
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of the cGAS-STING pathway by a mitochondrial DNA-targeted emissive rhodium(III) metallointercalator†
Yue
Zheng‡
 ,
Xiao-Xiao
Chen‡
,
Dong-Yang
Zhang
,
Wen-Jin
Wang
,
Kun
Peng
,
Zhi-Yuan
Li
,
Zong-Wan
Mao
* and
Cai-Ping
Tan
*
,
Xiao-Xiao
Chen‡
,
Dong-Yang
Zhang
,
Wen-Jin
Wang
,
Kun
Peng
,
Zhi-Yuan
Li
,
Zong-Wan
Mao
* and
Cai-Ping
Tan
*
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, State Key Laboratory of Oncology in South China, Sun Yat-Sen University, Guangzhou 510006, P. R. China. E-mail: cesmzw@mail.sysu.edu.cn; tancaip@mail.sysu.edu.cn
First published on 6th June 2023
Abstract
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon (STING) pathway is a key mediator of innate immunity involved in cancer development and treatment. The roles of mitochondrial DNA (mtDNA) in cancer immunotherapy have gradually emerged. Herein, we report a highly emissive rhodium(III) complex (Rh-Mito) as the mtDNA intercalator. Rh-Mito can specifically bind to mtDNA to cause the cytoplasmic release of mtDNA fragments to activate the cGAS-STING pathway. Moreover, Rh-Mito activates the mitochondrial retrograde signaling by disturbing the key metabolites involved in epigenetic modifications, which alters the nuclear genome methylation landscape to influence the expression of genes related to immune signaling pathways. Finally, we demonstrate that ferritin-encapsulated Rh-Mito elicits potent anticancer activities and evokes intense immune responses in vivo by intravenous injection. Overall, we report for the first time that small molecules targeting mtDNA can activate the cGAS-STING pathway, which gives insights into the development of biomacromolecule-targeted immunotherapeutic agents.
1 Introduction
As an important innate immune signal pathway, cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) provides great potential for cancer immunotherapy.1–3 Upon sensing of cytoplasmic DNA, cGAS is activated to catalyse the formation of 2′-3′-cyclic GMP-AMP (cGAMP).4,5 The activation of STING by cGAMP recruits and activates tank-binding kinase 1 (TBK1), leading to the phosphorylation of interferon regulatory factor 3 (IRF3).5,6 The phosphorylated IRF3 forms a dimer and reacts with nuclear factor-κB (NF-κB) for nuclear translocation, which induces the expression of type-I interferon (IFN-I) and various inflammatory factors.7At present, most STING activators are synthetic cyclic dinucleotides (CDNs). In 2015, Corrales et al. reported that CDN can bind to STING, thereby activating antigen-presenting cells (APCs) and CD8+ T cells.8 However, CDN-based STING agonists suffer from low metabolic stability, and they need to be administered by intratumoral injection. Several CDN analogues have entered clinical trials, but usually they require combined treatment with immune checkpoint inhibitors (ICIs).9 Non-nucleotide STING agonists have also attracted much attention due to their high specificity and effectiveness.2 In 2018, Ramanjulu et al. reported that amidobenzimidazole derivatives can act as STING agonists.10 Lairson’s group identified a non-nucleotide cGAMP mimetic SR-717 that can promote the activation of CD8+ T, natural killer and dendritic cells (DCs).11 Pan et al. developed a STING agonist MSA-2 amenable to oral administration.12 These non-CDN agonists provide possibilities for systemic administration, but the clinical data need further validation. Recently, Jiang's group demonstrated that manganese can promote the activation of the cGAS-STING pathway, and it shows synergistic antitumor immune effects with anti-PD-1 antibody.13 At the same time, delivery by nanomaterials also provides potential solutions for the in vivo applications of STING agonists.14–22
The discovery of other activators of the STING pathway has also shown potency in its immunopharmacological regulation.2 Mitochondria lie at the heart of immunity as they can regulate cancer immune responses from many aspects.23,24 The importance of mitochondrial retrograde signalling in cancer pathology has been gradually revealed, but its roles in cancer treatment are still unclear.25–27 Key intermediates in the Krebs cycle such as succinate, fumarate and citrate, are related to immunity and inflammation.28,29 Mitochondrial DNA (mtDNA) also acts as a danger-associated molecular pattern (DAMP) phagocytosed by DCs to release interferon (IFN) and activate effector T cells, e.g., cytotoxic T lymphocytes (CTLs).30–32 Meanwhile, fragmentation of mtDNA can activate the cGAS-STING pathway, which releases NF-κB-dependent transcription of cytokines to recruit CD8+ T cells.24
Besides platinum-based anticancer agents, non-platinum metallo-anticancer agents have drawn increasing attention, especially phosphorescent octahedral Ir(III), Ru(II), and Re(I) complexes.33–35 These complexes are characterized by their multiple functionalities due to the rich photophysical properties, e.g., high quantum yields, long-lived lifetimes, large Stokes shifts and good photostability.33 Based on the action mechanisms of platinum-based antitumor drugs, rhodium complexes are also considered to be potent anticancer candidates with high biological activities.33,36–38 Interestingly, kinetically inert rhodium complexes can specifically bind with mismatched DNA and attenuate protein–protein interaction by ligand tuning, and some rhodium complexes exhibit low nanomolar antitumor activity equivalent to or even better than ruthenium/iridium complexes.37–41
Considering these research backgrounds, we propose that specific mtDNA-intercalating agents may activate the cGAS-STING pathway for cancer immunotherapy. Moreover, the instinct luminescent properties of rhodium(III) complexes can be used to confirm their mtDNA-targeting specificity. Herein, we report an emissive mtDNA-targeted rhodium(III) complex (Rh-Mito) with antiproliferative activity in vitro. Rh-Mito can induce mtDNA damage to cause the cytoplasmic release of mtDNA fragments for activation of the cGAS-STING signaling pathway, which leads to the maturation of DCs for recruitment of CTLs to the tumor site in vivo (Scheme 1). Moreover, by adopting a multi-omics approach, we demonstrate that Rh-Mito disturbs mitochondrial metabolites especially those involved in DNA methylation, which alters the nuclear DNA methylation landscape and affects immune gene expression. In all, we present here the first mtDNA intercalator that activates the cGAS-STING pathway, which gives new insights into biomacromolecule-targeted chemo-immunotherapeutic agents.
 | ||
| Scheme 1 Schematic illustration of the mechanisms of chemo-immunotherapeutic responses evoked by Rh-Mito. SAM: S-adenosylmethionine, SAH: S-adenosyl homocysteine, FA: fumaric acid, α-KG: α-ketoglutarate, ROS: reactive oxygen species. | ||
2 Results
2.1 DNA binding properties in vitro and in situ
To investigate the structure–activity relationship, Rh1,42Rh2, Rh3 (ref. 43), and Rh-Mito were synthesised by refluxing the dimer precursor with ligands 1,10-phenanthroline (phen), pyrazino[2,3-f][1,10]phenanthroline (dpq), dipyrido[3,2-a:2′,3′-c]phenazine (dppz) and benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine (dppn) in CH2Cl2/CH3OH and purified by silica column chromatography (Scheme S1†). These compounds were characterized by ESI-MS, 1H NMR, 13C NMR (Fig. S1–S12†) and elemental analysis. The absorption spectra of rhodium(III) complexes are characterized by an intense band at 250–330 nm and a less intense band at 340–500 nm in CH3CN, CH2Cl2 and PBS (Fig. 1A and S13†), which can be attributed to intra-ligand charge transfer (ILCT) and metal-to-ligand charge transfer (MLCT), respectively.38,41,43 Upon excitation, only Rh-Mito displays solvent-dependent red emission with a maximum emission wavelength at 597–654 nm, indicating the imaging potential of Rh-Mito (Fig. 1A). The emission quantum yields and lifetimes of Rh-Mito fall between 0.07–1.07% and 4.61–8.37 ns, respectively (Table S1†). All of these rhodium(III) complexes show high stability in fetal bovine serum within 24 h according to the monitoring of the UV/Vis spectrum (Fig. S14†). UV/Vis spectroscopy measurement shows that the absorbance of the Rh(III) complexes is proportional to concentration up to 100 μM, indicating their sufficient solubility at working concentrations (Fig. S15†).44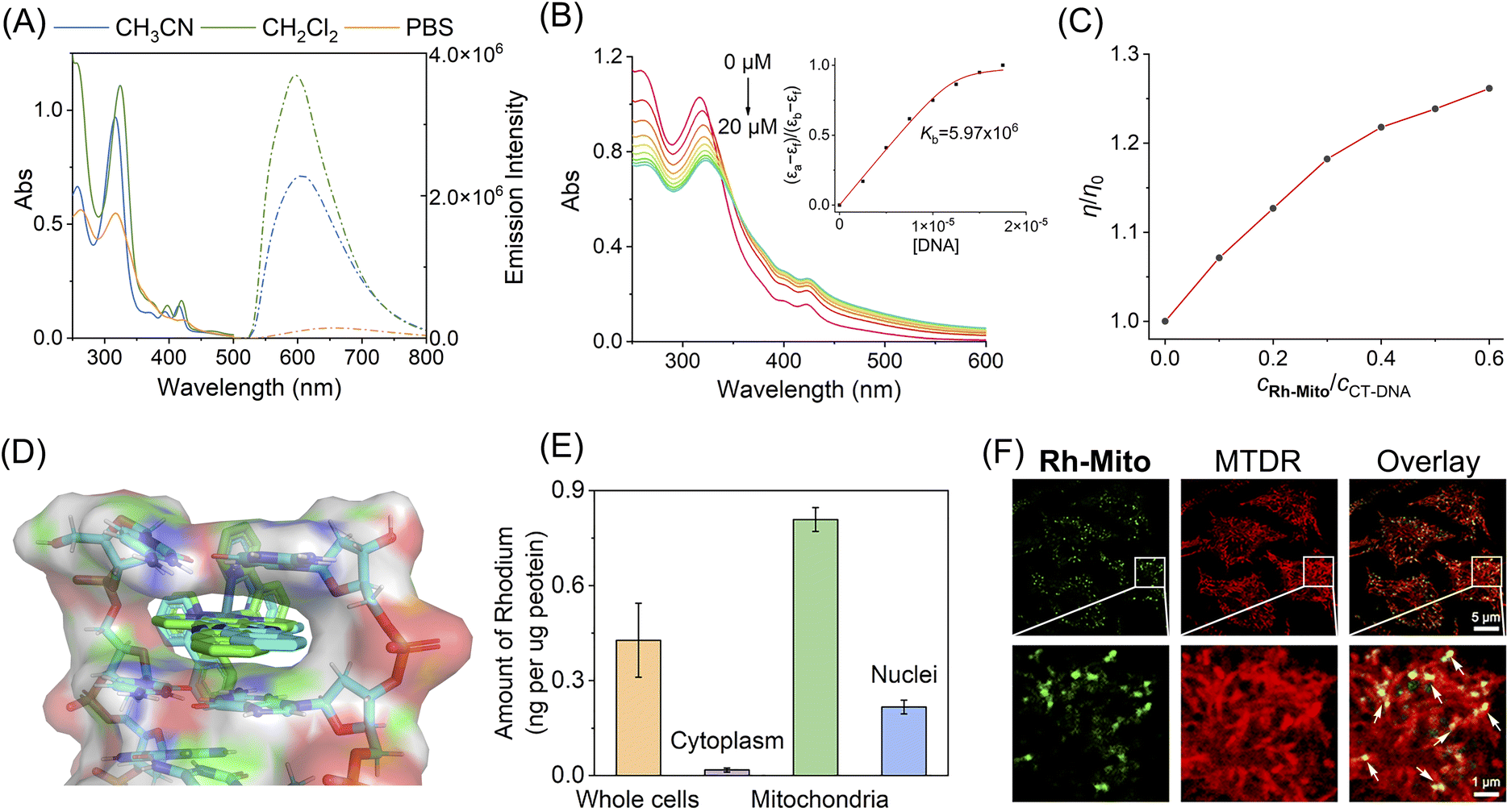 | ||
