
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multistep retrosynthesis combining a disconnection aware triple transformer loop with a route penalty score guided tree search†
David
Kreutter
 and
Jean-Louis
Reymond
*
and
Jean-Louis
Reymond
*
Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: david.kreutter@unibe.ch; jean-louis.reymond@unibe.ch
First published on 1st September 2023
Abstract
Computer-aided synthesis planning (CASP) aims to automatically learn organic reactivity from literature and perform retrosynthesis of unseen molecules. CASP systems must learn reactions sufficiently precisely to propose realistic disconnections, while avoiding overfitting to leave room for diverse options, and explore possible routes such as to allow short synthetic sequences to emerge. Herein we report an open-source CASP tool proposing original solutions to both challenges. First, we use a triple transformer loop (TTL) predicting starting materials (T1), reagents (T2), and products (T3) to explore various disconnection sites defined by combining systematic, template-based, and transformer-based tagging procedures. Second, we integrate TTL into a multistep tree search algorithm (TTLA) prioritizing sequences using a route penalty score (RPScore) considering the number of steps, their confidence score, and the simplicity of all intermediates along the route. Our approach favours short synthetic routes to commercial starting materials, as exemplified by retrosynthetic analyses of recently approved drugs.
Introduction
Retrosynthetic analysis consists in drafting a synthetic sequence to produce a desired product from available starting materials. This analysis is one of the most useful but also difficult tasks in organic chemistry because it requires to integrate the large and complex set of rules that have emerged from millions of reactions reported in almost 200 years of organic synthesis. Computer-aided synthesis planning (CASP), initially conceived by E. J. Corey in the 1960s,1 aims to harness the power of computers to automate retrosynthesis by exploiting data from experimental reactions collected in databases such as Reaxys2 or the open-access reaction dataset extracted from US patent office data.3,4 These databases list reactions of sets of starting materials (SM) and sets of reagents (R) to form one or several products (P).While expert systems based on hand-written rules such as Chematica/Synthia™ perform quite well for synthesis planning,5 CASP ultimately aims to exploit artificial intelligence to automatically learn organic synthesis from reaction examples and propose synthetic routes for new molecules without human intervention.6–10 Template-based approaches extract reaction rules in the form of substructure transformations and use machine learning to learn their applicability domain from the structure of P in the training data.11–14 On the other hand, transformer-based models use the linear SMILES15,16 notation of chemical reactions and learn to translate the character string of P into the character string of SM + R, or vice versa.17–27 The single-step predictions are then iterated to propose multistep retrosyntheses of target molecules from a selected set of building blocks (BB), which requires prioritizing possible routes using search algorithms such as Monte Carlo tree search,11,23,28 and-or trees,25,29 or a multistep graph exploration.24
Any CASP system must overcome two critical challenges to propose realistic retrosyntheses. First, the system must learn the context of reactions sufficiently well to propose reactions that make sense, but without overfitting such as to propose diverse retrosynthetic operations on previously unseen molecules. Second, the route-prioritizing algorithm must be designed to allow short sequences to emerge from the multitude of predicted possibilities.6 Herein, we report a transformer-based retrosynthesis tool which proposes original solutions to both challenges. For single-step retrosynthesis, we use three different transformer models assembled as a triple transformer loop (TTL, Fig. 1a). To broaden the scope of predicted disconnections on a given target molecule, the TTL explores multiple disconnections by using products with tagged reaction centers (P*) obtained by combining systematic, template-based and transformer-based tagging procedures. Compared to a transformer model trained on predicting SM + R directly from P*, the TTL achieves better round-trip accuracy for single-step retrosynthesis. For multistep retrosynthesis predictions, we integrate the TTL into a multistep tree search algorithm, here named TTLA, which selects reaction sequences using a new route penalty score (RPScore), which for a route of N steps, is the product of a step-penalty score SPN, the confidence scores of each single-step retrosynthesis (CS), and the simplicity scores24 of all SM along the route (Fig. 1b). This selection scheme favours short sequences and is exemplified with the prediction of synthetic routes for recently approved drugs.
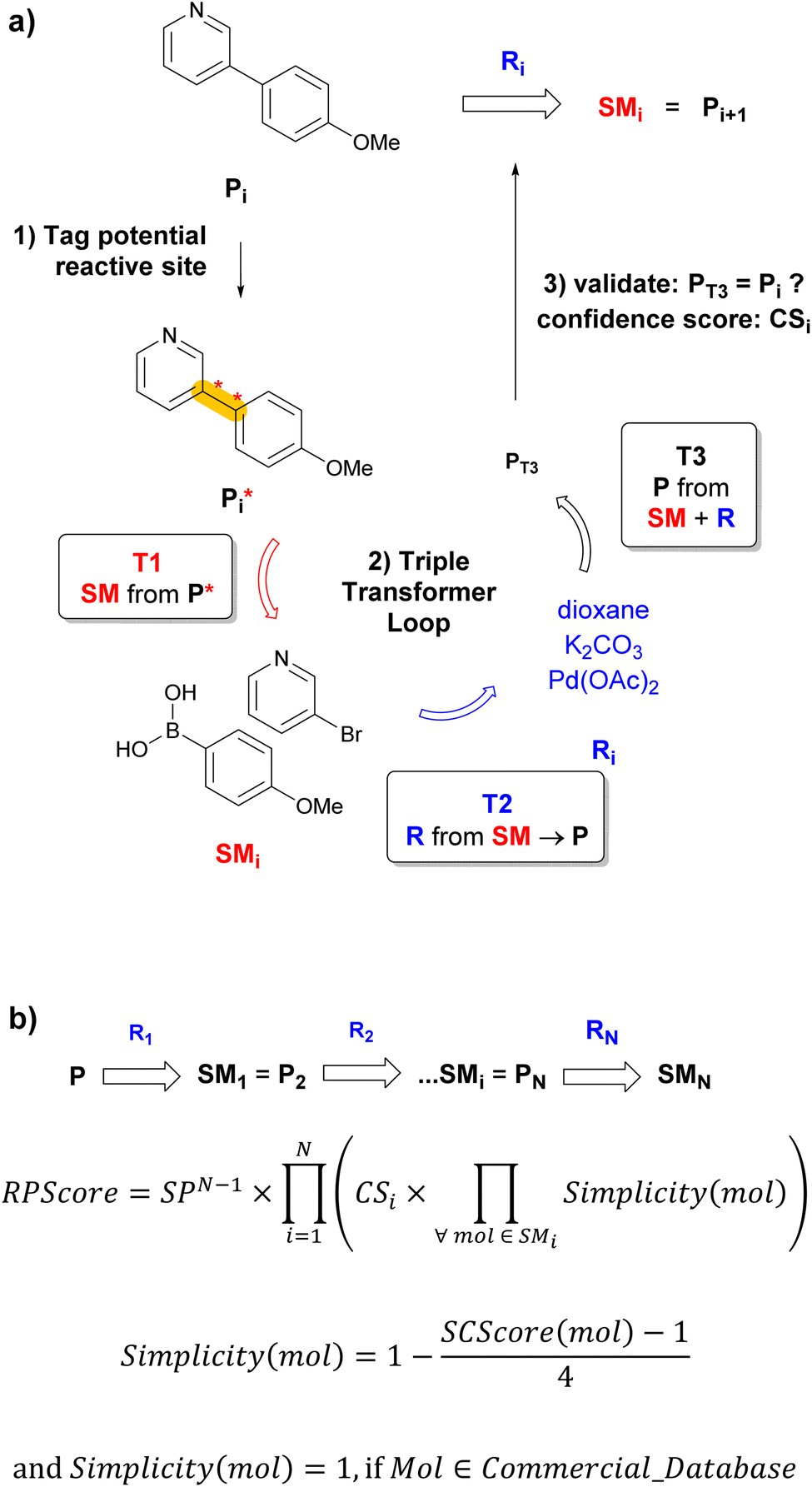 | ||
Fig. 1 Multistep retrosynthesis using TTLA. (a) Single-step retrosynthesis. At step i, each potential reactive site in Pi is identified systematically, using templates or a tagging transformer, and labelled to produce 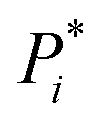 . Transformer T1 is applied to . Transformer T1 is applied to 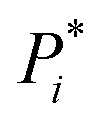 to predict SMi (one or more starting materials), transformer T2 is applied to the top-scoring SMi → Pi to predict Ri (one or more reagents), and finally transformer T3 is applied to the top-scoring SMi + Ri to produce PT3. The prediction is finally validated if PT3 = Pi with confidence score CSi of T3. Each molecule in the SMi set is then used as product Pi+1 for the next iteration. The route branches out if SMi contains multiple molecules. (b) TTLA sequence and route penalty scoring. All molecules in the SMi set of each step are used in the RPScore calculation of a linear sequence. See text for details. to predict SMi (one or more starting materials), transformer T2 is applied to the top-scoring SMi → Pi to predict Ri (one or more reagents), and finally transformer T3 is applied to the top-scoring SMi + Ri to produce PT3. The prediction is finally validated if PT3 = Pi with confidence score CSi of T3. Each molecule in the SMi set is then used as product Pi+1 for the next iteration. The route branches out if SMi contains multiple molecules. (b) TTLA sequence and route penalty scoring. All molecules in the SMi set of each step are used in the RPScore calculation of a linear sequence. See text for details. | ||
Methods
Dataset
The United States Patent and Trademark Office (USPTO) chemical reaction dataset version shared by Thakkar et al.30 was used for the single-step evaluation as well as for training all transformer models in this study. The dataset is derived from the version of Lowe3,4 and has been filtered by these authors to include reactions with a single product (P) and between 2 and 10 starting materials (SM) and reagents (R) only. In the present work, we removed the tagging information, and reactions were remapped and retagged using our new SMILES tagging strategy and syntax. The same dataset split for training, validation, and test (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5), as shared by Thakkar et al.30 was used across all models resulting in 1139608, 63672 and 63454 reactions respectively.
