DOI:
10.1039/D3SC01786A
(Edge Article)
Chem. Sci., 2023,
14, 8897-8904
Photocontrolled activation of doubly o-nitrobenzyl-protected small molecule benzimidazoles leads to cancer cell death†
Received
6th April 2023
, Accepted 20th July 2023
First published on 25th July 2023
Abstract
Artificial biomimetic chloride anionophores have shown promising applications as anticancer scaffolds. Importantly, stimuli-responsive chloride transporters that can be selectively activated inside the cancer cells to avoid undesired toxicity to normal, healthy cells are very rare. Particularly, light-responsive systems promise better applicability for photodynamic therapy because of their spatiotemporal controllability, low toxicity, and high tunability. Here, in this work, we report o-nitrobenzyl-linked, benzimidazole-based singly and doubly protected photocaged protransporters 2a, 2b, 3a, and 3b, respectively, and benzimidazole-2-amine-based active transporters 1a–1d. Among the active compounds, trifluoromethyl-based anionophore 1a showed efficient ion transport activity (EC50 = 1.2 ± 0.2 μM). Detailed mechanistic studies revealed Cl−/NO3− antiport as the main ion transport process. Interestingly, double protection with photocages was found to be necessary to achieve the complete “OFF-state” that could be activated by external light. The procarriers were eventually activated inside the MCF-7 cancer cells to induce phototoxic cell death.
Introduction
Naturally occurring chloride transporters like prodigiosin, tambjamine, etc. are known to perturb the chloride ion homeostasis inside the cancer cells to induce apoptosis.1,2 A wide variety of synthetic analogs in the form of ion channels and ion carriers have been developed that mimic natural systems and can lead cancer cells to undergo apoptosis.3–8 However, the undesired cytotoxicity towards the normal, healthy cells is one of the drawbacks associated with most of these ion transport systems, due to the lack of their selectivity towards the cancer cells. Various stimuli-responsive ion transporters have been reported to overcome this issue where external stimuli are used to activate them. The different kinds of stimuli that have been utilized include pH,9,10 light,11 voltage,12 ligand,13,14 enzymes,15–17 redox,18–20etc. However, very few stimuli-responsive systems have actually been activated inside the cancerous cells,21,22 and hence, efficient systems with better therapeutic applications are needed. Light is one of the versatile stimuli that has been employed because of the unique combination of features of spatiotemporal control, remote addressability, extreme tunability, and biocompatibility.23 The different photosystems that have been used to generate light-responsive ion transport systems include the use of photoswitches like azobenzene, acylhydrazone, phenylhydrazone, stiff-stilbene, etc.,24–34 and photocages, e.g., o-nitrobenzyl.35,36 It is still a challenging task to