| Fig. 1 (A) UV/Vis spectra and emission spectra of Rh-Mito (10 μM) measured in degassed CH3CN, CH2Cl2 and PBS at 298 K. (B) UV/Vis spectra of Rh-Mito (10 μM) titrated with CT-DNA (0–20 μM) in Tris–HCl buffer. The arrow shows the alteration in the absorption intensity of Rh-Mito upon the addition of CT-DNA. Insert: Plot of (εa − εf)/(εb − εf) against [DNA] for the binding of Rh-Mito to CT-DNA. (C) Effect of Rh-Mito and Rh-Mito on the relative viscosity of CT-DNA. (D) Molecular docking studies of Δ-Rh-Mito (green) and Λ-Rh-Mito (cyan) with double-stranded DNA (PDB: 5IP8). The DNA fragment is shown in colors as a cartoon representation. (E) Distribution of Rh-Mito (1.0 μM, 1 h) in cellular compartments of HeLa cells measured by ICP-MS. (F) Colocalization of Rh-Mito with MTDR analysed by confocal microscopy. The lower pictures are the enlarged parts of the red boxes in the upper pictures. Rh-Mito: λex = 405 nm; λem = 620 ± 20 nm. MTDR: λex = 633 nm; λem = 660 ± 20 nm. For (E) and (F), the cell viability is proved to be over 95% by MTT assay under the experimental conditions. | ||
The UV/Vis spectrum of Rh1–Rh3 and Rh-Mito exhibits hypochromism in the ILCT band and hyperchromism in the MLCT band upon the addition of calf thymus-DNA (CT-DNA) (Fig. 1B and S16†), and the binding constant (Kb) is calculated to be 5.97 × 106 M−1 for Rh-Mito, which is much higher than that for reference complexes (Rh1: 2.59 × 104 M−1; Rh2: 5.20 × 104 M−1; Rh3: 5.13 × 105 M−1). Meanwhile, an obvious fluorescence enhancement along with a blue shift of the maximum emission wavelength is observed for Rh-Mito upon the addition of CT-DNA (Fig. S17†). When the molar ratio of [DNA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Rh-Mito] reaches 8:1, the fluorescence of Rh-Mito is enhanced by about 2.7 times. In the presence of Rh-Mito, the viscosity of CT-DNA increases in a concentration-dependent manner, which proves that it interacts with DNA by intercalation (Fig. 1C). Molecular docking studies using a double-stranded DNA (PDB: 5IP8) show that the dppn ligand in Rh-Mito creates a complete intercalation cavity at the terminal C1C2/G9G10 step of duplex, affording a large π-overlap with nucleotide bases (Fig. 1D), and the binding energies are calculated to be −10.90 kcal mol−1 for Δ-Rh-Mito and −11.03 kcal mol−1 for Λ-Rh-Mito, which is higher than those obtained for reference complexes (Rh1: −7.34 kcal mol−1; Rh2: −8.99 kcal mol−1; Rh3: −9.90 kcal mol−1; Fig. S18†). These results indicate that the larger planar ligand in rhodium complexes provides higher DNA intercalating capabilities.
:[Rh-Mito] reaches 8:1, the fluorescence of Rh-Mito is enhanced by about 2.7 times. In the presence of Rh-Mito, the viscosity of CT-DNA increases in a concentration-dependent manner, which proves that it interacts with DNA by intercalation (Fig. 1C). Molecular docking studies using a double-stranded DNA (PDB: 5IP8) show that the dppn ligand in Rh-Mito creates a complete intercalation cavity at the terminal C1C2/G9G10 step of duplex, affording a large π-overlap with nucleotide bases (Fig. 1D), and the binding energies are calculated to be −10.90 kcal mol−1 for Δ-Rh-Mito and −11.03 kcal mol−1 for Λ-Rh-Mito, which is higher than those obtained for reference complexes (Rh1: −7.34 kcal mol−1; Rh2: −8.99 kcal mol−1; Rh3: −9.90 kcal mol−1; Fig. S18†). These results indicate that the larger planar ligand in rhodium complexes provides higher DNA intercalating capabilities.
Inductively coupled plasma mass spectrometry (ICP-MS) measurement in human cervical cancer (HeLa) cells indicates that the contents of rhodium in mitochondria are much higher than those obtained in cytosol and nuclei after Rh(III) treatment, and the mitochondrial-accumulating ability of Rh-Mito is higher than that of Rh1–Rh3 (Fig. 1E and S19†). Confocal microscopic observations show that the emission of Rh-Mito is well overlapped with that of MitoTracker Deep Red (MTDR, Fig. 1F). Moreover, gel electrophoresis analysis of proteins purified from HeLa cells shows minimal interaction of Rh-Mito with cellular proteins (Fig. S20†). These results collectively indicate that Rh-Mito can specifically bind to mtDNA in cells by the cascade targeting strategy.45
2.2 High capabilities to inhibit cancer cell proliferation and stemness
Lipophilicity (logPo/w, in which Po/w = octanol/water partition coefficient) can affect the anticancer mechanisms of metal complexes.46 As estimated, the logPo/w values are in the following order: Rh-Mito (2.54) > Rh3 (2.01) > Rh2 (1.99) > Rh1 (1.44), which positively correlates with the area of planar ligands. Cellular uptakes of these rhodium(III) complexes were investigated by ICP-MS in HeLa, human lung cancer (A549), human breast cancer (MCF-7), human liver cancer (HepG2) and human normal breast (MCF-10A) cells (Table S2†). The uptake capability of Rh-Mito is basically stronger than that of Rh1–Rh3. As expected, the uptake efficiency of Rh-Mito in cancer cells is also much higher than that in normal cells. After being incubated at 4 °C or pretreated with metabolic inhibitor carbonyl cyanide m-chlorophenyl hydrazone (CCCP),47 cellular rhodium contents are obviously decreased (Table S3†), and the pretreatment of the endocytosis inhibitor chloroquine doesn't show obvious influence on its cellular uptake level, which indicated that Rh-Mito penetrates into HeLa cells through energy-dependent active transportation.
The in vitro antiproliferative activities of Rh1–Rh3 and Rh-Mito were further assessed on HeLa, human breast cancer (MCF-7 and MDA-MB-231), human lung cancer (A549 and NCI-H1299), HepG2, MCF-10A and human lung fibroblast (HLF) cells. Rh-Mito exhibits low IC50 values ranging from 0.4 to 1.9 μM in cancer cells and relatively high IC50 values (3.2–4.7 μM) in normal cells, and the antiproliferative activity of Rh-Mito is about 7–31 times that of cisplatin in cancer cells (CDDP, Table 1). The IC50 values of Rh-Mito are also higher than that of Rh1–Rh3 in most cancer cells, which is consistent with their uptake efficacies (Table S4†). Trypan Blue assay48,49 also shows that Rh-Mito can successfully reduce the cell viability, and the 50% cytotoxicity concentration (CC50) values against the tested cell lines fall between 0.5 and 3.3 μM (Table S5†).
| Complexes | IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HeLa | HeLa-ρ0 (MTFs) | MCF-7 | MB-MDA-231 | A549 | NCI-H1299 | HepG2 | MCF-10A | HLF | |
| a Data are presented as the mean ± standard deviation (SD), and antiproliferative activity was assessed after 72 h of incubation. | |||||||||
| Rh-Mito | 0.4 ± 0.1 | 2.7 ± 0.2 (6.7) | 0.6 ± 0.1 | 0.8 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | 1.9 ± 0.2 | 3.2 ± 0.3 | 4.7 ± 0.4 |
| CDDP | 7.9 ± 0.6 | 7.1 ± 0.7 (0.9) | 8.6 ± 1.5 | 13.9 ± 2.1 | 10.2 ± 0.9 | 12.4 ± 1.9 | 12.9 ± 1.4 | 12.8 ± 1.6 | 8.4 ± 0.9 |
The mtDNA-targeting factor (MTF) defined as the ratios of the IC50 value in HeLa-ρ0 (mtDNA-less HeLa) cells to that in wild-type HeLa cells calculated for Rh-Mito is about 6.7, higher than that for the other complexes (Rh1: 0.8; Rh2: 2.3; Rh3: 5.7), which indicates that mtDNA damage caused by Rh-Mito plays an indispensable role in its antiproliferative capability. In the non-toxic nanomolar concentrations, Rh-Mito can inhibit the colony formation of HeLa cells in a concentration-dependent manner (Fig. S21†), which confirms its effectiveness in reducing cancer cell proliferation and stemness.