:5:5), as shared by Thakkar et al.30 was used across all models resulting in 1139608, 63672 and 63454 reactions respectively.
Tagging reaction centers
Training the disconnection-aware retrosynthesis model requires a training dataset where all product SMILES have tagged atoms. To tag reacting atoms, we use the atom-mapping tool shared by Schwaller et al.31 to identify the atoms having an environmental change during the reaction, defined as reacting atoms. These reacting atoms are then re-labelled with the atom mapping label “1” while all other atom mapping labels are removed, as described by Byekwaso et al..32 We then replace the atom tagging syntax by its unmapped SMILES notation, e.g. replacing “[C:1]” with “C”, and append the atom with another separated tagging token (“!”) using RDkit.33 This modification allows to maintain an invariant SMILES token usage independent of the neighbouring hydrogen count or stereochemistry.Single-step disconnection aware retrosynthesis (T1)
Being able to identify the reaction center of a given reaction, we apply our reaction tagging algorithm on USPTO to obtain a retrosynthesis-tagged training dataset. We remove reagents, catalysts, and solvents, which are identified as the unmapped species in atom-mapped reactions and train the retrosynthesis model to predict the starting materials given as input the tagged product. We use the transformer architecture18 and train it using the OpenNMT34,35 library with standard previously-reported hyperparameters for this type of task.22Automatic tagging of potentially reactive atoms
We use three complementary methods to maximize the tagging possibilities while maintaining a reasonable number of predictions. First, we systematically tag all possible single atoms, pairs of directly connected atoms, and triplets of adjacent atoms (chain or three-membered ring). Secondly, we use templates for tagging the reactive sets of atoms corresponding to the conditional substructure with a variable radius (typically from 1 to 3). Templates occurring more than once and having between 1 and 10 reactive atoms were identified by analyzing the original USPTO dataset. A given template can contain multiple disconnected sets of reactive atoms. Finally, the transformer model AutoTag reported by Thakkar et al.30 was trained from untagged SMILES to the corresponding tagged molecule to provide additional tagging examples.Reagent prediction (T2)
Transformer T2 is trained from the untagged USPTO training set to identify reagents (R) from the combination of SM and P using the same hyperparameters as for T1. Note that R often includes actual reagents and solvents.Forward validation (T3)
The third model of the triple-transformer loop is a forward reaction prediction model trained with untagged reactions (molecular transformer).22 We give this forward validation model the predicted SMi (from T1) and the predicted Ri (from T2) as input separated by the “>” token. If T3 predicts the correct Pi as its top-1 prediction, those SMi and Ri are stored for the tree search. The confidence score CSi for the T3 prediction is used as confidence score for the reaction. T3 serves to filter down a large number of predictions to retain feasible reactions only.Single-step TTL tagging strategies study
The performance of individual tagging methods was studied on 500 molecules randomly selected from the USPTO test set for single-step TTL retrosynthesis to which we varied the three strategies over various parameters, changing the template radius from 1 to 3 and the transformer tagging (AutoTag) beam size from 1 to 1000.Route penalty score (RPScore)
The RPScore is computed for each predicted linear retrosynthetic sequence of N steps leading from the final product P to starting materials SMN (Fig. 1b). To reduce the score of long sequences, we introduce a step penalty SP, with 0 < SP ≤ 1, extended to SPN for a sequence of N steps. The RPScore is the product of SPN with the product of all confidence scores CSi (from the T3 prediction) for each individual step and the Simplicity (mol) for all intermediates along the sequence of N steps. By default, the penalty value SP is set to 0.8, but this could be adapted for every search in the configuration file of the multistep exploration. Simplicity(mol)24 ranges from 0 for complex to 1 for simple molecules and is derived from the molecular synthetic complexity score (SCScore, ranging from 1 to 5) which describes molecular complexity taking synthetic accessibility into account.36 Here, we assign a value of 1 if the molecule occurs in the BB set of commercial starting materials. In contrast to Schwaller et al.,24 we exclude reagents Ri from the Simplicity calculation to avoid penalizing steps that use reagents with low calculated Simplicity, which is rarely a measure of their availability or ease of use.Multistep exploration strategy
We use a Heuristic Best-First Tree Search algorithm with beam search and iterative expansion to explore retrosynthetic routes as similarly reported for transformer-based retrosynthesis.24 Once predictions of an iteration are complete, the tree search updates and lists all possible routes, and computes the RPScore. Unsolved routes are sorted by decreasing RPScore. The top 20 unsolved routes, which lead to starting materials absent from the selected set of commercially available building blocks, are selected for expansion by defining them as products Pi and new SMi are predicted by applying a single-step retrosynthesis using TTL. The resulting set of predicted single-step retrosynthesis is updated back to the tree wherever those SMs were present. The tree is updated for the next iteration. The process stops when a chosen minimum number of solved routes or a maximum number of iterations has been reached.Building block (BB) set
We combined MolPort (https://www.molport.com) and Enamine (https://www.enamine.net) databases to build our database of 534058 commercially available compounds as the building block (BB) set.
Results and discussion
Training transformer T1 for single-step retrosynthesis
Initially, we use the atom-mapping transformer31 information to annotate reacting atoms in all products P in the training data, which results in a training dataset containing labelled P*. Our code is inspired by the recent report by Byekwaso et al.,32 however with a slightly simplified syntax for tagged atoms. Using the tagged P* data, we then train a transformer model T1 to predict SM from P*, a task which is simpler than predicting both SM and R from P* as done by Byekwaso et al.32Tagging potential reactive sites
To use T1 to predict possible SMi from a given product Pi at step i, one must first tag potentially reacting atoms in Pi. We do this using complementary methods. First, we tag all single atoms as well as pairs and triplets of adjacent atoms systematically in Pi. Second, we systematically apply templates extracted from tagged P* in the USPTO dataset. These templates with various conditional radiuses (from 1 to 3) are substructures containing up to ten tagged atoms, not necessarily connected. Although the most frequent templates are those with two or three connected atoms, which are tags also obtained in the systematic procedure, the templates also include many tags with disconnected atom pairs and triplets as well as tags with four or more atoms, which are missing from the systematic tagging procedure (Fig. 2a). As a third tagging option, we use the tagging approach recently reported by Thakkar et al.30 where reacting atoms are identified using a tagging transformer, here named AutoTag, trained to learn the detailed context from the tagged dataset. The number of predicted tags (sorted by confidence score, called beam size) of AutoTag can be varied to generate a given number of possible tags to extend the retrosynthesis options.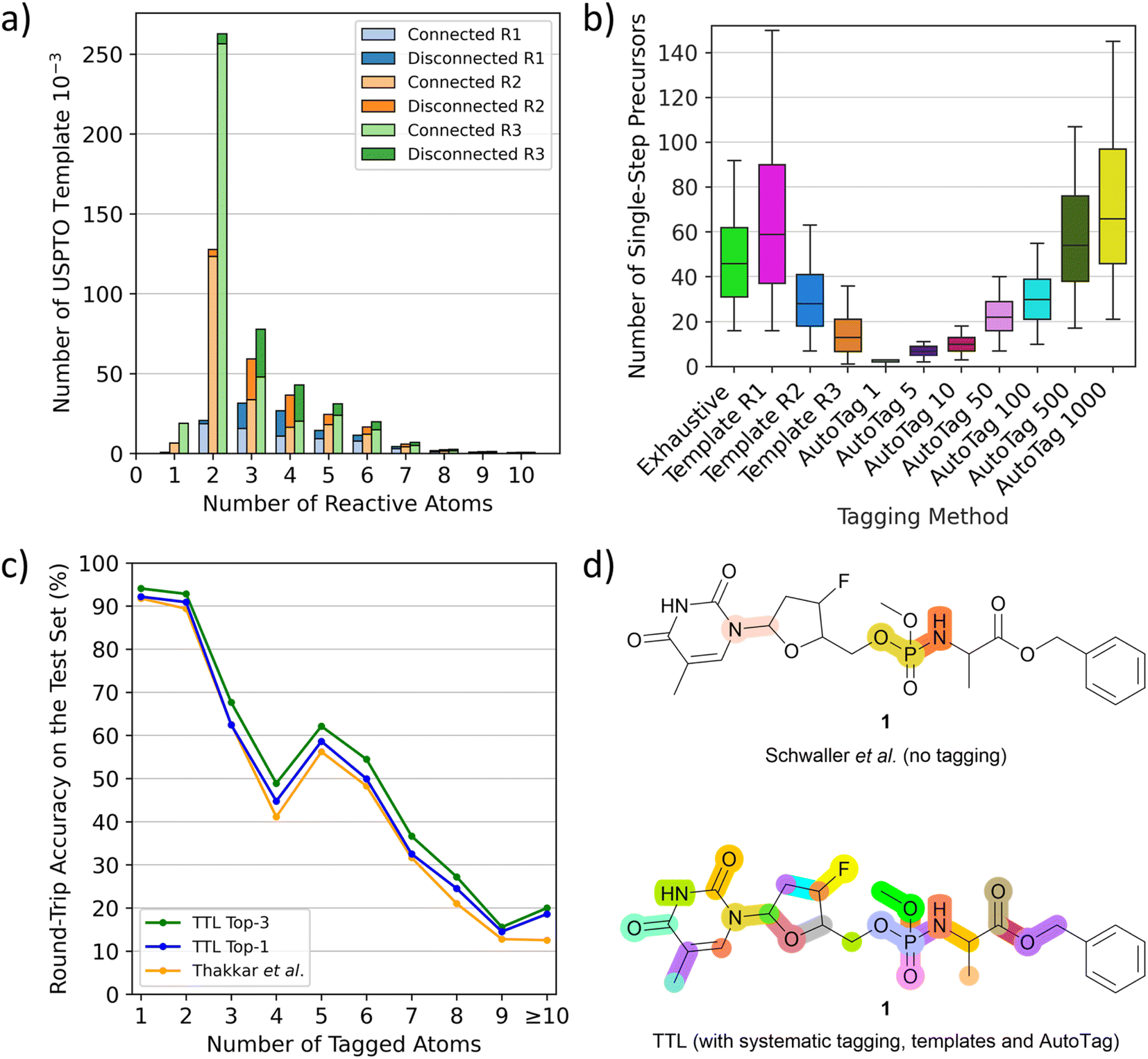 | ||
| Fig. 2 TTL and automatic atom tagging. (a) Distribution of the number of tagging templates extracted from USPTO depending on the number of atoms it tags, named “reactive atoms”. Triple bar plot to show the differences between conditional radiuses beyond tagged atoms from 1 to 3 (R1 to R3). Bars are split into a light-coloured part representing the fraction of templates that tags bond-connected atoms and dark-coloured for disconnected atoms. (b) Number of validated single-step starting materials (“precursors”) on the TTL generated depending on the automatic tagging strategy, tested over 500 molecules randomly selected from the TTL test set. (c) Round-trip accuracies of the TTL using the top-1 SM by T1 and the top-1 or top-3 R predicted by T2, compared to the disconnection-aware dual transformer of Thakkar et al.30 (d) Highlighted disconnection sites of the antiviral molecule 1 using the untagged retrosynthesis and forward validation models of Schwaller et al.,24 leading to four unique sets of starting materials among three reactive sites (top) and the TTL augmented by systematic tagging, template-based tagging (radius 2) and AutoTag (beam size 50) after forward validation leading to 231 unique sets of starting materials among 26 reactive sites (bottom). | ||
Analyzing the performance of the different tagging methods shows that less restrictive template radius or high AutoTag beam size both lead to an increased number of tagged atoms per molecule (Fig. S1†) as well as a much higher number of generated tagged SMILES (Fig. S2†) and significantly more single-step starting materials (Fig. 2b and S3†), but also to a lower number of high confidence predictions (Fig. S4†), indicating that most of the additionally obtained tags are less chemically meaningful (Fig. S5†). Moreover, the tagging efficiency, evaluated by dividing the number of successful retrosynthetic steps obtained by the number of TTL rounds (number of tags), drops for high AutoTag beam sizes and low radius templates (Fig. S6†). To obtain a good number of validated retrosynthetic steps at reasonable computing cost, we combine three strategies: the systematic tagging (1, 2 and 3 atoms), templates with a radius of 2, and the AutoTag transformer with a beam size of 50. A Venn diagram analysis of the number of unique retrosynthetic steps obtained shows that 17% of the steps (37.8% of high confidence steps) are predicted by all three methods, while 52.6% of the steps (25.5% of high confidence steps) are coming from only one of the three tagging methods, highlighting their complementarity (Fig. S7 and S8†).
Triple transformer loop (TTL)
To initiate a validated single-step retrosynthesis prediction for product Pi, we run T1 on all P*i obtained by the combined selected tagging procedures described above. The transformer outputs a series of possible SMi, which are sorted in order of the T1 confidence score. For the top-B SMi (beam size B = 1 or more), we then apply a second transformer (T2) trained to predict R from SM → P. For each SMi, T2 outputs a series of possible Ri, from which we retain the top-B′ (beam size B′ = 1 or more). The TTL is completed with a forward validation37 transformer (T3) trained to predict P from SM + R using the same training dataset used for T1 and T2. For all combinations of top SMi predicted by T1 and top Ri predicted by T2, we finally use T3 to predict the most likely product PT3. The TTL prediction is validated if the top-1 predicted PT3 is identical to the input product Pi (Fig. 1a). The T3 confidence scores CSi of the validated predictions SMi + Ri are used to select the best Ri if B′ > 1, and to calculate the route penalty score (RPScore, see below).Performance evaluation
The performance of TTL can be compared with previous single-step retrosynthesis models at three different levels. First, transformer T1, which predicts SM from the tagged product P*, can be compared with other single-step retrosynthesis models predicting SM from P, both transformer-based and template-based.17,19,21,23–27 While these models perform between 40% and 55% top-1 accuracy, our tagged T1 achieves 66% top-1 accuracy, which shows that tagging provides a significant advantage for this task.Second, the performance of the TTL loop can be compared with the disconnection-aware retrosynthesis model of Thakkar et al.30 in terms of single-step round-trip prediction accuracy from the tagged product P*, which is the accuracy of predicting P from the SM + R initially predicted from P*. TTL using only the top-1 predictions for T1 and T2 performs comparably to Thakkar's disconnection-aware retrosynthesis model (80.44% vs. 79.09% accuracy). The TTL performance increases to 83.04% when considering the top-1 prediction of T1 and the top-3 predictions of T2. Similar to the observation by Thakkar et al.,30 we furthermore find that the prediction accuracy strongly decreases as a function of the number of tagged atoms (Fig. 2c). Subsequently to our preprinted report, a separate study has investigated the performance of the reagent prediction transformer.38
Thirdly, one can compare the single-step round trip accuracy of TTL with that of the non-tagged retrosynthesis model of Schwaller et al.,24,37 who evaluated if a forward prediction model predicted the correct product P from the SM + R predicted by their model from the non-tagged P. As discussed by Thakkar et al.,30 the untagged transformer may sometimes choose a different and easier to predict disconnection than that recorded in the test set, and therefore performs slightly better (82.4% top-1 accuracy) than the tagged transformer, which is forced by tagging to apply the retrosynthesis of the test set. Here, we find that the top-1 round-trip prediction accuracy (P → P), obtained by applying our multiple tagging procedure followed by the TTL, reaches 99.9%, which means that our approach is almost always able to propose at least one forward-validated possible retrosynthetic step from any product molecule.
Furthermore, a critical feature of any single-step retrosynthesis model in view of multi-step retrosynthesis concerns the diversity of possible disconnections proposed. We find that this diversity is greatly enhanced by the multiple tagging approach. For instance, when tested on unseen molecules, the TTL combined multiple tagging provides validated disconnections at several possible reactive sites. By contrast, the baseline transformer, trained as reported by Schwaller et al.24 to produce SM directly from P using the unannotated data for training, chooses fewer disconnection points, as exemplified here for the pro-nucleotide 1 (Fig. 2d).39
Multistep retrosynthesis
By integrating the single-step retrosynthesis TTL into a multistep tree search, we obtain a multistep retrosynthesis algorithm, here named TTLA. In each retrosynthesis iteration, TTLA runs the TTL exhaustively on all SM of the preceding iteration, newly defined as P, and ranks the routes to the newly predicted SM using a composite route penalty score RPScore (Fig. 1b, see Methods for details).When prioritizing multiple retrosynthesis options during the tree search, TTLA uses the RPScore to rank the different routes leading to the SM produced in the latest iteration of TTL, and only extends retrosynthesis on a small number (typically 20) of SM taken from the top RPScoring routes. Because each additional step imposes a penalty (usually P = 0.8), lengthy routes and unproductive loops involving protection/deprotection cycles of the same functional group are rapidly falling down the RPScore priority list, which leads the algorithm to explore alternative routes, so that short synthetic sequences are eventually prioritized even if their first retrosynthetic steps were initially not top scoring.
As commonly observed with CASP tools as well as with transformer models in general, the top-scoring outputs of TTLA must be inspected to identify relevant predictions. While the RPScore is used in the tree search, we find relevant routes by inspecting both the top-RPScoring route and the top-CScoring routes (CScore (route) = the product of CSi for all steps) in the TTLA output, as discussed below with examples.
TTLA is exemplified here for predicting the synthesis of two drug molecules approved in 2020, namely fostemsavir (2, Fig. 3), a prodrug which upon phosphatase cleavage releases the antiretroviral agent temsavir as HIV entry inhibitor,40 and ozanimod (10, Fig. 4), a sphingosine-1-phosphate receptor antagonist used as an immunomodulatory agent to treat multiple sclerosis.41 The commercial process for both drugs was recently reviewed.42 None of the synthetic steps involved in these two processes occur in the USPTO dataset used for training TTL, making them a good test case for TTLA. For these examples, we challenged TTLA to predict synthetic routes starting from a list of 534058 commercially available BB.
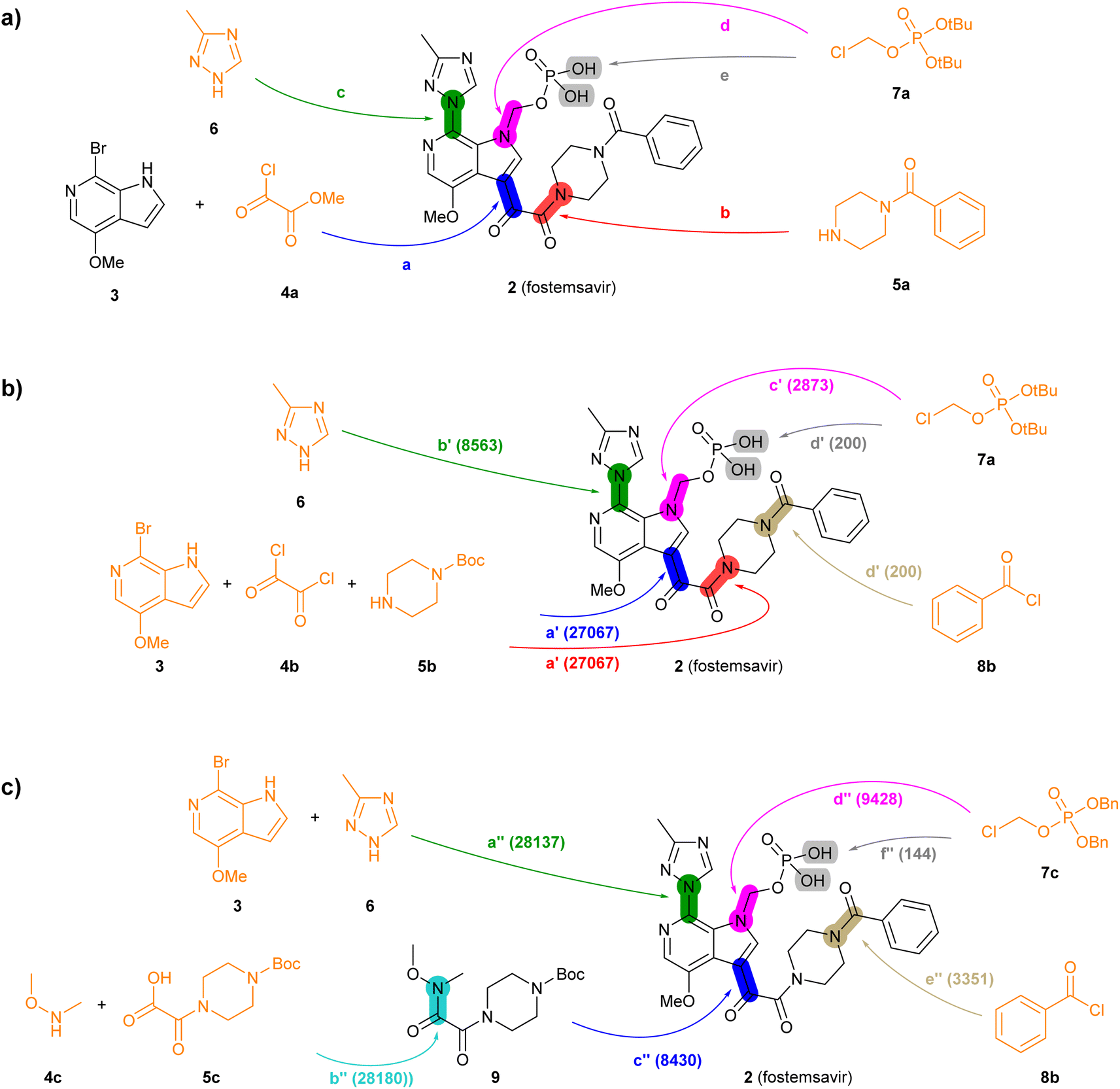 | ||
| Fig. 3 Summary of reported and TTLA predicted routes for fostemsavir 2. Bonds formed in each step are highlighted in colour. Numbers in parenthesis correspond to the order in which the multistep tree search prioritized predictions. The full retrosynthesis routes are drawn out in the ESI† Fig. S9–S11.† (a) Commercial process. Reported reagents: (a) AlCl3, Bu4NHSO4, CH2Cl2, then KOH, then H3PO4; (b) Ph2POCl, NMM, NMP; (c) KOH, CuI, then KOH, EtOH, LiI; (d) Et4NI, K2CO3, CH3CN/H2O; (e) AcOH, H2O. (b) Highest TTLA RPScoring route. Predicted reagents: (a′) Et3N, CH2Cl2; (b′) K2CO3, CuI, toluene; (c′) K2CO3, DMF; (d′) HCl, N,N-diisopropylethylamine, H2O, dioxane. (c) Highest TTLA CScoring route. Predicted reagents: (a′′) (2S)-pyrrolidine-2-carboxylic acid, K2CO3, CuI, EtOAc, DMSO; (b′′) no reagent predicted; (c′′) n-BuLi, THF; (d′′) K2CO3, DMF; (e′′) TFA, DMAP, CH2Cl2, (f′′) Pd, EtOH. | ||
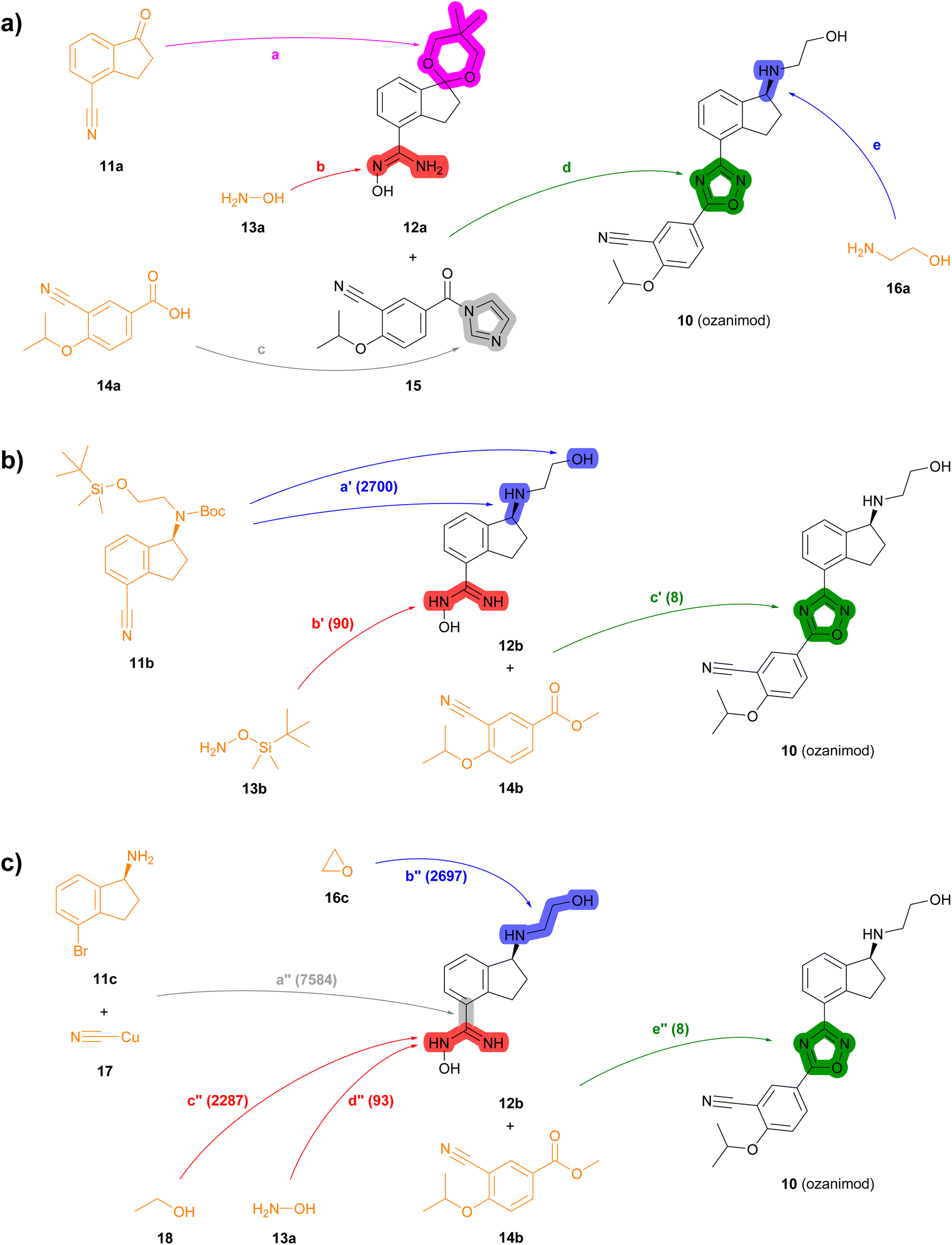 | ||
| Fig. 4 Summary of reported and TTLA predicted routes for ozanimod 10. Bonds formed in each step are highlighted in colour. Numbers in parenthesis correspond to the order in which the multistep tree search prioritized predictions. The full retrosynthesis routes are drawn out in the ESI† Fig. S12–S14.† (a) Commercial process. Reported reagents: (a) HC(Ome)3, p-TsOH, PhCH3; (b) NH2OH·HCl, Et3N; (c) carbonyl diimidazole; (d) NaOH; (e) (i) p-TsOH, acetone, (ii) NH2CH2CH2OH, p-TsOH, PhCH3, (iii) chiral Ru-complex, Et3N/HCO2H. (b) Highest TTLA RPScoring route. Predicted reagents: (a′) HCl, dioxane; (b′) ZnCl2, AcOEt, toluene; (c′) HCl, t-BuOK, THF. (c) Highest TTLA CScoring route. Predicted reagents: (a′′) 1-Methylpyrrolidin-2-one; (b′′) no reagent predicted; (c′′) HCl, Et2O; (d′′) HCl, NaHCO3, EtOH; (e′′) HCl, t-BuOK, THF. | ||
The reported commercial process for the antiviral drug fostemsavir (2, Fig. 3a, details in Fig. S9†) is a linear sequence involving the sequential C-acylation of pyrrolopyridine 3 with oxalyl monochloride 4a (step a) and benzoylpiperazine (5a, step b), followed by coupling of with triazole 6 (step c), N-alkylation of the pyrrole with the protected chloromethylphosphate 7a (step d), and finally deprotection of the tert-butyl ester protecting groups (step e).
When challenged with 2, TTLA proposes many possible routes from similar starting materials as the commercial process, but in a different order. The highest RPScoing route is a linear sequence starting from the double C- and N-alkylation of oxalyl chloride (4b) with pyrrolopyridine (3) and 1-boc-piperazine (5b) in one pot (step a′, Fig. 3b, details in Fig. S10†). The aryl bromide of the resulting intermediate is then substituted with triazole 6 (step b′), and its pyrrole NH group is alkylated with tert-butyl chloromethyl phosphate 7a, similarly to the commercial route (step c′). In the final step, the phosphate and the piperazine groups are deprotected with acid, followed by benzoylation of the free piperazine with benzoylchloride (8b) to form fostemsavir 2 (step d′).
On the other hand, the highest CScoring route is a convergent sequence starting with alkylation of triazole 6 with pyrrolopyridine 3 on the one hand (step a′′, Fig. 3c, details in Fig. S11†), and the preparation of the Weinreb amide 9 from boc-oxalylpiperazine 5c and N,O-dimethylhydroxylamine 4c on the other hand (step b′′). The resulting intermediates are then coupled (step c′′), and the product is N-alkylated on the pyrrole nitrogen with benzyl-protected chloromethyl phosphate 7c (step d′′). Deprotection of the piperazine group allows the acylation with benzoylchloride (8b, step e′′). Reductive deprotection of the benzyl phosphate esters finally gives the product 2 (step f′′).
In the second example, the drug ozanimod 10 is synthesized commercially in a convergent sequence of 7 steps from ketone 11a and benzoic acid 14a (Fig. 4a, details in Fig. S12†). After initial protection of ketone 11a as an acetal (step a), its nitrile group is reacted with hydroxylamine 13a to form the N-hydroxyamidine intermediate 12a (step b). In parallel, benzoic acid 14a is activated to the corresponding benzoyl imidazole 15 (step c). Intermediates 12a and 15 are then condensed to form the oxazole ring (step d). The acetal group of the resulting intermediate is then deprotected and condensed with ethanolamine (16a) to the corresponding imine, which is reduced enantioselectively using a chiral ruthenium catalyst to form 10 (step e).
Many of the high-scoring routes identified with TTLA are extremely short sequences starting with commercially available close analogs of the drug and were removed from the list of top-scoring routes. Interestingly, TTLA also proposes routes that resemble the commercial process but start from chiral starting materials such as aminoindanes 11b and 11c, which avoids the enantioselective reaction used for the commercial process. For example, the best RPScoring route is a linear synthesis from 11b starting with the removal of the Boc and TBS protecting groups of the ethanolamine side chain and conversion of the cyano group to the corresponding N-hydroxyamidine by reaction with TBS-hydroxylamine (13b) to form intermediate 12b (steps a′ and b′, Fig. 4b, details in Fig. S13†). The third and final step of this short sequence is the condensation of N-hydroxyamidine 12b with cyanobenzoate 14b yielding ozanimod 10 (step c′).
The best CScoring route is a somewhat longer linear sequence employing the same condensation of 12b and 14b as the final step (step e′′, Fig. 4c, details in Fig. S14†). In this proposed sequence however, intermediate 12b requires four steps from the chiral aminobromoindane 11c as follows. First, the cyano group is installed by reaction of the aryl bromide with copper cyanide (step a′′). Second, the primary amine reacts with ethylene oxide 16c to form the N-hydroxyethyl side chain (step b′′). Third, the cyano group introduced in step a′′ reacts with ethanol (18) to form an ethyl imidate intermediate (step c′′), which further reacts with ethanolamine (13a) in a fourth step to form the N-hydroxyamidine group in 12b (step d′′).
Analyzing the details of the TTLA collective output shows that, although TTLA did not formulate routes identical to the commercial processes, the set of commercial starting materials used by TTLA are very similar to those used in the reported commercial processes for both drugs (Fig. S15 and S16†). In fact, all starting materials used in the commercial process for fostemsavir are present in the set for this drug.
In terms of individual reaction steps, we find that TTLA explores a large number of single reactions to arrive at the top-scoring short routes proposed in the above retrosynthesis. In the case of fostemsavir, the key retrosynthetic C- and N-acylation of the oxalyl starting material is discovered after 27067 single predicted steps (Fig. 3b, step a′), probably because this step is rather complex and unusual. In the case of ozanimod, TTLA performed 7594 individual single-step predictions to arrive at the proposed retrosyntheses, with the best scoring route being discovered after 2700 iterations. Interestingly, the formation of the oxadiazole ring is discovered already at iteration 8 (Fig. 4b, step c′). It should be noted that the order of iterations and therefore the number of attempts necessary to identify high-scoring routes depends on the scoring function used to prioritize node expansion, here the RPScore, which takes the simplicity and number of steps into account.
The output of TTLA can be visualized by representing the collective predicted single steps in a TMAP43 computed using the differential reaction fingerprint (DRFP)44 as a similarity measure. As illustrated for ozanimod, colour-coding by step iteration number indicates that TTLA explores a broad diversity of steps directly from the beginning of the retrosynthesis exploration, which we attribute to our diverse reaction center tagging approach used (Fig. 5a). This diversity is also visible when colour-coding all steps involving the final product, corresponding to the initial retrosynthesis, which are broadly distributed on the map (Fig. 5b). A similar pattern is visible in the TMAP of the predicted single steps for fostemsavir (Fig. S17†).
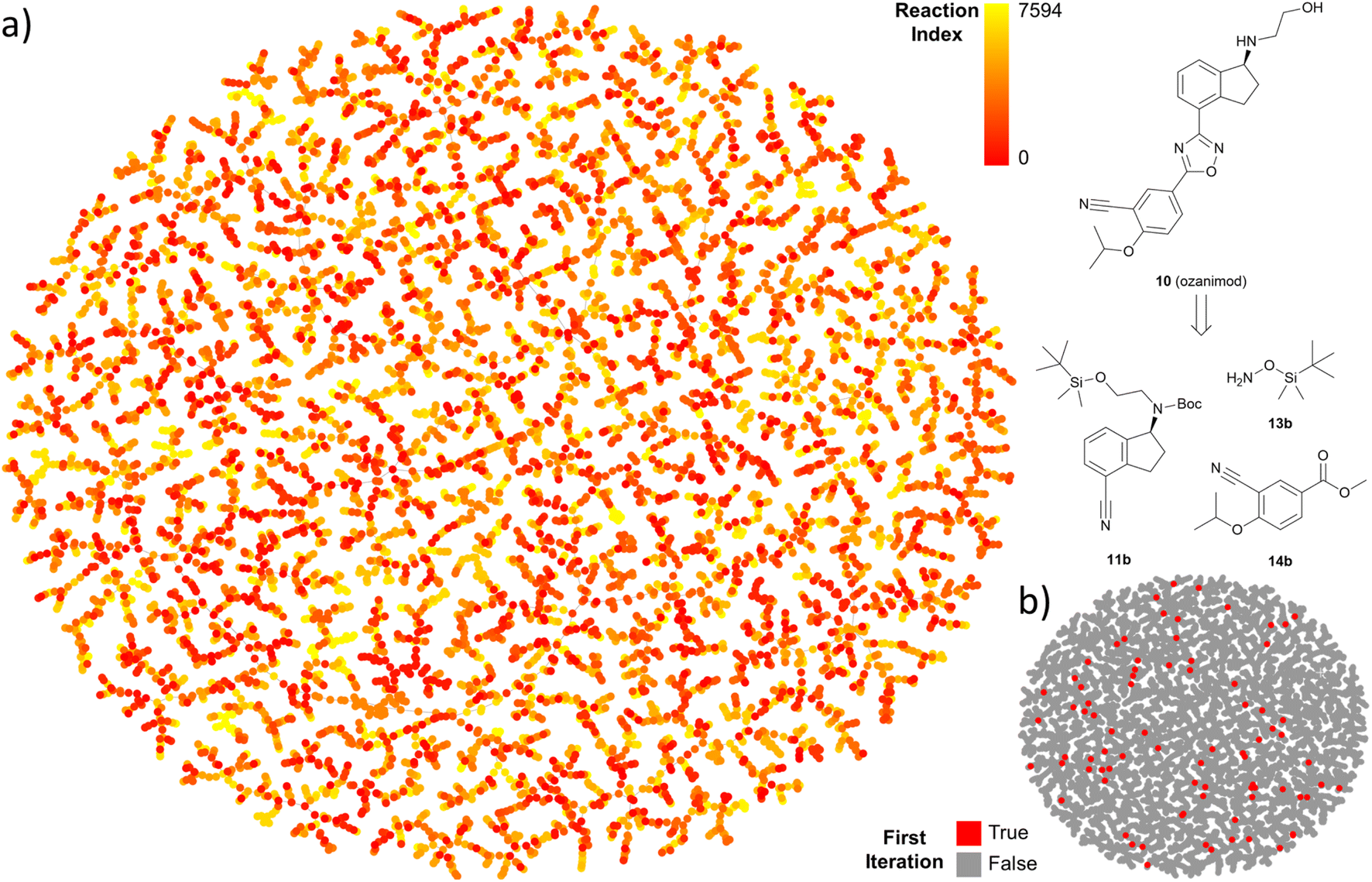 | ||
| Fig. 5 TMAP representation of iterated predictions for the multistep search of ozanimod. (a) Predicted reactions from the target molecule (low indexes) to end nodes. (b) Highlighted first iteration of the TTLA search. Interactive map available at https://tm.gdb.tools/TTLA/ozanimod. | ||
Comparing TTLA with other retrosynthesis tools
Previous retrosynthesis tools, template-based or transformer-based, predict starting material from products by applying the most probable retrosynthetic operation according to a training set. Here we combined exhaustive and template-based methods to label many potential reactive sites, which lead us to test many possible disconnections (Fig. 2d). These potential reactive sites were then challenged with the TTL, which produced detailed predictions including starting materials and reagents. In the examples discussed above TTLA identified short routes comparable to the reported processes, which were all examples of optimized production routes.By comparison, a currently available version of AiZynthFinder (v3.7.0),14 a templated-based retrosynthesis tool, fails to propose a synthesis for fostemsavir due to its inability to find a synthesis for a bis-tert-butyl phosphate starting material (Fig. S18†). AizynthFinder furthermore proposes a short route similar to TTLA for ozanimod, although including somewhat less realistic steps, for example, an alkylation of a primary amine with 2-bromoethyl acetate which would probably rather lead to acetyl transfer, and no indication of reagents (Fig. S19†). On the other hand, the online portal of IBM RXN for chemistry,45 which uses a transformer model, predicts essentially the same route as TTLA for fostemsavir (Fig. S20†). For ozanimod however, this tool settles on an eight-step route which, although containing realistic steps, is simply much longer than the commercial process or the route proposed by TTLA (Fig. S21†). For both of these retrosynthesis tools, whether the routes are part of their training sets is not known.
To statistically evaluate our TTLA, we selected target molecules from the retrosynthesis benchmark dataset shared by Genheden et al. which were absent from our training dataset.46 Due to the high computing time of our method, a random subset of 240 target molecules was selected. Solved routes involving reaction steps present in our training dataset were removed from the evaluation. TTLA proposed retrosyntheses to commercially available starting materials for 97.5% of the target molecules, which is comparable to the performance of other retrosynthetic tools reported in the original paper.46 Selected examples are shown in Fig. S22–S31.†
Conclusion
In summary, our data shows that a triple transformer loop (TTL) operating on products with tagged reactive atoms achieves efficient single-step retrosynthesis predictions. TTL was integrated into a tree-exploration strategy using a route penalty scoring scheme to form the multistep retrosynthesis tool TTLA, which can predict short synthetic routes for drug molecules. Since our approach uses transformer models, it should be possible to specialize TTLA for specific reaction classes by transfer learning similar to transformer models for forward prediction.47 Furthermore, predicting SM from P and R from SM + P separately might be potentially adapted to reactions with more complex reagents such as enzymes48–50 and help expand the scope of CASP systems. It should however be noted that the use of multiple transformer models and the detailed analysis of many possible disconnections renders our approach relatively slow, requiring up to several hours of computing time for a full retrosynthetic analysis. Efficiency increases might be possible in the future by fine-tuning the selection of potential disconnections and improving the tree search.Data availability
Code and instructions to compute multistep retrosynthesis as well as the code to tag reactive sites can be found on our GitHub repository: https://github.com/reymond-group/MultiStepRetrosynthesisTTL. The original USPTO dataset can be found at https://doi.org/10.6084/m9.figshare.5104873.v1. The derived version of USPTO of Thakkar et al. could be found in their Zenodo repository.30,51Author contributions
DK designed and carried out the study and wrote the paper, JLR designed and supervised the study and wrote the paper.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported financially by Novartis. We would like to thank Dr Thierry Schlama, Dr Daniel Kaufmann, Dr Radka Snajdrova, Dr John Lopez, Dr Frederic Stanger, Dr Fabrice Gallou and Dr Thomas Ruch, for helpful discussions. Calculations were performed on UBELIX (http://www.id.unibe.ch/hpc), the HPC cluster at the University of Bern.References
- E. J. Corey and W. T. Wipke, Computer-Assisted Design of Complex Organic Syntheses, Science, 1969, 166(3902), 178–192, DOI:10.1126/science.166.3902.178.
- A. J. Lawson, J. Swienty-Busch, T. Géoui and D. Evans, The Making of Reaxys—Towards Unobstructed Access to Relevant Chemistry Information, in The Future of the History of Chemical Information, ACS Symposium Series, American Chemical Society, 2014, vol. 1164, pp. 127–148, DOI:10.1021/bk-2014-1164.ch008.
- D. M. Lowe, Extraction of Chemical Structures and Reactions from the Literature, PhD thesis, University of Cambridge, 2012, DOI:10.17863/CAM.16293.
- D. M. Lowe, Chemical Reactions from US Patents (1976-Sep2016), Figshare Dataset, 2017 DOI:10.6084/m9.figshare.5104873.v1.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Computer-Assisted Synthetic Planning: The End of the Beginning, Angew. Chem., Int. Ed., 2016, 55(20), 5904–5937, DOI:10.1002/anie.201506101.
- C. W. Coley, W. H. Green and K. F. Jensen, Machine Learning in Computer-Aided Synthesis Planning, Acc. Chem. Res., 2018, 51(5), 1281–1289, DOI:10.1021/acs.accounts.8b00087.
- F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler and F. Glorius, Machine Learning the Ropes: Principles, Applications and Directions in Synthetic Chemistry, Chem. Soc. Rev., 2020, 49(17), 6154–6168, 10.1039/C9CS00786E.
- A. Thakkar, S. Johansson, K. Jorner, D. Buttar, J.-L. Reymond and O. Engkvist, Artificial Intelligence and Automation in Computer Aided Synthesis Planning, React. Chem. Eng., 2021, 6(1), 27–51, 10.1039/D0RE00340A.
- K. Molga, S. Szymkuć and B. A. Grzybowski, Chemist Ex Machina: Advanced Synthesis Planning by Computers, Acc. Chem. Res., 2021, 54(5), 1094–1106, DOI:10.1021/acs.accounts.0c00714.
- P. Schwaller, A. C. Vaucher, R. Laplaza, C. Bunne, A. Krause, C. Corminboeuf and T. Laino, Machine Intelligence for Chemical Reaction Space, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12(5), e1604, DOI:10.1002/wcms.1604.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI, Nature, 2018, 555(7698), 604–610, DOI:10.1038/nature25978.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Prediction of Organic Reaction Outcomes Using Machine Learning, ACS Cent. Sci., 2017, 3(5), 434–443, DOI:10.1021/acscentsci.7b00064.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, Datasets and Their Influence on the Development of Computer Assisted Synthesis Planning Tools in the Pharmaceutical Domain, Chem. Sci., 2019, 11(1), 154–168, 10.1039/C9SC04944D.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, AiZynthFinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning, J. Cheminf., 2020, 12(1), 70, DOI:10.1186/s13321-020-00472-1.
- D. Weininger, SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005.
- D. Weininger, A. Weininger and J. L. Weininger, SMILES. 2. Algorithm for Generation of Unique SMILES Notation, J. Chem. Inf. Comput. Sci., 1989, 29(2), 97–101, DOI:10.1021/ci00062a008.
- J. Nam and J. Kim, Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions, arXiv, 2016, preprint, arXiv:1612.09529, DOI:10.48550/arXiv.1612.09529.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser and I. Polosukhin, Attention Is All You Need, in Advances in Neural Information Processing Systems 30, ed. I. Guyon, U. V. Luxburg, S. Bengio, H. Wallach, R. Fergus, S. Vishwanathan and R. Garnett, Curran Associates, Inc., 2017, pp. 5998–6008 Search PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models, ACS Cent. Sci., 2017, 3(10), 1103–1113, DOI:10.1021/acscentsci.7b00303.
- P. Schwaller, T. Gaudin, D. Lányi, C. Bekas and T. Laino, “Found in Translation”: Predicting Outcomes of Complex Organic Chemistry Reactions Using Neural Sequence-to-Sequence Models, Chem. Sci., 2018, 9(28), 6091–6098, 10.1039/C8SC02339E.
- P. Karpov, G. Godin and I. V. Tetko, A Transformer Model for Retrosynthesis, in Artificial Neural Networks and Machine Learning – ICANN 2019: Workshop and Special Sessions, ed. I. V. Tetko, V. Kůrková, P. Karpov and F. Theis, Lecture Notes in Computer Science; Springer International Publishing, Cham, 2019, pp. 817–830, DOI:10.1007/978-3-030-30493-5_78.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583, DOI:10.1021/acscentsci.9b00576.
- K. Lin, Y. Xu, J. Pei and L. Lai, Automatic Retrosynthetic Route Planning Using Template-Free Models, Chem. Sci., 2020, 11(12), 3355–3364, 10.1039/C9SC03666K.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting Retrosynthetic Pathways Using Transformer-Based Models and a Hyper-Graph Exploration Strategy, Chem. Sci., 2020, 11(12), 3316–3325, 10.1039/C9SC05704H.
- S. Zheng, T. Zeng, C. Li, B. Chen, C. W. Coley, Y. Yang and R. Wu, Deep Learning Driven Biosynthetic Pathways Navigation for Natural Products with BioNavi-NP, Nat. Commun., 2022, 13(1), 3342, DOI:10.1038/s41467-022-30970-9.
- I. V. Tetko, P. Karpov, R. Van Deursen and G. Godin, State-of-the-Art Augmented NLP Transformer Models for Direct and Single-Step Retrosynthesis, Nat. Commun., 2020, 11(1), 5575, DOI:10.1038/s41467-020-19266-y.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Chemformer: A Pre-Trained Transformer for Computational Chemistry, Mach. learn.: sci. technol., 2022, 3(1), 015022, DOI:10.1088/2632-2153/ac3ffb.
- X. Wang, Y. Qian, H. Gao, W. C. Coley, Y. Mo, R. Barzilay and K. F. Jensen, Towards Efficient Discovery of Green Synthetic Pathways with Monte Carlo Tree Search and Reinforcement Learning, Chem. Sci., 2020, 11(40), 10959–10972, 10.1039/D0SC04184J.
- B. Chen, C. Li, H. Dai and L. Song, Retro*: Learning Retrosynthetic Planning with Neural Guided A* Search, in Proceedings of the 37th International Conference on Machine Learning, PMLR, 2020, pp. 1608–1616 Search PubMed.
- A. Thakkar, A. C. Vaucher, A. Byekwaso, P. Schwaller, A. Toniato and T. Laino, Unbiasing Retrosynthesis Language Models with Disconnection Prompts, ACS Cent. Sci., 2023, 9(7), 1488–1498, DOI:10.1021/acscentsci.3c00372.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of Organic Chemistry Grammar from Unsupervised Learning of Chemical Reactions, Sci. Adv., 2021, 7(15), eabe4166, DOI:10.1126/sciadv.abe4166.
- A. Byekwaso, A. C. Vaucher, P. Schwaller, A. Toniato and T. Laino, A Sequence-to-Sequence Transformer Model for Disconnection Aware Retrosynthesis, 2021, DOI:10.26434/chemrxiv-2021-7hp1s.
- G. Landrum, RDKit: Open-Source Cheminformatics, 2006 Search PubMed.
- G. Klein, Y. Kim, Y. Deng, J. Senellart and A. Rush, OpenNMT: Open-Source Toolkit for Neural Machine Translation, in Proceedings of ACL 2017, System Demonstrations, Association for Computational Linguistics, Vancouver, Canada, 2017, pp. 67–72 Search PubMed.
- OpenNMT/OpenNMT-py. GitHub, https://github.com/OpenNMT/OpenNMT-py accessed 2020-07-28.
- C. W. Coley, L. Rogers, W. H. Green and K. F. S. C. S. Jensen, Synthetic Complexity Learned from a Reaction Corpus, J. Chem. Inf. Model., 2018, 58(2), 252–261, DOI:10.1021/acs.jcim.7b00622.
- P. Schwaller, R. Petraglia, V. H. Nair and T. LainoEvaluation Metrics for Single-Step Retrosynthetic Models, Second Workshop on Machine Learning and the Physical Sciences (NeurIPS 2019), 2019 Search PubMed.
- M. Andronov, V. Voinarovska, N. Andronova, M. Wand, D.-A. Clevert and J. Schmidhuber, Reagent Prediction with a Molecular Transformer Improves Reaction Data Quality, Chem. Sci., 2023, 14(12), 3235–3246, 10.1039/D2SC06798F.
- W. Velanguparackel, N. Hamon, J. Balzarini, C. McGuigan and A. D. Westwell, Synthesis, Anti-HIV and Cytostatic Evaluation of 3′-Deoxy-3′-Fluorothymidine (FLT) pro-Nucleotides, Bioorg. Med. Chem. Lett., 2014, 24(10), 2240–2243, DOI:10.1016/j.bmcl.2014.03.092.
- T. Wang, Y. Ueda, Z. Zhang, Z. Yin, J. Matiskella, B. C. Pearce, Z. Yang, M. Zheng, D. D. Parker, G. A. Yamanaka, Y.-F. Gong, H.-T. Ho, R. J. Colonno, D. R. Langley, P.-F. Lin, N. A. Meanwell and J. F. Kadow, Discovery of the Human Immunodeficiency Virus Type 1 (HIV-1) Attachment Inhibitor Temsavir and Its Phosphonooxymethyl Prodrug Fostemsavir, J. Med. Chem., 2018, 61(14), 6308–6327, DOI:10.1021/acs.jmedchem.8b00759.
- F. L. Scott, B. Clemons, J. Brooks, E. Brahmachary, R. Powell, H. Dedman, H. G. Desale, G. A. Timony, E. Martinborough, H. Rosen, E. Roberts, M. F. Boehm and R. J. Peach, Ozanimod (RPC1063) Is a Potent Sphingosine-1-Phosphate Receptor-1 (S1P1) and Receptor-5 (S1P5) Agonist with Autoimmune Disease-Modifying Activity, Br. J. Pharmacol., 2016, 173(11), 1778–1792, DOI:10.1111/bph.13476.
- A. C. Flick, C. A. Leverett, H. X. Ding, E. L. McInturff, S. J. Fink, S. Mahapatra, D. W. Carney, E. A. Lindsey, J. C. DeForest, S. P. France, S. Berritt, S. V. Bigi-Botterill, T. S. Gibson, R. B. Watson, Y. Liu and C. J. O'Donnell, Synthetic Approaches to the New Drugs Approved During 2020, J. Med. Chem., 2022, 65(14), 9607–9661, DOI:10.1021/acs.jmedchem.2c00710.
- D. Probst and J.-L. Reymond, Visualization of Very Large High-Dimensional Data Sets as Minimum Spanning Trees, J. Cheminf., 2020, 12(1), 12, DOI:10.1186/s13321-020-0416-x.
- D. Probst, P. Schwaller and J.-L. Reymond, Reaction Classification and Yield Prediction Using the Differential Reaction Fingerprint DRFP, Digital Discovery, 2022, 1(2), 91–97, 10.1039/D1DD00006C.
- IBM RXN for Chemistry, https://rxn.res.ibm.com accessed 2022-09-05.
- S. Genheden and E. PaR. Bjerrum, Towards a Framework for Benchmarking Retrosynthesis Route Predictions, Digital Discovery, 2022, 1(4), 527–539, 10.1039/D2DD00015F.
- G. Pesciullesi, P. Schwaller, T. Laino and J.-L. Reymond, Transfer Learning Enables the Molecular Transformer to Predict Regio- and Stereoselective Reactions on Carbohydrates, Nat. Commun., 2020, 11(1), 4874, DOI:10.1038/s41467-020-18671-7.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, RetroBioCat as a Computer-Aided Synthesis Planning Tool for Biocatalytic Reactions and Cascades, Nat. Catal., 2021, 4(2), 98–104, DOI:10.1038/s41929-020-00556-z.
- D. Kreutter, P. Schwaller and J.-L. Reymond, Predicting Enzymatic Reactions with a Molecular Transformer, Chem. Sci., 2021, 12(25), 8648–8659, 10.1039/D1SC02362D.
- D. Probst, M. Manica, Y. G. Nana Teukam, A. Castrogiovanni, F. Paratore and T. Laino, Biocatalysed Synthesis Planning Using Data-Driven Learning, Nat. Commun., 2022, 13(1), 964, DOI:10.1038/s41467-022-28536-w.
- A. Thakkar, A. Vaucher, A. Byekwaso, P. Schwaller, A. Toniato and T. Laino, Disconnection Labelled Reaction Data, Zenodo, 2022, DOI:10.5281/zenodo.7101695.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01604h |
| This journal is © The Royal Society of Chemistry 2023 |