design photoresponsive ion transport systems possessing a fully inactive ‘OFF’ state and a fully active ‘ON’ state. One promising approach to achieve this involves the use of photocleavable groups connected to block the anion binding sites in the active ion transporters that can be removed using external electromagnetic radiation. The o-nitrobenzyl (ONB) group is one such kind of photocage that has been utilized for the activation or delivery of anticancer drugs.37–39 Our group demonstrated the photoactivation of ONB-linked indole-2-carboxamide-based procarriers inside the cancer cells to induce phototoxic cell death.40 The active transporter, however, itself did not show any cell death and hints that systems with better biological activities need to be developed for improved therapeutic applications.
Benzimidazole is one of the interesting moieties that is known to bind anions through hydrogen bonding interactions and has been used for ion transport purposes.41 A wide variety of benzimidazole-based anion transporters have shown potential anticancer applications.42 Recently, our group developed pyridyl-linked benzimidazole-based HCl cotransporters that were found to have five-fold higher efficiency compared to prodigiosin (a naturally occurring HCl cotransporter), and exhibited significant Cl− mediated cell death in MCF-7 breast cancer cells.43
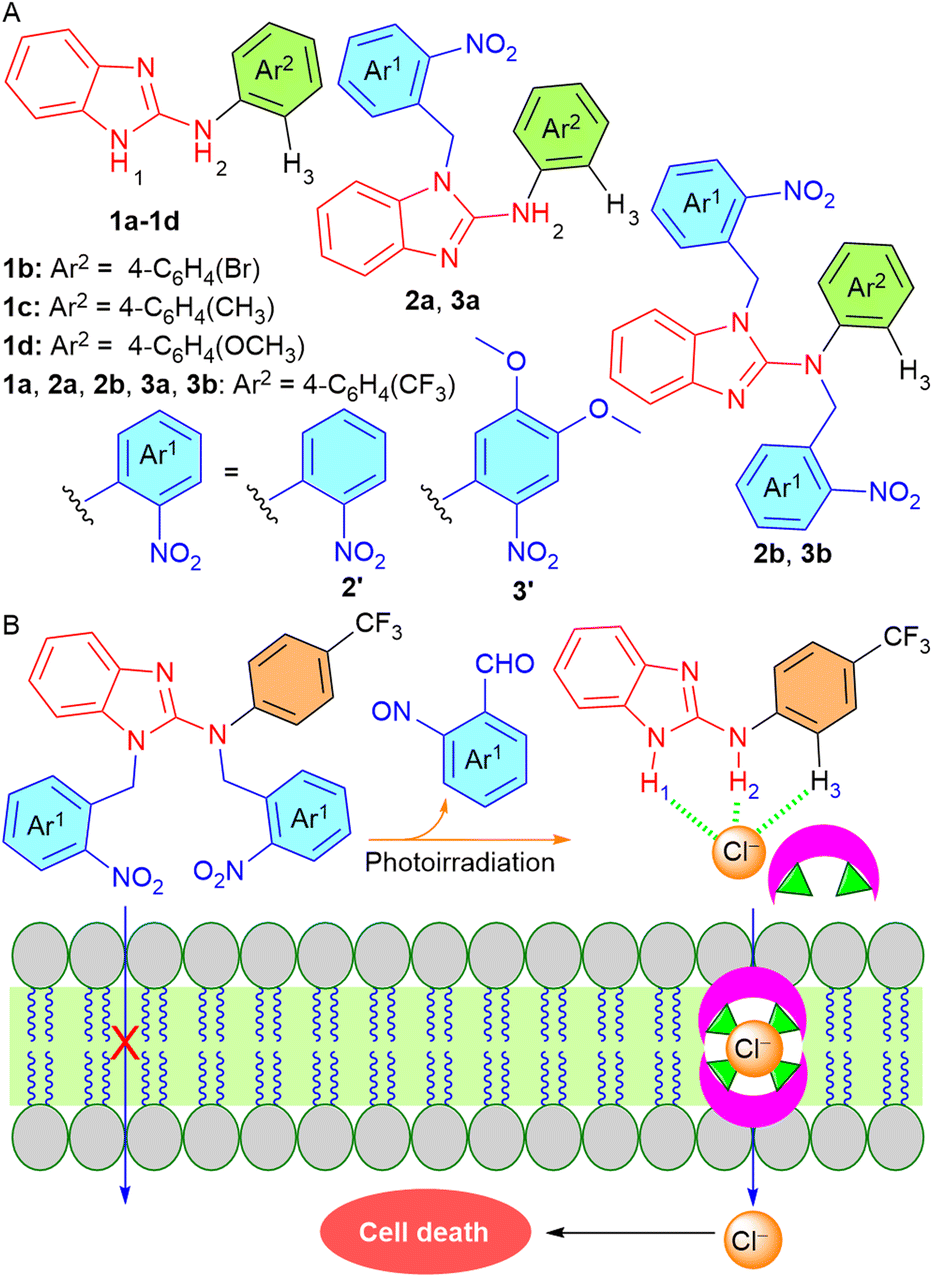
Such findings prompted us to utilize this efficient ion transport moiety by selectively activating it inside the cancer cells without affecting the normal cells. Here, we developed ONB-linked benzimidazole-2-amine procarriers 2a, 2b, 3a, and 3b that were photoactivated inside the artificial liposomes and MCF-7 breast cancer cells. We used benzimidazole-2-amines 1a–1d as the active transporters that were expected to interact with the anions through imidazole and amine N–H hydrogen bonding interactions (Fig. 1A). Furthermore, different substituents were connected to the aromatic ring in order to fine-tune the membrane permeability and binding affinity of these anion transporters.7,44 We anticipated that protection of the anion binding N–H protons with a photoresponsive ONB group would render them inactive for anion transport in the lipid membrane. Eventually, the photocaged procarriers would be activated inside the artificial liposomes and cancer cells to induce cell death by utilizing external electromagnetic radiation (Fig. 1B). Moreover, the incorporation of methoxy groups into the ONB group was expected to affect the photolytic properties of the procarriers.45
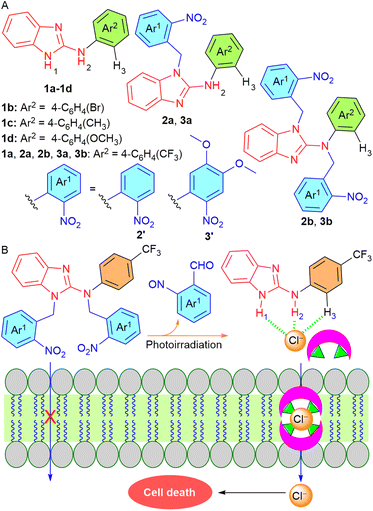 |
| | Fig. 1 (A) Chemical structures of the procarrier molecules 2a, 2b, 3a, 3b and active transporters 1a–1d. (B) Working principle of phototriggered activation inside the lipid membrane. | |
Results and discussion
Synthesis
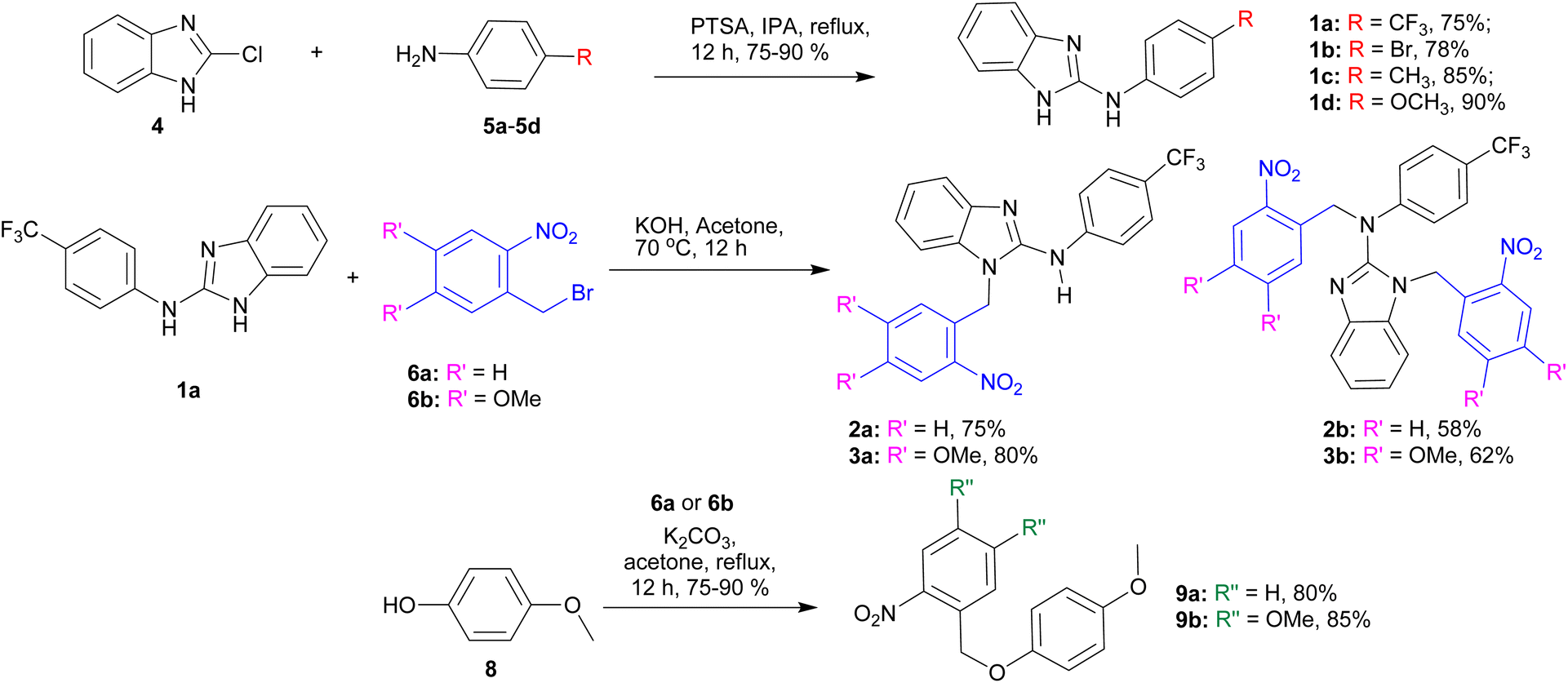