2.3 MtDNA damage-induced mitochondrial dysfunction and necroptosis
Rh-Mito causes an obviously enhanced immunofluorescent signal of 8-oxoguanine (8-OG; the most common form of oxidative in DNA)50 mainly in the cytoplasm (Fig. 2A). Compared with that of nuclear DNA (nucDNA), the copy number of mtDNA is decreased by about 75% after Rh-Mito treatment at 2.0 μM (Fig. 2B; Table S6†). Moreover, Rh-Mito treatment reduces the amplification of mtDNA segments significantly and its impact on nucDNA is unobvious (Fig. 2C). Gel electrophoresis shows that Rh-Mito treatment causes obvious fragmentation of mtDNA in HeLa cells (Fig. 2D).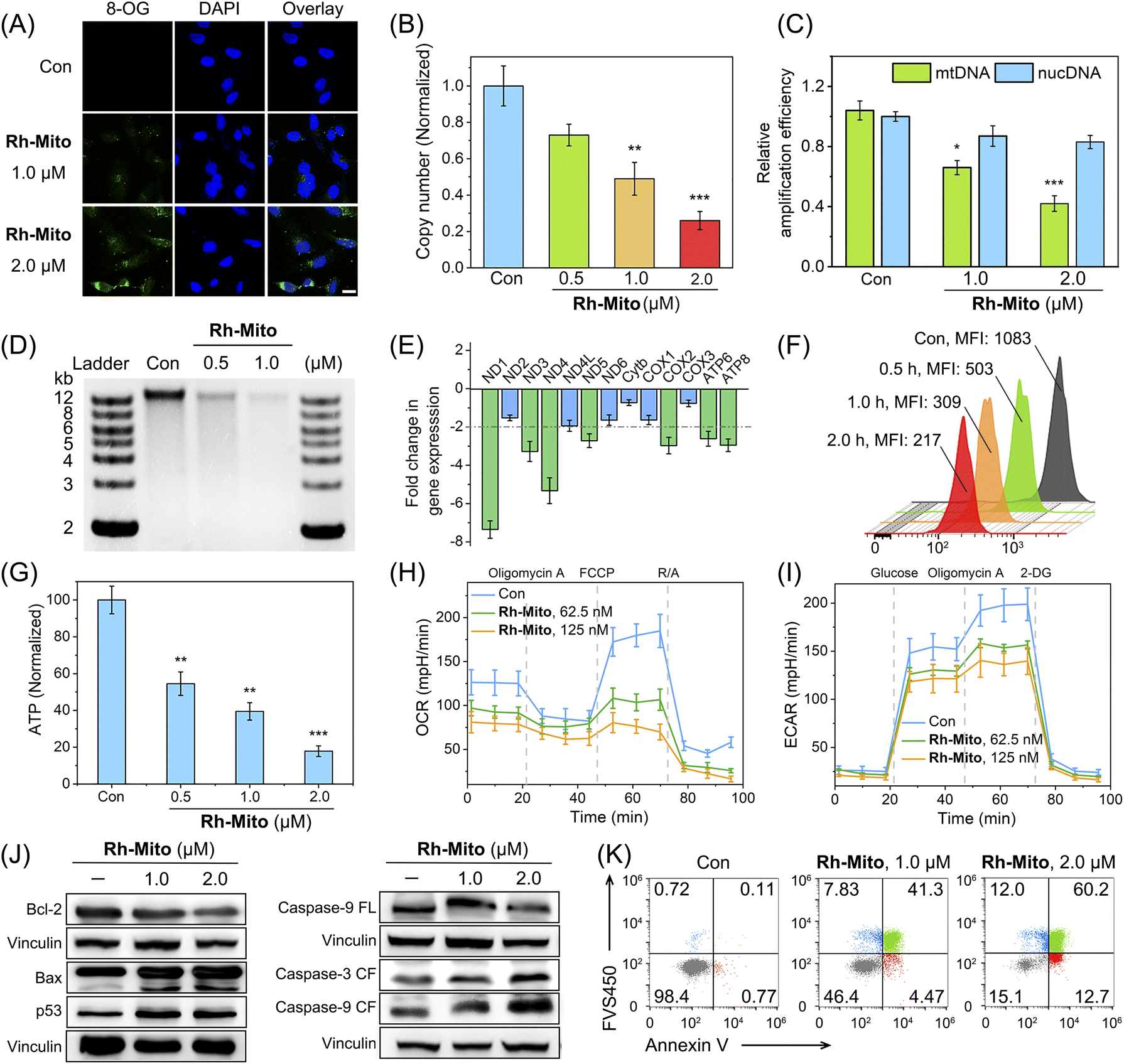 | ||
| Fig. 2 (A) Immunofluorescence staining of 8-OG in HeLa cells treated with Rh-Mito at the indicated concentrations for 6 h. 8-OG: λex = 488 nm; λem = 518 ± 20 nm. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI): λex = 405 nm; λem = 461 ± 20 nm. Scale bar: 10 μm. (B and C) Impact of Rh-Mito on mtDNA copy number and amplification. HeLa cells were treated with Rh-Mito for 6 h. (D) Gel electrophoresis showing mtDNA damage in HeLa cells incubated with Rh-Mito for 2 h. (E) Impact of Rh-Mito (1.0 μM, 6 h) on the expression of 13 genes encoded by the mitochondrial genome. (F) Impact of Rh-Mito (1.0 μM) on MMP by Rh123 staining. λex = 488 nm, λem = 530 ± 20 nm. (G) Impact of Rh-Mito (6 h) on the intracellular ATP levels. OCR (H) and ECAR (I) profiles of HeLa cells treated with Rh-Mito for 2 h. R/A: a mixture of rotenone and antimycin A. 2-DG: 2-deoxyglucose. (J) Western blotting analysis of the effects of Rh-Mito on the expression of the proteins. HeLa cells were treated with Rh-Mito for 6 h. FL: full length. CF: cleaved form. Vinculin was used as the internal reference. (K) Annexin V-APC and FVS450-BV421 double staining of HeLa cells treated with Rh-Mito for 12 h. Annexin V-APC: λex = 637 nm, λem = 670 ± 20 nm. FVS450-BV421: λex = 405 nm, λem = 440 ± 20 nm. *p < 0.05, **p < 0.01 and ***p < 0.001 by the Student's two-tailed t test. | ||
Human mitochondrial genome encodes 37 genes, of which 13 genes are essential components of the oxidative phosphorylation (OXPHOS) system.51 Real-time quantitative polymerase chain reaction (RT-qPCR) shows that the expression of ND1, ND3, ND4, ND5 COX2, ATP6 and ATP8 is decreased in HeLa cells treated with Rh-Mito (fold change > 2, Fig. 2E). These genes encode subunits of nicotinamide adenine dinucleotide (NADH) dehydrogenase, cytochrome c oxidase and ATP synthase belonging to the complexes of the respiratory chain complex.52 Consistently, rhodamine 123 (Rh123) staining shows that Rh-Mito can decrease the mitochondrial membrane potential (MMP) in a time-dependent manner (Fig. 2F). In cells treated with Rh-Mito (2.0 μM, 6 h), the intracellular ATP level is also notably suppressed by about 83% (Fig. 2G).
The impact of Rh-Mito on mitochondrial respiration and glycolysis was then evaluated by measuring the oxygen consumption rate (OCR, Fig. 2H) and extracellular acidification rate (ECAR, Fig. 2I), respectively. Rh-Mito induces a dose-dependent decrease in basal respiration, ATP production, maximum respiration and non-mitochondrial respiration (Fig. S22†), along with a dose-dependent inhibition of glycolysis, glycolytic reserve and glycolytic capacity (Fig. S23†). These results suggest that Rh-Mito can suppress the transcription of mtDNA and impair mitochondrial functions.
The expression of Bcl-2 is decreased upon Rh-Mito treatment, accompanied by an increased expression in Bax, resulting in a decreased Bcl-2/Bax ratio (Fig. 2J). The expression of the guardian of genome p53 is increased, and the cleavage of two regulatory proteins of apoptosis, caspase-3 and -9, is also detected. Accordingly, Rh-Mito induces a dose-dependent increase in the proportion of cells in the late apoptosis/necrosis phase (Fig. 2K). Moreover, pretreatment of z-VAD-fmk (50 μM; a pan-caspase inhibitor) and Necrostatin-1 (50 μM; an inhibitor of receptor-interacting protein 1 regulating programmed necrosis) diminishes the antiproliferative activity of Rh-Mito (Fig. S24†), while minimal effects are detected for the other cell death inhibitors tested. These results suggest that Rh-Mito induces necroptosis through a mitochondrial-mediated pathway by damaging mtDNA.
2.4 cGAS-STING pathway activation in both HeLa cells and DCs
Since Rh-Mito can damage mtDNA, we next evaluated whether Rh-Mito can elevate mitochondrial ROS levels and cause cytoplasmic DNA release. 2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) staining shows that Rh-Mito treatment markedly elevates mitochondrial ROS, as the punctate fluorescence of DCF well colocalizes with that of MTDR (Fig. 3A). After Rh-Mito treatment, the fluorescence of PicoGreen gradually separates from that of MTDR, indicating the cytoplasmic release of mtDNA, while MTDR and PicoGreen show a well colocalization pattern in the control cells. Moreover, giant and bright nucleoids (green spots) are formed in Rh-Mito-treated HeLa cells after 1 h treatment, indicating the remodeling of mitochondrial nucleoids as a result of mtDNA damage. Finally, the mitochondrial morphology changes from tightly connected filaments to loosely dispersion dots, accompanied by the gradually disappeared fluorescence of PicoGreen, which implies that mitochondria are damaged and mtDNA is degraded gradually (Fig. 3B).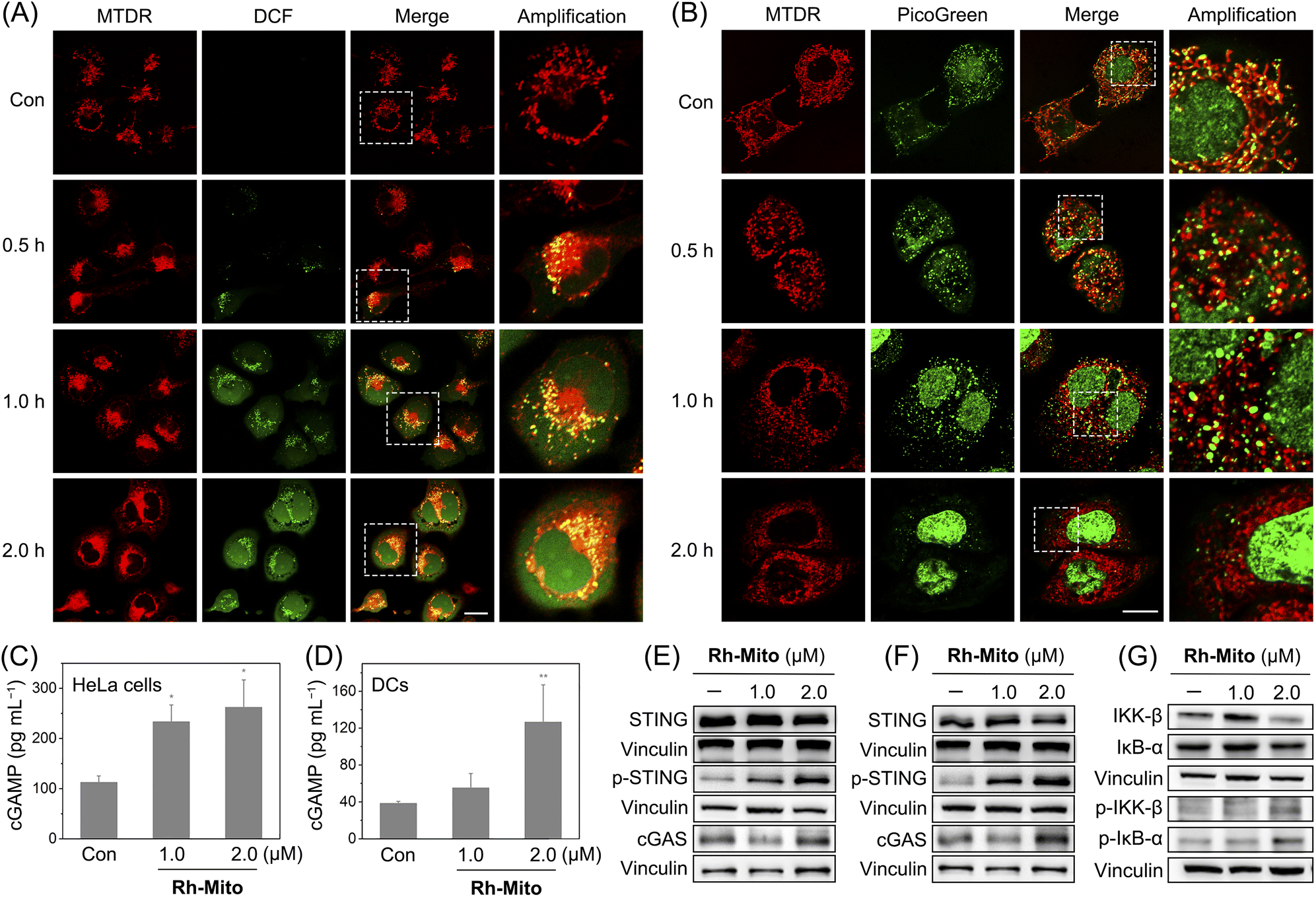 | ||
| Fig. 3 Confocal images of HeLa cells double-labeled with H2DCFDA/MTDR (A) or PicoGreen/MTDR (B). HeLa cells were treated with Rh-Mito (0.5 μM) for different time intervals. Con: control. DCF: λex = 488 nm; λem = 520 ± 20 nm. MTDR: λex = 633 nm; λem = 660 ± 20 nm. PicoGreen: λex = 488 nm; λem = 525 ± 20 nm. The images in the right column are the magnified views of those in the left boxes. Scale bar: 10 μm. The impact of Rh-Mito treatment on cGAMP levels in HeLa cells (C) and DCs (D) measured by ELISA. The impact of Rh-Mito treatment (6 h) on the expression of proteins measured by western blotting in HeLa cells (E) and DCs (F). For (D) and (F), DCs were cultured with the supernate of Rh-Mito-treated HeLa cells for 6 h. (G) The impact of Rh-Mito treatment (6 h) on the phosphorylation of IKK and IκB-α in HeLa cells. *p < 0.05, **p < 0.01 and ***p < 0.001 by the Student's two-tailed t test. | ||
The cGAS-STING pathway activates NF-κB to induce cytokines to promote cell death, which further leads to the release of DNA fragments as tumor-derived antigens.3 Uptake of DNA fragments by DCs elicits a complimentary cGAS-STING-dependent type I IFN-mediated antitumor immune response.53 The content of cGAMP upon Rh-Mito treatments is significantly increased in a dose-dependent manner in HeLa cells (Fig. 3C), along with elevated expression levels of cGAS and p-STING (Fig. 3E). Similar phenomena are found in DCs incubated with the supernate from Rh-Mito-treated HeLa cells (Fig. 3D and F). As expected, the phosphorylation of IKKβ and IκB-α is markedly increased in HeLa cells upon Rh-Mito treatment (Fig. 3G), which indicates that Rh-Mito causes the elevation of cGAMP leading to activation of STING and its downstream factor NF-κB.
2.5 Mitochondrial metabolism and key epigenetic metabolite dysregulation
As mitochondrial bioenergetics and cancer metabolism are closely connected, we then measured the impact of Rh-Mito on cancer cell metabolism by gas chromatography-time of-flight mass spectrometry (GC-TOF-MS). The data were analyzed by principal component analysis (PCA) to find outliers (Fig. S25†). Orthogonal partial least squares discriminant analysis (OPLS-DA) score plots show a distinct metabolic profile for Rh-Mito-treated samples compared with the control (Fig. S26†). Based on a criterion of VIP > 1 and p < 0.05, 715 (up-regulated: 55; down-regulated: 660) and 299 (up-regulated: 102; down-regulated: 197) differential metabolites are detected for the positive and negative ion modes in Rh-Mito-treated samples, respectively (Fig. S27 and ESI Data S1 and S2†). These biomarkers are selected and further analyzed with MetaboAnalyst to investigate the potential metabolic pathways (Fig. 4A and S28†). Rh-Mito treatment influences several metabolic pathways including alanine, aspartate and glutamate metabolism, pyrimidine metabolism, purine metabolism, pentose phosphate and tricarboxylic acid (TCA) pathways. As a mtDNA intercalator, Rh-Mito strongly disturbs the nucleic acid-related metabolic pathways, including the content of bases (adenine, uracil and cytosine; Fig. 4B) and nucleosides (guanosine, cytidine, thymidine and uridine, Fig. 4C).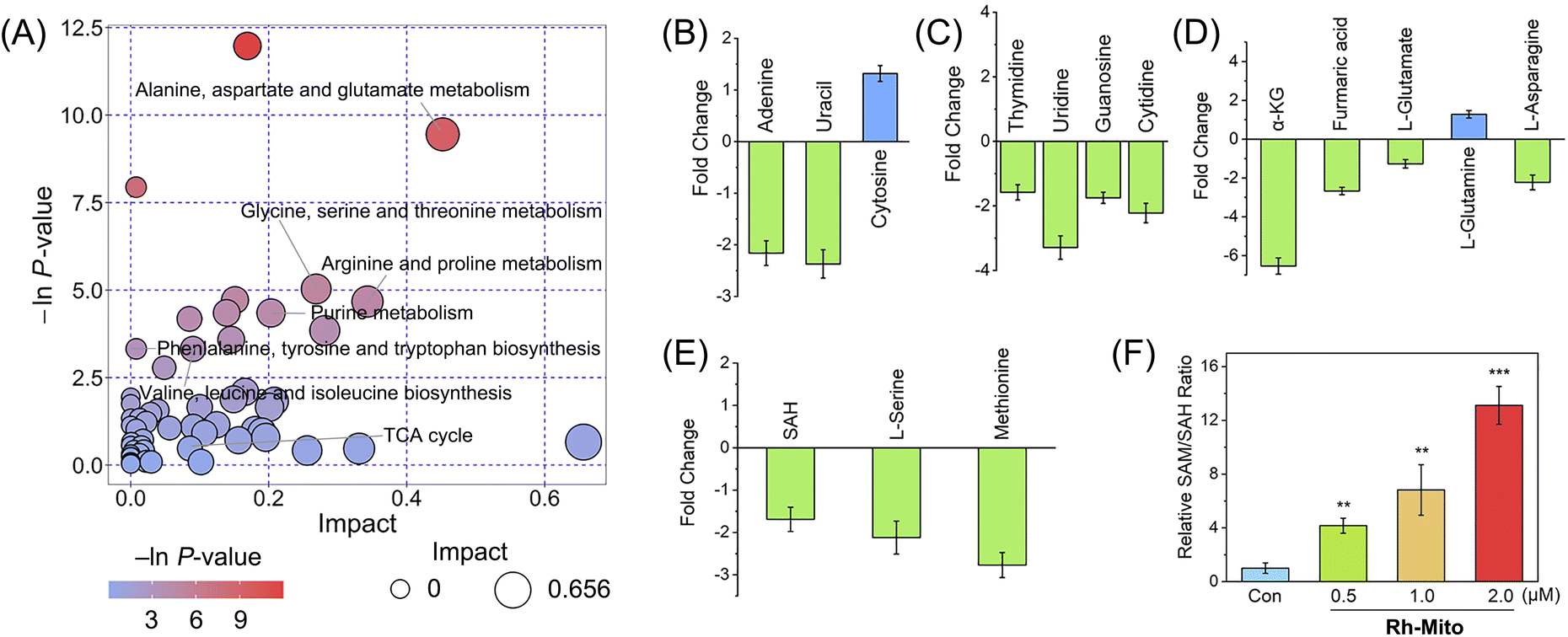 | ||
| Fig. 4 (A) Alternations in the metabolic pathways in HeLa cells treated with Rh-Mito (1.0 μM, 6 h). These metabolites are analysed by GC-TOF-MS in the positive mode. The color of the circles means the p-value, and the area of the circles means pathway impact. The impact of Rh-Mito treatment on the cellular levels of base metabolites (B), nucleoside metabolites (C), TCA cycle and glutaminolysis-related metabolites (D), and one-carbon metabolism-related metabolites (E). The relative content is normalized by the peak area of adonitol (internal standard). (F) The impact of Rh-Mito (6 h) on the SAM/SAH ratio detected with ELISA. *p < 0.05, **p < 0.01 and ***p < 0.001 by the Student's two-tailed t test. | ||
Interestingly, several metabolites in one-carbon metabolism and the TCA cycle involved in epigenetic modifications are influenced by Rh-Mito treatment. DNA demethylation reactions utilize α-kG as the cofactor, while FA acts as the inhibitor of these reactions.54 SAM is the methyl group donor for DNA methyltransferases, while SAH is the reaction product and a competitive inhibitor of them (Fig. S29†).55 After treatment with Rh-Mito, α-kG (ca. 6.54-fold), fumaric acid (ca. 2.67-fold), L-serine (ca. 2.12-fold), methionine (ca. 2.77-fold) and SAH (ca. 1.69-fold) are down-regulated (Fig. 4D and E). A concentration-dependent increase in the ratio of SAM/SAH is detected by enzyme-linked immunosorbent assay (ELISA, Fig. 4F). The SAM/SAH ratio increases by about 12-fold in HeLa cells treated with Rh-Mito (2.0 μM, 6 h). These results indicate that Rh-Mito can influence cancer metabolome and may alter the epigenetic methylation status.
2.6 DNA-methylated status alteration and immuno-pathway regulation
As Rh-Mito affects the intracellular metabolites related to methylation (SAM/SAH) and demethylation (α-kG) reactions, we first investigated its impact on DNA methylation levels by dot blotting. Rh-Mito treatment causes an obvious increase in 5-methylcytosine (5mC) in wild-type HeLa cells (Fig. 5A) but makes it invariantly in mtDNA-free HeLa-ρ0 cell (Fig. S30†). Accordingly, as the demethylated product, 5-hydroxymethylcytosine (5hmC) is markedly decreased by Rh-Mito treatment (Fig. S31†), which is consistent with the variation trend of α-kG. | ||
| Fig. 5 (A) The impact of Rh-Mito (6 h) on the 5mC levels determined by dot blotting assay. Methylene blue (MB) represents the loading control of DNA samples. (B) Average methylated levels in DMRs between the control and Rh-Mito-treated HeLa cells (1.0 μM, 6 h). (C) KEGG enrichment analysis shows pathways influenced by Rh-Mito treatment (1.0 μM, 6 h) in RRBS. (D) Volcano plots showing the DEGs in HeLa cells treated with Rh-Mito (1.0 μM, 6 h). Standard: p-value < 0.05; VIP > 1; fold change > 2. (E) KEGG enrichment analysis of pathways influenced by Rh-Mito treatment (1.0 μM, 6 h). (F and G) GSEA reveals negative and positive enrichment of Rh-Mito-altered genes in various cellular processes. NES: normalized enrichment score. FDR: false-positive rate. (H) Overlapping of DEGs with DMRs. UR: up-regulated genes, DR: down-regulated genes. (I) Validation of DEGs regulated by DMRs related to oncogenes or immunomodulators using RT-qPCR. Relative fold changes in gene expression were normalized according to the average expression of the housekeeping gene β-actin. (J) KEGG enrichment analysis was performed to identify pathways mediated by Rh-Mito treatment (1.0 μM, 6 h) in anchoring promoter regions. *p < 0.05, **p < 0.01 and ***p < 0.001 by the Student's two-tailed t test. | ||
The impact of Rh-Mito on 5mC methylation at the genomic level was further analyzed by reduced representation bisulphite sequencing (RRBS) and the number of methylated C sites in different sequence contexts (CG, CHH, CHG, where H represents A, C, T) was counted. The methylated status in the CG sequence context where most of the methylated C sites locate (>98.9%) was dominantly analyzed (Fig. S32†). The cancer genome always shows hypomethylation generally but hypermethylation of CpG islands in the promoter region of tumor suppressor genes.56 The genome methylation status is different in various biological samples, and differentially methylated regions (DMRs) are thought to be involved in the regulation of the gene transcription level.57 1537 hypermethylated DMRs (hyper-DMRs) and 6022 hypomethylated DMRs (hypo-DMRs) are screened out in HeLa cells (Fig. 5B and ESI Data S3†). After being treated with Rh-Mito, the average methylated level in hyper-DMR is decreased, while the hypo-DMR level is increased. Gene Ontology (GO) analysis shows that DMR-related gene categories including the nucleus, metabolic process and DNA binding and transcription are significantly influenced by Rh-Mito treatment (Fig. S33†). Consistent with the previous results, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis shows that Rh-Mito mainly influences signaling pathways closely associated with mitochondrial metabolism, cancer progression and immunity including metabolic, cGMP, cAMP, Ras-associated protein-1 (Rap1) and mitogen-activated protein kinase (MAPK) signaling pathways (Fig. 5C).
DNA methylation plays pivotal roles in gene transcription,; we then use RNA-sequencing (RNA-Seq) to study the impact of Rh-Mito on the transcriptome. The correlation coefficients between every two individual samples from the same group are above 0.97 (Fig. S34A†), implying the reproducibility of the experiment. The overall Q30 percentage is above 93.6%. More than 94.1% of readings are mapped to reference genes in all samples, and 78.3% of readings are located in exons (Fig. S34B†). The expression levels of 742 and 347 genes are found to be up-regulated and down-regulated, respectively (Fig. 5D and ESI Data S4†). The heatmap of RNA-Seq shows that expression patterns are very similar across each treatment group (Fig. S35†). GO analysis shows that Rh-Mito causes significant overall changes in gene categories including the nucleus, DNA binding and transcription (Fig. S36†). The KEGG pathway annotation indicates that various pathways involved in cancer for immunity and progression including p53, NF-κB, T cell receptor, Toll-like receptor (TLR) and anus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway are influenced by Rh-Mito treatment (Fig. 5E). Gene set enrichment analysis (GSEA) shows that alternation of genes expression is negatively related to mitochondria-associated metabolism (Fig. 5F and S37A†) and positively related to apoptosis and immune signaling pathways (Fig. 5G and S37B†).
Finally, we correlated the result of methylome with transcriptome to screen out those differentially expressed genes (DEGs) with DMRs induced by Rh-Mito treatment. Generally, the gene expression level is negatively correlated with the DNA methylated level in promoter and gene body regions (Fig. S38†). The correlation between DEGs and DMRs in promoter regions is much higher than that in gene body regions (Fig. S39†). Therefore, these DEGs with DMRs in promoter regions are investigated, as shown in Fig. 5H and ESI Data S5.† Among them, the expression of oncogenes and immune modulators including THSD1, FOXQ1, SOX17, FOXO3, NPTX1, PRICKLE1, TMEM88, TOLLIP, CD24, and FJX1 is influenced by Rh-Mito treatment (Table S7†), which is further validated by RT-qPCR (Fig. 5I). GO analysis shows that Rh-Mito causes significant overall changes in gene categories including the nucleus, DNA binding and transcription (Fig. S40†). The KEGG pathway annotation indicates that various pathways involved in cancer development and immunity including antigen presentation are influenced by Rh-Mito (Fig. 5J). These results suggest that Rh-Mito can affect the signaling pathways related to cancer growth and immunity by regulating the DNA methylation status.
2.7 cGAS-STING pathway activation to evoke antitumor immunity in vivo
Finally, an immunocompetent cervical cancer subcutaneous bearing mouse model was established to evaluate the antitumor potency of Rh-Mitoin vivo. Mouse cervical cancer (U14) cells were inoculated to the BALB/c mice as primary tumors. 6 days later, U14 cells were planted into the contralateral as distant tumors (Fig. 6A). Rh-Mito was intratumorally injected into the primary tumors and the tumor volumes were monitored for 14 days. At the end of the treatment, no significant weight change (Fig. S41†) nor organ damage of mice is found (Fig. S42†). The growth of both the primary and distant tumors is successfully inhibited upon Rh-Mito treatment (Fig. 6B and C and S43†). The in vivo inhibitory effects of Rh-Mito are better than those of CDDP, especially for distant tumors.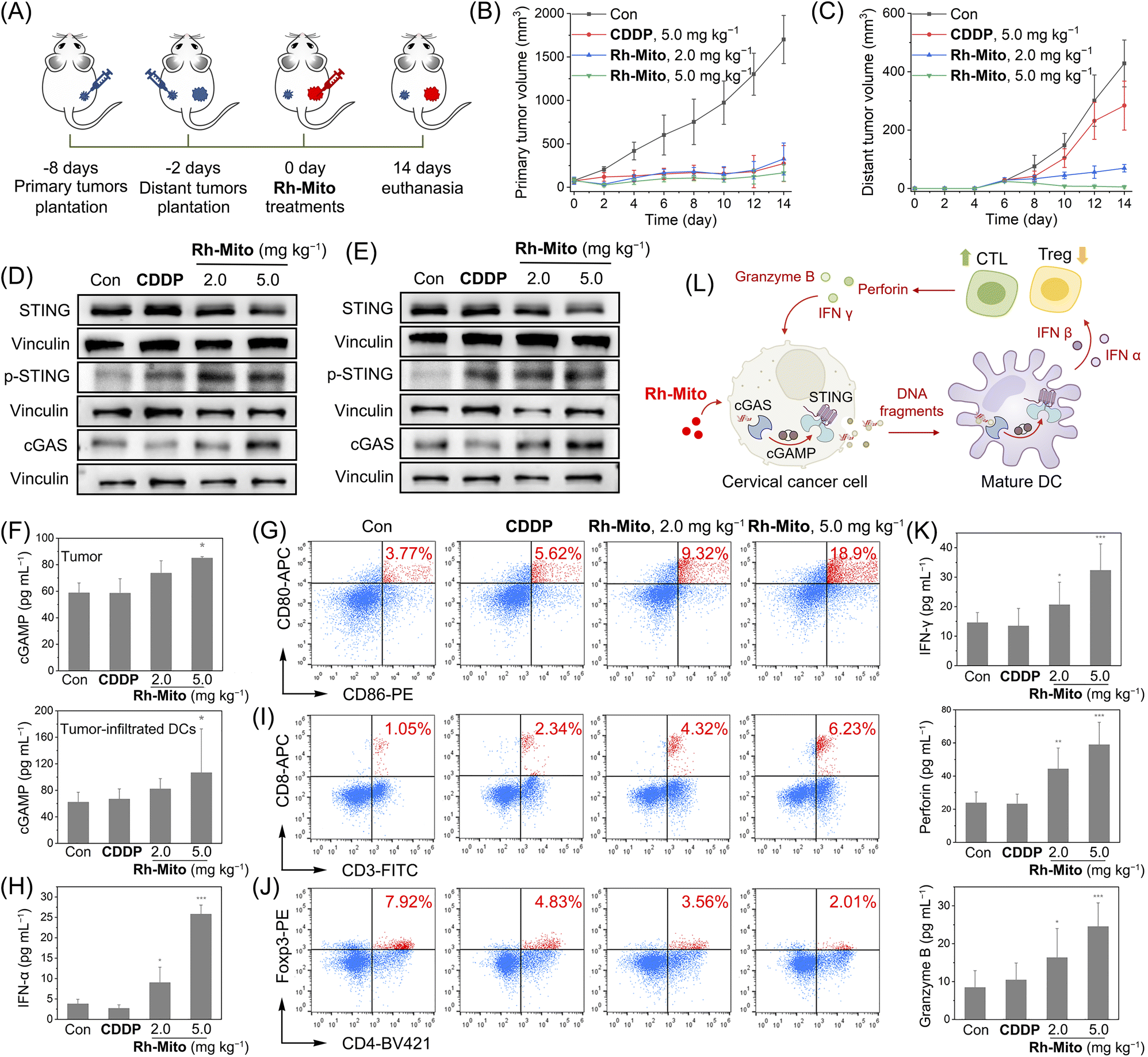 | ||
| Fig. 6 (A) Schematic illustration of the in vivo therapeutic protocol. (B and C) The volume curves of primary tumors (B) and distant tumors. (D–F) Western blots showing elevated expression of cGAS and p-STING in tumor (D) and tumor-infiltrated DCs (E). (F and H) ELISA showing increased cGAMP in tumor and tumor-infiltrated DCs (F), and type I IFNs (IFN-α) in serum (H). Quantitative detection of populations of CD86+CD80+ DCs (gating on CD11c+, G), CD8+ (I) and Foxp3+ T cells (J) in tumors after different treatments (n = 3). (K) Secretion of IFN-γ, perforin and granzyme B in serum measured by ELISA assay (n = 3). (L) Schematic illustration of the modulation of anticancer immunity by Rh-Mitoin vivo. *p < 0.05, **p < 0.01 and ***p < 0.001 by the Student's two-tailed t test. | ||
Compared with the control mice, the cGAMP content as well as the expression levels of cGAS and p-STING levels are markedly elevated in the Rh-Mito-treated group in both tumor and tumor-infiltrated DCs (Fig. 6D–F). The proportion of CD86+CD80+ cells, hallmarks for DC maturation, also increases from about 3.77% to 18.9% in mice treated with Rh-Mito at the higher dose (Fig. 6G). Both IFN-α and IFN-β in serum are elevated after Rh-Mito treatment (Fig. 6H and S44†). Accordingly, the percentage of CTLs (CD8+ T cells) in the tumor section of Rh-Mito-treated samples increases from about 1.05% to 6.23% (Fig. 6I), and CD4+ T cells are also increased from about 13.7% to 32.0% (Fig. S45†). Moreover, the regulatory T cells (Foxp3+ T cells) are decreased from 7.92% to 2.01% (Fig. 6J). CTLs can release IFN-γ, perforin and granzyme B to directly kill tumor cells, and all of these secreta are increased upon Rh-Mito treatments (Fig. 6K). Finally, obvious increases in pro-inflammatory cytokines (interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α)) in serum are also detected (Fig. S46†). These results show that Rh-Mito can activate the CTLs and regulatory T cells, cause the maturation of DCs, and improve the cancer immune microenvironments by activating the cGAS-STING pathway in vivo (Fig. 6L).
In order to improve the solubility and bioavailability of Rh-Mito, we use the recombination of human heavy-chain ferritin (HFn) to deliver it for intravenous injection (Fig. 7A). Ferritin is an iron storage protein comprising 24 subunits that can assemble into a spherical cage for the encapsulation of drugs.58 HFn can bind to transferrin receptor 1 (TfR1) overexpressing in many cancer types so as to achieve tumor-targeted delivery.59 Transmission electron microscope (TEM) observation shows that Rh-Mito@HFn nanoparticles (NPs) display a pseudo spherical nanostructure with an average diameter of 10.5 nm (Fig. 7B). Dynamic light scattering (DLS) measurement shows that the average hydrodynamic diameter of Rh-Mito@HFn NPs is about 24.0 nm with a narrow distribution pattern (Fig. 7C). The release rates of Rh-Mito from Rh-Mito@HFn NPs are about 63% and 75% at pH 6.0 and 7.4 after 12 h, respectively (Fig. 7D). Confocal microscopic observation shows that Rh-Mito@HFn NPs first localize in lysosomes in HeLa cells after 1 h incubation, and the emission of Rh-Mito displays a high colocalization with MTDR after 4 h (Fig. S47†), which indicates that Rh-Mito can be successfully released and retain its mtDNA targeting properties.
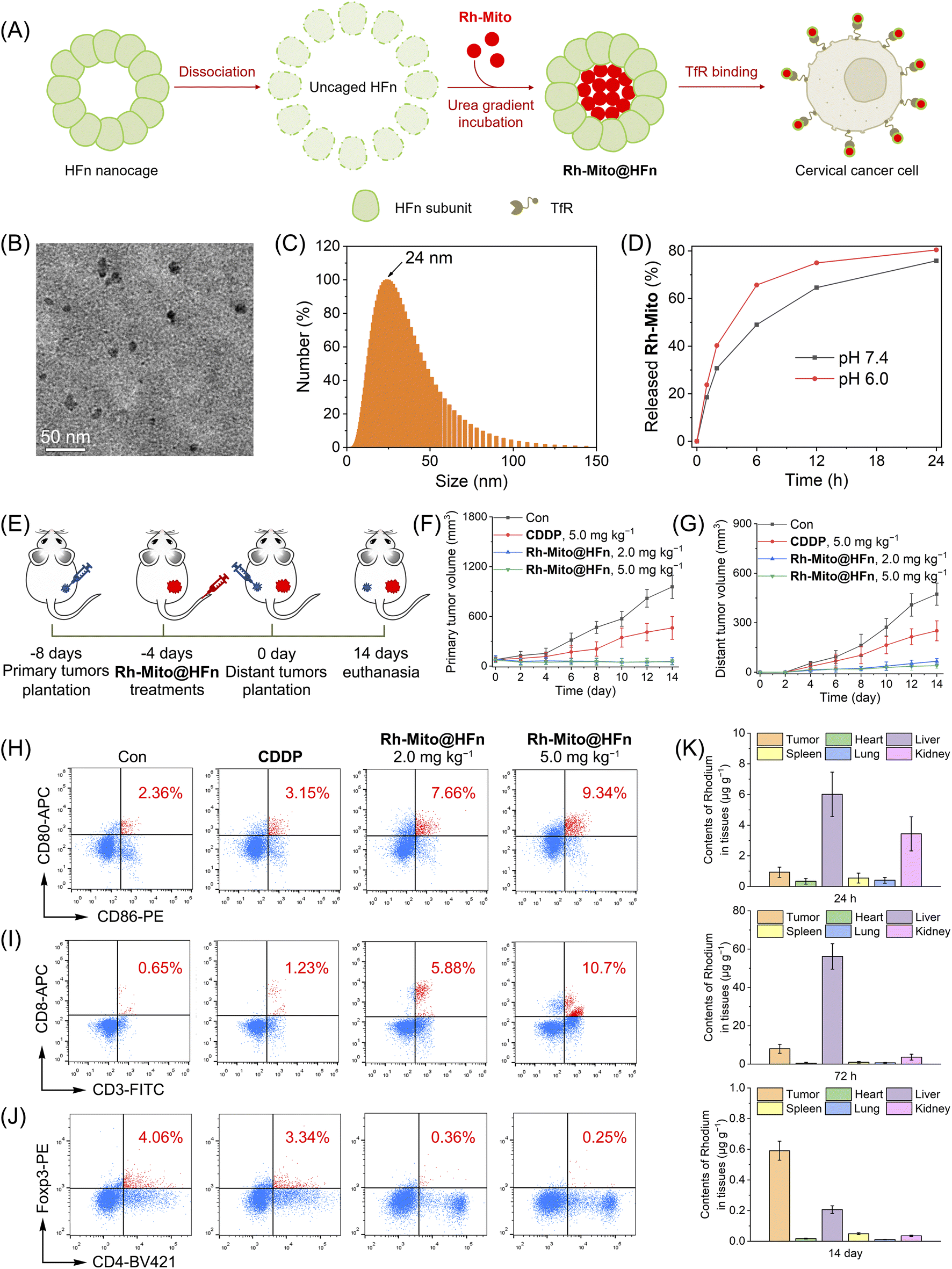 | ||
| Fig. 7 (A) The construction of Rh-Mito@HFn NPs. (B) The TEM image of Rh-Mito@HFn NPs. (C) Size distribution of Rh-Mito@HFn NPs characterized by DLS. (D) Time-dependent release of Rh-Mito from Rh-Mito@HFn NPs in buffer with different pH values. (E) Schematic illustration of in vivo therapeutic protocol. The volume curves of primary tumors (F) and distant tumors (G) upon different treatments. Quantitative detection of populations of CD86+CD80+ DCs (H, gating on CD11c+), CD8+ (I), and Foxp3+ T cells (J) in tumors after different treatment (n = 3) by flow cytometry. (K) Contents of rhodium in diverse organs of tumor-bearing mice at different time points. | ||
The antitumor efficacy of Rh-Mito@HFn NPs was evaluated on U14 cancer and the therapeutic protocol is illustrated in Fig. 7E. U14 cells were inoculated to the BALB/c mice as primary tumors. 4 days later, Rh-Mito@HFn NPs were intravenously injected. After another 4 days, U14 cells were planted into the contralateral as distant tumors. After 14 days of treatment, the inhibitory rates of Rh-Mito@HFn NPs on primary and distant tumors are about 95.4% and 86.1%, respectively (Fig. 7F and G and S48†). Rh-Mito@HFn NP treatment increases the content of CD86+CD80+ cells from about 2.36% to 9.34%, which indicates that it can increase the maturation of DCs (Fig. 7H). Moreover, Rh-Mito@HFn NPs increase the percentage of CTLs (CD8+ T cells) from about 0.65% to 10.7% (Fig. 7I) and downregulate the regulatory T cells (Foxp3+ T) cells from about 4.06% to 0.25% (Fig. 7J). The CD4+ T cells are also increased from 6.19% to 31.9% (Fig. S49†).
The content of Rh in different organs was measured by ICP-MS to investigate the distribution and metabolism of Rh-Mito@HFn NPs (Fig. 7K). At 24 h and 72 h post-injection, Rh mainly concentrates in the liver, kidney and tumor, which indicates that Rh-Mito@HFn NPs possess good tumor-targeting properties. On day 14, the Rh content in all organs, including tumor tissue, was very low (<0.6 μg g−1), which proves that Rh-Mito@HFn NPs can be metabolized in the body. Moreover, Rh-Mito@HFn NPs did not cause body weight loss in mice at the end of the treatment (Fig. S50†).
3 Discussion
The cGAS-STING pathway plays important roles in the innate immune responses triggered by viruses, bacteria, parasite infections as well as cancer immunity by regulating protein synthesis and cytokine expression through phosphorylation, ubiquitination and dimerization.3,60 cGAS-STING can also be activated by sensing endogenous DNA, including extranuclear chromatin and DNA fragments released from mitochondria, which makes it an important regulator in autoimmunity, inflammation and cell senescence.5,61 The release of damaged mtDNA into cytosol can activate the cGAS-STING pathway, which induces cytokine expression leading to the recruitment and activation of immune cells.7,32 Traditional therapies, e.g., radiotherapy and chemotherapy, can cause cell death by inducing DNA damage, while they can also activate the cGAS-STING pathway to enhance DC-mediated antigen presentation and T cell response.62 For example, cisplatin is reported to activate the cGAS/STING axis and enhance the anticancer immune response.63 Recent studies have shown that STING is a potent target in cancer therapy, and agonists of STING have been developed rapidly.2,64 Moreover, non-CDN STING agonists, small molecules, and metal ions have also been proven to be effective in activating the STING pathway.10–13,65 In this study, we report a metal complex Rh-Mito with an octahedral three-dimensional structure as the mtDNA intercalating agent. Due to its cationic lipophilic nature, Rh-Mito can accumulate in mitochondria to cause mtDNA damage resulting in its cytosolic release, which activates the cGAS-STING pathway.Recent studies have shown that mitochondrial metabolism is closely related to the epigenetic status of cells.29 SAM, an intermediate metabolite of one-carbon metabolism, is the most important donor of methylation reactions, while its downstream product SAH can inhibit the activity of methyl transferase.66 Another important intermediate metabolite of the TCA cycle, α-KG, is a cofactor for histone/DNA/RNA demethylases.54 Furthermore, epigenetic modifications play key roles in immunogenic activation, tumor antigen presentation, T-cell infiltration, and tumor immunosuppression.67–70 Most solid tumors show loss of overall methylation and hypermethylation of CpG island in the tumor suppressor gene promoter region, which silences antitumor immunity genes and impairs antigen recognition and presentation.71 In this work, we find that Rh-Mito can interfere with cellular metabolic pathways by damaging mtDNA, especially nucleotide anabolism, and metabolites related to DNA methylation modifications. Combined with the results of RRBS and RNA-seq, we show that Rh-Mito can affect the 5mC DNA modification to regulate the immune pathways, including the cGAS-STING pathway. The regulation of cancer immunity through epigenetic modifications caused by attenuation in mitochondrial metabolome reflects the importance of mitochondrial retrograde signaling in shaping the immune microenvironments. We propose that the metabolic modulation may be an effective complement to the cGAS-STING pathway activated by the cytoplasmic release of mtDNA.
The activation of the cGAS-STING signaling pathway is considered to be a very effective strategy for tumor immunotherapy, and the treatment by intratumoral injection of CDNs has shown significant efficacy in preclinical studies and is undergoing human clinical trials.72 However, CDNs are hydrophilic molecules, and they are not easy to penetrate the membrane. CDNs are also easy to hydrolyze, making them not feasible for systematic administration.2 With the development of structural modification strategies and non-CDN agonists, the stability of STING agonists has been improved, but they also face toxicity problems.2,64 The unsatisfactory initial clinical trial results of STING agonists trigger an urgent need for efficient delivery strategies, including liposomes and inorganic NPs.14–16 In our work, we use HFn nanocages as the carrier for Rh-Mito. We demonstrate that Rh-Mito@HFn NPs possess good tumor-targeting capability and high biocompatibility, and they can successfully evoke potent antitumor immune responses by intravenous injection.
4 Conclusion
In summary, we report an mtDNA-targeting rhodium(III) complex, Rh-Mito, as an effective cancer immunotherapeutic agent. Rh-Mito can damage mtDNA to cause mitochondrial damage and the cytosolic leakage of mtDNA fragments, which elicits the cGAS-STING-dependent type I IFN-mediated antitumor immune responses. Moreover, Rh-Mito disturbs the mitochondrial metabolic status and alters several key metabolites involved in epigenetic modification to reshape the nucDNA methylation landscape. Moreover, we also prove that Rh-Mito encapsulated in HFn nanocages can activate tumor immunotherapeutic responses in vivo by intravenous administration. Overall, we demonstrate that mtDNA-targeted small molecules can activate cGAS-STING signaling, which provides new insight into the design of potential immune-modulating anticancer agents.Data availability
RNA-sequencing data are deposited and available on the Gene Expression Omnibus under accession ID GSE221227 with secure token ijkncqqexbebvwr (https://www.ncbi.nlm.nih.gov/geo/). RRBS data for DNA methylation are deposited and available on Sequence Read Archive under accession ID PRJNA913445 with link https://dataview.ncbi.nlm.nih.gov/object/PRJNA913445?reviewer=jtm1nu53im54t3vt8oonohbn2p.Author contributions
YZ and XXC contributed equally to this work. YZ, ZWM, and CPT conceived this study and designed the experiments. YZ, DYZ, KP, and ZYL completed material synthesis and characterization and analyzed the data. WJW performed the docking assay and analyzed computational data. YZ, DYZ, and XXC carried out cellular studies and analyzed the data. YZ and XXC conducted the in vivo studies and analyzed the data. YZ, XXC, ZWM, and CPT analyzed the results and wrote the manuscript. YZ, ZWM, and CPT provided the funds to support this article. All authors revised this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 22022707, 22107124, 22177142, 21837006 and 91953117), China Postdoctoral Science Foundation (No. 2021M693604), and the Fundamental Research Funds for the Central Universities.References
- G. N. Barber, Nat. Rev. Immunol., 2015, 15, 760–770 CrossRef CAS PubMed.
- K. M. Garland, T. L. Sheehy and J. T. Wilson, Chem. Rev., 2022, 122, 5977–6039 CrossRef CAS PubMed.
- J. Kwon and S. F. Bakhoum, Cancer Discovery, 2020, 10, 26–39 CrossRef CAS PubMed.
- X. Cai, Y. H. Chiu and Z. J. Chen, Mol. Cell, 2014, 54, 289–296 CrossRef CAS PubMed.
- K. P. Hopfner and V. Hornung, Nat. Rev. Mol. Cell Biol., 2020, 21, 501–521 CrossRef CAS PubMed.
- Y. Tanaka and Z. J. Chen, Sci. Signaling, 2012, 5, ra20 CrossRef PubMed.
- A. Decout, J. D. Katz, S. Venkatraman and A. Ablasser, Nat. Rev. Immunol., 2021, 21, 548–569 CrossRef CAS PubMed.
- L. Corrales, L. H. Glickman, S. M. McWhirter, D. B. Kanne, K. E. Sivick, G. E. Katibah, S. R. Woo, E. Lemmens, T. Banda, J. J. Leong, K. Metchette, T. W. Dubensky Jr and T. F. Gajewski, Cell Rep., 2015, 11, 1018–1030 CrossRef CAS PubMed.
- K. Luo, N. Li, W. Ye, H. Gao, X. Luo and B. Cheng, Molecules, 2022, 27, 4638 CrossRef CAS PubMed.
- J. M. Ramanjulu, G. S. Pesiridis, J. Yang, N. Concha, R. Singhaus, S. Y. Zhang, J. L. Tran, P. Moore, S. Lehmann, H. C. Eberl, M. Muelbaier, J. L. Schneck, J. Clemens, M. Adam, J. Mehlmann, J. Romano, A. Morales, J. Kang, L. Leister, T. L. Graybill, A. K. Charnley, G. Ye, N. Nevins, K. Behnia, A. I. Wolf, V. Kasparcova, K. Nurse, L. Wang, A. C. Puhl, Y. Li, M. Klein, C. B. Hopson, J. Guss, M. Bantscheff, G. Bergamini, M. A. Reilly, Y. Lian, K. J. Duffy, J. Adams, K. P. Foley, P. J. Gough, R. W. Marquis, J. Smothers, A. Hoos and J. Bertin, Nature, 2018, 564, 439–443 CrossRef CAS PubMed.
- E. N. Chin, C. Yu, V. F. Vartabedian, Y. Jia, M. Kumar, A. M. Gamo, W. Vernier, S. H. Ali, M. Kissai, D. C. Lazar, N. Nguyen, L. E. Pereira, B. Benish, A. K. Woods, S. B. Joseph, A. Chu, K. A. Johnson, P. N. Sander, F. Martínez-Peña, E. N. Hampton, T. S. Young, D. W. Wolan, A. K. Chatterjee, P. G. Schultz, H. Michael Petrassi, J. R. Teijaro and L. L. Lairson, Science, 2020, 369, 993–999 CrossRef CAS PubMed.
- B. S. Pan, S. A. Perera, J. A. Piesvaux, J. P. Presland, G. K. Schroeder, J. N. Cumming, B. W. Trotter, M. D. Altman, A. V. Buevich, B. Cash, S. Cemerski, W. Chang, Y. Chen, P. J. Dandliker, G. Feng, A. Haidle, T. Henderson, J. Jewell, I. Kariv, I. Knemeyer, J. Kopinja, B. M. Lacey, J. Laskey, C. A. Lesburg, R. Liang, B. J. Long, M. Lu, Y. Ma, E. C. Minnihan, G. O'Donnell, R. Otte, L. Price, L. Rakhilina, B. Sauvagnat, S. Sharma, S. Tyagarajan, H. Woo, D. F. Wyss, S. Xu, D. J. Bennett and G. H. Addona, Science, 2020, 369, 935 CrossRef PubMed.
- M. Lv, M. Chen, R. Zhang, W. Zhang, C. Wang, Y. Zhang, X. Wei, Y. Guan, J. Liu, K. Feng, M. Jing, X. Wang, Y. C. Liu, Q. Mei, W. Han and Z. Jiang, Cell Res., 2020, 30, 966–979 CrossRef CAS PubMed.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS PubMed.
- E. L. Dane, A. Belessiotis-Richards, C. Backlund, J. Wang, K. Hidaka, L. E. Milling, S. Bhagchandani, M. B. Melo, S. Wu, N. Li, N. Donahue, K. Ni, L. Ma, M. Okaniwa, M. M. Stevens, A. Alexander-Katz and D. J. Irvine, Nat. Mater., 2022, 21, 710–720 CrossRef CAS PubMed.
- X. Lu, L. Miao, W. Gao and Z. Chen, Sci. Transl. Med., 2020, 12, eaaz6606 CrossRef CAS PubMed.
- F. Wang, H. Su, D. Xu, W. Dai, W. Zhang, Z. Wang, C. F. Anderson, M. Zheng, R. Oh, F. Wan and H. Cui, Nat. Biomed. Eng., 2020, 4, 1090–1101 CrossRef CAS PubMed.
- Z. Luo, X. Liang, T. He, X. Qin, X. Li, Y. Li, L. Li, X. J. Loh, C. Gong and X. Liu, J. Am. Chem. Soc., 2022, 144, 16366–16377 CrossRef CAS PubMed.
- T. Luo, G. T. Nash, X. Jiang, X. Feng, J. Mao, J. Liu, A. Juloori, A. T. Pearson and W. Lin, Adv. Mater., 2022, 34, e2110588 CrossRef PubMed.
- K. Yang, W. Han, X. Jiang, A. Piffko, J. Bugno, C. Han, S. Li, H. Liang, Z. Xu, W. Zheng, L. Wang, J. Wang, X. Huang, J. P. Y. Ting, Y. X. Fu, W. Lin and R. R. Weichselbaum, Nat. Nanotechnol., 2022, 17, 1322–1331 CrossRef CAS PubMed.
- W. H. Li, J. Y. Su and Y. M. Li, Acc. Chem. Res., 2022, 55, 2660–2671 CrossRef CAS PubMed.
- E. L. Mills, B. Kelly and L. A. J. O'Neill, Nat. Immunol., 2017, 18, 488–498 CrossRef CAS PubMed.
- C. R. Harapas, E. Idiiatullina, M. Al-Azab, K. Hrovat-Schaale, T. Reygaerts, A. Steiner, P. Laohamonthonkul, S. Davidson, C. H. Yu, L. Booty and S. L. Masters, Nat. Rev. Immunol., 2022, 22, 535–549 CrossRef CAS PubMed.
- M. G. Vizioli, T. Liu, K. N. Miller, N. A. Robertson, K. Gilroy, A. B. Lagnado, A. Perez-Garcia, C. Kiourtis, N. Dasgupta, X. Lei, P. J. Kruger, C. Nixon, W. Clark, D. Jurk, T. G. Bird, J. F. Passos, S. L. Berger, Z. Dou and P. D. Adams, Genes Dev., 2020, 34, 428–445 CrossRef CAS PubMed.
- M. Tigano, D. C. Vargas, S. Tremblay-Belzile, Y. Fu and A. Sfeir, Nature, 2021, 591, 477–481 CrossRef CAS PubMed.
- M. Rigon, A. R. Townley and M. Campanella, Cell Metab., 2021, 33, 853–855 CrossRef CAS PubMed.
- M. D. Buck, R. T. Sowell, S. M. Kaech and E. L. Pearce, Cell, 2017, 169, 570–586 CrossRef CAS PubMed.
- I. Martinez-Reyes and N. S. Chandel, Nat. Commun., 2020, 11, 102 CrossRef CAS PubMed.
- L. S. Huang, Z. Hong, W. Wu, S. Xiong, M. Zhong, X. Gao, J. Rehman and A. B. Malik, Immunity, 2020, 52, 475–486 CrossRef CAS PubMed.
- C. H. Yu, S. Davidson, C. R. Harapas, J. B. Hilton, M. J. Mlodzianoski, P. Laohamonthonkul, C. Louis, R. R. J. Low, J. Moecking, D. De Nardo, K. R. Balka, D. J. Calleja, F. Moghaddas, E. Ni, C. A. McLean, A. L. Samson, S. Tyebji, C. J. Tonkin, C. R. Bye, B. J. Turner, G. Pepin, M. P. Gantier, K. L. Rogers, K. McArthur, P. J. Crouch and S. L. Masters, Cell, 2020, 183, 636–649 CrossRef CAS PubMed.
- K. W. Chung, P. Dhillon, S. Huang, X. Sheng, R. Shrestha, C. Qiu, B. A. Kaufman, J. Park, L. Pei, J. Baur, M. Palmer and K. Susztak, Cell Metab., 2019, 30, 784–799 CrossRef CAS PubMed.
- C. C. Lee Lawrence and K. M. Lo Kenneth, J. Am. Chem. Soc., 2022, 144, 14420–14440 CrossRef PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- Z. Y. Pan, C. P. Tan, L. S. Rao, H. Zhang, Y. Zheng, L. Hao, L. N. Ji and Z. W. Mao, Angew. Chem., Int. Ed., 2020, 59, 18755–18762 CrossRef CAS PubMed.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- K. M. Boyle and J. K. Barton, J. Am. Chem. Soc., 2018, 140, 5612–5624 CrossRef CAS PubMed.
- D. L. Ma, L. J. Liu, K. H. Leung, Y. T. Chen, H. J. Zhong, D. S. Chan, H. M. Wang and C. H. Leung, Angew. Chem., Int. Ed., 2014, 53, 9178–9182 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, J. Am. Chem. Soc., 2014, 136, 14160–14172 CrossRef CAS PubMed.
- J. M. Bailis, A. G. Weidmann, N. F. Mariano and J. K. Barton, PNAS, 2017, 114, 6948–6953 CrossRef CAS PubMed.
- G. J. Yang, W. Wang, S. W. F. Mok, C. Wu, B. Y. K. Law, X. M. Miao, K. J. Wu, H. J. Zhong, C. Y. Wong, V. K. W. Wong, D. L. Ma and C. H. Leung, Angew. Chem., Int. Ed., 2018, 57, 13091–13095 CrossRef CAS PubMed.
- T.-S. Kang, W. Wang, H.-J. Zhong, J.-X. Liang, C.-N. Ko, J.-J. Lu, X.-P. Chen, D.-L. Ma and C.-H. Leung, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 256–263 CrossRef CAS PubMed.
- H. Liang, T. Hao, C. Yin, X. Yang, H. Fu, X. Zheng, R. Li, D. Xiao and H. Chen, Eur. J. Inorg. Chem., 2017, 2017, 4149–4157 CrossRef CAS.
- Y.-Y. Ling, Y.-J. Kong, L. Hao, Z.-Y. Pan, Z.-W. Mao and C.-P. Tan, Inorg. Chem. Front., 2023, 10, 3284–3292 RSC.
- K. Peng, Y. Zheng, W. Xia and Z.-W. Mao, Chem. Soc. Rev., 2023, 52, 2790–2832 RSC.
- K. K.-W. Lo, B. T.-N. Chan, H.-W. Liu, K. Y. Zhang, S. P.-Y. Li and T. S.-M. Tang, Chem. Commun., 2013, 49, 4271–4273 RSC.
- Y. Zheng, L. He, D.-Y. Zhang, C.-P. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2017, 46, 11395–11407 RSC.
- R. Mirzayans, B. Andrais and D. Murray, Int. J. Mol. Sci., 2017, 18, 1679 CrossRef PubMed.
- A. Eastman, Oncotarget, 2017, 8, 8854–8866 CrossRef PubMed.
- X. Cheng, C. Kelso, V. Hornak, C. d. l. Santos, A. P. Grollman and C. Simmerling, J. Am. Chem. Soc., 2005, 127, 13906–13918 CrossRef CAS PubMed.
- E. A. Schon, S. DiMauro and M. Hirano, Nat. Rev. Genet., 2012, 13, 878–890 CrossRef CAS PubMed.
- C. Yan, X. Duanmu, L. Zeng, B. Liu and Z. Song, Cells, 2019, 8, 379 CrossRef CAS PubMed.
- M. Motwani, S. Pesiridis and K. A. Fitzgerald, Nat. Rev. Genet., 2019, 20, 657–674 CrossRef CAS PubMed.
- M. Xiao, H. Yang, W. Xu, S. Ma, H. Lin, H. Zhu, L. Liu, Y. Liu, C. Yang, Y. Xu, S. Zhao, D. Ye, Y. Xiong and K. L. Guan, Genes Dev., 2012, 26, 1326–1338 CrossRef CAS PubMed.
- M. Yang and K. H. Vousden, Nat. Rev. Cancer, 2016, 16, 650–662 CrossRef CAS PubMed.
- Z. D. Smith and A. Meissner, Nat. Rev. Genet., 2013, 14, 204–220 CrossRef CAS PubMed.
- F. Song, J. F. Smith, M. T. Kimura, A. D. Morrow, T. Matsuyama, H. Nagase and W. A. Held, PNAS, 2005, 102, 3336–3341 CrossRef CAS PubMed.
- N. Song, J. Zhang, J. Zhai, J. Hong, C. Yuan and M. Liang, Acc. Chem. Res., 2021, 54, 3313–3325 CrossRef CAS PubMed.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS PubMed.
- X. Zhang, X. C. Bai and Z. J. Chen, Immunity, 2020, 53, 43–53 CrossRef CAS PubMed.
- K. N. Miller, S. G. Victorelli, H. Salmonowicz, N. Dasgupta, T. Liu, J. F. Passos and P. D. Adams, Cell, 2021, 184, 5506–5526 CrossRef CAS PubMed.
- S. Yum, M. Li and Z. J. Chen, Cell Res., 2020, 30, 639–648 CrossRef CAS PubMed.
- S. Grabosch, M. Bulatovic, F. Zeng, T. Ma, L. Zhang, M. Ross, J. Brozick, Y. Fang, G. Tseng, E. Kim, A. Gambotto, E. Elishaev, R. P. Edwards and A. M. Vlad, Oncogene, 2019, 38, 2380–2393 CrossRef CAS PubMed.
- Q. Zhang, C. Chen, B. Xia and P. Xu, Curr. Opin. Chem. Biol., 2022, 69, 102170 CrossRef CAS PubMed.
- D. Lu, G. Shang, J. Li, Y. Lu, X. C. Bai and X. Zhang, Nature, 2022, 604, 557–562 CrossRef CAS PubMed.
- J. W. Locasale, Nat. Rev. Cancer, 2013, 13, 572–583 CrossRef CAS PubMed.
- E. Dai, Z. Zhu, S. Wahed, Z. Qu, W. J. Storkus and Z. S. Guo, Mol. Cancer, 2021, 20, 171 CrossRef CAS PubMed.
- Z. Zhou, K. Van der Jeught, Y. Fang, T. Yu, Y. Li, Z. Ao, S. Liu, L. Zhang, Y. Yang, H. Eyvani, M. L. Cox, X. Wang, X. He, G. Ji, B. P. Schneider, F. Guo, J. Wan, X. Zhang and X. Lu, Nat. Biomed. Eng., 2021, 5, 1320–1335 CrossRef CAS PubMed.
- Q. Duan, H. Zhang, J. Zheng and L. Zhang, Trends Cancer, 2020, 6, 605–618 CrossRef CAS PubMed.
- W. Sheng, M. W. LaFleur, T. H. Nguyen, S. Chen, A. Chakravarthy, J. R. Conway, Y. Li, H. Chen, H. Yang, P. H. Hsu, E. M. Van Allen, G. J. Freeman, D. D. De Carvalho, H. H. He, A. H. Sharpe and Y. Shi, Cell, 2018, 174, 549–563 CrossRef CAS PubMed.
- X. Pan and L. Zheng, Cell. Mol. Immunol., 2020, 17, 940–953 CrossRef CAS PubMed.
- A. Guilherme, L. A. Rowland, H. Wang and M. P. Czech, Trends Cell Biol., 2023, 33, 340–354 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01737k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |