
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenated PETN derivatives: interplay between physical and chemical factors in explosive sensitivity†
Nicholas
Lease
 *a,
Kyle D.
Spielvogel
a,
Jack V.
Davis
a,
Jeremy T.
Tisdale
a,
Lisa M.
Klamborowski
a,
M. J.
Cawkwell
b and
Virginia W.
Manner
a
*a,
Kyle D.
Spielvogel
a,
Jack V.
Davis
a,
Jeremy T.
Tisdale
a,
Lisa M.
Klamborowski
a,
M. J.
Cawkwell
b and
Virginia W.
Manner
a
aHigh Explosives Science and Technology, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: nlease@lanl.gov
bTheoretical Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
First published on 3rd May 2023
Abstract
Determining the factors that influence and can help predict energetic material sensitivity has long been a challenge in the explosives community. Decades of literature reports identify a multitude of factors both chemical and physical that influence explosive sensitivity; however no unifying theory has been observed. Recent work by our team has demonstrated that the kinetics of “trigger linkages” (i.e., the weakest bonds in the energetic material) showed strong correlations with experimental drop hammer impact sensitivity. These correlations suggest that the simple kinetics of the first bonds to break are good indicators for the reactivity observed in simple handling sensitivity tests. Herein we report the synthesis of derivatives of the explosive pentaerythritol tetranitrate (PETN) in which one, two or three of the nitrate ester functional groups are substituted with an inert group. Experimental and computational studies show that explosive sensitivity correlates well with Q (heat of explosion), due to the change in the number of trigger linkages removed from the starting material. In addition, this correlation appears more significant than other observed chemical or physical effects imparted on the material by different inert functional groups, such as heat of formation, heat of explosion, heat capacity, oxygen balance, and the crystal structure of the material.
Introduction
Energetic materials, which include explosives and propellants, store fuel and oxidizers on the same molecule. Upon reaction, energetic materials rapidly release large quantities of heat and gas. For example, the power output at the shock front from a detonating secondary explosive is about 1010 W cm−2.1 Understanding the sensitivity of explosives is vital to their proper usage and the prevention of any potential accidents. In the early design stage, the handling safety of new energetic materials is assessed by exposing the material to a range of stimuli, most notably temperature, shock, impact, friction, or electrostatic discharge, and determining if a violent event has occurred. Decades of research have been devoted to understanding the factors that determine the sensitivity of energetic materials. For example, multiple microscopic factors are thought to affect the sensitivity, including the crystal structure,2–4 free volume within the unit cell,5,6 intermolecular hydrogen bonding,7–14 and electrostatic potentials.15,16 Other studies have identified the importance of oxygen balance,17,18 bond dissociation energies,19 reaction rates20,21 and the heat of explosion.22,23Most sensitivity tests convolute those intrinsic properties of energetic molecules and materials that control their sensitivity, such as their functional groups, stoichiometry and oxygen balance, and decomposition kinetics with meso- and macroscopic properties including the phase, impurities, and particle shape and size, such that it is often difficult to understand which properties of a new material are responsible for a particular result. To simplify these analyses, we have explored the use of a core chemical scaffold to which systematic modifications can be made to better assess how these specific functional changes influence explosive sensitivity. Recent studies focusing on pentaerythritol tetranitrate (PETN)24–28 and erythritol tetranitrate (ETN)29 have been conducted looking at substituting functional groups on their core scaffold to more systematically modulate explosive sensitivity.
The weakest bond in an energetic molecule, its “trigger linkage”, has been shown to be of particular importance to sensitivity because the cascade of exothermic reactions that occur during a deflagration or detonation ultimately begins with the thermally-activated rupture of the weakest bonds. Recent theoretical work has confirmed that the kinetics of trigger linkage rupture is correlated with experimental drop weight impact sensitivity.22,30 These correlations suggest that the simple kinetics of the first bonds to break are good indicators for the reactivity observed in simple handling sensitivity tests. In addition, handling sensitivity was shown to depend sensitively on the total energy release, or enthalpy of explosion, Q, upon the formation of product species.20,21,31–33 This result is consistent with the observation that the explosives with the best performance also tend to be among the most sensitive.
Several models for the connection between sensitivity and energy release have been proposed in the literature34 but recent work by this team found that a reduction in the activation enthalpy for decomposition via the Bell–Evans–Polanyi principle gave the best fit to a set of sensitivity data for explosives with diverse functional groups.22 This model was validated experimentally by a study on a series of PETN-related explosives where nitrate esters were systematically replaced with inert hydroxyl groups. The decrease in the heat of explosion that occurs when energetic groups are replaced by inert ones led to a precipitous decrease in sensitivity, which can be rationalized if the effective activation energy for decomposition in an Arrhenius rate itself depends on Q. Hence, this study highlighted the importance of kinetic and thermodynamic influences on handling sensitivity. The purpose of this study was to investigate a wider range of substituents on a simple chemical scaffold to evaluate if the correlations with trigger linkage strength and energy release override other chemical, structural, and physical influences such as oxygen balance or the crystal structure. Herein, we report the synthesis of PETN derivatives in which either one, two or three nitrate esters are replaced with other functional groups. Novel syntheses of PETN derivatives functionalized with halogen atoms are reported and are rare examples of aliphatic energetic materials with halogen functional groups. While halogen-based explosives are known primarily as agent defeat weapons (ADWs)35,36 most of these materials are aromatic37–42 and known aliphatic materials commonly use fluorine as the primary halogen.43–47 To compare halogen PETN derivatives, materials in which the nitrate ester of PETN is substituted with methyl and hydroxyl groups were also prepared. The sensitivities of PETN derivatives to impact, friction, and electrostatic discharge were evaluated. Their melting and decomposition onset temperatures were measured by differential scanning calorimetry (DSC). Thermochemical properties including heat of formation 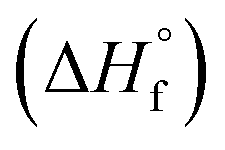 and heat of explosion (Q) were evaluated using a new, extended version of the density functional theory (DFT) atom-equivalent energy scheme developed originally by Byrd and Rice (SI). In addition, room temperature heat capacities (Cv) and oxygen balance were calculated for each PETN derivative using DFT and stoichiometry, respectively. Crystal structures were obtained for all the derivatives using single crystal X-ray diffraction in order to analyze the effects of substitution on crystal structures with the aim of identifying any correlations with the measured explosive sensitivities. Our analyses showed that the dominant influence on the PETN derivatives derived from the systematic decrease in the enthalpy of explosion (Q) when energetic groups were substituted with inert halogen, methyl, and hydroxyl groups, while crystal structures have little to no influence on sensitivity.
and heat of explosion (Q) were evaluated using a new, extended version of the density functional theory (DFT) atom-equivalent energy scheme developed originally by Byrd and Rice (SI). In addition, room temperature heat capacities (Cv) and oxygen balance were calculated for each PETN derivative using DFT and stoichiometry, respectively. Crystal structures were obtained for all the derivatives using single crystal X-ray diffraction in order to analyze the effects of substitution on crystal structures with the aim of identifying any correlations with the measured explosive sensitivities. Our analyses showed that the dominant influence on the PETN derivatives derived from the systematic decrease in the enthalpy of explosion (Q) when energetic groups were substituted with inert halogen, methyl, and hydroxyl groups, while crystal structures have little to no influence on sensitivity.
Results
Synthesis of PETN derivatives
Pentaerythritol tetranitrate (PETN) is composed of a neo-pentyl scaffold with a nitrate ester attached to each of the four CH2 groups. Due to the neo-pentyl structure, substitution on the CH2 groups can be difficult due to the increased steric bulk at the substitution position.48 The derivatives described in this paper focus on mono (pentaerythritol trinitrate, PETriN), di (pentaerythritol dinitrate, PEDN) and tri (pentaerythritol mononitrate, PEMN) substitution of the nitrate esters by the halogen atoms, chlorine, bromine, and iodine. While the synthesis of some of these materials has been reported, in many cases there has been limited or no characterization of the materials and no sensitivity testing has been performed.49–51 Due to the complicated and extensive synthesis pathways and need for hazardous reagents the fluorine derivatives of PETN were not examined.All the starting materials for the bromide-based PETN derivatives were commercially available. Synthesis of the mixed bromide/nitrate ester derivatives of PETN was carried out using a standard mixed acid synthesis of fuming nitric acid and sulfuric acid (Scheme 1). The three derivatives PETriN-Br, PEDN-Br2 and PEMN-Br3 were isolated as white crystalline solids. Differential scanning calorimetry (DSC) showed melting temperatures of 91.6, 73.1 and 56.1 °C for PETriN-Br, PEDN-Br2, and PEMN-Br3 respectively. The onset of thermal decomposition temperatures for the three derivatives are 169.2, 172.6 and 181.0 °C for PETriN-Br, PEDN-Br2, and PEMN-Br3. Bromine derivatization of PETN (melt 140.9/decomposition 166.8 °C) led to a decrease in the melting point of each derivative and a slight increase in the decomposition temperature.
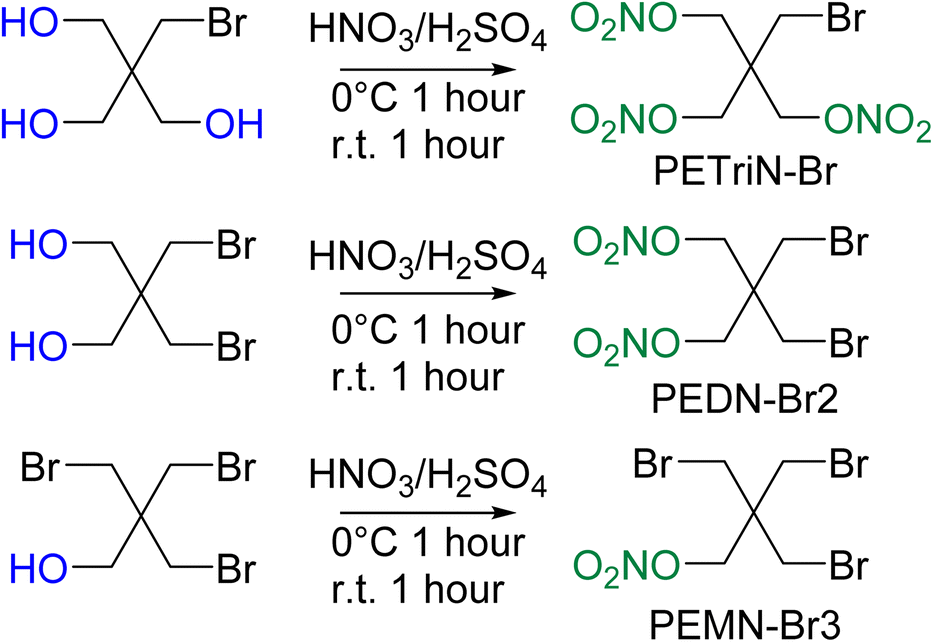 | ||
| Scheme 1 Synthesis of Br derivatives of PETN. | ||
Precursor materials for the iodine PETN derivatives were not commercially available and were synthesized through a salt metathesis reaction of the bromine starting materials with sodium iodide in DMF using a modified literature procedure52 (Scheme 2). Heating the solutions to temperatures over 100 °C led to the formation of iodine substituted pentaerythritols. The disubstituted iodine material was isolated as a white powder while the trisubstituted pentaerythritol was isolated as a sticky solid.
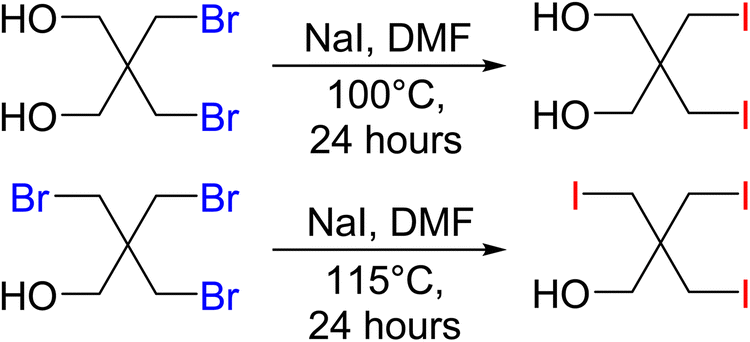 | ||
| Scheme 2 Synthesis of precursors for iodine derivatives of PETN. | ||
Initial attempts at nitration of the iodo-substituted derivatives using a mixed acid nitration were unsuccessful. Nitration methods using 70% nitric acid with trifluoroacetic anhydride (TFAA) resulted in iodo-substituted PETN derivatives, though with the formation of multiple products. A reddish-brown solution formed almost immediately upon addition of the iodo precursor materials to TFAA and nitric acid. After pouring the solution into ice water, a dark colored solid was isolated. Washing the solid with sodium thiosulfate removed the color leaving a white powdery solid. NMR spectroscopy of the product revealed two products in solution, one identified as di-iodo pentaerythritol dinitrate (PEDN-I2) and the other as mono-iodo pentaerythritol trinitrate (PETriN-I) (Scheme 3). The two products, PETRIN-I and PEDN-I2, were isolated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The presence of PETriN-I implies that one of the iodide functional groups was displaced during the nitration. This displacement would be consistent with the dark color formed during the reaction (most likely I2 formation, and removal of color with thiosulfate). The CH2–I bond is likely hydrolyzed from the water present in 70% nitric acid to form the corresponding alcohol which is then nitrated in the presence of mixed acids. This reactivity was unexpected as substitutions at neopentyl carbons are notoriously difficult. Attempts to mitigate this side product formation using more concentrated nitric acid (fuming) produced pure PEDN-I2; however the yield from this reaction was below ten percent so this synthesis was deemed unfeasible. Purification of the 1:1 material was conducted using a centrifugal chromatography system which isolated both materials as pure white crystalline solids.
:1 ratio. The presence of PETriN-I implies that one of the iodide functional groups was displaced during the nitration. This displacement would be consistent with the dark color formed during the reaction (most likely I2 formation, and removal of color with thiosulfate). The CH2–I bond is likely hydrolyzed from the water present in 70% nitric acid to form the corresponding alcohol which is then nitrated in the presence of mixed acids. This reactivity was unexpected as substitutions at neopentyl carbons are notoriously difficult. Attempts to mitigate this side product formation using more concentrated nitric acid (fuming) produced pure PEDN-I2; however the yield from this reaction was below ten percent so this synthesis was deemed unfeasible. Purification of the 1:1 material was conducted using a centrifugal chromatography system which isolated both materials as pure white crystalline solids.
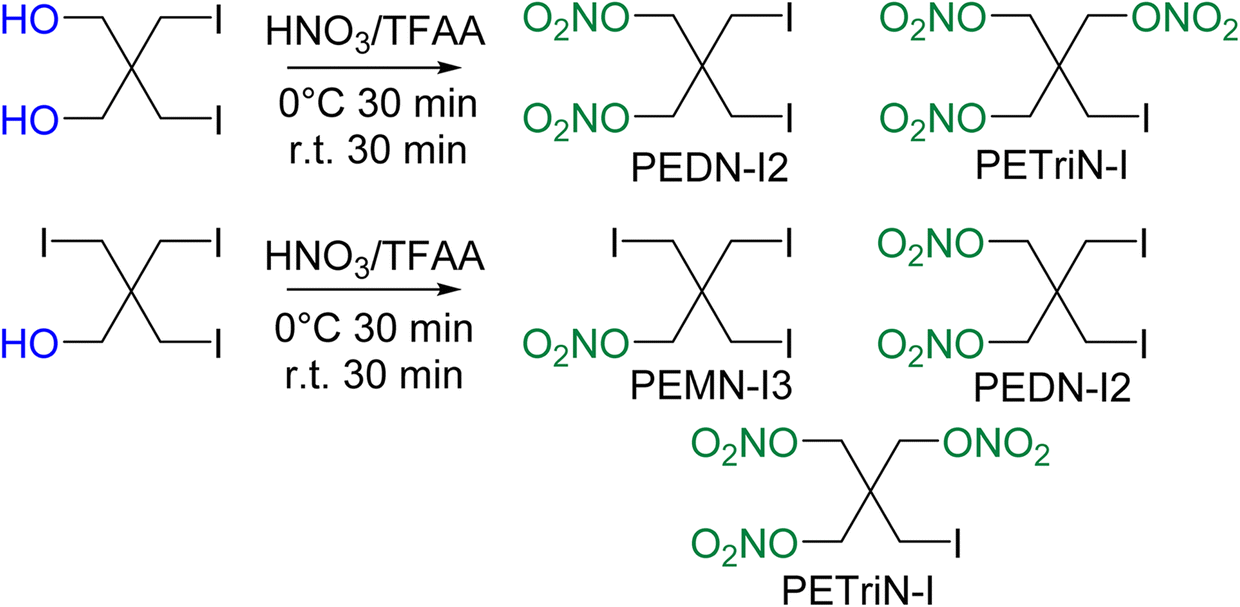 | ||
| Scheme 3 Synthesis of the iodine derivatives of PETN. | ||
Nitration of the triiodo pentaerythritol precursor similarly led to multiple products, PETRIN-I, PEDN-I2, and PEMN-I3 (Scheme 3). NMR spectroscopy showed the synthesis products to be in a ratio of 2:3:1 for PETRIN-I, PEDN-I2 and PEMN-I3 respectively. Centrifugal chromatography was used to isolate the three materials as pure white crystalline solids. DSC analysis of the three iodo derivatives showed similar results to the bromine derivatives, in which increased halide functionalization led to a decrease in melting points as well as an increase in the onset of thermal decomposition temperature. Melting points of 107.2, 81.9 and 61.5 °C and decomposition onset temperatures of 167.1, 178.3, and 182.4 °C were observed for PETRIN-I, PEDN-I2, and PEMN-I3 respectively.
Precursor chlorine derivatives of PETN were synthesized from pentaerythritol using thionyl chloride following literature procedures.53 The reaction of pentaerythritol with thionyl chloride resulted in the formation of the di and trichloro substituted pentaerythritol precursors (Scheme 4). The mono-chloride material could not be isolated and was purchased commercially. Nitration of the chloro-substituted pentaerythritol precursors was analogous to the bromine substituted materials using a mixed acid synthesis of fuming nitric and sulfuric acids (Scheme 5). PETriN-Cl, PEDN-Cl2 and PEMN-Cl3 were isolated as crystalline white solids. DSC analysis of the three chloro derivatives demonstrated similar results to the other halide derivatives with respect to melting points, but an inverse relationship was observed with the onset decomposition temperatures as increased halide functionalization led to a decrease in both melting points and onset decomposition temperature. Melting points of 66.2, 58.7 and 46.5 °C and onset decomposition temperatures of 166.3, 161.3 and 154.2 °C were observed for PETriN-Cl, PEDN-Cl2, and PEMN-Cl3 respectively.
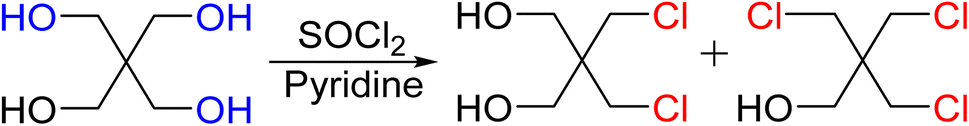 | ||
| Scheme 4 Synthesis of precursors for chlorine derivatives of PETN. | ||
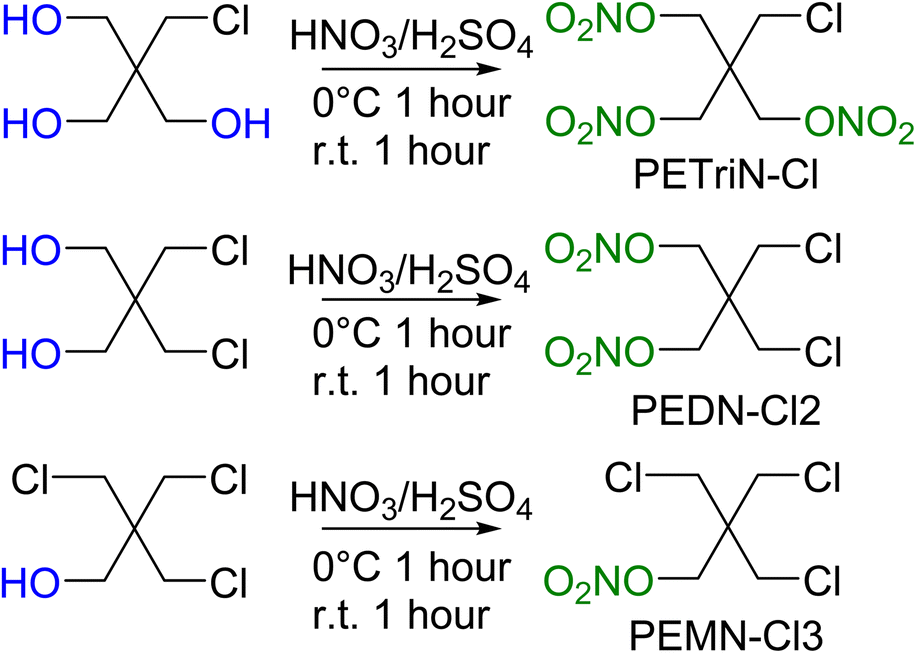 | ||
| Scheme 5 Synthesis of chlorine derivatives of PETN. | ||
In addition to the halogenated PETN derivatives, other substituted derivatives of PETN were synthesized to compare with the halogenated materials (Scheme 6). Derivatives of PETN in which the NO2 group was substituted with a methyl group (CH3) were prepared from commercially available starting materials. Nitration using a mixed acid synthesis produced the mixed nitrate ester methyl derivatives PETriN-Me54 and PEDN-Me2. The derivative with three substituted methyl groups was not prepared. While PETriN-Me was obtained as a white crystalline solid, PEDN-Me2 could only be isolated as a clear oil. PETriN-Me had a melting point of 51.5 °C and a onset decomposition temperature of 163.4 °C. PEDN-Me2 as a liquid did not show a melting point on DSC but showed a thermal onset decomposition temperature of 170.5 °C. Hydroxyl derivatives of PETN, PETriN-OH and PEDN-OH2 and PEMN-OH3, were prepared following known literature procedures.55,56 PETriN-OH can only be isolated as a clear oil while PEDN-OH2 and PEMN-OH3 can be isolated as clear oils or amorphous solids. No melting points were observed for either PETriN-OH or PEDN-OH2, though PEMN-OH3 melted at 57.7 °C. Onset thermal decomposition temperatures of 163.9, 166.4, and 162.4 °C were observed for PETriN-OH, PEDN-OH2 and PEMN-OH3 respectively.
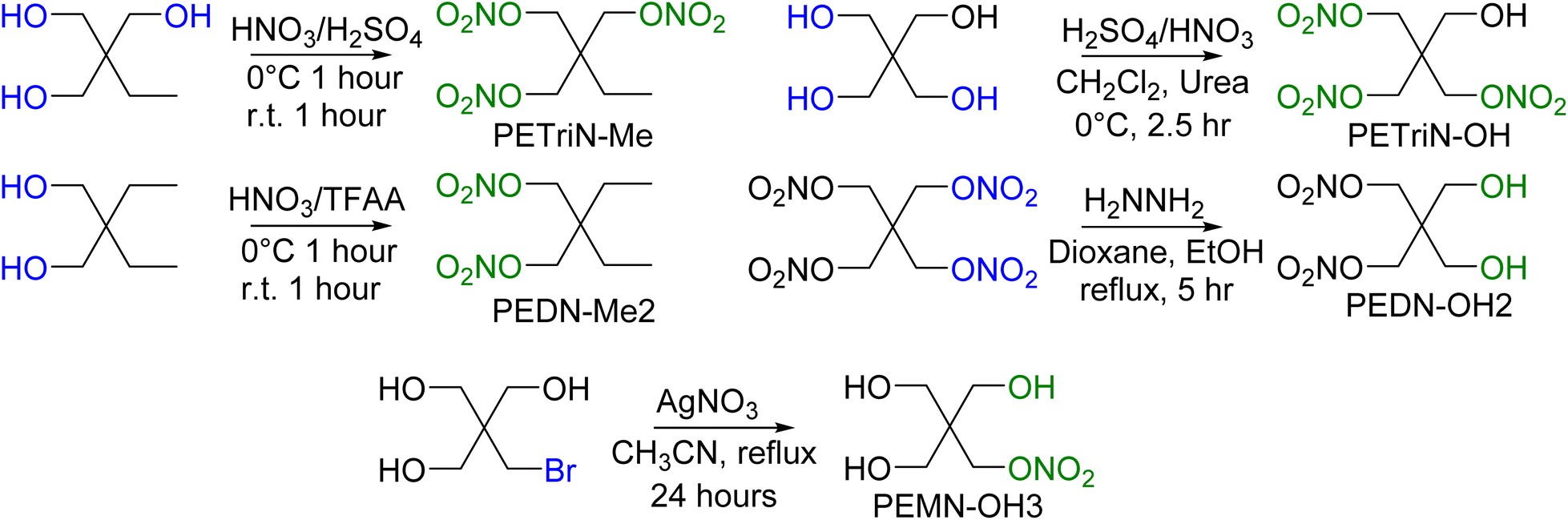 | ||
| Scheme 6 Synthesis of methyl and hydroxyl derivatives of PETN. | ||
Crystal structure results
Single crystal X-ray diffraction (XRD) analysis was conducted on the substituted PETN derivatives, except for PEDN-Me2, PETriN-OH, PEDN-OH2 and PEMN-OH3 as those materials were not isolated as crystalline solids. Crystals of the mono-substituted PETN derivatives PETriN-Cl, PETriN-Br, PETRIN-I, and PETriN-Me were all grown through slow evaporation from an acetone solution. Single crystal XRD analysis shows that all the mono-substituted derivatives of PETN have very similar crystal structures when compared to one another (Fig. 1). Small differences exist between the crystals, most notably with PETriN-Cl and PETriN-Me having monoclinic structures, while the PETriN-Br and PETRIN-I crystals have orthorhombic structures. This difference could be attributed to the smaller atomic sizes of the chlorine and methyl groups. The smaller sizes of the chlorine and methyl functional groups create small asymmetries in the molecular packing structures. The bromine and iodine derivatives are closer in size to a nitrate ester functional group, resulting in higher symmetry. Overall, Fig. 2 shows that PETN exhibits a fairly similar packing arrangement to all of the mono-substituted PETN derivatives.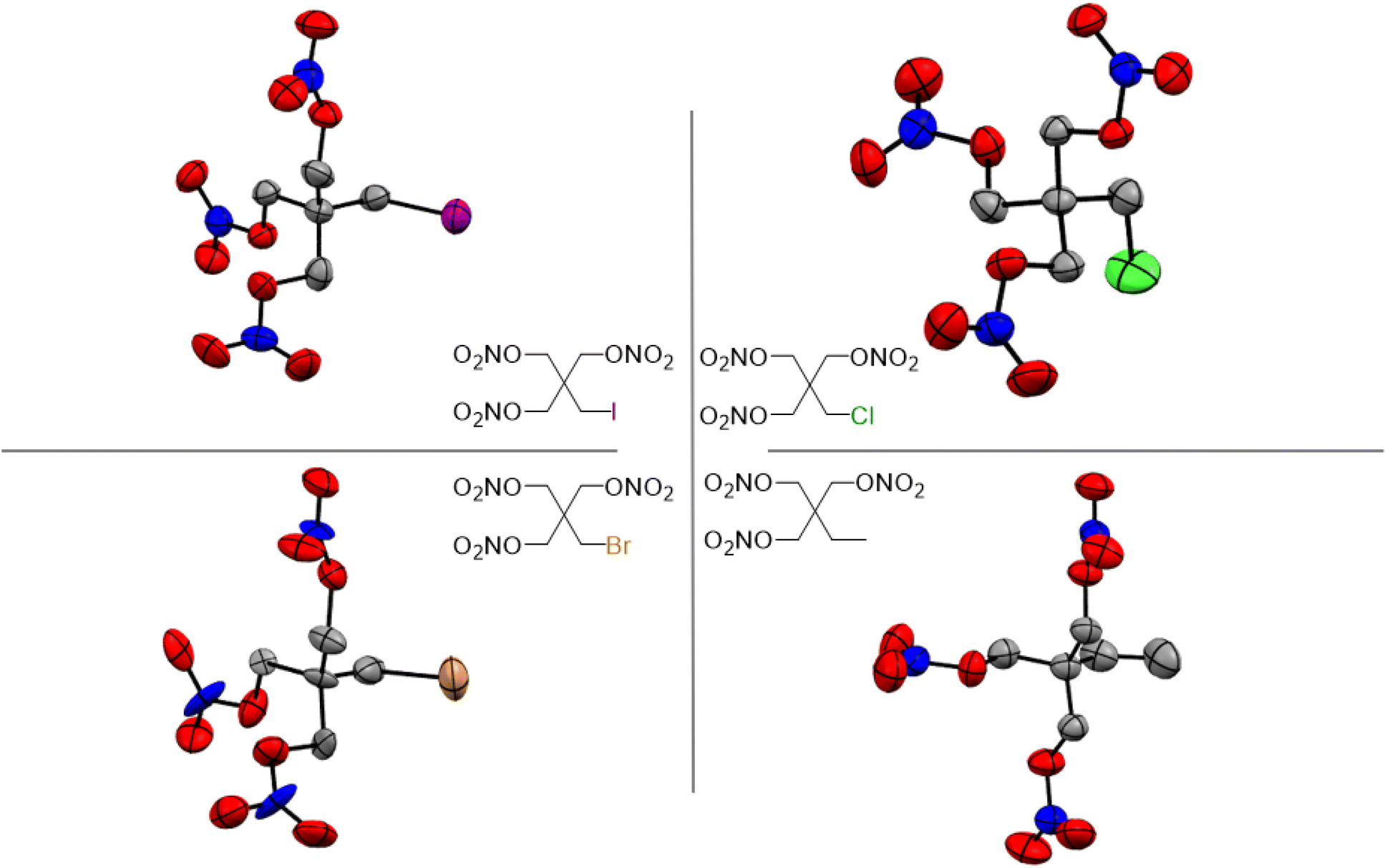 | ||
| Fig. 1 Molecular structures within single crystals of PETriN-Cl, PETriN-Br, PETriN-I and PETriN-Me. | ||
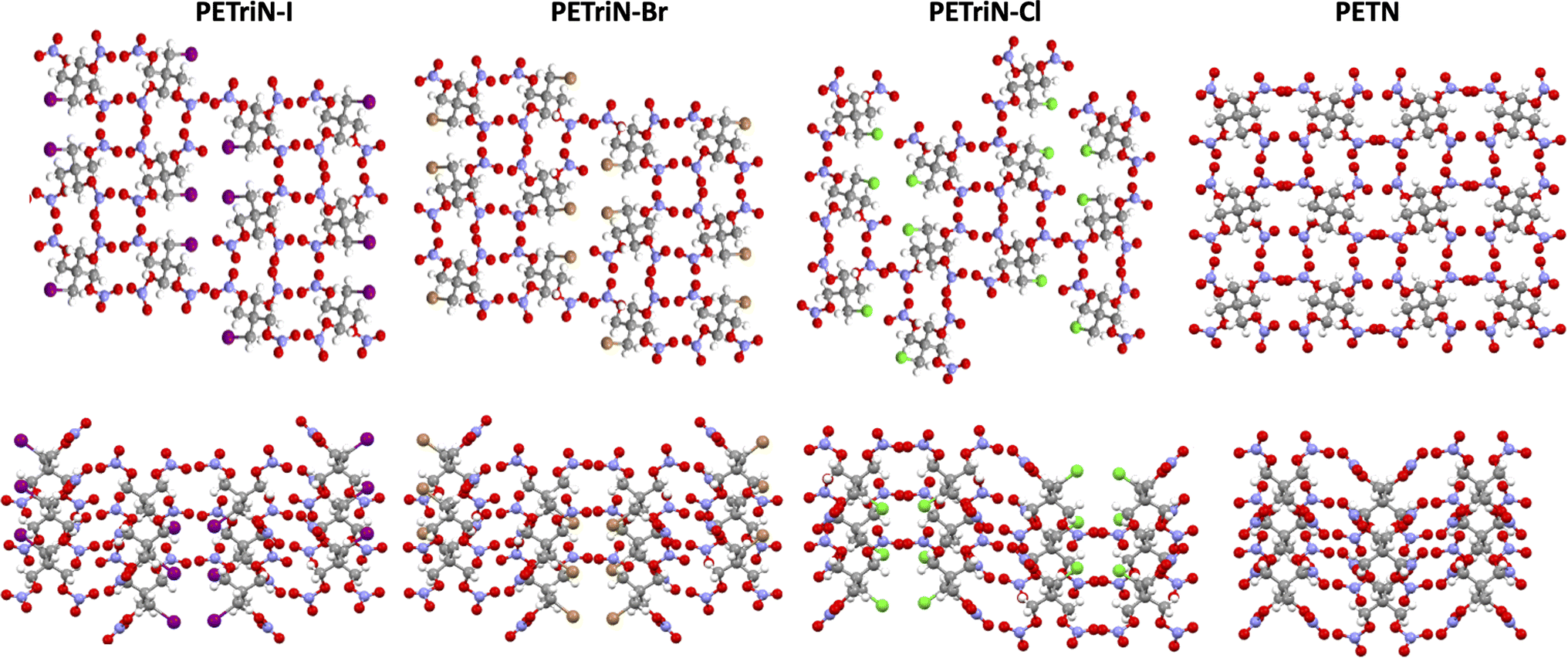 | ||
| Fig. 2 Crystal packing of PETriN-Cl, PETriN-Br, PETRIN-I and PETN. | ||
In addition to the similarity of the packing of the mono-substituted molecules, prominent bond angles and bond distances were very consistent among all the mono derivatives and PETN (Table 1). Bond distances and angles of the nitrate ester groups were consistent among all the materials with bond angles of approximately 128° (Oterm–N–Oterm) and 115° (Obridge–N–Oterm) and bond distances of 1.19 Å (Obridge–N) and 1.4 Å (N–Oterminal). The main difference between the monosubstituted materials is the bond length of the carbon–X (Cl, Br, I, CH3) bond with the larger substituents having longer bond distances.
| Bonds and angles | PETriN-Cl | PETriN-Br | PETriN-I | PETriN-Me | PETN |
|---|---|---|---|---|---|
| N–Oterminal | 1.188 | 1.195 | 1.193 | 1.194 | 1.219 |
| Obridge–N | 1.404 | 1.401 | 1.406 | 1.389 | 1.400 |
| X–C–C (deg.) | 113.28 | 113.85 | 116.19 | 115.93 | N/A |
| C–O–N (deg.) | 113.32 | 113.27 | 113.44 | 114.27 | N/A |
| C–X | 1.790 | 1.960 | 2.133 | N/A | N/A |
| C–C | N/A | N/A | N/A | 1.506 | N/A |
| Oterm–N–Oterm | 129.40 | 129.59 | 129.62 | 129.17 | 127.79 |
| Obridge–N–Oterm | 115.30 | 115.20 | 115.18 | 115.41 | 115.95 |
| ρ calc (g cm−3) [temp. (K)] | 1.723 | 1.957 | 2.158 | 1.525 | 1.845 |
| Crystal system/space group | Monoclinic/P121/c1 | Orthorhombic/P212121 | Orthorhombic/P212121 | Monoclinic/P121/n1 | Tetragonal/P42(1)c |
Like the mono-halogenated derivatives, the crystal structures of PEDN-Cl2, PEDN-Br2, and PEDN-I2 were very similar to one another. Unfortunately, these structures could not be compared to those of the explosive di-substituted PETN derivatives, PEDN-OH2 and PEDN-Me2, due to the inability to obtain single crystals of those materials. Crystal structures of the tri-halogenated materials were also obtained, showing similar patterns as observed with the other halogenated PETN derivatives. For full crystal information on all the substituted PETN derivative materials, see the ESI.†
Hirschfield surfaces and two-dimensional fingerprint analysis are used to study prominent intermolecular interactions in crystal structures.57 Information gathered from these two-dimensional fingerprints provides insight into the most prominent close contacts in the crystal structures as well as physiochemical properties of the molecules. The two spikes seen in the fingerprint plot in Fig. 3 correspond to the strongest interactions along the de (exterior interactions) and di (interior interactions) axes. Resemblance in the overall shape of the fingerprint plots compared to one another shows the similarity in the crystal structures of the derivatives. Analysis of the plots shows that in every case the dominant intermolecular and intramolecular contact is between oxygen and hydrogen, with it making up 48.3, 47.6, 42.5 and 66.7% of all close contacts for PETriN-Cl, PETriN-Br, PETRIN-I and PETriN-Me respectively. The second largest close contact contributors for each derivative were the O–O close contacts which were approximately 24% for all the mono-halogen derivatives and 16% for PETriN-Me. The main difference between the structures lies in the close contacts between the halogen X and hydrogen. As the size of the halogen increases from Cl to Br to I, the number of close contacts between X and hydrogen increases from 7.9 to 8.8 to 15.9%.
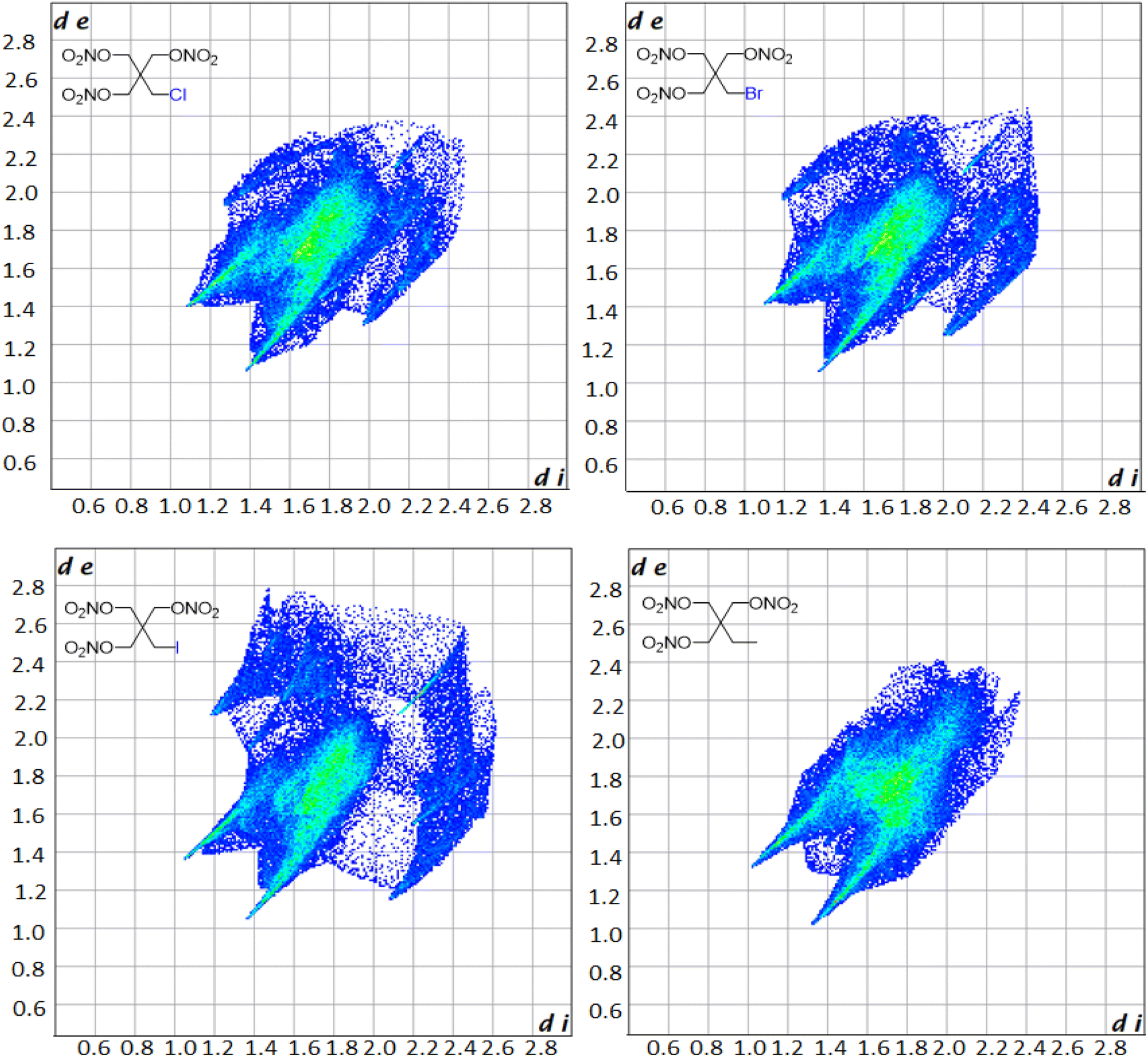 | ||
| Fig. 3 Hirschfield fingerprint plots for PETriN-Cl, PETriN-Br, PETriN-I and PETriN-Me. | ||
Discussion
Sensitivity measurements of PETN derivatives
Explosive sensitivity testing was performed on the PETN derivatives focusing on impact, electrostatic discharge (ESD) and friction sensitivity. Impact testing is measured on a standard type 12 ERL drop hammer, using 40 milligram samples placed on high melting temperature grit paper (150-grit) on a steel anvil. A 0.8 kilogram striker was placed gently over the sample being tested, and a 2.5 kilogram weight was dropped on the sample from various heights. A “go” or energetic event was determined using decibel levels measured with sound meters located behind and to the side of the apparatus.58 A set of fifteen tests were conducted, using the Neyer statistical59 method to determine a DH50 value (the drop height corresponding to a 50% chance of a “go”). Friction sensitivity analysis was performed on a BAM friction apparatus in which 20 milligrams of material was placed on a porcelain plate while a porcelain plug was rubbed against the material with varying amounts of force. Electrostatic discharge sensitivity was performed using a SMS ABL electrostatic discharge machine in which a sample of material was placed between two pieces of tape and a discharge of varying energy was applied to the sample. If the tape was broken or a popping sound was observed, a “go” was determined to have occurred after statistical analysis of 15 samples.Drop weight impact experiments were performed on the nine halogen-substituted derivatives of PETN in order to examine how increased halogen functionalization would affect the impact sensitivity (Table 2). PETN is a sensitive secondary explosive with an impact sensitivity of 10.2 ± 0.9 cm. The three mono-substituted halogen derivatives, PETriN-Cl, PETriN-Br and PETRIN-I, exhibited very similar impact drop heights of 30.8 ± 2.5, 30.7 ± 2.4 and 26.6 ± 7.1 cm respectively. It was surprising that the sensitivities for all the mono-halogen PETN derivatives were within error of each other despite the slightly different oxygen balances, molecular weights and molecular sizes of the halogen used. To examine if this similarity in sensitivity extended to other mono-substituted PETN derivatives, two additional PETN compounds were prepared in which a single nitrate ester was substituted with either a methyl or hydroxyl group. Recently reported work22 studying the impact sensitivity of hydroxyl-based PETN derivatives on the same apparatus showed that the derivative PETriN-OH had an impact sensitivity of 27.0 ± 2.2 cm, again very similar to those of the three mono-halogen substituted PETN derivatives. Likewise, PETriN-Me exhibited a DH50 value of 31.6 ± 1.8 cm, within error of the other derivatives studied.
| Material | Impact DH50 (cm) | Friction DH50 (N) | Electrostatic discharge DH50 (J) | DSC (°C) melt/decomp. onset |
|---|---|---|---|---|
| a Material was a liquid and tested on a bare anvil without the use of grit paper. b Material was tested five times on impact and friction apparatus at the highest setting without a reaction; no further testing was conducted as these materials were deemed inert. | ||||
| PETriN-Cl | 30.8 ± 2.5 | >360 | 0.0625 | 66.2/166.3 |
| PEDN-Cl2b | >320 | >360 | 0.25 | 59.5/161.3 |
| PEMN-Cl3b | >320 | >360 | 0.25 | 46.5/154.2 |
| PETriN-Br | 30.7 ± 2.4 | >360 | 0.125 | 91.6/169.2 |
| PEDN-Br2b | >320 | >360 | 0.125 | 73.1/172.6 |
| PEMN-Br3b | >320 | >360 | 0.125 | 56.1/181.0 |
| PETRIN-I | 26.6 ± 7.1 | >360 | 0.0625 | 107.2/167.1 |
| PEDN-I2b | >320 | >360 | 0.0625 | 81.9/178.3 |
| PEMN-I3b | >320 | N/A | N/A | 61.5/182.4 |
| PETriN-Me | 31.6 ± 1.8 | >360 | 0.0625 | 51.5/163.4 |
| PEDN-Me2a | 90.2 ± 5.5 | >360 | 0.025 | N/A/161.4 |
| PETriN-OHa | 27 ± 2.0 | >360 | 0.0625 | N/A/163.95 |
| PEDN-OH2 | 112.1 ± 6.1 | >360 | 0.0625 | N/A/166.4 |
| PEMN-OH3 | >320 | >360 | 0.0625 | 57.7/162.4 |
| PETN | 10.2 ± 0.9 | 59.5 | 0.0625 | 140.9/166.8 |
The mono-halogen PETN derivatives display similar sensitivities to conventional explosives such as RDX and HMX. However, the di- and tri-substituted halogen PETN derivatives exhibited no reactions on the drop hammer impact apparatus, even at the maximum height (320 cm). However, the impact sensitivity of the di-hydroxyl PETN derivative (PEDN-OH2) is 112.1 ± 6.1 cm, less sensitive than the mono-hydroxy derivative (PETriN-OH) but still energetic. Likewise, the dimethyl PETN derivative PEDN-Me2 has an impact sensitivity of 90.2 ± 5.5 cm. For all tri-substituted PETN derivatives, including PEMN-Me3 and PEMN-OH3, no “go”s were recorded on our apparatus up to a maximum drop height of 320 cm.
Friction and electrostatic discharge sensitivities of all PETN derivatives were also examined (Table 2). For all derivatives, once any functional group was substituted onto the PETN framework, all friction sensitivity was lost. This result was unexpected as PETN is a relatively friction sensitive molecule (59.5 N) and it was surprising that the replacement of one nitrate ester with a single inert substituent would completely remove all responses in the test. In some cases, friction sensitivity can be affected by the phase of the material,60 as liquid explosives can be less friction sensitive; however in this case most of the materials were crystalline solids. Electrostatic discharge sensitivity (Table 2) did not follow any observable pattern or trend.
Discussion: trends observed with impact sensitivity
All of the mono-substituted PETN derivatives have almost identical impact sensitivities, even though the substituents used to functionalize PETN varied in basic molecular properties such as atomic weight and electronegativity. Literature studies have reported the importance of crystal structures on explosive sensitivity primarily through inter- and intramolecular interactions. In our system, all of the mono-substituted PETN materials exhibited similar crystal packing characteristics such as bond angles and distances. Though the similarities between the handling sensitivity properties and the crystal structures may be related, it is not possible to draw strong conclusions on the influence of crystal packing in this system. In addition, Pospisil et al. observed that H50 is approximately inversely proportional to the free volume per unit cell.5,6 Additional density functional theory calculations, which are beyond the scope of this work, would be required to evaluate the free volume for the mono-, di-, and tri-substituted derivatives to better understand any correlations with free volume, but the relatively weak trends reported by Pospisil et al. are unlikely to account for the precipitous changes in H50 with sequential substitutions seen in this work.In order to better understand why all of the mono-substituted PETN derivatives exhibited similar sensitivity but the di-substituted materials varied in sensitivity with the type of substitution, we investigated properties such as oxygen balance,61 heat of formation, heat of explosion, and room temperature heat capacity. A molecule with perfect oxygen balance will release all hydrogen as H2O and all carbon as CO or CO2. Therefore, molecules with a better oxygen balance tend to have better performance and a higher heat of explosion because the reactants are converted completely to highly exothermic product molecules. Molecules with a negative oxygen balance will form soot and H2 while molecules with a positive oxygen balance will liberate excess oxygen as O2. Multiple studies have shown correlations between oxygen balance and explosive sensitivity.62 Notably, Kamlet investigated correlations between impact sensitivity and properties such as oxygen balance as early as the 1960s.63,64 More recently, we and others confirmed that oxygen balance is one of the more important properties that is correlated with trends in sensitivity.61Table 3 shows oxygen balance for the PETN derivatives, and Fig. 4 plots the trend with the number of energetic substituents. The mono-substituted derivatives exhibit mostly similar oxygen balance values, with the exception of PETriN-Me, which shows a significantly lower value. As energetic substituents are removed, the PETriN-Me oxygen balance decreases more dramatically than the halide substituents, though it retains a measurable sensitivity on the impact test. Surprisingly, PEDN-OH2 and PEDN-Me2 have similar impact test values, even though their oxygen balance values differ significantly (−49.56% vs. −108.1%). Overall, the trends in oxygen balance do not account for the observed differences in the di-substituted materials in this system.
| Material | Δ°Hf (kJ mol−1) | Q (kJ mol−1) | Oxygen balance (%) | C v (J mol−1 K−1) |
|---|---|---|---|---|
| a Calculated at the B3LYP/6-31+G(d,p) level and iodine atoms are calculated at the def2svp level.68,69 | ||||
| PETriN-Cl | −385 | 1248 | −21 | 259 |
| PEDN-Cl2 | −357 | 885 | −28.7 | 224 |
| PEMN-Cl3 | −314 | 663 | −34.5 | 188 |
| PETriN-Br | −335 | 1241 | −21.6 | 260 |
| PEDN-Br2 | −258 | 872 | −32.0 | 227 |
| PEMN-Br3 | −168 | 601 | −41.4 | 192 |
| PETriN-I | −276 | 1244 | −24.9 | 262 |
| PEDN-I2 | −140 | 864 | −42.8 | 230 |
| PEMN-I3 | 8 | 588 | −64.7 | 198 |
| PETriN-Me | −385 | 1352 | −50.5 | 268 |
| PEDN-Me2 | −324 | 1150 | −108.1 | 242 |
| PEMN-Me3 | −252 | 485 | −196.4 | 216 |
| PETriN-OH | −520 | 1288 | −26.6 | 263 |
| PEDN-OH2 | −561 | 977 | −49.56 | 232 |
| PEMN-OH3 | −620 | 787 | −83.95 | 202 |
| PETN | −444 | 1872 | −10.1 | 294 |
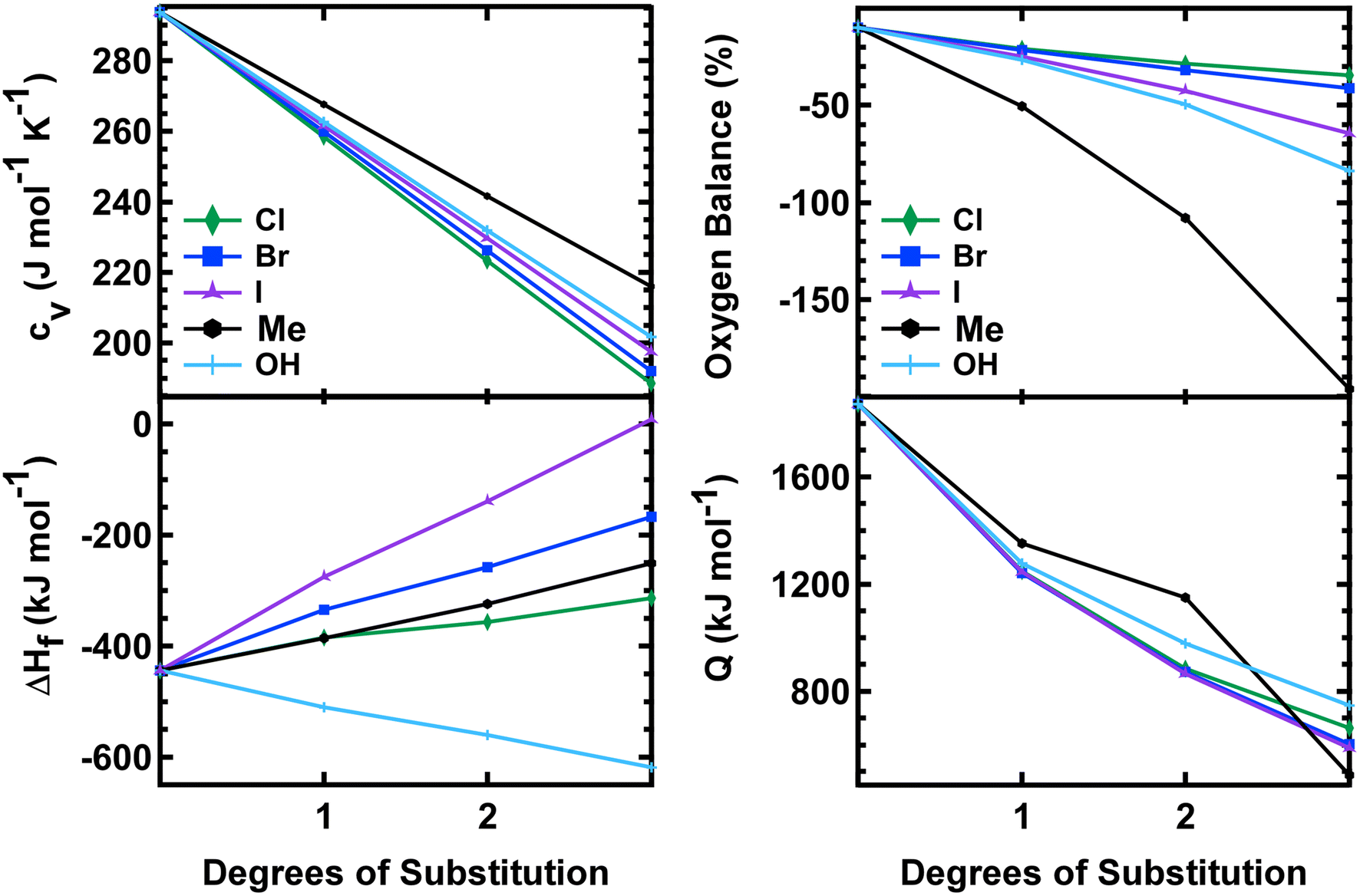 | ||
| Fig. 4 Heat capacity, oxygen balance, heat of formation, and heat of explosion vs. degrees of substitution on the PETN scaffold. | ||
The initiation of energetic materials is a complex chemical event involving a multitude of chemical reactions behaving simultaneously to propagate an energetic event. These reactions generally begin through formation of hot spots65 which generate temperatures high enough to break the weakest chemical bonds, or trigger linkages, which forms radicals or other highly reactive species that result in further chemical reactions propagating through the material. We and others have observed that impact sensitivity correlates well with the nature and number of energetic functional groups and their weakest bonds, or trigger linkages, along with the heat of explosion, Q.21,22 An earlier systematic study of the rates of trigger linkage rupture in a set of 24 explosives, including nitrate esters, nitramines, nitro aromatic compounds, azoxyfuraxans, azides, and nitramines, suggested that impact sensitivity is (i) inversely proportional to the rate of trigger linkage rupture and (ii) that the heat of explosion tends to reduce the effective activation barrier, Ea, consistent with the Bell–Evans–Polanyi principle,
| H50 ∝ 1/(Aexp(−(Ea − αQ)/kBT)) | (1) |
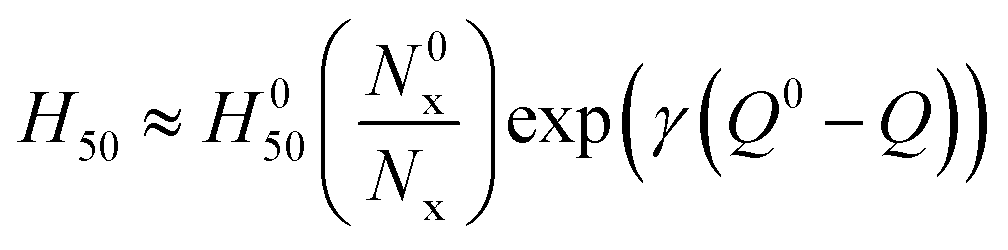 | (2) |
To understand how the sensitivities of the PETN derivatives depend on changes in their heats of explosion, we have calculated Q for each of the derivatives using the standard result,
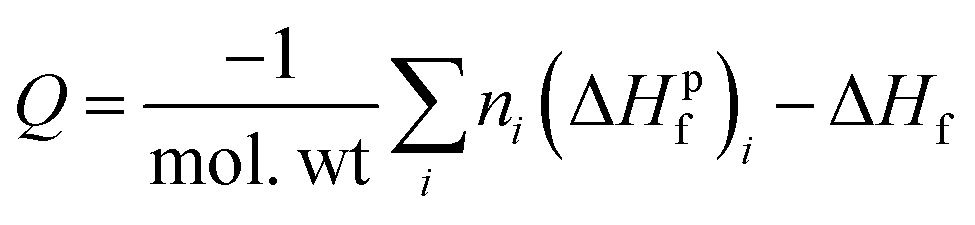 | (3) |
The heat of formation, Δ°Hf, heat of explosion, Q, and room temperature heat capacity, Cv, of the PETN derivatives are presented in Table 3 and Fig. 4. The calculated Q values for all mono-substituted derivatives are similar, falling between 1250 and 1350 kJ mol−1. All of the di-substituted PETN derivatives also fall within a fairly similar range of values, between 880 and 1150 kJ mol−1. For the tri-substituted derivatives, values fall between 485 and 787 kJ mol−1. Overall, as shown in Fig. 4, the heats of explosion for all derivatives typically decrease as the number of energetic groups decrease, as expected. The heat of explosion values therefore correlate well with the decrease in impact sensitivity as the energetic functional groups are removed, which is consistent with the simple model presented in eqn (2). However, the observed decrease in sensitivity with the di- and tri-substituted halogen substituents is even more dramatic than that observed with the hydroxyl and ethyl substituted derivatives.
Heat of formation Δ°Hf and heat capacity were also evaluated (Fig. 3). All derivatives exhibited a positive shift in their heat of formation values as energetic groups were removed except for the hydroxyl substituted PETN derivatives, with each substitution resulting in a more negative Δ°Hf value (from −520 to −620 kJ mol−1). Heat capacity calculations showed almost identical values for each substituted PETN derivative (for example, each PETriN-X had a value of 259–268 J mol−1 K−1). The plot in Fig. 4 shows that heat capacity decreases after every additional substitution on the PETN backbone, which correlates with the loss in impact sensitivity observed with every additional substitution.
As in ref. 21, eqn (2) captures the rapid increase in H50 for the methyl derivatives as ONO2 groups are lost. However, unlike the di-methyl and di-hydroxyl derivatives, all of the di-substituted halogen derivatives appear to be completely inert under drop weight testing. We propose that the loss of impact sensitivity of the di-halogenated materials could be caused by the increased presence of the halogen atoms themselves. Halocarbons such as bromochloromethane, CF3Br and CF2BrCl have long been used in a variety of fire suppressants and fire extinguishers.70 Halogenated materials are known to inhibit fires by interfering with the radical chain mechanism in a combustion reaction. The halogen radicals generated during combustion can react with the active chain carriers generating a more inert material slowing the reaction rate and energy release, thereby halting the overall reaction.71 Despite the combustion inhibiting properties of the halogen atoms many explosives are known that include halogen atoms, some with large halogen content (over 60% of the molecules molecular weight).72 However, it should be noted that most of the reported materials are aromatic with the few aliphatic materials using fluorine, which is the least inhibiting halogen. Studies have also shown that aromatic bound halogens are weaker inhibitors of combustion reactions compared to aliphatic bound halogens,70 due to the higher relative stability and melting points of the aromatic compounds compared to aliphatic ones. For the materials in this study, the increased halogenation lowers both the melting and decomposition temperatures, potentially increasing each material's ability to inhibit energetic chemical reactions. The insensitivity of the di-halogenated PETN derivatives is therefore most likely a result of the increased halogen content of the materials, relative to the hydroxy and methyl PETN derivatives.
Conclusions
We have reported the synthesis of derivatives of PETN in which the nitrate ester functional groups are substituted with halogen atoms, hydroxy or methyl groups. Crystals of ten of the derivatives were grown and studied by single crystal XRD and Hirschfield surface analyses. Decreases in the number of energetic functional groups and heat of explosion of the molecules are correlated with decreasing impact sensitivity. For the mono-substituted materials, almost identical impact sensitivities were observed among all derivatives regardless of the inert substituted functional group. Removing two energetic functional groups resulted in a further decrease in sensitivity. However, the di-halogenated materials decreased in sensitivity to a much larger extent than the methyl and hydroxy derivatives, which is hypothesized to be caused by the combustion-inhibiting nature of halogen atoms. Overall, we have demonstrated that the number of energetic functional groups and heat of explosion account for the explosive sensitivity of a material, consistent with recent previous work by our group and others.21,22Data availability
The data and datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Nicholas Lease – conceptualization, investigation, writing – original draft, visualization, resources, supervision, formal analysis. Kyle D. Spielvogel – investigation, formal analysis. Jack V. Davis – investigation, formal analysis. Jeremy T. Tisdale – investigation. Lisa M. Klamborowski – investigation. M. J. Cawkwell – formal analysis, investigation, supervision, funding acquisition, writing – review and editing. Virginia W. Manner – supervision, funding acquisition, writing-review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
MJC thanks Ed Kober for fruitful discussions. This work was supported by the Laboratory Directed Research and Development program of Los Alamos National Laboratory under project number 20220068DR. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy (Contract No. 89233218CNA000001). This work has been approved for unlimited release by Los Alamos National Laboratory (LANL), LA-UR-23-21991 and LA-UR-23-22896.References
- W. Fickett and W. C. Davis, Detonation Theory and Experiment. Dover Publications, 2011 Search PubMed.
- V. W. Manner, M. J. Cawkwell, E. M. Kober, T. W. Myers, G. W. Brown, H. Tian, C. J. Snyder, R. Perriot and D. N. Preston, Examining the Chemical and Structural Properties that Influence the Sensitivity of Energetic Nitrate Esters, Chem. Sci., 2018, 9, 3649–3663 RSC.
- Q.-L. Yan, S. Zeman, A. Elbeih, Z.-W. Song and J. Malek, The effect of crystal structure on the thermal reactivity of CL-20 and its C4 bonded explosives (I): thermodynamic properties and decomposition kinetics, J. Therm. Anal. Calorim., 2013, 112, 823–836 CrossRef CAS.
- K. F. Mueller, R. H. Renner, W. H. Gilligan, H. G. Adolph and M. J. Kamlet, Thermal Stability/Structure Relations of Some Polynitroaliphatic Explosives, Combust. Flame, 1983, 50, 341–349 CrossRef CAS.
- V. Pospisil, P. Vavra, M. C. Concha, J. S. Murray and P. Politzer, A possible crystal volume factor in impact sensitivities of some energetic compounds, J. Mol. Model., 2010, 16, 895–901 CrossRef PubMed.
- V. Pospisil, P. Vavra, M. C. Concha, J. S. Murray and P. Politzer, Sensitivity and the available free space per molecule in the unit cell, J. Mol. Model., 2011, 17, 2569–2574 CrossRef PubMed.
- H. Zhang, C. Guo, X. Wang, J. Xu, X. He, Y. Liu, X. Liu, H. Huang and J. Sun, Five Energetic Cocrystals of BTF by Intermolecular Hydrogen Bond and π-Stacking Interactions, Cryst. Growth Des., 2013, 13, 679–687 CrossRef CAS.
- Y. Cheng, X. Chen, N. Yang, Y. Zhang, H. Ma and Z. Guo, Sandwich-like low-sensitive nitroamine explosives stabilized by hydrogen bonds and π-π stacking interactions, CrystEngComm, 2021, 23, 1953 RSC.
- C. Ye and J. M. Shreeve, New Atom/Group Volume Additivity Method to Compensate for the Impact of Strong Hydrogen Bonding on Densities of Energetic Materials, J. Chem. Eng. Data, 2008, 53(2), 520–524 CrossRef CAS.
- R. Bu, Y. Xiong, X. Wei, H. Li and C. Zhang, Hydrogen Bonding in CHON-Containing Energetic Crystals: A Review, Cryst. Growth Des., 2019, 19(10), 5981–5997 CrossRef CAS.
- H. H. Cady and A. C. Larson, The Crystal Structure of 1,3,5-Triamino-2,4,6-Trinitrobenzene, Acta Crystallogr., 1965, 18, 485–496 CrossRef CAS.
- E. A. Zhurova, A. I. Stash, V. G. Tsirelson, V. V. Zhurov, E. V. Bartashevich, V. A. Potemkin and A. A. Pinkerton, Atoms- in Molecules Study of Intra- and Intermolecular Bonding in the Pentaerythritol Tetranitrate Crystal, J. Am. Chem. Soc., 2006, 128, 14728–14734 CrossRef CAS PubMed.
- A. D. Yau, E. F. C. Byrd and B. M. Rice, An Investigation of KS-DFT Electron Densities used in Atoms-in-Molecules Studies of Energetic Molecules, J. Phys. Chem. A, 2009, 113, 6166–6171 CrossRef CAS PubMed.
- J. Evers, M. Göbel, B. Krumm, F. Martin, S. Medvedyev, G. Oehlinger, F. X. Steemann, I. Troyan, T. M. Klapötke and M. I. Eremets, Molecular Structure of Hydrazoic Acid with Hydrogen-Bonded Tetramers in Nearly Planar Layers, J. Am. Chem. Soc., 2011, 133, 12100–12105 CrossRef CAS PubMed.
- B. M. Rice and J. J. Hare, A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules, J. Phys. Chem. A, 2002, 106, 1770–1783 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, Effects of strongly electron-attracting components on molecular surface electrostatic potentials: application to predicting impact sensitivities of energetic materials, Mol. Phys., 1998, 93, 187–194 CrossRef CAS.
- W. S. Wilson, D. E. Bliss, S. L. Christian and D. J. Knight, Explosive Properties of Polynitroaromatics, Naval Weapons Center, NWC TP 7073, 1990 Search PubMed.
- D. E. Bliss, S. L. Christian and W. S. Wilson, Impact Sensitivity of Polynitroaromatics, J. Energy Mater., 1991, 9, 319–344 CrossRef CAS.
- A. C. Landerville, I. I. Oleynik and C. T. White, Reactive Molecular Dynamics of Hypervelocity Collisions of PETN Molecules, J. Phys. Chem. A, 2009, 113, 12094–12104 CrossRef CAS PubMed.
- D. Mathieu and T. Alaime, Predicting Impact Sensitivities of Nitro Compounds on the Basis of a Semi-Empirical Rate Constant, J. Phys. Chem. A, 2014, 118, 9720–9726 CrossRef CAS PubMed.
- M. J. Cawkwell, J. Davis, N. Lease, F. W. Marrs, A. Burch, S. Ferreira and V. W. Manner, Understanding Explosive Sensitivity with Effective Trigger Linkage Kinetics, ACS Phys. Chem. Au, 2022, 2, 448–458 CrossRef CAS PubMed.
- N. Lease, L. M. Klamborowski, R. Perriot, M. J. Cawkwell and V. W. Manner, Identifying the Molecular Properties that Drive Explosive Sensitivity in a Series of Nitrate Esters, J. Phys. Chem. Lett., 2022, 13(40), 9422–9428 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, High Performance, Low Sensitivity: Conflicting or Compatible?, Propellants, Explos., Pyrotech., 2016, 41, 414–425 CrossRef CAS.
- T. M. Myers, C. J. Snyder and V. W. Manner, Reduction of Mechanical Sensitivity in Alkyl Nitrate Explosives through Efficient Crystal Packing, Cryst. Growth Des., 2017, 17, 3204–3209 CrossRef CAS.
- V. W. Manner, T. W. Myers, M. J. Cawkwell, E. M. Kober, G. W. Brown, H. Tian, C. J. Snyder and D. N. Preston, Understanding and Manipulating the Sensitivity of Nitrate Ester Explosives, AIP Conf. Proc., 2018, 1979, 150027 CrossRef.
- D. E. Chavez, S. K. Hanson, J. M. Veauithier and D. A. Parrish, Electroactive Explosives: Nitrate Ester-Functionalized 1,2,4,5-Tetrazines, Angew. Chem., 2013, 125, 7014–7017 CrossRef.
- T. Lenz, T. M. Klapotke, M. Muhlemann and J. Stierstorfer, About the Azido Derivatives of Pentaerythritol Tetranitrate, Propellants, Explos., Pyrotech., 2021, 46, 723–731 CrossRef CAS.
- M. Benz, T. M. Klapotke, N. Kolbl, J. Kuch, T. Lenz, E. Parigi and J. Stierstorfer, Metal Castable Derivatives of Pentaerythritol Tetranitrate, Chem.–Eur. J., 2023, e20224013 Search PubMed.
- N. Lease, L. M. Kay, G. W. Brown, D. E. Chavez, D. Robbins, E. F. C. Byrd, G. H. Imler, D. A. Parrish and V. W. Manner, Synthesis of Erythritol Tetranitrate Derivatives: Functional Group Tuning of Explosive Sensitivity, J. Org. Chem., 2020, 85(7), 4619–4626 CrossRef CAS PubMed.
- V. W. Manner, M. J. Cawkwell, E. M. Kober, T. W. Myers, G. W. Brown, T. Hongzhao, C. J. Synder, R. Perriot and D. N. Preston, Examining the chemical and structural properties that influence the sensitivity of energetic nitrate esters, Chem. Sci., 2018, 9(15), 3649–3663 RSC.
- S. Zeman and M. Jungova, Sensitivity and Performance of Energetic Materials, Propellants, Explos. Pyrotech., 2016, 41, 426–451 CrossRef CAS.
- S. Zeman, Influence of the Energy Content and Its Outputs on Sensitivity of Polynitroarenes, J. Energ. Mater., 2019, 37, 445 CrossRef CAS.
- S. Zeman, M. Dimun and S. Truchlik, The relationship between kinetic data of the low-temperature thermolysis and the heats of explosion of organic polynitro compounds, Thermochim. Acta, 1984, 78, 181–209 CrossRef CAS.
- D. Mathieu and T. Alaime, Predicting Impact Sensitivities of Nitro Compounds on the Basis of a Semi-empirical Rate Constant, J. Phys. Chem. A, 2014, 118, 9720–9726 CrossRef CAS PubMed.
- R. D. Chapman, Halogenated Explosives to Defeat Biological Agents, Defense Threat Reduction Agency, 2015 Search PubMed.
- R. D. Chapman, D. Thompson, G. Ooi, D. Wooldridge, P. N. Cash and R. A. Hollins, N,N-Dihaloamine Explosives to Defeat Biological Weapons, in Presented at the Joint 66th Southwest and 62nd Southeast Regional Meeting of the American Chemical Society, New Orleans, LA, 2012 Search PubMed.
- C. He, J. Zhang and J. M. Shreeve, Dense Iodine-Rich Compounds with Low Detonation Pressures as Biocidal Agents, Chem.–Eur. J., 2013, 19, 7503–7509 CrossRef CAS PubMed.
- D. Chand, C. He, J. P. Hooper, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, Mono-and diiodo-1,2,3-triazoles and their mono nitro derivatives, Dalton Trans., 2016, 45, 9684–9688 RSC.
- Y. Tang, H. Gao, D. A. Parrish and J. M. Shreeve, 1,2,4-Triazole linkers and N-azo bridges yield energetic compounds, Chem.–Eur. J., 2015, 21, 11401–11407 CrossRef CAS PubMed.
- Y. Tang, C. He, H. Gao and J. M. Shreeve, Energized nitrosubstituted azoles through ether bridges, J. Mater. Chem. A, 2015, 3, 15576–15582 RSC.
- D. Kumar, C. He, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, Connecting Energetic Nitropyrazole and Aminotetrazole Moieties with N,N′-Ethylene Bridges: A Promising Approach for Fine Tuning Energetic Properties, J. Mater. Chem. A, 2016, 4, 9220–9228 RSC.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, Resolving Synthetic Challenges Faced in the Syntheses of Asymmetric N,N-Ethylene-Bridged Energetic Compounds, New J. Chem., 2017, 41, 4040–4047 RSC.
- R. D. Chapman, Halogenated Explosives to Defeat Biological Agents, Technical Report, Defense Threat Reduction Agency, 2015 Search PubMed.
- E. E. Toops Jr, J. Phys. Chem., 1956, 60, 304 CrossRef.
- V. A. Ginsburg, A. N. Medvedev, M. F. Lebedeva, M. N. Vasil'eva and L. L. Martynova, Zh. Obshch. Khim., 1967, 37, 611 CAS.
- H. J. Marcus, Preprints of Papers – American Chemical Society, Division of Fuel Chemistry, 1968, vol. 12, issue 2, p. 56 Search PubMed.
- L. T. Eremenko, G. V. Oreshko and M. A. Fadeev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 101 CrossRef.
- G. Caldwell, T. F. Magnera and P. Kebarle, SN2 reaction in the gas phase. Temperature dependence of the rate constants and energies of transition states. Comparison with solution, J. Am. Chem. Soc., 1984, 106, 959–966 CrossRef CAS.
- D. E. Elrick, W. H. Gardner, N. S. Marans and R. F. Preckel, Preparation of halides of pentaerythritol trinitrate, J. Am. Chem. Soc., 1954, 76, 1374–1375 CrossRef CAS.
- W. Kalow and J. Schunk, The nitrate esters of pentaerythritol, Naunyn-Schmiedebergs Arch. Pharmakol. Exp. Pathol., 1950, 211, 388–391 CAS.
- J. C. Bottaro, R. J. Schmitt and C. D. Bedford, Nonacidic Nitration of Secondary Amines, J. Org. Chem., 1987, 52, 2294–2297 CrossRef.
- Y. Zou, Y. Wei, G. Wang, F. Meng, M. Gao, G. Storm and Z. Zhong, Nanopolymersomes with an Ultrahigh Iodine Content for High Performance X-Ray Computed Tomography Imaging In Vivo, Adv. Mater., 2017, 29, 1603997 CrossRef PubMed.
- K. Mondanaro Lynch and W. P. Dailey, Improved Preparations of 3-Chloro-2-(chloromethyl)-1-propene and 1,1-Dibromo-2,2-bis(chloromethyl)-cyclopropane: Intermediates in the Synthesis of [1.1.1] Propellane, J. Org. Chem., 1995, 60, 4666 CrossRef.
- J. Cui, W. Guo, D. Cao and C. Xu, Synthesis and Characterization of 1,1,1-Trimethylolpropane Trinitrate, Huozhayao Xuebao, 2005, 28, 78–79 CAS.
- H. A. Rolewicz, C. D. Grimes Jr and K. Stevenson Jr, Preparation of Pentaerythritol Trinitrate, US Pat., US3408383A, 1968 Search PubMed.
- K. Lange, A. Koenig, C. Roegler, A. Seeling and J. Lehmann, NO Donors. Part 18: Bioactive Metabolites of GTN and PETN-Synthesis and Vasorelaxant Properties, Bioorg. Med. Chem. Lett., 2009, 19, 3141–3144 CrossRef CAS PubMed.
- J. J. Mckinnon, M. A. Spackman and A. S. Mitchell, Novel tools for vizualizing and exploring intermolecular interaction in molecular crystals, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- F. W. Marrs, V. W. Manner, A. C. Burch, J. D. Yeager, G. W. Brown, L. M. Kay, R. T. Buckley, C. M. Anderson-Cook and M. J. Cawkwell, Sources of Variation in Drop-Weight Impact Sensitivity Testing of the Explosive Pentaerythritol Tetranitrate, Ind. Eng. Chem. Res., 2021, 60, 5024–5033 CrossRef CAS.
- B. T. Neyer, A D-Optimality-Based Sensitivity Test, Technometrics, 1994, 36, 61 CrossRef.
- M. M. Sandstrom, G. W. Brown, D. N. Preston, C. J. Pollard, K. F. Warner, D. N. Sorensen, D. L. Remmers, J. J. Phillips, T. J. Shelley, J. A. Reyes, P. C. Hsu and J. G. Reynolds, Variation of Methods in Small-Scale Safety and Thermal Testing of Improvised Explosives, Propellants, Explos., Pyrotech., 2015, 40, 109–126 CrossRef CAS.
- F. W. Marrs, J. V. Davis, A. C. Burch, G. W. Brown, N. Lease, P. L. Huestis, M. J. Cawkwell and V. W. Manner, Chemical Descriptors for a Large-Scale Study on Drop-Weight Impact Sensitivity of High Explosives, J. Chem. Inf. Model., 2023, 63(3), 753–769 CrossRef CAS PubMed.
- P. W. Cooper, Explosives Engineering, John Wiley & Sons, 2018 Search PubMed.
- M. J. Kamlet Sensitivity Relationships, in Proceedings of the 3rd Symposium on Detonation, Office of Naval Research, 1960, p. 671 Search PubMed.
- M. J. Kamlet The Relationship of Impact Sensitivity with Structure of Organic High Explosives., 1. Polynitroaliphatic Explosives, in Proceedings of the 6th Symposium (International) on Detonation, 1976, p. 312 Search PubMed.
- C. M. Tarver, S. K. Chidester and A. L. Nichols III, Critical Conditions for Impact- and Shock-Induced Hot Spots in Solid Explosives, J. Phys. Chem., 1996, 100, 5794–5799 CrossRef CAS.
- D. R. Burgess Jr, Thermochemical Data, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD Search PubMed.
- M. J. Cawkwell, A. C. Burch, S. R. Ferreira, N. Lease and V. W. Manner, Atom Equivalent Energies for the Rapid Estimation of the Heat of Formation of Explosive Molecules from Density Functional Tight Binding Theory, J. Chem. Inf. Model., 2021, 61, 337–3347 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. G. Gann, A Millennial View of Fire Suppression, Halon Options Technical Working Conference. Proceedings HOTWC, 2000 Search PubMed.
- J. Green, Mechanisms for Flame Retardancy and Smoke Suppression-A Review, J. Fire Sci., 1996, 14, 426–442 CrossRef CAS.
- J. Chang, G. Zhao, X. Zhao, C. He, S. Pang and J. M. Shreeve, New Promises from an Old Friend: Iodine-Rich Compounds as Prospective Energetic Biocidal Agents, Acc. Chem. Res., 2021, 54, 332–343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. NMR spectra, synthesis details for novel materials, sensitivity data for all novel PETN derivatives, DSC data for all PETN derivatives, crystal structures and computation analysis details. Cif files for crystal structures have been uploaded to the CCDC with the following deposition numbers: 2252052–2252061. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01627g |
| This journal is © The Royal Society of Chemistry 2023 |