The synthesis of the active transporters 1a–1d was achieved by reacting 2-chlorobenzimidazole 4 with different aromatic amines 5a–5d in the presence of p-toluene sulfonic acid (Scheme 1). In order to synthesize the protected compounds 2a, 2b, 3a, and 3b, the most active transporter 1a was coupled with the corresponding o-nitrobenzyl bromide derivatives 6a and 6b, respectively. The reaction of 1a with one equivalent of 6a and 6b led to the formation of mono-ONB-protected compounds 2a and 3a, and on the other hand, the reaction of 1a with the excess (3 equivalents) of 6a and 6b led to the formation of di-ONB protected compounds 2b and 3b, respectively. For the synthesis of the corresponding control compounds, 4-methoxyphenol 8 was reacted with the corresponding o-nitrobenzyl bromide derivatives 6a and 6b in the presence of potassium carbonate to furnish 9a and 9b in excellent yields.
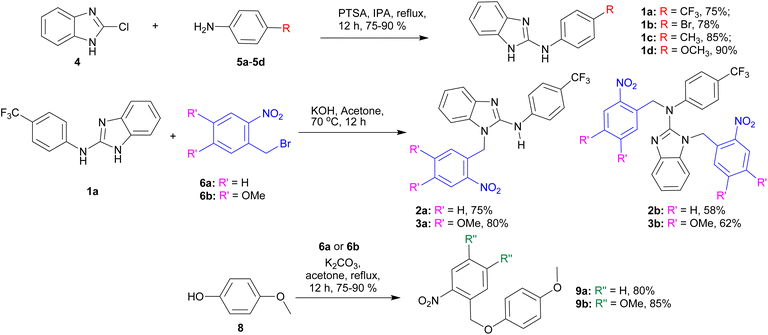 |
| | Scheme 1 Synthesis of active carriers 1a–1d, ONB-protected procarriers 2a, 2b, 3a, and 3b, and control compounds 9a and 9b. | |
Anion binding studies
After synthesizing compounds 1a–1d, the chloride binding studies were performed by 1H NMR titrations in acetonitrile-d3 using TBACl (a Cl− ion source). The addition of TBACl to the receptors led to a significant downfield shift of imidazole N–H1, amine N–H2, and (Ar)C–H3 protons, indicating their involvement in the chloride binding through N–H⋯Cl− and C(Ar)–H⋯Cl− hydrogen bonding interactions (Fig. S22, S24, S26, and S28†). The Bindfit program46 revealed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 receptor:Cl− binding stoichiometry and the binding constant values (Ka) were observed to be 2100 ± 185 M−1 for 1a, 1200 ± 175 M−1 for 1b, 500 ± 71 M−1 for 1c, and 300 ± 43 M−1 for 1d, respectively (Table 1, Fig. S23, S25, S27, and S29†). The evidence of 1:1 receptor:Cl− binding stoichiometry was further clarified by electrospray ionization mass spectroscopic studies (ESI-MS). The data provided the peaks at m/z = 330.0620 and 332.0597, which correspond to the complex 1a·Cl− (Fig. S30†).
:1 receptor:Cl− binding stoichiometry and the binding constant values (Ka) were observed to be 2100 ± 185 M−1 for 1a, 1200 ± 175 M−1 for 1b, 500 ± 71 M−1 for 1c, and 300 ± 43 M−1 for 1d, respectively (Table 1, Fig. S23, S25, S27, and S29†). The evidence of 1:1 receptor:Cl− binding stoichiometry was further clarified by electrospray ionization mass spectroscopic studies (ESI-MS). The data provided the peaks at m/z = 330.0620 and 332.0597, which correspond to the complex 1a·Cl− (Fig. S30†).
Table 1 Summary of logP, chloride binding Ka (M−1) EC50 (μM), and Hill coefficient (n) values of compounds 1a–1d
| Comp |
R |
logP |
K
a
(M−1)/Cl− |
EC50b (μM) |
n
|
|
Association constants obtained using the Bindfit Program based on the 1:1 binding model for NH2 protons.
Effective concentration to reach 50% of maximal activity across EYPC-LUVs entrapped with 1 mM HPTS, NaCl (100 mM), buffered at pH 7.0 using 10 mM HEPES.
Hill coefficient. logP values were calculated using MarvinSketch software.
|
|
1a
|
CF3 |
4.26 |
2100 ± 185 |
1.2 ± 0.2 |
2.3 ± 0.3 |
|
1b
|
Br |
4.15 |
1200 ± 75 |
3.2 ± 0.2 |
2.0 ± 0.2 |
|
1c
|
CH3 |
3.90 |
500 ± 71 |
13.5 ± 2.5 |
1.9 ± 0.6 |
|
1d
|
OCH3 |
3.23 |
300 ± 43 |
26.9 ± 3.8 |
2.9 ± 0.8 |
Ion transport studies
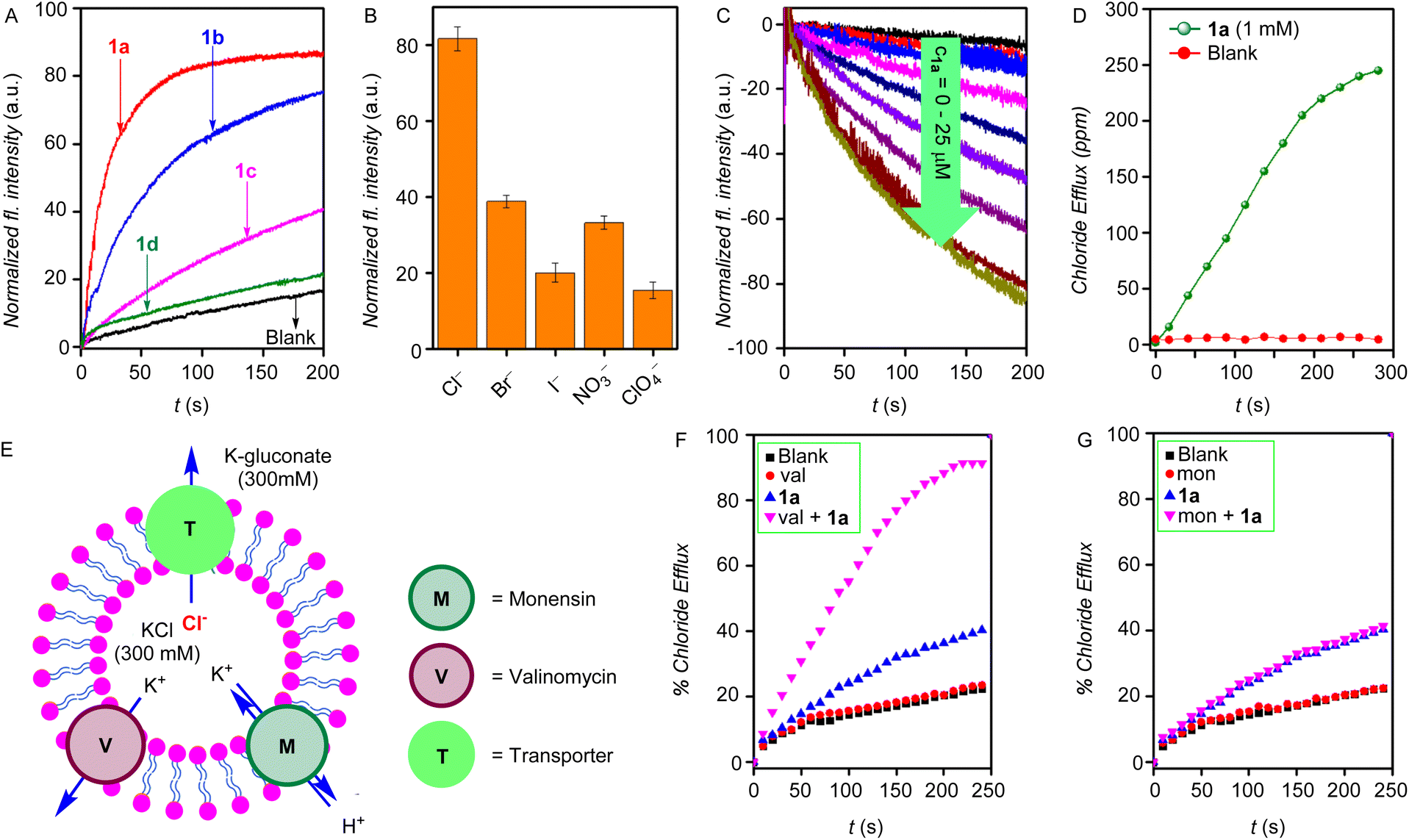
The ion transport properties of the compounds 1a–1d were determined across egg yolk phosphatidylcholine unilamellar vesicles (EYPC-LUVs) entrapped with 8-hydroxy pyrene-1,3,6-trisulfonic acid trisodium salt (HPTS, a pH-sensitive dye) (EYPC-LUVs⊃HPTS) and 100 mM NaCl buffered at pH = 7.0.47 Subsequently, a pH gradient was created across the vesicular membrane by adding NaOH to the extravesicular buffer. The dissipation of the pH gradient by the transporter molecules 1a–1d was determined by monitoring the time-dependent change in the HPTS fluorescence (λem = 510 nm and λex = 450 nm). Triton X-100 was added at the end of each experiment for calibration to get 100% transport activity. The ion transport activity monitored at c = 7.5 μM showed the ion transport sequence of 1a > 1b > 1d > 1c (Fig. 2A). The Hill analysis of the dose–response curves furnished the EC50 values (effective concentration to reach the 50% activity) of 1.2 ± 0.2 μM (1.9 mol%) for 1a, 3.2 ± 0.2 μM (5.1 mol%) for 1b, 13.5 ± 2.5 μM (21.6 mol%) for 1c, and 26.9 ± 3.8 μM (43.0 mol%) for 1d, respectively (Fig. S32–S35†). The Hill coefficient of ∼2 suggests the involvement of two receptor molecules to drive the ion transport across the lipid membrane. Furthermore, upon varying the cations in the external buffer by using different MCl salt solutions (M+ = Li+, Na+, K+, Rb+, and Cs+), no significant change in the ion transport rate was observed, indicating no role of cations in the transport process (Fig. S37†). However, upon varying the anions in the external buffer by using different NaX salts (X = Cl−, Br−, I−, ClO4−, and NO3−), a significant change in the ion transport was observed, indicating the active role of anions in the transport process (Fig. 2B).
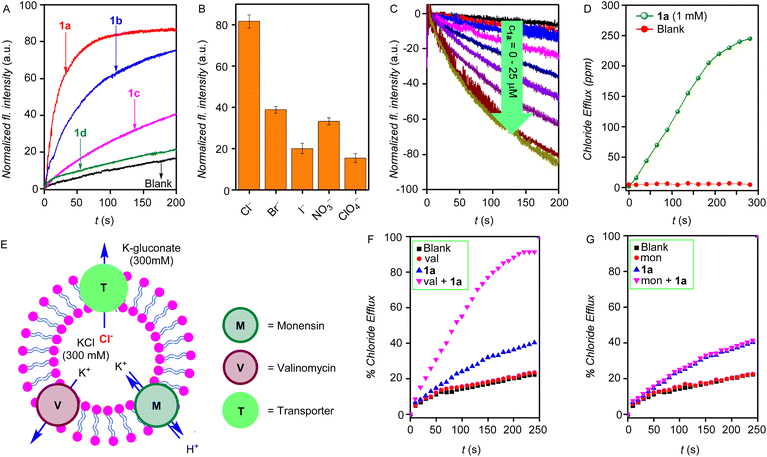 |
| | Fig. 2 (A) Activity comparison of 1a–1d (7.5 μM) across EYPC–LUVs⊃HPTS. (B) Anion selectivity of 1a (5.0 μM) by varying external anions across EYPC–LUVs⊃HPTS, each bar graph represents mean ion transport activity, calculated from three independent experiments. (C) Concentration-dependent activity of compound 1a across EYPC–LUVs⊃Lucigenin. (D) The chloride transport in the U-tube experiment in the presence and absence of transporter molecule 1a. (E) Schematic representation of ISE-based valinomycin and monensin assays. Normalized chloride efflux of 1a, (F) in the presence and absence of monensin, and (G) the presence and absence of valinomycin. | |
Chloride leakage studies
The Cl− leakage for 1a was monitored across lucigenin-based vesicles. Vesicles were prepared by entrapping lucigenin dye (1 mM) in 100 mM NaNO3 buffered at pH 7.0.48 Subsequently, a Cl−/NO3− gradient was created by adding a Cl− pulse in the external buffer, and the transport of Cl− ions was determined by measuring the time-dependent change in the lucigenin fluorescence (λex = 455 nm and λem = 535 nm). The concentration-dependent fluorescent study of 1a is shown in Fig. 2C. Hill analysis of the dose–responsive curve furnished the EC50 value of 13.9 ± 3.5 μM (Fig. S39†).
Ion selectivity and mechanism of ion transport
Mechanistically speaking, the ion transport in the above studies can occur through one of the modes of (a) H+/X− symport, (b) OH−/X− antiport, (c) H+/M+ antiport, or (d) OH−/M+ symport. However, no change in the ion transport activity in the presence of cations in the HPTS-based studies rules out the possibilities of H+/M+ antiport and OH−/M+ symport modes. To validate the antiport mode, the chloride efflux of 1a across LUVs containing 500 mM of KCl and suspended in a 500 mM potassium gluconate solution was monitored in the presence and absence of valinomycin (highly selective K+ antiporter) and monensin (an H+/K+ antiporter) using a chloride sensitive ion-selective electrode (Fig. 2E). A significant enhancement in the chloride efflux was observed in the presence of valinomycin (Fig. 2F), while on the other hand, no change was observed in the presence of monensin (Fig. 2G). This cooperative effect of valinomycin and 1a is a proof of the antiport mode of ion transport.49–51
Additionally, the classic U-tube experiment was performed to confirm the carrier mode of ion transport. Significant chloride transport from the source arm of the U-tube was observed into the receiver arm only in the presence of the transporter molecule 1a (Fig. 2D), which indicates the carrier mode of ion transport.52–54
Geometry optimization and binding energy calculation
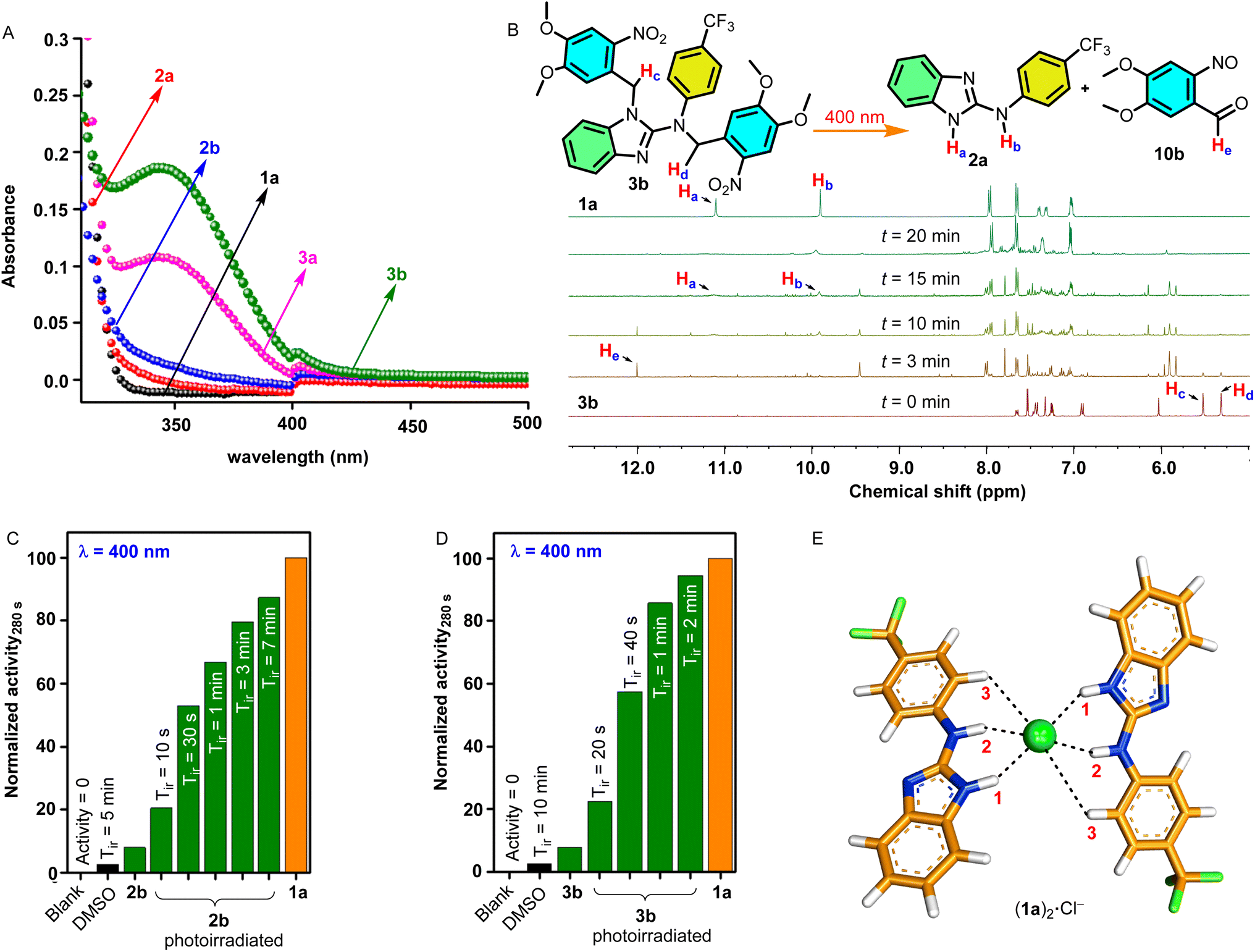
To obtain insights into the geometry of the [(1a)2+Cl−] complex, DFT calculations were performed. The [(1a)2+Cl−] complex was chosen based on the experimentally determined Hill coefficient value, which was found to be ∼2, indicating that two receptor molecules are involved in the formation of an active complex during the ion transport process. Initially, the CONFLEX 8 software program55,56 provides several conformational structures with almost equal populations (Fig. S51†). The highest Boltzmann populated structure was then optimized using the Gaussian 09 program57 utilizing a B3LYP functional and 6-31G (d, p) basis set.58 The geometry-optimized analysis of the [(1a)2+Cl−] complex showed the involvement of two receptor molecules interacting orthogonally with the chloride anion interacting through benzimidazole-N–H1⋯Cl− (H1⋯Cl− = 2.312 Å), amine-N–H2⋯Cl− (H2⋯Cl− = 2.274 Å), and C(Ar)–H3⋯Cl− (H3⋯Cl− = 3.620 Å) hydrogen bonding interactions (Fig. 3E). The binding energy was predicted to be −68.46 kcal mol−1.
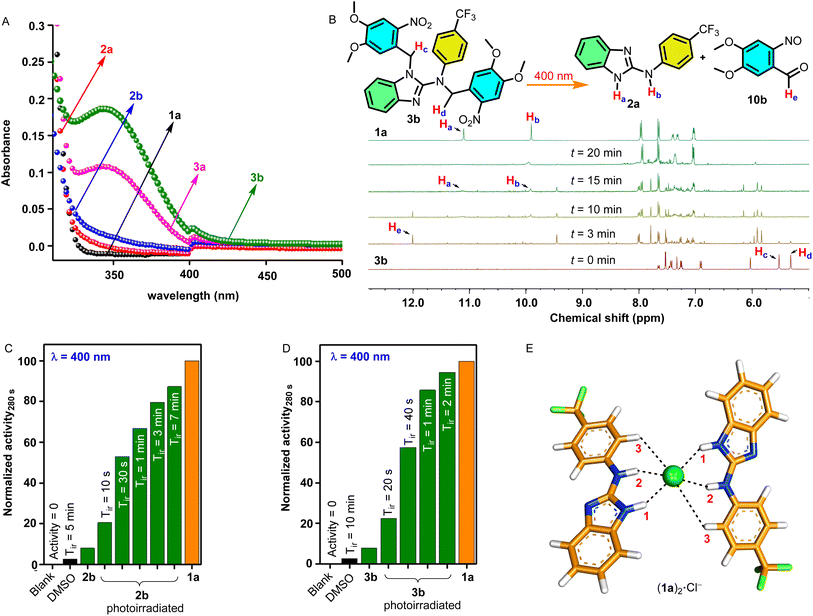 |
| | Fig. 3 (A) UV-Visible absorption spectra of 1a and ONB-protected procarrier compounds 2a, 2b, 3a, and 3b. (B) Phototriggered release of an active carrier 1a from ONB-protected procarrier 3b monitored by 1H NMR studies recorded in DMSO-d6 photoirradiated using a 400 nm LED at different time intervals. Normalized ion transport activity data upon photoirradiation at 400 nm for (C) 2b and (D) 3b, respectively. (E) The geometry-optimized structure of the [(1a)2+Cl−] complex. | |
Photolytic studies
After successfully achieving the ion transport studies, the photolytic studies of the procarriers 2a, 2b, 3a, and 3b were performed. Initially, The UV-Vis absorption studies of these compounds were performed in acetonitrile. The absorption spectra of all compounds were found to be extended up to 400 nm of wavelength with strong absorption bands at λ = 350 nm for dimethoxy-ONB substituted compounds 3a and 3b compared to the weaker absorption bands for the compounds 2a and 2b, respectively (Fig. 3A). After UV-Vis absorption studies, the photolytic studies were performed through 1H NMR analysis. Initially, the 1H NMR spectrum of each of the procarriers was recorded in DMSO-d6. Subsequently, each sample was subjected to photoirradiation at 400 nm using LEDs (1 × 3.8 Watt) at different time intervals, and the 1H NMR spectrum was recorded after each photoirradiation process. Finally, 1H NMR spectra of procarrier molecules (2a, 2b, 3a, and 3b) before irradiation, photoirradiated samples, and as-synthesized active carrier 1a were stacked (Fig. S43–S45†). The photoirradiation process of compound 3b is shown in Fig. 3B. Upon photoirradiation, Hc and Hd protons corresponding to the ONB group of the procarriers at 5.52 ppm and 5.31 ppm disappeared with the appearance of a new signal at 12.01 ppm corresponding to proton He of the o-nitrosobenzaldehyde byproduct 10b. Similarly, new proton signals Ha and Hb appeared at 11.11 ppm and 9.92 ppm, respectively, which correspond to the proton signals of as-synthesized benzimidazole carrier 1a. These changes in the IH NMR spectrum of the 3b upon photoirradiation confirmed the release of the active transporter 1a along with the o-nitrosobenzaldehyde byproduct 11. Similarly, compounds 2a, 2b, and 3a furnish the formation of active transporter 1a and the o- nitrosobenzaldehyde byproducts 10a and 10b, respectively. Noticeably, the dimethoxy-based compounds 3a and 3b showed efficient photocleavage (20 minutes for complete deprotection) as compared to the corresponding simple ONB-based compounds 2a and 2b (40 minutes for complete deprotection).
Photocleavable ion transport studies
Soon after the photolytic studies, the phototriggered ion transport studies were carried out, across HPTS entrapped EYPC-LUVs. Initially, the ion transport activities for the protected compounds 2a, 2b, 3a, and 3b were monitored. Subsequently, the vesicles containing these compounds were photoirradiated at different time intervals using a LED of 400 nm wavelength, and ion transport activity was monitored after each photoirradiation process. The ion transport activity after the photoirradiation process was compared with that of the as-synthesized active transporter 1a. The ion transport activity of the blank (only with DMSO) was taken as 0%, and for active transporter 1a as 100%. The procarriers 2b and 3b showed excellent enhancement in the ion transport activity upon photoirradiation (Fig. 3C and D), and on the other hand, compounds 2a and 3a did not furnish any change in the transport activity before and after the photoirradiation (Fig. S49†). These results indicate that the protection of both imidazole N–H and amine N–H protons is necessary to render the procarrier molecules inactive that are then successfully activated using external electromagnetic radiation. In the monoprotected compounds (2a and 3a), the amine N–H2 and the aryl C–H3 protons (Fig. 1A) are capable of cooperatively binding to the anion. Hence 2a and 3a exhibited substantial ion transport activity through probably the formation of transport complexes with the anion involving single or multiple host molecules. In the case of the doubly protected 2b and 3b, the absence of both benzimidazole (N–H1) and amine (N–H2) protons renders them incapable of anion transport as the aryl (C–H3) proton alone offers very weak anion binding and transport. The vesicles containing only DMSO did not furnish any transport activity while applying the photoirradiation for 10 min, confirming that light does not disrupt the integrity of the lipid membrane. Furthermore, the dimethoxy-ONB-based procarrier 3b furnishes efficient photoactivation (complete activation within 2 min of photoirradiation) compared to simple ONB-based procarrier 2b (complete activation after 7 min of photoirradiation) due to the photolytic properties of dimethoxy-based ONB systems as evident from NMR (Fig. 3B, S46†) and vesicle transport (Fig. 3C and D) studies.
Cellular viability and phototriggered cell death
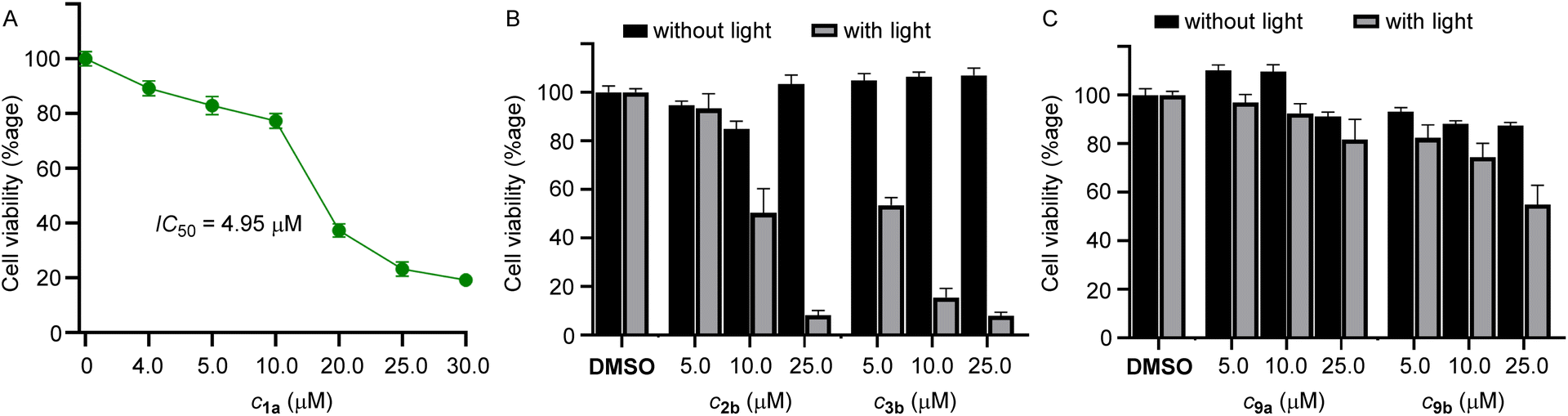
Among the compounds 1a–1d, we chose 1a for the biological studies because of its efficient ion transport properties. The MCF-7 cells (human breast cancer cell line) were incubated with 1a (0–25 μM) for 24 h, and cell viability was monitored using the standard MTT assay.59 The compound furnished efficient cytotoxicity with an IC50 value of 4.5 μM (Fig. 4A).
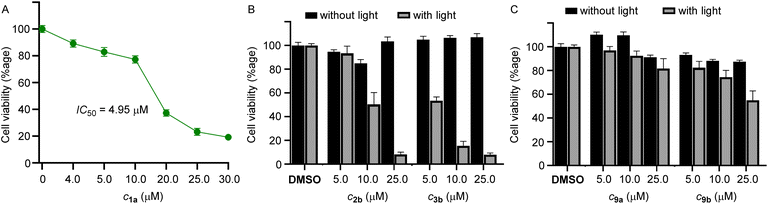 |
| | Fig. 4 (A) Dose-dependent cell viability of MCF-7 cells incubated with 1a (0–25 μM each) for 24 hours. (B) Cell viability obtained from the MTT assay upon addition of 2b and 3b for 24 h in MCF-7 cells in the absence of light and upon photoirradiation at 400 nm for 20 min using (1 × 3.8 W LED). Mean cell viability was obtained from three independent experiments. (C) Cell viability obtained from the MTT assay upon addition of 9a and 9b for 24 h in MCF-7 cells in the absence of light and upon photoirradiation at 400 nm for 20 min using (1 × 3.8 W LED). Mean cell viability was obtained from three independent experiments. | |
Subsequently, for phototriggered cell death, MCF-7 cells were incubated with procarriers 2b and 3b, respectively, in two different 96-well plates. One of the plates containing the pro carriers 2b and 3b was photoirradiated at 400 nm (1 × 3.8 W LED, 20 min), and other plates containing the same compounds were not photoirradiated in order to observe the effect of photoirradiation in the cytotoxicity process. The cell plates were then kept in the incubator for 24 h and monitored using the standard MTT assay. Photoirradiation leads to significant cell death, likely due to the formation of the active transporter 1a (Fig. 4B). Cells that do not contain the procarrier molecules do not furnish any cell death, which indicates that cells are not destroyed by light. Furthermore, in order to analyze the effect of in situ generated o-nitrosobenzaldehyde byproducts 10a and 10b, control compounds 9a and 9b were used. These compounds generate the same byproduct upon photoirradiation as is generated by the photoirradiation of procarrier compounds 2b and 3b. Photoirradiation of these control compounds did not furnish any significant cytotoxicity, which rules out any role of the nitrobenzaldehyde byproducts in the cytotoxicity process (Fig. 4C).
Conclusions
In conclusion, we developed benzimidazole amine-based active anionophores 1a–1d and doubly protected ONB-linked photocaged procarriers 2b and 3b, respectively. The chloride binding studies of receptors were confirmed by 1H NMR titration and ESI-MS studies. Among different active compounds, trifluoromethyl-based anionophore 1a showed efficient ion transport activity with an EC50 value of 1.2 ± 0.2 μM. The Hill coefficient was found to be ∼2, which indicates the involvement of two receptor molecules in driving the ion transport process. Detailed mechanistic studies confirmed Cl−/NO3− antiport as the primary ion transport process. The double protection of the active anionophore 1a with ONB-based photocages to give procarriers 2b and 3b renders them inactive due to the blockage of anion-binding sites. These procarriers were photoactivated inside the artificial liposomes using external electromagnetic radiation of 400 nm. Eventually, the photoactivation of the procarriers 2b and 3b inside the MCF-7 cancer cells leads to photoinduced cell death.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI,† and raw data files are available from the corresponding author upon request.
Author contributions
P.T. conceived the project and directed the experimental studies. N. J. R. performed the biological studies. A. S. performed the anion binding studies. M. A. synthesized and characterized the compounds, and performed the ion transport studies. D. M. performed the computational studies. A. M. performed photoirradiation studies. T. V. performed the characterization of some of the compounds. M. A. and P. T. wrote the paper. All authors approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Science and Engineering Research Board (SERB), Govt. of India (EMR/2016/001897, DIA/2018/000017), and Department of Biotechnology (DBT), Govt. of India (BT/HRD/NBA/38/06/2018). M. A. and D. M. thank UGC, Govt. of India for research fellowships. N. J. R. thanks CSIR, Govt. of India for a research fellowship. A. S. thanks DST-Inspire for the fellowship. A. M. thanks IISER Pune and Prime Minister's Research Fellowship.
References
- S. Cheung, D. Wu, H. C. Daly, N. Busschaert, M. Morgunova, J. C. Simpson, D. Scholz, P. A. Gale and D. F. O'Shea, Chem, 2018, 4, 879–895 CAS.
- J. L. Sessler, L. R. Eller, W. S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989–5992 CrossRef CAS PubMed.
- T. Saha, M. S. Hossain, D. Saha, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 7558–7567 CrossRef CAS PubMed.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885 CrossRef CAS PubMed.
- N. Busschaert, S.-H. Park, K.-H. Baek, Y. P. Choi, J. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannidis, I. Marques, V. Félix, W. Namkung, J. L. Sessler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS PubMed.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS PubMed.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- S. J. Moore, C. J. E. Haynes, J. González, J. L. Sutton, S. J. Brooks, M. E. Light, J. Herniman, G. J. Langley, V. Soto-Cerrato, R. Pérez-Tomás, I. Marques, P. J. Costa, V. Félix and P. A. Gale, Chem. Sci., 2013, 4, 103–117 RSC.
- E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS PubMed.
- S. V. Shinde and P. Talukdar, Angew. Chem., Int. Ed., 2017, 56, 4238–4242 CrossRef CAS PubMed.
- L. Lien, D. C. J. Jaikaran, Z. Zhang and G. A. Woolley, J. Am. Chem. Soc., 1996, 118, 12222–12223 CrossRef CAS.
- Y. Kobuke and K. Morita, Inorg. Chim. Acta, 1998, 283, 167–174 CrossRef CAS.
- M. M. Tedesco, B. Ghebremariam, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 1999, 38, 540–543 CrossRef CAS PubMed.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, Chem.–Eur. J., 2005, 11, 6525–6532 CrossRef CAS PubMed.
- Y. R. Choi, B. Lee, J. Park, W. Namkung and K.-S. Jeong, J. Am. Chem. Soc., 2016, 138, 15319–15322 CrossRef CAS PubMed.
- E. B. Park and K.-S. Jeong, Chem. Commun., 2015, 51, 9197–9200 RSC.
- N. Akhtar, N. Pradhan, A. Saha, V. Kumar, O. Biswas, S. Dey, M. Shah, S. Kumar and D. Manna, Chem. Commun., 2019, 55, 8482–8485 RSC.
- A. Docker, T. G. Johnson, H. Kuhn, Z. Zhang and M. J. Langton, J. Am. Chem. Soc., 2023, 145, 2661–2668 CrossRef CAS PubMed.
- M. Fares, X. Wu, D. Ramesh, W. Lewis, P. A. Keller, E. N. W. Howe, R. Pérez-Tomás and P. A. Gale, Angew. Chem., Int. Ed., 2020, 59, 17614–17621 CrossRef CAS PubMed.
- B. Zhou and F. P. Gabbaï, Chem. Sci., 2020, 11, 7495–7500 RSC.
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed.
- J. A. Malla, R. M. Umesh, S. