
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalysed enantio- and regioselective (3 + 2) cycloaddition reactions of sulfamidate imine-derived 1-azadienes towards spirocyclic cyclopentanes†
Quoc Hoang
Pham
 a,
Andrew J.
Tague
a,
Christopher
Richardson
a,
Michael G.
Gardiner
b,
Stephen G.
Pyne
*a and
Christopher J. T.
Hyland
*a
a,
Andrew J.
Tague
a,
Christopher
Richardson
a,
Michael G.
Gardiner
b,
Stephen G.
Pyne
*a and
Christopher J. T.
Hyland
*a
aSchool of Chemistry and Molecular Bioscience, Molecular Horizons Research Institute, University of Wollongong, Wollongong, 2522, New South Wales, Australia. E-mail: chrhyl@uow.edu.au; spyne@uow.edu.au
bResearch School of Chemistry, The Australian National University, Canberra, 2601, Australia
First published on 18th April 2023
Abstract
An enantio- and diastereoselective Pd-catalysed (3 + 2) cycloaddition of bis(trifluoroethyl) 2-vinyl-cyclopropane-1,1-dicarboxylate (VCP) with cyclic sulfamidate imine-derived 1-azadienes (SDAs) has been developed. These reactions provide highly functionalized spiroheterocycles having three contiguous stereocentres, including a tetrasubstituted carbon bearing an oxygen functionality. The two geminal trifluoroethyl ester moieties can be manipulated in a facially selective manner to afford more diversely decorated spirocycles with four contiguous stereocentres. In addition, diastereoselective reduction of the imine moiety can also afford a fourth stereocentre and exposes the important 1,2-amino alcohol functionality.
Introduction
The sterically congested and highly functionalised cyclopentane scaffold is among the most encountered architectural features in natural products, bioactive compounds, and medicinally relevant molecules (Fig. 1).1–3 Due to their importance, synthetic protocols enabling efficient access to this privileged motif with high stereocontrol are indispensable and highly sought after. Among these, a commonly employed strategy concerns the use of donor–acceptor cyclopropanes (DACs) as C3 synthons in (3 + 2) cycloaddditions, which allow the effective construction of these five-membered carbocycles4–8 as well as their heterocyclic counterparts9–13 in a stereoselective and atom economical fashion. Within the array of structurally diverse DACs lies the vinylcyclopropane (VCP) scaffold, which readily ring-open in the presence of a transition metal catalyst such as palladium to afford a metal-bound zwitterionic π-allyl dipole that can undergo (3 + 2) cycloaddition with an array of dipolarophiles to furnish five-membered rings bearing a synthetically valuable vinyl handle.14–20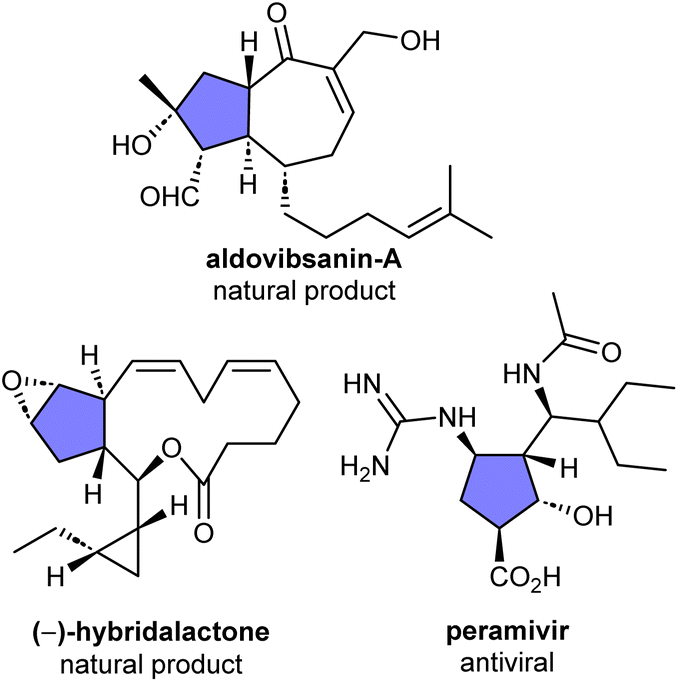 | ||
| Fig. 1 Selected examples of naturally occurring and medicinally relevant compounds containing a densely functionalized cyclopentane core. | ||
A wide range of dipolarophiles have been utilised in conjunction with VCP-derived 1,3-dipoles,21–43 enabling the synthesis of fused-23–26,30,43 and spirocyclic cyclopentanes and 5-membered heterocycles via (3 + 2) cycloaddition reactions.33,37,39,41,42 Among these, benzofuran-derived azadienes (BDAs) are an intriguing potential class of dipolarophiles for Pd-catalysed (3 + 2) reactions for cyclopentane synthesis.31,34 However, BDAs often act as four-atom synthons in (4 + n) cycloadditions to give larger ring systems via an aromatic benzofuran intermediate I (Fig. 2a).44–55 In comparison, their employment as two-atom synthons has been scarcer,31,34,47,50 among which there are only two examples of (3 + 2) cycloaddition reactions with BDAs known, with chemoselectivity for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N versus CC addition being a potential challenge.31,34 The preference for (4 + n) reactions of BDAs is due to the favoured attack of the intermediate sulfonamide anion on the pendant Pd-allyl complex to preserve the aromatic benzofuran moiety (highlighted in grey). We therefore designed a new azadiene dipolarophile that would completely preclude (4 + n) reactivity and enable reliable (3 + 2) processes with VCPs to be realised. From our experience with cyclic sulfamidate imines,56 we envisaged that sulfamidate imine-derived azadienes (SDAs) would be an intriguing alternative to the BDA system that would completely preclude the (4 + n) reaction mode and give highly functionalised cyclopentanes (Fig. 2b).57,58 Such (3 + 2) cycloadditions would proceed via a non-aromatic intermediate II, where attack by the nitrogen on the pendant π-allyl palladium complex would be unlikely as it would result in a strained bicyclic product. Furthermore, in contrast to BDA cycloaddition products, the oxygen substituent in the SDA-derived cyclopentanes would not be attached to an aromatic ring. We envision that this method will open up new (3 + 2) reaction manifolds for aza-dienes and find wide application in target synthesis of densely functionalised bioactive cyclopentanes.
N versus CC addition being a potential challenge.31,34 The preference for (4 + n) reactions of BDAs is due to the favoured attack of the intermediate sulfonamide anion on the pendant Pd-allyl complex to preserve the aromatic benzofuran moiety (highlighted in grey). We therefore designed a new azadiene dipolarophile that would completely preclude (4 + n) reactivity and enable reliable (3 + 2) processes with VCPs to be realised. From our experience with cyclic sulfamidate imines,56 we envisaged that sulfamidate imine-derived azadienes (SDAs) would be an intriguing alternative to the BDA system that would completely preclude the (4 + n) reaction mode and give highly functionalised cyclopentanes (Fig. 2b).57,58 Such (3 + 2) cycloadditions would proceed via a non-aromatic intermediate II, where attack by the nitrogen on the pendant π-allyl palladium complex would be unlikely as it would result in a strained bicyclic product. Furthermore, in contrast to BDA cycloaddition products, the oxygen substituent in the SDA-derived cyclopentanes would not be attached to an aromatic ring. We envision that this method will open up new (3 + 2) reaction manifolds for aza-dienes and find wide application in target synthesis of densely functionalised bioactive cyclopentanes.
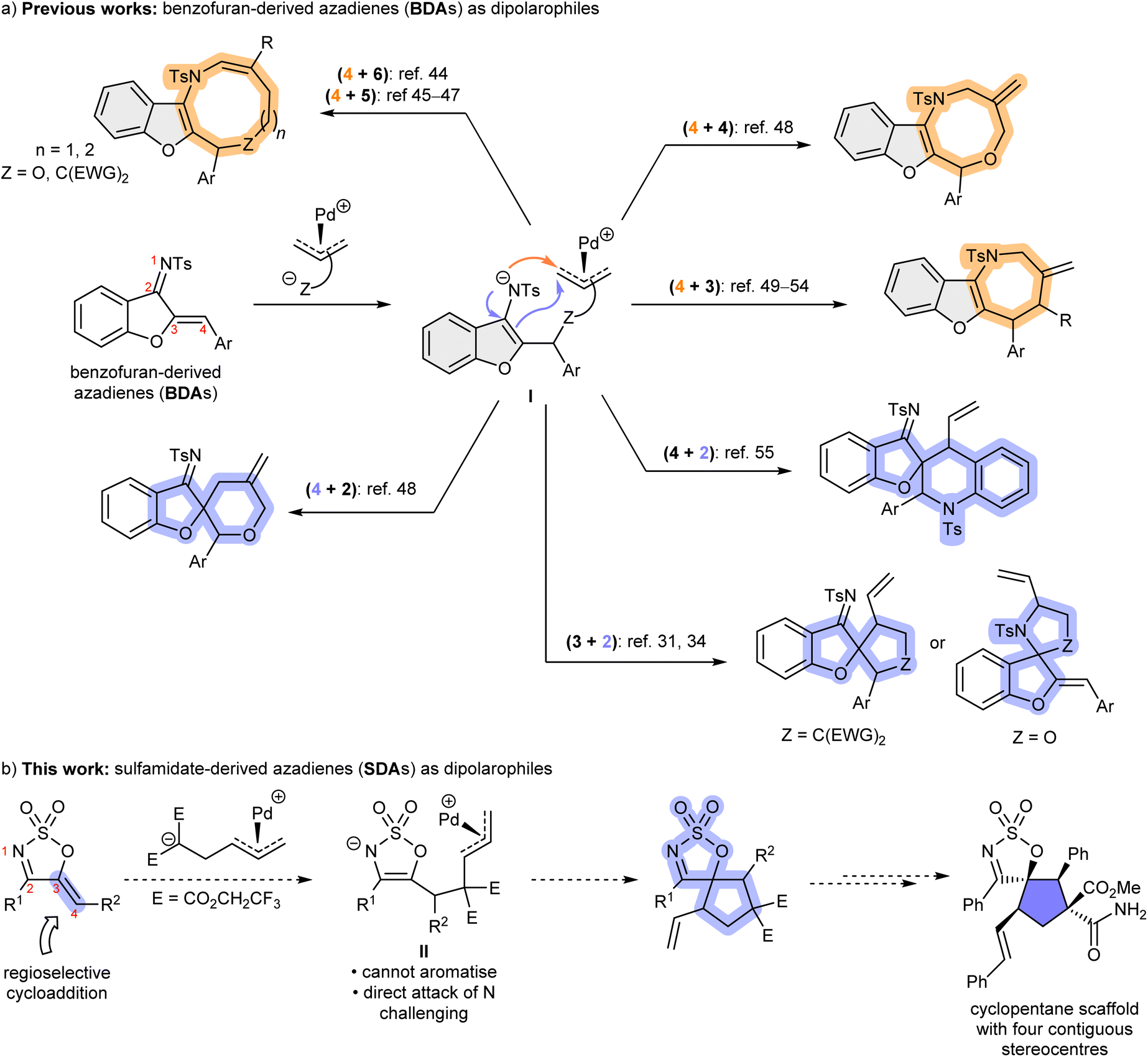 | ||
| Fig. 2 (a) Prior work on the different reaction modes of BDAs as dipolarophiles in Pd-catalysed cycloadditions. (b) This work: SDA scaffolds as dipolarophiles in Pd-catalysed (3 + 2) cycloadditions with VCP-derived 1,3-dipoles. | ||
Herein, we report this new class of dipolarophiles successfully undergoing chemo- and enantioselective (3 + 2) cycloaddition with the exocyclic CC bond to give exclusively stereodefined and densely functionalised spirocyclic cyclopentanes containing three-contiguous stereogenic centres. In addition, we can demonstrate that these cycloadducts—having a structurally rigid skeleton decorated with useful synthetic handles such as the imine, vinyl, and geminal diester moieties—can undergo further stereoselective transformations to create an additional stereogenic centre.
Results and discussion
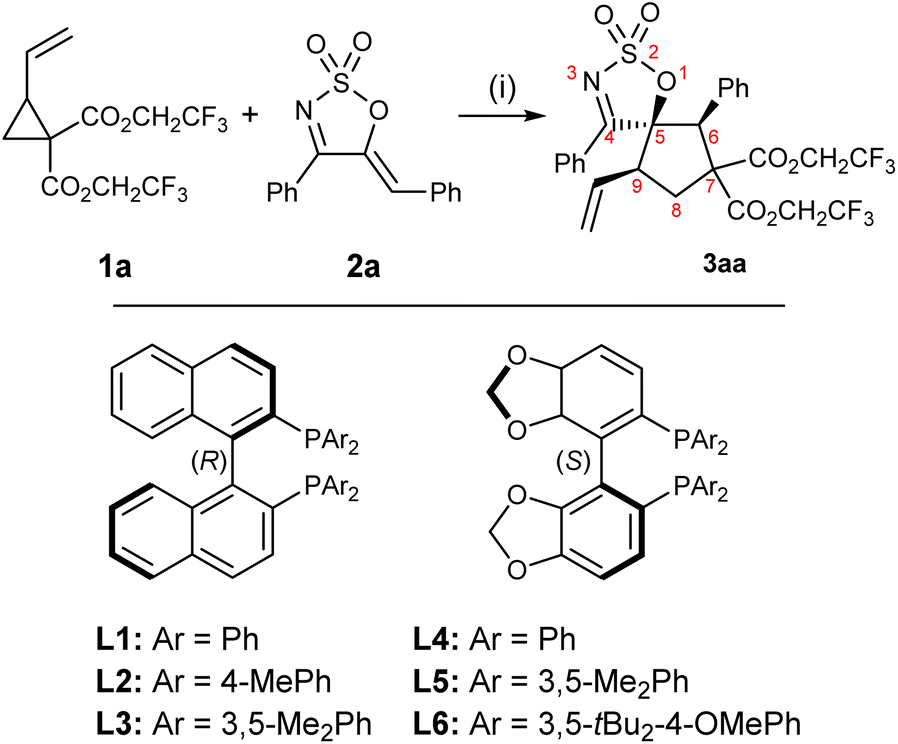
A preliminary optimisation, carried out using VCP 1a59 and cyclic 1-azadiene 2a as model substrates, identified toluene as the solvent of choice (see the ESI† for full optimisation details). Azadiene 2a was readily accessible from the corresponding C5-unsubstituted cyclic sulfamidate imine via a Knoevenagel condensation.60 A selection of C2 symmetric chiral ligands were then screened in toluene (Table 1). BINAP analogues L2 and L3 both performed well, furnishing the product in high yields and drs, but slightly lower ers compared to L1 (entries 1–3). Among the three SEGPHOS-type ligands L4–6 tested (entries 4–6), (S)-DM-SEGPHOS L5 was found to be the most suitable candidate, affording the desired product in high yield, good dr and high er (entry 5). Encouraged by the results obtained with L5, further fine tuning of other reaction conditions was attempted with this ligand. Performing the transformation at 0 °C or −10 °C led to a significant decline in dr, er and yield (entries 7 and 8). The diastereoselectivity of the reaction was found to be concentration-dependent: performing the reaction at a higher concentration resulted in a significant decline in dr (entry 9), whereas a lower reaction concentration allowed for better diastereocontrol (entry 10). Gratifyingly, a slight increase in the amount of VCP (from 1.2 to 1.5 equivalents), to account for any potential polymerisation, allowed the reaction to achieve near quantitative yield (entry 11). The results obtained by variations of the Pd0 source and catalytic loadings were all inferior to that outlined in entry 11 (see the ESI†).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Yieldb | drc | erd |
| a Reaction conditions: 1a (0.15 mmol, 1.2 equiv.), 2a (0.1 mmol, 1.0 equiv.), Pd2dba3·CHCl3 (2.5 mol%), ligand (7.5 mol%), PhMe (0.067 M w.r.t. 2a), rt, 24 h. b Calculated yields are provided in parentheses and were determined by 1H NMR integration against an internal standard (1,3,5-trimethoxybenzene). c Determined by 1H NMR analysis of the crude reaction mixture. d Enantiomeric ratio was determined by chiral HPLC. e Reaction was carried out at 0 °C. f Reaction was carried out at −10 °C. g Reaction concentration was doubled (0.134 M w.r.t. 2a). h Reaction concentration was halved (0.034 M w.r.t. 2a). i 1.5 Equiv. of 1a used. | ||||
| 1 | L1 | 36% (71%) | 7.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
7:93 |
| 2 | L2 | 81% (87%) | 8.7:1 |
11:89 |
| 3 | L3 | 74% (91%) | 7.1:1 |
12:88 |
| 4 | L4 | 76% (80%) | 7.1:1 |
90:10 |
| 5 | L5 | 84% (98%) | 7.3:1 |
94:6 |
| 6 | L6 | 34% (31%) | 1:1.2 |
94:6 |
| 7e | L5 | 92% (84%) | 3.8:1 |
90:10 |
| 8f | L5 | 85% (81%) | 2.7:1 |
81:91 |
| 9g | L5 | 84% (88%) | 5.4:1 |
93:7 |
| 10h | L5 | 77% (83%) | 8.5:1 |
94:6 |
| 11h,i | L5 | 99% (99%) | 8.0:1 |
95:5 |
The optimum VCP was then examined (Scheme 1). The styryl derivative 1b was found to have comparable reactivity to 1a, affording the product in excellent yield, although in significantly poorer dr and er. The reaction of the diethyl malonate substrate 1c was remarkably lower yielding compared to its bis(trifluoro)ethyl counterparts 1a and 1b, affording 3ca in a modest 43% yield, 2.3:1 dr and 82:12 er. Interestingly, when the malononitrile derivative 1d was tested, the trans-diastereoisomer 3da′ was formed preferentially.59 Among the three spirocyclic VCPs 1e–g subjected to the optimised reaction conditions, only 1e reacted to afford the desired product (3ea) but in moderate yield, poor dr, and modest er. No reaction took place when either 1f or 1g was employed as the reactant, and the decomposition of these VCPs was noted in both cases. These results highlight the VCP as the optimum reactant.
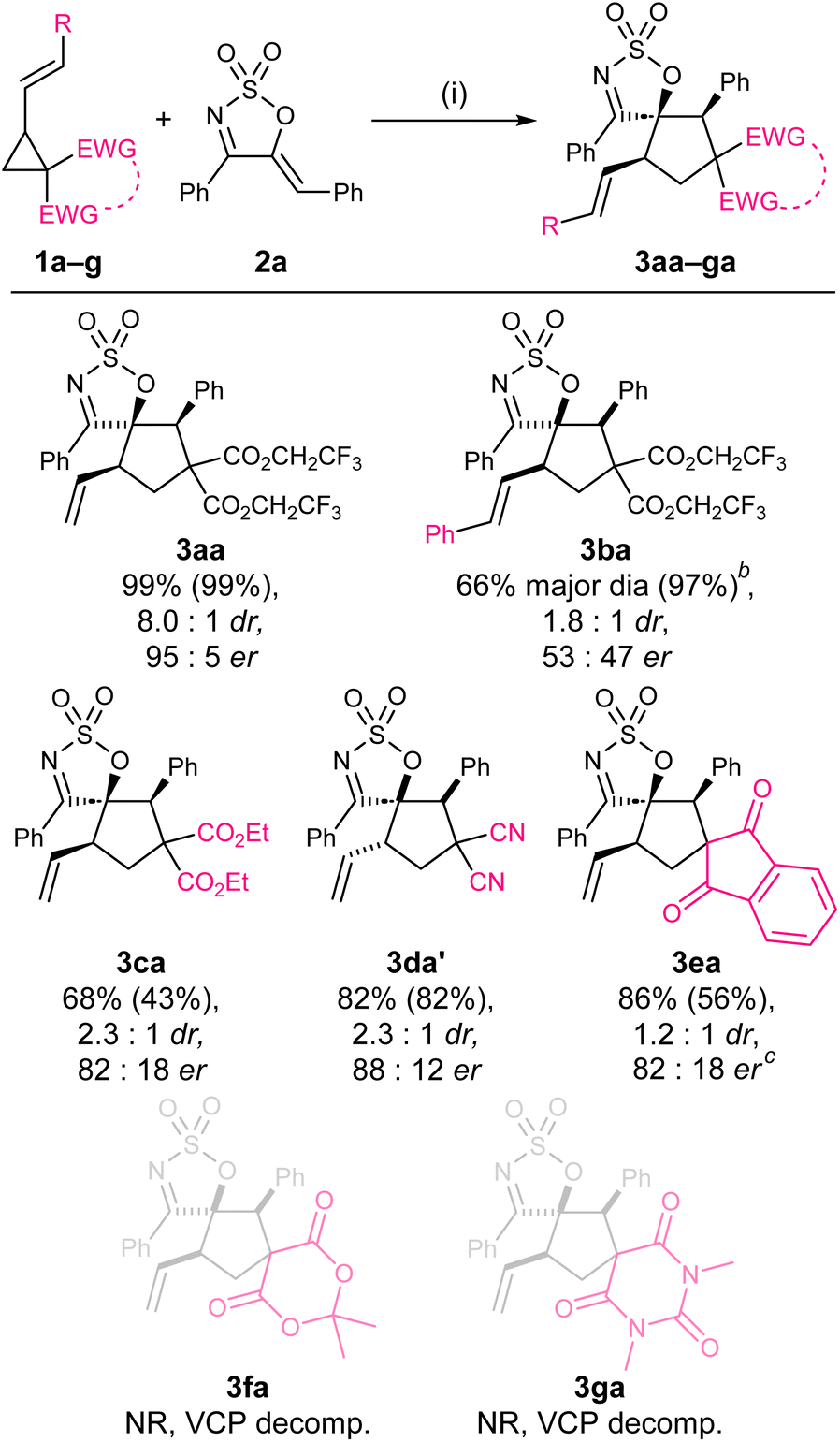 | ||
| Scheme 1 Screening of various VCPs. aReaction conditions: (i) 1a (0.15 mmol, 1.5 equiv.), 2a (0.1 mmol, 1.0 equiv.), Pd2dba3·CHCl3 (2.5 mol%), L5 (7.5 mol%), PhMe (0.034 M w.r.t. 2a), rt, 24 h. Calculated yields are provided in parentheses and were determined by 1H NMR integration against an internal standard (1,2,3-trimethoxybenzene). The diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. The enantiomeric ratio was determined by chiral HPLC. bThe two diastereoisomers are separable by chromatography. cLowest estimation due to HPLC traces of the two diastereoisomers being undistinguishable. | ||
With the optimised conditions and VCP in hand, the scope of cyclic sulfamidate imine-derived 1-azadiene was next investigated (Scheme 2). 1-Azadienes with electronically diverse aryl substituents at the C4 position (R1) were first examined, and were generally well tolerated. Almost all of these substrates (with the exception of 2f) furnished the desired (3 + 2) cycloaddition product in lower drs compared to the parent substrate 2a. While electron rich 1-azadiene 2b reacted smoothly under the optimised reaction conditions, the reactions of 2c and 2d—each bearing an electron deficient C4 aryl moiety—were both sluggish at rt; therefore, heating under reflux conditions was required. In stark contrast to the low reactivity of 2d, the reactions of its analogues 2e and 2f were significantly more efficient, furnishing the desired products in good yields, drs, and ers. The excellent diastereocontrol achieved with substrate 2f was particularly remarkable, which is hypothesised to be due to the sterically demanding ortho-bromophenyl moiety being twisted out of the plane with the cyclic sulfamidate imine ring, exerting additional facial selectivity that favours the formation of the major diastereoisomer. A single 4-alkyl substrate, the tert-butyl derivative 2g was tested and was poorly tolerated under the optimised reaction conditions, as shown by the poor dr and er of the product 3ag.
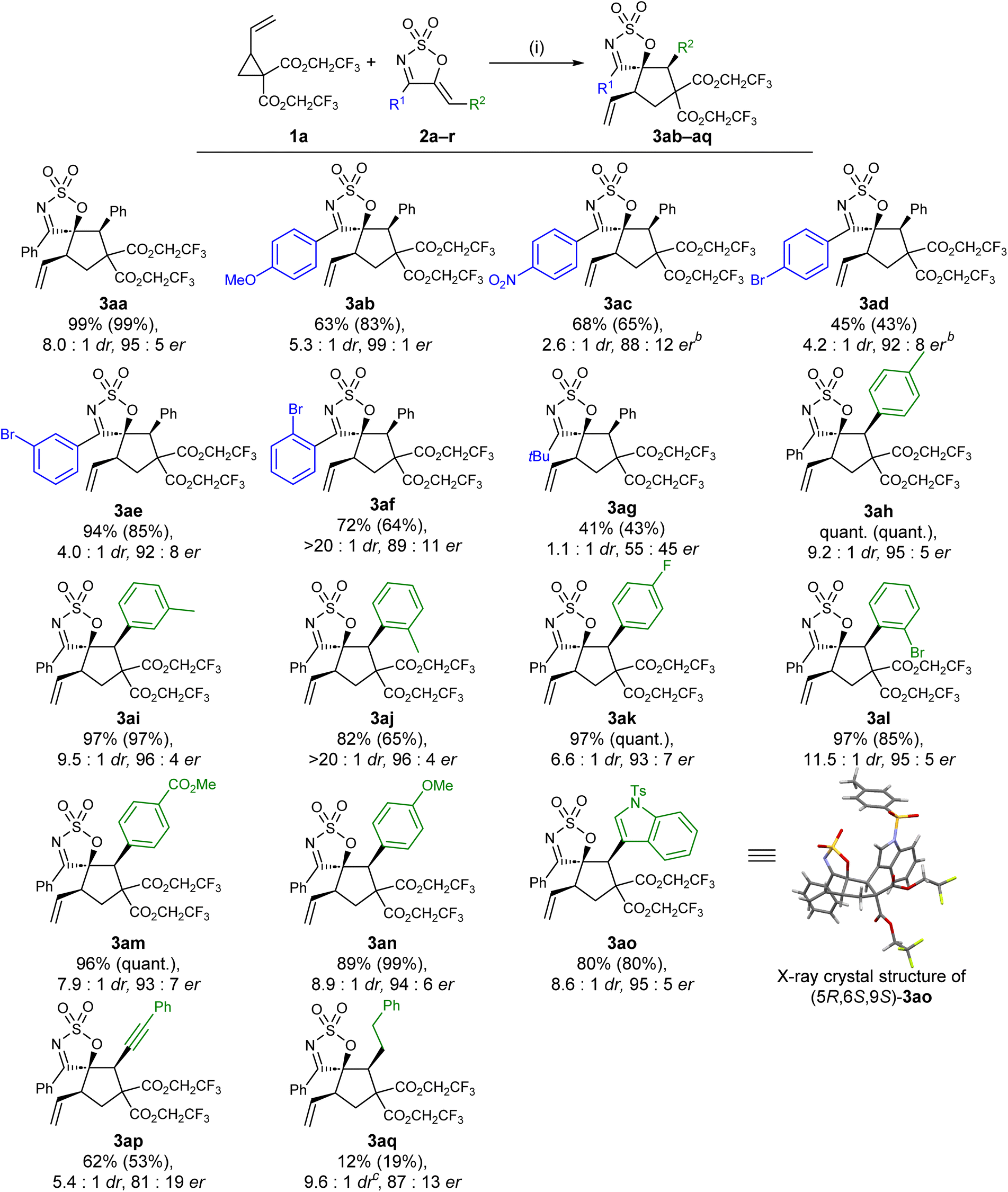 | ||
| Scheme 2 Substrate scope of cyclic 1-azadienes. aReaction conditions: (i) 1a (0.15 mmol, 1.5 equiv.), 2a (0.1 mmol, 1.0 equiv.), Pd2dba3·CHCl3 (2.5 mol%), L5 (7.5 mol%), PhMe (0.034 M w.r.t. 2a), rt, 24 h. Calculated yields are provided in parentheses and were determined by 1H NMR integration against an internal standard 1,2,3-trimethoxybenzene. The diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. The enantiomeric ratio was determined by chiral HPLC. bReaction was carried out at reflux. cDiastereomeric ratio was determined by 1H NMR analysis of the purified product. | ||
Cyclic sulfamidate 1-azadienes with a range of electronically and sterically diverse phenyl and heteroaryl groups at the olefinic terminal (2h–o) were next subjected to the optimised reaction conditions (Scheme 2). Gratifyingly, all the tested substrates underwent the transformation efficiently, affording the desired (3 + 2) products in good to high yields and in high ers, suggesting that the impact of either electronic or steric effects on both the efficiency and the enantioselectivity of the transformation was minor. The diastereocontrol of the reaction seemed to be more prone to these variations in the electronic and steric properties of the substrate, as illustrated by the minimal erosion in diastereoselectivity observed for substrates with electron deficient aryl groups (2k and 2m), as well as the notable increase in dr noted with substrates bearing more sterically demanding aryl moieties (2j and 2l). The alkynyl substituted 1-azadienyne 2p was found to be a competent substrate, furnishing the desired product 3ap in respectable yield, dr, and er. In comparison, the reaction of its saturated analogue 2q gave the product 3aq in poor yield, but in slightly higher dr and er.
The absolute configuration of the major diastereomeric cycloadduct was determined via X-ray crystallographic analysis of the enantiopure crystal of 3ao, which allowed the absolute configuration of other substrates to be assigned by analogy.
To demonstrate the synthetic viability of our optimised protocol, the reaction of VCP 1a and 1-azadiene 2a was carried out on a 1 mmol scale (Scheme 3a). Delightfully, the cycloadduct 3aa was obtained in comparably high yield and enantioselectivity to that observed on a smaller scale, although a significant decline in dr was observed. Although direct manipulation of a diastereo-enriched mixture of 3aa/3aa′ was possible, it was soon realised that the resulting diastereomeric mixture—which would likely consist of more than two species—might pose a significant analytical challenge. Aiming to avoid the challenging task of analysing these complex mixtures, cycloadduct 3aa was first converted to its styryl analogue via olefin cross-metathesis, allowing the major diastereomer 3ba to be isolated in 72% isolated yield without any erosion in er. With a substantial amount of 3ba in hand, several post-synthetic transformations were then attempted to illustrate the versatility of this scaffold (Scheme 3b).
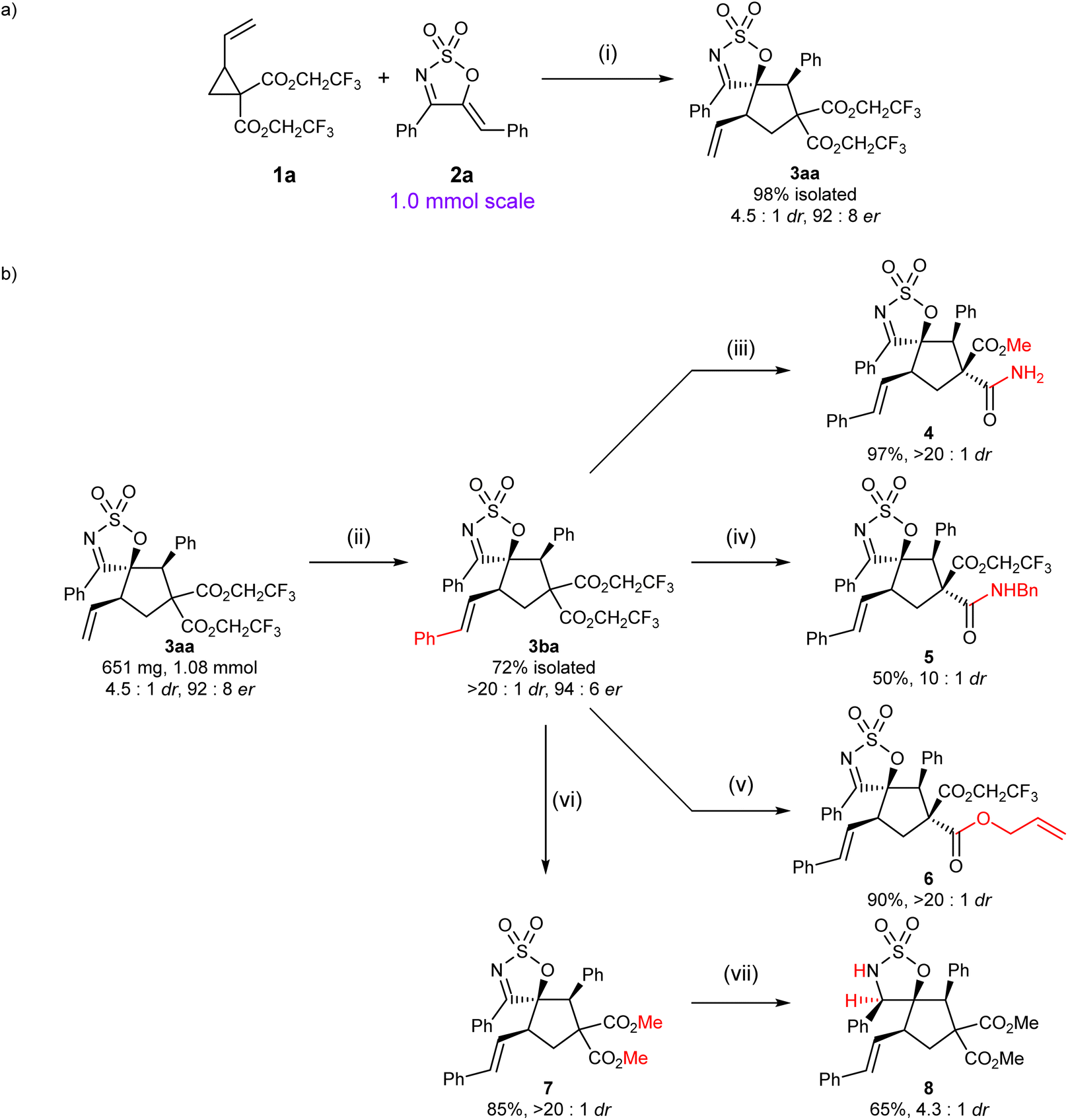 | ||
| Scheme 3 Scale up and derivatisation of products 3aa and 3ba. aReaction conditions: (i) 1a (0.15 mmol, 1.5 equiv.), 2a (0.1 mmol, 1.0 equiv.), Pd2dba3·CHCl3 (2.5 mol%), L5 (7.5 mol%), PhMe (0.034 M w.r.t. 2a), rt, 24 h. (ii) Styrene (200 equiv.), Grubbs catalyst 2nd generation (3.5 mol%), CH2Cl2, reflux, overnight. (iii) NH3 (7 N in MeOH), 40 °C, 88 h. (iv) BnNH2 (1.9 equiv.), THF, reflux, 24 h. (v) K2CO3 (3.1 equiv.), allyl alcohol, rt, 66 h. (vi) Na(s) (2.4 equiv.), MeOH, rt, 23 h. (vii) NaBH3CN (8.4 equiv.), 1:1 MeOH/CH2Cl2, rt, 65 h. | ||
The distinction in the reactivity of the geminal esters provided opportunities for some chemoselective reactions to be conducted and allowed a fourth stereogenic centre to be readily created. Treatment of spirocyclic cyclic imine 3ba with ammonia in methanol allowed a double transesterification to take place, followed by a regioselective amidation of the more reactive ester moiety trans- to the SO2 bridge, furnishing amide 4 in high yield and dr (relative configuration of 4 was determined by 2D NOESY, see the ESI†). These differences in reactivity of the two ester moieties of 3ba were further exploited for the preparation of amide 5 and diester 6—via chemoselective amidation and transesterification, respectively. These reactions occurred in moderate to high yields and good to high drs, with that of 5 being diminished relative to that of 4, presumably due to the higher reaction temperature (reflux in THF). In addition to the selective functionalisation of the gem diesters, construction of an additional stereogenic centre could be otherwise achieved via the functionalisation of the reactive imine moiety. Reduction of 7—readily obtained by a double transesterification of 3ba with NaOMe—by NaBH3CN afforded cyclic sulfamidate 8 in moderate yield and dr (relative stereochemistry of the major diastereoisomer was established by 2D NOESY analysis, see the ESI†). Attempts to subject 3ba to a variety of other common hydride reducing agents were complicated by the highly reactive nature of the bis(trifluoroethyl) diester moieties.
The stereochemical outcomes of the transformation can be rationalised using a quadrant analysis, generated based on X-ray data for analogous Pd–Segphos complexes (Fig. 3).61,62 Ionisation of the racemic VCP by the chiral [(S)-DM-Segphos]Pd0 complex can afford either diastereomeric complex A or B, which can interconvert via π–σ–π isomerisation. Of these two, complex A is likely more favoured due to the lower steric interaction between the dipole fragment and the equatorial xylyl group from the ligand. In line with established literature, the Michael addition of the dipole to the dipolarophile is likely a reversible process, whereas the ring-closure occurs irreversibly and under the control of the ligand to determine the configuration [stereochemistry] of the cycloadduct. Specifically, addition of A to the Re-face of 1-azadiene viaC should form the pre-cyclisation conformer E—where the R2 moiety is placed in the pseudo-equatorial position—followed by irreversible ring closure to furnish the major diastereomeric product with the observed absolute configuration. In contrast, addition of A to the Si face of 1-azadiene viaD gives pre-cyclisation conformer F—with the R2 substituent similarly occupying the pseudo-equatorial position—and then ring closure of F provides an enantiomer of the minor diastereomeric product. For pre-cyclised conformer E, it is likely that the more favourable disposition of a significant portion of the intermediate in the open quadrant of the chiral ligand space may have allowed the cyclisation to proceed faster than that of F, thus leading to high stereocontrol.
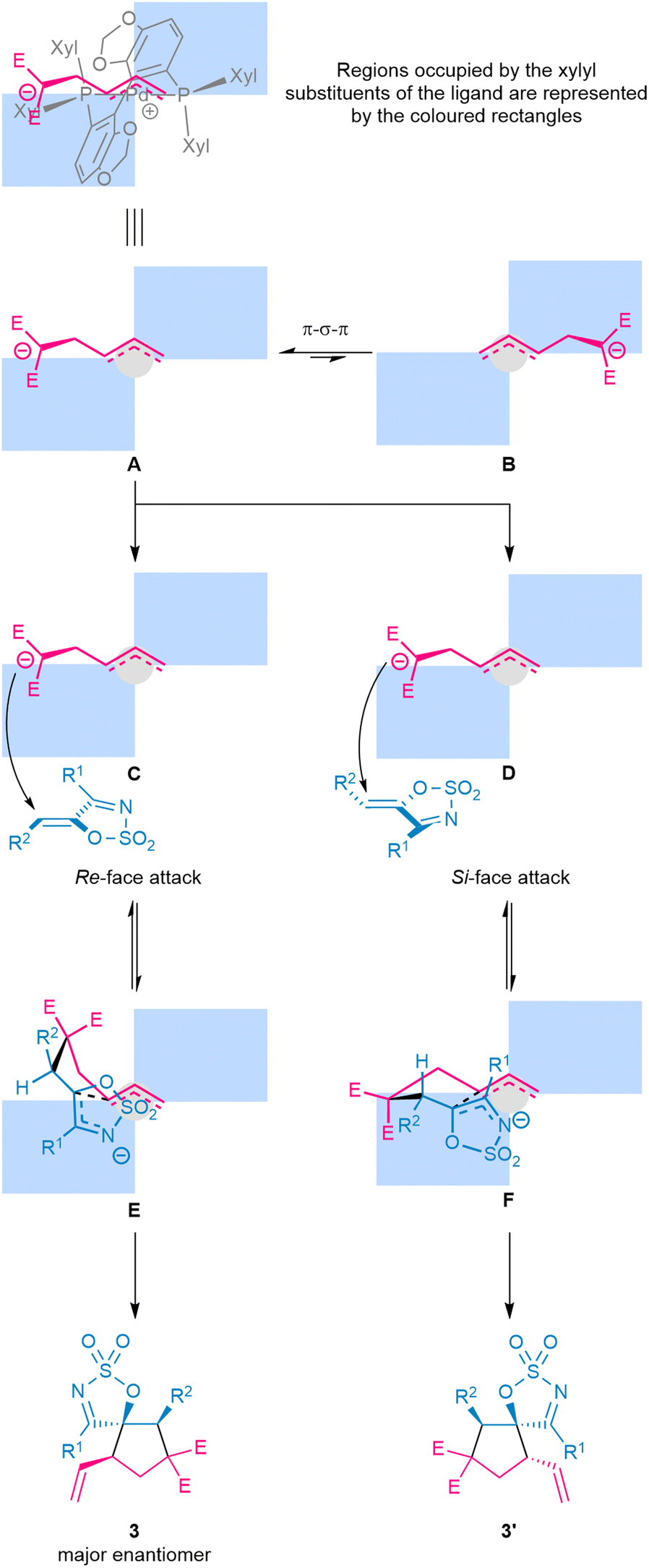 | ||
| Fig. 3 Rationalisation of the observed stereochemical outcomes using a quadrant analysis. For clarity, in complexes A–F, the chiral ligand scaffold is omitted and the cationic metal centre is represented by the light grey circle. | ||
Conclusions
In conclusion, SDAs have been demonstrated to be competent partners in stereo- and regioselective Pd-catalysed (3 + 2) cycloaddition reactions with vinylcyclopropane derived 1,3-dipoles. The ability of this dipolarophile to avoid (4 + n) reaction manifolds has implications for reactions with a myriad of other Pd-stabilised dipoles and in organocatalysed processes,63,64 which can also be utilised with BDAs but are far less explored, with no (3 + 2) processes known. The cycloaddition provides a rapid assembly of highly substituted and stereodefined cyclopentanes as part of a spirocyclic system, with sp3-rich systems being privileged structures in pharmaceutical development. The transformation is scalable and post-synthetic modifications targeting different functional handles demonstrate the synthetic utility of the cycloadduct. Many of these transformations allow an additional stereocentre to be readily installed via either chemoselective manipulations of geminal diester functionalities or via diastereoselective reduction of the imine moiety.Data availability
Data for all compounds reported in this manuscript are available in the ESI,† which includes experimental details, characterisation, copies of 1H and 13C NMR spectra as well as HPLC traces. Crystallographic data for compounds has been deposited at the CCDC under CCDC 2224300.Author contributions
HP, CH, and SP conceptualised the project. CH and SP were involved with supervision, funding acquisition and writing – reviewing and editing. HP carried out the investigation and formal analysis of the data and was involved with writing the original draft of the manuscript. AT was involved in writing – reviewing and editing and formal analysis of data. MGG and CR carried out the X-ray crystallography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Australian Research Council Discovery Grant DP180101332 for funding of this research. QHP is the recipient of a UOW UPA and IPTA PhD Scholarship.Notes and references
- B. Heasley, Eur. J. Org. Chem., 2009, 1477–1489 CrossRef CAS.
- B. Heasley, Curr. Org. Chem., 2014, 18, 641–686 CrossRef CAS.
- A. J. Ferreira and C. M. Beaudry, Tetrahedron, 2017, 73, 965–1084 CrossRef CAS.
- G. Archer, P. Cavalère, M. Médebielle and J. Merad, Angew. Chem., Int. Ed., 2022, 61, 1–6 CrossRef PubMed.
- Z. Ding, Z. Liu, Z. Wang, T. Yu, M. Xu, J. Wen, K. Yang, H. Zhang, L. Xu and P. Li, J. Am. Chem. Soc., 2022, 144, 8870–8882 CrossRef CAS PubMed.
- S. Agasti, N. A. Beattie, J. J. W. McDouall and D. J. Procter, J. Am. Chem. Soc., 2021, 143, 3655–3661 CrossRef CAS PubMed.
- N. L. Ahlburg, P. G. Jones and D. B. Werz, Org. Lett., 2020, 22, 6404–6408 CrossRef CAS PubMed.
- X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454–5458 CrossRef CAS PubMed.
- N. L. Ahlburg, O. Hergert, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2023, 62, e202214390 CrossRef CAS PubMed.
- G. A. Oliver, M. N. Loch, A. U. Augustin, P. Steinbach, M. Sharique, U. K. Tambar, P. G. Jones, C. Bannwarth and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 25825–25831 CrossRef CAS PubMed.
- V. Pirenne, E. G. L. Robert and J. Waser, Chem. Sci., 2021, 12, 8706–8712 RSC.
- Z. B. Zheng, W. F. Cheng, L. Wang, J. Zhu, X. L. Sun and Y. Tang, Chin. J. Chem., 2020, 38, 1629–1634 CrossRef CAS.
- G. Mlostoń, M. Kowalczyk, A. U. Augustin, P. G. Jones and D. B. Werz, Beilstein J. Org. Chem., 2020, 16, 1288–1295 CrossRef PubMed.
- L. Jiao and Z. X. Yu, J. Org. Chem., 2013, 78, 6842–6848 CrossRef CAS PubMed.
- V. Ganesh and S. Chandrasekaran, Synthesis, 2016, 48, 4347–4380 CrossRef CAS.
- M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479–2490 RSC.
- D. K. Brownsey, E. Gorobets and D. J. Derksen, Org. Biomol. Chem., 2018, 16, 3506–3523 RSC.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- Y. Xia, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192–9204 CrossRef CAS PubMed.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- I. Shimizu, Y. Ohashi and J. Tsuji, Tetrahedron Lett., 1985, 26, 3825–3828 CrossRef CAS.
- A. T. Parsons, M. J. Campbell and J. S. Johnson, Org. Lett., 2008, 10, 2541–2544 CrossRef CAS PubMed.
- Y. S. Gee, D. J. Rivinoja, S. M. Wales, M. G. Gardiner, J. H. Ryan and C. J. T. Hyland, J. Org. Chem., 2017, 82, 13517–13529 CrossRef CAS PubMed.
- J. Q. Zhang, F. Tong, B. B. Sun, W. T. Fan, J. B. Chen, D. Hu and X. W. Wang, J. Org. Chem., 2018, 83, 2882–2891 CrossRef CAS PubMed.
- J. Ling, M. Laugeois, V. Michelet, V. Ratovelomanana-Vidal and M. R. Vitale, Synlett, 2018, 29, 928–932 CrossRef CAS.
- Q. Wang, C. Wang, W. Shi, Y. Xiao and H. Guo, Org. Biomol. Chem., 2018, 16, 4881–4887 RSC.
- J. Liu, M. M. Li, B. Le Qu, L. Q. Lu and W. J. Xiao, Chem. Commun., 2019, 55, 2031–2034 RSC.
- X. B. Huang, X. J. Li, T. T. Li, B. Chen, W. D. Chu, L. He and Q. Z. Liu, Org. Lett., 2019, 21, 1713–1716 CrossRef CAS PubMed.
- W. P. Ding, G. P. Zhang, Y. J. Jiang, J. Du, X. Y. Liu, D. Chen, C. H. Ding, Q. H. Deng and X. L. Hou, Org. Lett., 2019, 21, 6805–6810 CrossRef CAS PubMed.
- Q. Cheng, J. H. Xie, Y. C. Weng and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed.
- B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2021, 60, 5806–5810 CrossRef CAS PubMed.
- M. Faltracco, K. N. A. van de Vrande, M. Dijkstra, J. M. Saya, T. A. Hamlin and E. Ruijter, Angew. Chem., Int. Ed., 2021, 60, 14410–14414 CrossRef CAS PubMed.
- B. M. Trost and P. J. Morris, Angew. Chem., Int. Ed., 2011, 50, 6167–6170 CrossRef CAS PubMed.
- K. Liu, J. Yang and X. Li, Org. Lett., 2021, 23, 826–831 CrossRef CAS PubMed.
- M. A. Drew, A. J. Tague, C. Richardson, S. G. Pyne and C. J. T. Hyland, Org. Lett., 2021, 23, 4635–4639 CrossRef CAS PubMed.
- Z. X. Yang, Y. Q. Wei, C. Yang, Y. F. Li, C. H. Ding and B. Xu, Asian J. Org. Chem., 2022, 11, e202100683 CAS.
- B. M. Trost, P. J. Morris and S. J. Sprague, J. Am. Chem. Soc., 2012, 134, 17823–17831 CrossRef CAS PubMed.
- L. Y. Mei, Y. Wei, Q. Xu and M. Shi, Organometallics, 2012, 31, 7591–7599 CrossRef CAS.
- L. Y. Mei, Y. Wei, Q. Xu and M. Shi, Organometallics, 2013, 32, 3544–3556 CrossRef CAS.
- W. K. Li, Z. S. Liu, L. He, T. R. Kang and Q. Z. Liu, Asian J. Org. Chem., 2015, 4, 28–32 CrossRef CAS.
- Z. S. Liu, W. K. Li, T. R. Kang, L. He and Q. Z. Liu, Org. Lett., 2015, 17, 150–153 CrossRef CAS PubMed.
- Z. Yuan, W. Wei, A. Lin and H. Yao, Org. Lett., 2016, 18, 3370–3373 CrossRef CAS PubMed.
- M. Laugeois, J. Ling, C. Férard, V. Michelet, V. Ratovelomanana-Vidal and M. R. Vitale, Org. Lett., 2017, 19, 2266–2269 CrossRef CAS PubMed.
- Y. N. Wang, L. C. Yang, Z. Q. Rong, T. L. Liu, R. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1596–1600 CrossRef CAS PubMed.
- L. C. Yang, Z. Q. Rong, Y. N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 2927–2931 CrossRef CAS PubMed.
- Y. Z. Liu, Z. Wang, Z. Huang, W. L. Yang and W. P. Deng, Org. Lett., 2021, 23, 948–952 CrossRef CAS PubMed.
- S. N. Fairuz Binte Sheikh Ismail, B. Yang and Y. Zhao, Org. Lett., 2021, 23, 2884–2889 CrossRef CAS PubMed.
- Z. Q. Rong, L. C. Yang, S. Liu, Z. Yu, Y. N. Wang, Z. Y. Tan, R. Z. Huang, Y. Lan and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 15304–15307 CrossRef CAS PubMed.
- A. Scuiller, A. Karnat, N. Casaretto and A. Archambeau, Org. Lett., 2021, 23, 2332–2336 CrossRef CAS PubMed.
- Q. Li, R. Pan, M. Wang, H. Yao and A. Lin, Org. Lett., 2021, 23, 2292–2297 CrossRef CAS PubMed.
- P. Kumari, W. Liu, C. J. Wang, J. Dai, M. X. Wang, Q. Q. Yang, Y. H. Deng and Z. Shao, Chin. J. Chem., 2020, 38, 151–157 CrossRef CAS.
- B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2020, 59, 1243–1247 CrossRef CAS PubMed.
- Y. Z. Liu, Z. Wang, Z. Huang, X. Zheng, W. L. Yang and W. P. Deng, Angew. Chem., Int. Ed., 2020, 59, 1238–1242 CrossRef CAS PubMed.
- B. M. Trost, Z. Zuo, Y. Wang and J. E. Schultz, ACS Catal., 2020, 10, 9496–9503 CrossRef CAS.
- B. M. Trost, Z. Zuo and Y. Wang, Org. Lett., 2021, 23, 979–983 CrossRef CAS PubMed.
- Q. H. Pham, A. J. Tague, C. Richardson, C. J. T. Hyland and S. G. Pyne, Chem. Sci., 2021, 12, 12695–12703 RSC.
- Q. H. Pham, C. J. T. Hyland and S. G. Pyne, Org. Biomol. Chem., 2020, 18, 7467–7484 RSC.
- S. Guin, D. Majee and S. Samanta, Chem. Commun., 2021, 57, 9010–9028 RSC.
- A similar switch in diastereoselectivity between malonitrile and malonate-derived vinylcyclopropanes has been observed in the cycloaddition to 3-nitroindoles (see references 23 and 25). The smaller size of the cyano group compared to the ester groups of the other derivatives may contribute to this switch by reducing 1,3-diaxial-like interactions in pre-cyclisation conformers such as E and F in Fig. 3.
- D. Majee, A. Srivastava, S. M. Mobin and S. Samanta, RSC Adv., 2013, 3, 11502–11506 RSC.
- K. Van Hecke, T. R. Benton, M. Casper, D. Mauldin, B. Drake and J. B. Morgan, Org. Lett., 2021, 23, 7916–7920 CrossRef CAS PubMed.
- C. Dubs, Y. Hamashima, N. Sasamoto, T. M. Seidel, S. Suzuki, D. Hashizume and M. Sodeoka, J. Org. Chem., 2008, 73, 5859–5871 CrossRef CAS PubMed.
- Z. Q. Rong, M. Wang, C. H. E. Chow and Y. Zhao, Chem.–Eur. J., 2016, 22, 9483–9487 CrossRef CAS PubMed.
- H. Ni, X. Tang, W. Zheng, W. Yao, N. Ullah and Y. Lu, Angew. Chem., Int. Ed., 2017, 56, 14222–14226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2224300. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01510f |
| This journal is © The Royal Society of Chemistry 2023 |