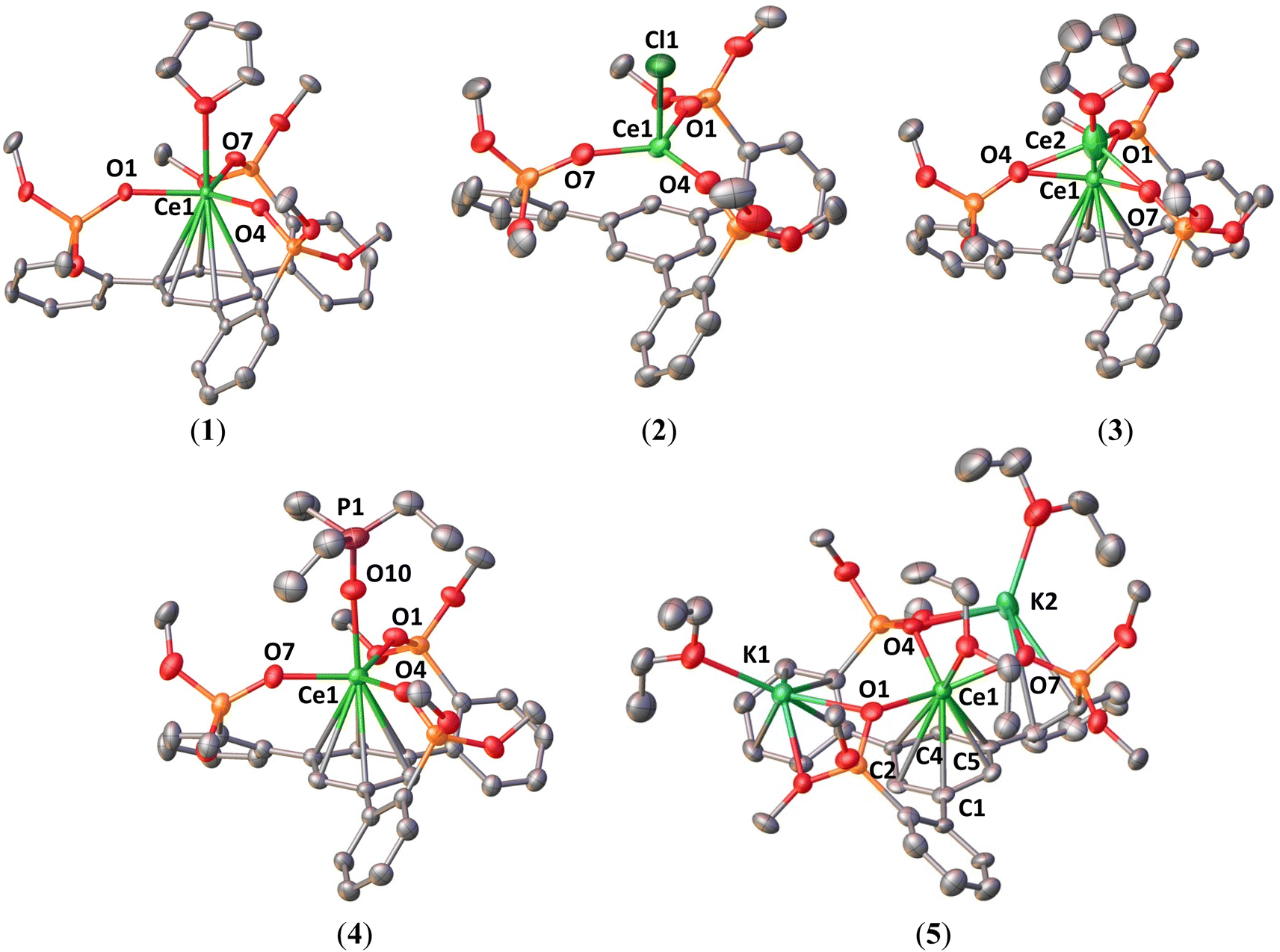 | ||
| Fig. 5 Molecular structure of 1–5, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and methyl groups on the –OSi(OtBu)2 ligands and [K(2.2.2-cryptand)] (3 and 4) have been omitted for clarity. | ||
| Compound | L(OH)3 | [(K(crypt))(L(OK)3)]2 | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|---|
| Ce–O (avg.) | — | — | 2.237(3) | 2.102(6) | 2.258(5) | 2.277(5) | 2.363(9) |
| Ce–Ccentroid | — | — | 2.730(1) | 2.821(2) | 2.569(2) | 2.632(2) | 2.394(3) |
| C–Carene (avg.) | 1.39(2) | 1.411(5) | 1.402(7) | 1.402(15) | 1.415(15) | 1.417(12) | 1.43(3) |
| Torsion angle (°) (central arene) | 0.3(7)–3.6(7) | 0.2(2)–2.4(2) | 0.0(3)–0.2(3) | 0.1(7)–0.4(7) | 0.2(6)–5.3(6) | 1.0(6)–7.3(5) | 6.9(12)–18.0(11) |
Complex 1 displays a mononuclear structure where the cerium cation is bound by three siloxide oxygens of the tripodal ligand and one THF molecule and display an additional η6-arene interaction with the arene ring. The mean value of the Ce–Osil distances of 2.237(3) Å is comparable to that reported for the Ce(III) complex [Ce(OSi(OtBu)3)3(THF)2] (2.246(2) Å).21 The average values of Ce–C distances (3.068(5) Å) and the Ce–Ccentroid distance (2.730(1) Å) are in the range of previously reported π-arene–cerium(III) interactions (average Ce–C = 2.95(4)–3.07(7) Å and Ce–Ccentroid = 2.607–2.734 Å).22 The solid-state molecular structure of 2 was determined by X-ray diffraction studies and shows a mononuclear Ce(IV) complex. The Ce center is four-coordinated by three –OSi(OtBu)2 groups of the tripodal ligand and one axial chloride ligand in a pseudo-tetrahedron geometry. The average value of the Ce–Osil distances (2.102(6) Å) is significantly shorter than those found in the Ce(III) precursor (1) and reduced species (3–5), indicating the presence of a Ce(IV) center. This value is also comparable to those found for the previously reported Ce(IV) complex [Ce(OSi(OtBu)3)4] (2.11(3) Å).10c The Ce–Ccentroid distance (2.821(2) Å) is longer than in complexes 1 and 3–5 (2.394(3)–2.730(1) Å) indicating the absence of any cerium–arene interaction. The average C–C bond length in the centroid arene (1.40(1) Å) is comparable to those found in the free ligand (L(OH)3) (1.39(2) Å) and in complex 1 (1.402(7) Å). The Ce–Cl distance (2.5709(15) Å) is similar to that found in the Ce(IV) precursor [CeCl(N(SiMe3)2)3] (2.597(2) Å).17
The solid-state structure of 3 shows the same coordination geometry and ligand environment as 1, and the structure is completed by an outer-sphere [K(2.2.2-cryptand)]+ cation, revealing that one-electron reduction has occurred. The structure contains two different disordered metal locations. The predominant position (Ce1) of the Ce cation (89%) is situated below the plane of the three oxygen atoms and the minor one (Ce2) is above the plane (11%).
In the following metrical data analysis, the data on the 89% components of 3 are reported. The average Ln–ligand distances were used in particular by the Evans group to differentiate the reduction of 4fn Ln3+ complexes to 4fn5d1 Ln2+ complexes, resulting only in small increases in distances (0.02–0.04 Å), leading to larger differences (0.10–0.20 Å).2f,13b The average Ce–Osil distance of 2.258(5) Å is only 0.02 Å longer than that found in the precursor 1 which points either to reduction to a 4fn5d1 Ce2+ complex or to an unchanged metal oxidation state.2f,13b The average values of the Ce–C distances (2.933(10) Å) and the Ce–Ccentroid distance (2.569(2) Å) are significantly shorter than those found in 1 suggesting that the Ce–arene interaction is stronger, but they remain longer than those reported for the complex [K(2.2.2-cryptand)][((Ad,MeArO)3Mes)Ce] containing Ce(III) and a reduced arene tripodal ligand (average Ce–C = 2.804 Å and Ce–Ccentroid = 2.413 Å).13b
It is noteworthy that the central arene is almost unchanged in 3 with minimal torsion angles (0.2(6)–5.3(6)°) with an average C–C bond length of 1.41(1) Å, which is comparable to that observed for the Ce(III) precursor (1.402(7) Å).
Overall, the degree of reduction of the metal cannot be unambiguously established from structural parameters.
The molecular structure of complex 4 shows a similar structure to 3, but the axial THF ligand is replaced by a Et3OP ligand. The average value of the Ce–Osil distances (2.277(5) Å), O–Ce–O bond angles (118.64(11)°) and the C–C bond distances (1.417(12) Å) are comparable to that of 3. Conversely, the average Ce–C bond lengths (2.989(10) Å) and Ce–Ccentroid distance (2.632(2) Å) are longer than those in 3, suggesting that the interaction between the arene and metal center becomes weaker due to the presence of the strong electron-donating Et3OP ligand in the axial position. The Ce–Ophosphine distance (2.439(3) Å) is in the range of Ce(III) complexes with neutral phosphine oxide ligands (2.319(3)–2.5165(9) Å).23
In complex 5, the Ce center is coordinated by the three siloxide groups of the tripodal ligand ligands, the distorted arene anchor and one Et2O molecule. One K+ cation is bound by two siloxide ligands and a second K+ cation is held by a siloxide and by interaction with one arene side-arm.
The central arene moiety η6-bound to the cerium is significantly bent, with values of the torsion angles ranging from 6.9(12)° to 18.0(11)° suggesting the presence of a reduced arene moiety. The highest values of the torsion angles are similar to those found in the reduced arene moiety of the recently reported Th(IV) [K[2.2.2]cryptand][Th(TDA)3(THF)] (TDA = N-(2,6-diisopropylphenyl)pivalamido) complex (torsion angles: 17.4(4)° and 24.2(4)°).24 The average values of Ce–C distances (2.78(2) Å) and the Ce–Ccentroid distance (2.394(3) Å) are significantly shorter than those found in the mono-reduced complex 3 revealing a stronger interaction between the arene and metal center. The average value of the Ce–Osil distances (2.363(9) Å) is longer than that found in the Ce(III) precursor (1), and it is comparable to Ce–Osil bond lengths (2.367(4)–2.376(4) Å) in the reduced [K(2.2.2-cryptand)]2[{(K(OSi(OtBu)3)3Ce)(μ-η6:η6-C7H8)}2Ce], and could suggest the presence of a reduced Ce ion, despite the effect of potassium binding to siloxides.
Computational studies
To get some insights into the bonding and concomitantly into the oxidation state of the cerium atom, DFT calculations (B3PW91) including dispersion corrections (see the ESI† for details) were carried out on complexes 1–5. The effect of alkali ion coordination on 5 was also investigated. The optimized geometries are found to be in good agreement with the experimental ones (see Tables S5, S11, S17, S24, S30 and S38 in the ESI†). The calculations were first carried out on complex 2, which is found to have a closed-shell singlet ground state, in line with Ce(IV). For complex 1, a doublet spin state was computed and the geometry is in good agreement with the experimental ones with the Ce–O distances reproduced with a 0.03 Å accuracy as well as the average C–C distances in the arene (1.40 Å). The SOMO of the system is a pure f orbital on Ce (see Table S10†) and the unpaired spin density is localized only at the cerium. This is in line with the presence of a classical Ce(III) system. Interestingly, the LUMO of the system displays a small f–arene π* interaction so that one can anticipate that reducing complex 1 may lead to mainly the arene reduction. The reduction of 1 was carried out experimentally and it forms either complex 3 or 5 depending on the number of electrons involved in the reduction (respectively 1 or 2). Two different spin states were considered for complex 3, namely a singlet and a triplet. As expected, the triplet is 17.1 kcal mol−1 more stable than the singlet. The optimized geometry is in good agreement with the experimental one with the Ce–O and Ce–C distances well reproduced with a maximum deviation of 0.06 Å. The average C–C bond distance as well as Ce–centroid distances are also nicely reproduced computationally (1.41 and 2.55 Å). In this complex a Ce–arene interaction is found as reflected by both the C–C distances which are elongated and by the Ce–C Wiberg bond index of 0.1 (to be compared with a WBI of 0.4 for the Ce–O bonds) (Table S20†). It is interesting to note that the two SOMOs (Fig. 6) a pure f (Fig. 6a) and a Ce–arene δ-bond (Fig. 6c) are similar to the frontier orbitals of complex 1 (Table S10†) but with a larger metal contribution in the Ce–arene π*, which is better described as a Ce–arene δ bond. Both f and d orbitals of cerium participate in the δ bonding. The unpaired electron density plot shows that the electron density is mainly located at the cerium atom with some residues on the arene (Fig. 6b). Therefore, the cerium oxidation state is in between +II and +III with an arene that is in between neutral and singly reduced. This is somewhat similar to that observed by the Meyer group for the U(II) complex of arene(tris-aryloxide ligand)12c where unpaired electrons were located in the δ-bond. CASSCF calculations indicate that the triplet ground state is 85% Ce(III) and 15% Ce(II). TDDFT calculations on complex 3 indicate that the absorption bands at 375, 545 and 770 nm involve excitation from the δ-bond to ligand orbitals (Fig. S82, S83 and Tables S43, S44†).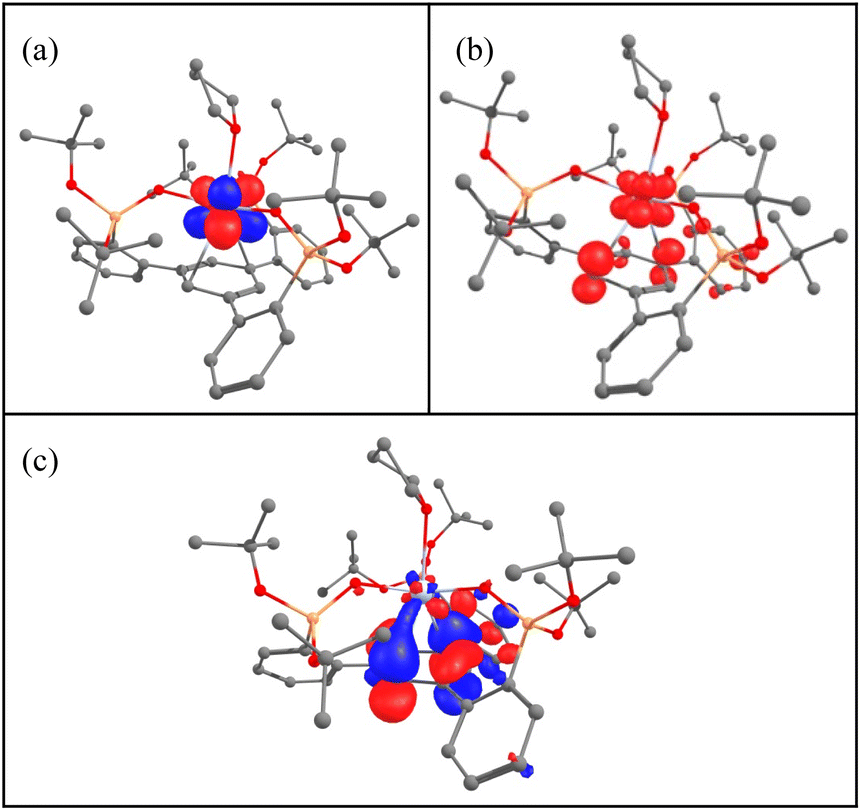 | ||
| Fig. 6 (a) and (c) SOMO and (b) unpaired spin density plot of complex 3. | ||
It is noteworthy that the replacement of a THF molecule by a phosphine oxide, complex 4, only slightly modifies the bonding situation. Indeed, the triplet is also the ground-state with some slightly longer Ce–C distances than in 3. This longer distance, also reflected by the Ce–centroid one (2.68 comp. vs. 2.63 exp), implies that the Ce–arene π* interaction in the SOMO (Table S29†) in complex 4 is weaker so that the unpaired spin density is more distributed between the cerium and the arene. Therefore, in complex 4, the oxidation state of Ce is close to +III with a singly reduced arene. The observed difference compared to complex 3 suggests that it should be possible to identify axial ligands that would favor a pure Ce(II) configuration.
In the case of the two-electrons reduction of complex 1, forming complex 5, the situation is different. Indeed, two different spin states, namely a quartet and a doublet, were considered and are found to be very close in energy. The doublet is slightly more stable than the quartet by 5.3 kcal mol−1 but interestingly both structures are in good agreement with the experimental one (see Table S31 in the ESI†). This can be easily explained by checking the frontier orbitals in both spin states (Table S36†). In fact, in the doublet ground state, a singly occupied f orbital at the cerium center is found and the δ-bonding interaction is doubly occupied. Therefore, the arene is doubly reduced with the presence of a Ce(III) center. Interestingly, removing the potassium atoms from complex 5 results in a ground state switch, with the quartet becoming the ground state (Table S37†). In the quartet, 2 unpaired electrons are located on two different δ bonds and the unpaired spin density is now more localized on the cerium center (Table S42†) so that one would conclude that the cerium is in the oxidation state +II rather than +III. This is in line with the experimental observation. However, the storage of the electrons onto the δ-interaction allows complexes 3, 4 and 5 to be described as either masked Ce(II) complexes for the two former and as masked Ce(I) for the latter.
Redox reactivity
With the low-valent tripodal complexes in hand, we performed preliminary reactivity studies to investigate if the electrons stored in the complexes 3 and 5 could become available for the reduction of substrates (Scheme 4).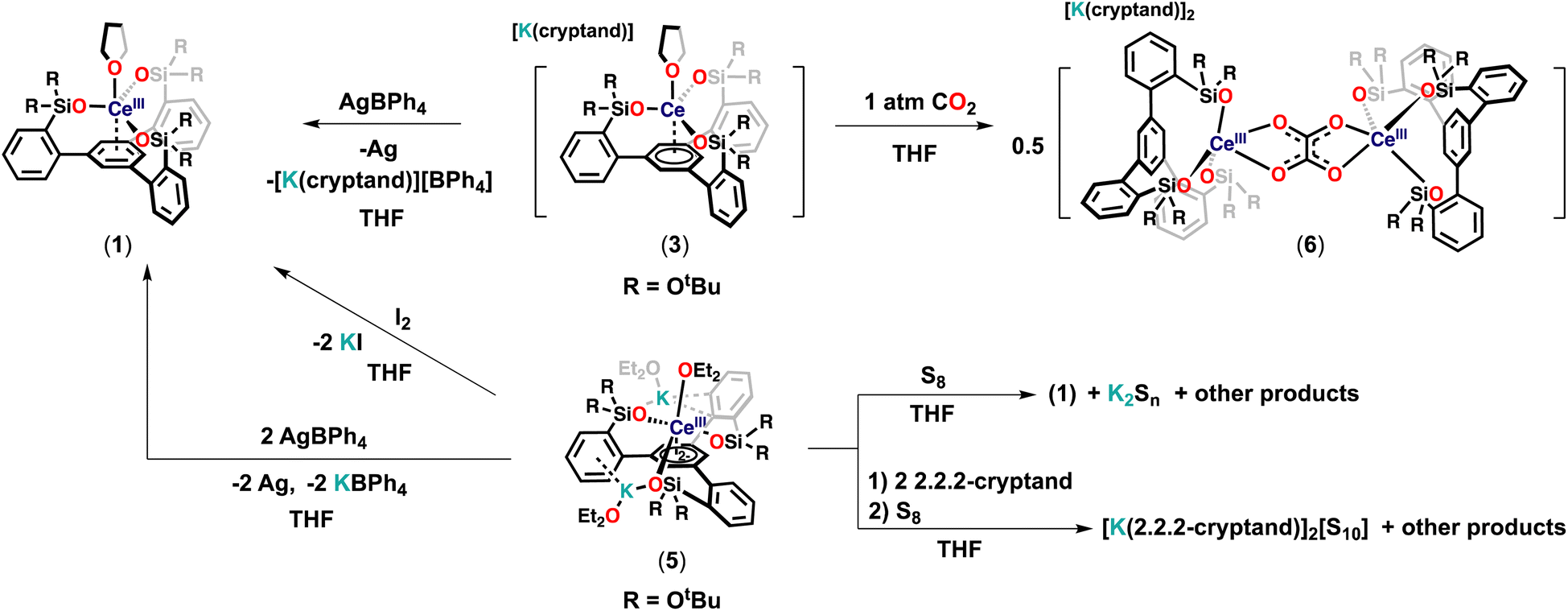 | ||
| Scheme 4 Reactivity of complexes 3 and 5. | ||
At first, we reacted complex 3 with 1 equivalent of AgBPh4, leading immediately to a color change from dark-blue to light-yellow. The 1H NMR spectrum of the reaction mixture revealed the clean formation of 1 (Fig. S46†). The observed reactivities showed that the electron stored in the complex 3 can be transferred to oxidizing substrates.
Then, we investigated the reduction of CO2 as an example of a less reactive substrate.
The addition of 1 atm CO2 to a THF solution of 3 led to an immediate color change from dark-blue to colorless, full consumption of the starting material and the formation of a new species as indicated by 1H NMR studies (Fig. S47†). Colorless crystals of the complex [K(2.2.2-cryptand)]2[{((LO3)Ce)2(μ-C2O4)}] 6 were isolated in 52% yield from concentrated THF at room temperature. The conversion, as measured by 1H NMR spectroscopy (using TMS2O as the internal standard), is 73% (Fig. S48†). The solid-state structure of 6 (Fig. 7), determined by X-ray diffraction studies showed the presence of a dinuclear complex consisting of two equivalent Ce(III) ions, bridged by one oxalate ligand. Each Ce(III) center is penta-coordinated in a distorted square pyramidal geometry and is bound by three –OSi(OtBu)2R (R = C6H4) ligands and one bridging oxalate.
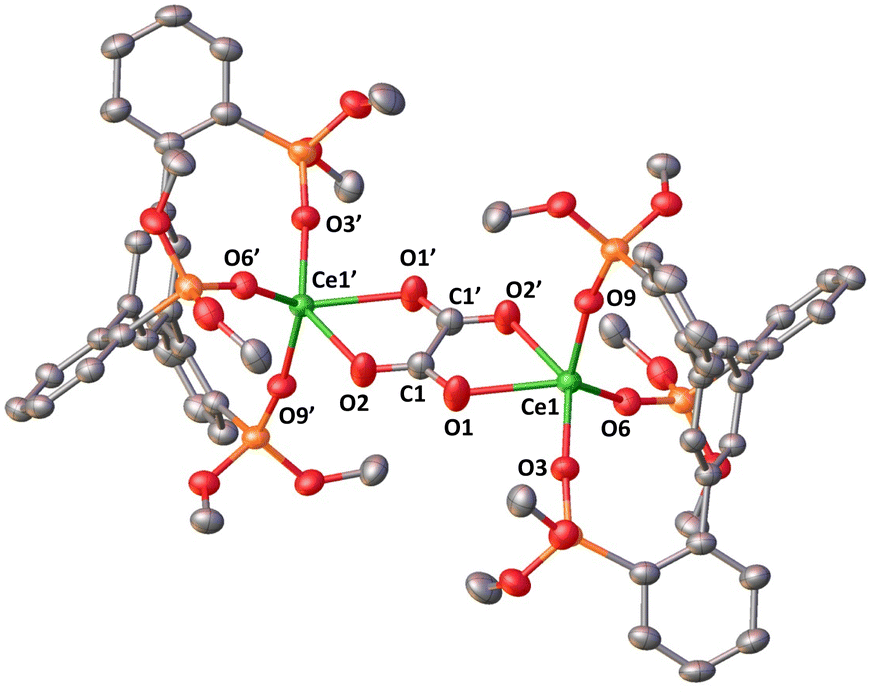 | ||
| Fig. 7 Molecular structure of 6, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and methyl groups on the –OSi(OtBu)2 ligands and [K(2.2.2-cryptand)] have been omitted for clarity. | ||
Two [K(2.2.2-cryptand)] cations complete the structure. The average Ce–Osil distance of 2.235(2) Å is comparable to that found in the Ce(III) precursor (1). The Ce–Ooxalate (2.465(2) Å) distances are shorter than those found in previously reported oxalate-bridged cerium(III) polymeric complexes (2.515(13)–2.62(5) Å) prepared from the reaction of oxalate with cerium salts.25
The C1–C1′ bond length is 1.536(4) Å which is consistent with the formation of a single C–C bond (1.54 Å in ethane). Moreover, the Ce–Ccentroid distance (3.547(1) Å) is significantly longer than that of complex 1 with a Ce–Ccentroid distance of 2.730(1) Å, indicating that there is no interaction between the arene and metal center. The formation of complex 6 indicates that a CO2 coupling reaction occurs after the reduction of two molecules of CO2 by two molecules of complex 3. Notably, complex 6 is the only isolated product and the major product in the reaction mixture. This is the first example of CO2 reduction by a low valent cerium complex and only the third example of CO2 reduction to oxalate by a low-valent lanthanide complex.26 In previous studies the reduction of CO2 by Sm(II) Yb(II) or Eu(II) complexes led either to carbonate alone27 or to a mixture of carbonate and oxalate depending on the supporting ligand.28
Following isolation of complex 5, the addition of 2 equivalents of AgBPh4 to 5 led to a clean regeneration of 1, with concomitant formation of KBPh4 and Ag (Fig. S50†) suggesting that 5 could potentially act as a two-electron reductant. Moreover, complex 5 reacted with 1 equivalent of I2, leading immediately to a color change from dark-purple to light-yellow and the precipitation of KI. The 1H NMR spectrum of the reaction mixture revealed the clean formation of 1 (Fig. S51†). The conversion, as measured by 1H NMR spectroscopy (using TMS2O as the internal standard), is 99% (Fig. S52†).
Furthermore, the reaction of 1 equiv. of S8 with a THF solution of complex 5 at room temperature resulted immediately in a light-yellow solution. 1H NMR studies showed that complex 1 is formed (Fig. S53†) but the reduced sulfur products could not be isolated.
However, when the same reaction was performed in the presence of 2 equiv. of 2.2.2-cryptand at room temperature, it resulted immediately in a dark-red reaction mixture. Full consumption of the starting material and the formation of the multiple species were observed in the 1H NMR spectrum (Fig. S54†). The recrystallization of the reaction mixture from concentrated THF at room temperature afforded few orange crystals of the S10 dianion [K(2.2.2-cryptand)]2[S10]29 as confirmed by X-ray diffraction. These results show that complex 5 can act as a two-electron reductant masked “Ce(I)”. Complex 5 was found to react also with less oxidizing substrates such as azobenzene and phenylacetylene but the isolation of the resulting products was not successful so far.
Conclusions
In conclusion, we synthesized a new tripodal tert-butoxysiloxide ligand that incorporates an arene ring as part of a potentially redox-active chelating ligand and showed that this ligand allows the promotion of multielectron redox reactivity in cerium complexes and the stabilization of cerium complexes in four different redox states.Remarkably the one-electron reduction and the unprecedented two-electron reduction of the tripodal Ce(III) complex are easily achieved to yield complexes that are formally “Ce(II)” and “Ce(I)” analogues. Structural, spectroscopic and computational studies indicated that the neutral complexes 1 and 2 show the presence of cerium in +3 and +4 oxidation states with respectively little and no interaction of the metal center with the arene ring. The assignment of the formal state of cerium is more ambiguous for the mono-reduced complex 3 and is better described as an intermediate between +2 and +3 with partial localization of electrons on the ligand and a Ce–arene δ-interaction. In complex 5 the arene is doubly reduced with the presence of a Ce(III) center, but the removal of potassium results in a Ce(II)(arene−) species. However, the storage of electrons onto the δ-interaction allows the description of complexes 3 and 5 as masked “Ce(II)” and “Ce(I)” complexes respectively. Preliminary reactivity studies show that these complexes can act as masked Ce(II) and Ce(I) effecting both one- and two-electron transfer that were not accessible in classical cerium chemistry. These results demonstrate that the high stability of the arene tripodal ligand under reducing and oxidizing conditions is well poised to promote the development of f element multielectron redox chemistry.
Data availability
Synthetic details and analytical data including depictions of all spectra and coordinate data of all computationally optimised species are documented in the ESI. Crystallographic data are made available via the CCDC.† The data that support the findings of this study are openly available in the Zenodo repository at https://doi.org/10.5281/zenodo.7869094.Author contributions
F.-C. H. designed and carried out all the experiments and analyzed the data; M. M. designed and supervised the project; T. R. and L. M. carried out the computational study; R. S. measured and analyzed the X-ray data; A. S. measured the EPR data. F.-C. H., L. M, and M. M. wrote the manuscript with contributions of all authors, and all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200020_212723 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Farzaneh Fadaei-Tirani for contributions to the X-ray single crystal structure analyses. We thank Maxime Tricoire for help with EPR analysis and Anne-Sophie Chauvin and Romain Aeschlimann for the synthesis of batches of the ligand. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.Notes and references
- J. C. Wedal and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 18354–18367 CrossRef CAS PubMed.
- (a) L. M. Anderson-Sanchez, J. M. Yu, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chem., 2023, 62, 706–714 Search PubMed; (b) W. J. Evans, N. T. Allen and J. W. Ziller, Angew. Chem., Int. Ed. Engl., 2002, 41, 359–360 CrossRef CAS PubMed; (c) P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed. Engl., 2008, 47, 1488–1491 CrossRef CAS PubMed; (d) F. Jaroschik, A. Momin, F. Nief, X. F. Le Goff, G. B. Deacon and P. C. Junk, Angew. Chem., Int. Ed. Engl., 2009, 48, 1117–1121 CrossRef CAS PubMed; (e) M. R. MacDonald, J. E. Bates, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 8420–8423 CrossRef CAS PubMed; (f) M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2015, 137, 369–382 CrossRef CAS PubMed; (g) K. R. Meihaus, M. E. Fieser, J. F. Corbey, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2015, 137, 9855–9860 CrossRef CAS PubMed; (h) M. E. Fieser, C. T. Palumbo, H. S. La Pierre, D. P. Halter, V. K. Voora, J. W. Ziller, F. Furche, K. Meyer and W. J. Evans, Chem. Sci., 2017, 8, 7424–7433 RSC; (i) A. J. Ryan, L. E. Darago, S. G. Balasubramani, G. P. Chen, J. W. Ziller, F. Furche, J. R. Long and W. J. Evans, Chem.–Eur. J., 2018, 24, 7702–7709 CrossRef CAS PubMed; (j) D. N. Huh, S. R. Ciccone, S. Bekoe, S. Roy, J. W. Ziller, F. Furche and W. J. Evans, Angew. Chem., Int. Ed. Engl., 2020, 59, 16141–16146 CrossRef CAS PubMed; (k) D. N. Huh, J. P. Bruce, S. G. Balasubramani, S. R. Ciccone, F. Furche, J. C. Hemminger and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 16610–16620 CrossRef CAS PubMed.
- P. L. Arnold, F. G. N. Cloke, P. B. Hitchcock and J. F. Nixon, J. Am. Chem. Soc., 1996, 118, 7630–7631 CrossRef CAS.
- L. Barluzzi, S. R. Giblin, A. Mansikkamaki and R. A. Layfield, J. Am. Chem. Soc., 2022, 144, 18229–18233 CrossRef CAS PubMed.
- J. D. Martin and J. D. Corbett, Angew. Chem., Int. Ed. Engl., 1995, 34, 233–235 CrossRef CAS.
- (a) X. Chen, T. T. Chen, W. L. Li, J. B. Lu, L. J. Zhao, T. Jian, H. S. Hu, L. S. Wang and J. Li, Inorg. Chem., 2019, 58, 411–418 CrossRef CAS PubMed; (b) W. L. Li, T. T. Chen, W. J. Chen, J. Li and L. S. Wang, Nat. Commun., 2021, 12, 6467 CrossRef CAS PubMed.
- (a) E. J. Schelter, Nat. Chem., 2013, 5, 348 CrossRef PubMed; (b) N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS; (c) J. A. Bogart, C. A. Lippincott, P. J. Carroll, C. H. Booth and E. J. Schelter, Chem.–Eur. J., 2015, 21, 17850–17859 CrossRef CAS PubMed; (d) L. Mathey, M. Paul, C. Coperet, H. Tsurugi and K. Mashima, Chem.–Eur. J., 2015, 21, 13454–13461 CrossRef CAS PubMed; (e) T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed; (f) J. R. Robinson, Y. S. Qiao, J. Gu, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Sci., 2016, 7, 4537–4547 RSC; (g) H. L. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984–5993 CrossRef CAS PubMed; (h) R. Anwander, M. Dolg and F. T. Edelmann, Chem. Soc. Rev., 2017, 46, 6697–6709 RSC; (i) R. L. Halbach, G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Inorg. Chem., 2018, 57, 7290–7298 CrossRef CAS PubMed; (j) Q. M. Yang, Y. H. Wang, Y. S. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS PubMed.
- M. A. Angadol, D. H. Woen, C. J. Windorff, J. W. Ziller and W. J. Evans, Organometallics, 2019, 38, 1151–1158 CrossRef CAS.
- T. F. Jenkins, D. H. Woen, L. N. Mohanam, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2018, 37, 3863–3873 CrossRef CAS.
- (a) Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Organometallics, 2000, 19, 2832–2834 CrossRef; (b) C. M. Kotyk, M. E. Fieser, C. T. Palumbo, J. W. Ziller, L. E. Darago, J. R. Long, F. Furche and W. J. Evans, Chem. Sci., 2015, 6, 7267–7273 RSC; (c) R. P. Kelly, L. Maron, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2017, 56, 15663–15666 CrossRef CAS PubMed.
- (a) C. Camp, V. Guidal, B. Biswas, J. Pecaut, L. Dubois and M. Mazzanti, Chem. Sci., 2012, 3, 2433–2448 RSC; (b) I. L. Fedushkin, O. V. Maslova, A. G. Morozov, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed. Engl., 2012, 51, 10584–10587 CrossRef CAS PubMed; (c) E. J. Coughlin, M. Zeller and S. C. Bart, Angew. Chem., Int. Ed. Engl., 2017, 56, 12142–12145 CrossRef CAS PubMed; (d) I. L. Fedushkin, A. N. Lukoyanov and E. V. Baranov, Inorg. Chem., 2018, 57, 4301–4309 CrossRef CAS PubMed; (e) S. S. Galley, S. A. Pattenaude, R. F. Higgins, C. J. Tatebe, D. A. Stanley, P. E. Fanwick, M. Zeller, E. J. Schelter and S. C. Bart, Dalton Trans., 2019, 48, 8021–8025 RSC; (f) N. Jori, D. Toniolo, B. C. Huynh, R. Scopelliti and M. Mazzanti, Inorg. Chem. Front., 2020, 7, 3598–3608 RSC; (g) G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed.
- (a) S. C. Bart, F. W. Heinemann, C. Anthon, C. Hauser and K. Meyer, Inorg. Chem., 2009, 48, 9419–9426 CrossRef CAS PubMed; (b) O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC; (c) H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed. Engl., 2014, 53, 7158–7162 CrossRef CAS PubMed.
- (a) D. P. Halter, C. T. Palumbo, J. W. Ziller, M. Gembicky, A. L. Rheingold, W. J. Evans and K. Meyer, J. Am. Chem. Soc., 2018, 140, 2587–2594 CrossRef CAS PubMed; (b) C. T. Palumbo, D. P. Halter, V. K. Voora, G. P. Chen, A. K. Chan, M. E. Fieser, J. W. Ziller, W. Hieringer, F. Furche, K. Meyer and W. J. Evans, Inorg. Chem., 2018, 57, 2823–2833 CrossRef CAS PubMed.
- R. R. Thompson, M. E. Rotella, P. Du, X. Zhou, F. R. Fronczek, R. Kumar, O. Gutierrez and S. Lee, Organometallics, 2019, 38, 4054–4059 CrossRef CAS.
- (a) V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862–1891 CrossRef CAS PubMed; (b) A. H. Hu, J. J. Guo, H. Pan and Z. W. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- Y. M. So and W. H. Leung, Coord. Chem. Rev., 2017, 340, 172–197 CrossRef CAS.
- O. Eisenstein, P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and L. Maron, Chem. Commun., 2001, 1560–1561 RSC.
- T. A. Albright, W. J. Freeman and E. E. Schweizer, J. Org. Chem., 1975, 40, 3437–3441 CrossRef CAS.
- N. T. Rice, J. Su, T. P. Gompa, D. R. Russo, J. Telser, L. Palatinus, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS PubMed.
- J. Mayans, L. Tesi, M. Briganti, M. E. Boulon, M. Font-Bardia, A. Escuer and L. Sorace, Inorg. Chem., 2021, 60, 8692–8703 CrossRef CAS PubMed.
- J. Friedrich, C. Maichle-Mossmer and R. Anwander, Chem. Commun., 2017, 53, 12044–12047 RSC.
- (a) G. B. Deacon, T. C. Feng, C. M. Forsyth, A. Gitlits, D. C. R. Hockless, S. Qi, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2000, 961–966 RSC; (b) M. Gorlov, L. L. Hussami, A. Fischer and L. Kloo, Eur. J. Inorg. Chem., 2008, 5191–5195 CrossRef CAS; (c) A. N. Selikhov, A. V. Cherkasov, K. A. Lyssenko and A. A. Trifonov, Organometallics, 2022, 41, 820–828 CrossRef CAS.
- (a) J. C. Berthet, M. Nierlich and M. Ephritikhine, Polyhedron, 2003, 22, 3475–3482 CrossRef CAS; (b) A. Bowden, P. N. Horton and A. W. G. Platt, Inorg. Chem., 2011, 50, 2553–2561 CrossRef CAS PubMed; (c) J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed; (d) R. D. Bannister, W. Levason and G. Reid, Chemistry, 2020, 2, 947–959 CrossRef CAS.
- M. D. Straub, E. T. Ouellette, M. A. Boreen, R. D. Britt, K. Chakarawet, I. Douair, C. A. Gould, L. Maron, I. Del Rosal, D. Villarreal, S. G. Minasian and J. Arnold, J. Am. Chem. Soc., 2021, 143, 19748–19760 CrossRef CAS PubMed.
- L. Vella-Zarb and U. Baisch, Z. Anorg. Allg. Chem., 2017, 643, 1712–1716 CrossRef CAS.
- (a) W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770–776 CrossRef CAS; (b) W. J. Evans, J. M. Perotti, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 5204–5212 CrossRef CAS PubMed.
- (a) N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Chem. Commun., 2006, 4853–4855 RSC; (b) A. R. Willauer, A. M. Dabrowska, R. Scopelliti and M. Mazzanti, Chem. Commun., 2020, 56, 8936–8939 RSC.
- (a) J. Andrez, J. Pecaut, P.-A. Bayle and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2014, 53, 10448–10452 CrossRef CAS PubMed; (b) M. Xemard, V. Goudy, A. Braun, M. Tricoire, M. Cordier, L. Ricard, L. Castro, E. Louyriac, C. E. Kefalidis, C. Clavaguera, L. Maron and G. Nocton, Organometallics, 2017, 36, 4660–4668 CrossRef CAS; (c) A. R. Willauer, D. Toniolo, F. Fadaei-Tirani, Y. Yang, M. Laurent and M. Mazzanti, Dalton Trans., 2019, 48, 6100–6110 RSC; (d) A. R. Willauer, F. Fadaei-Tirani, I. Zivkovic, A. Sienkiewicz and M. Mazzanti, Inorg. Chem., 2022, 61, 7436–7447 CrossRef CAS PubMed.
- M. K. Mondal, L. Zhang, Z. T. Feng, S. X. Tang, R. Feng, Y. Zhao, G. W. Tan, H. P. Ruan and X. P. Wang, Angew. Chem., Int. Ed. Engl., 2019, 58, 15829–15833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2239278–2239286 and 2240543. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01478a |
| This journal is © The Royal Society of Chemistry 2023 |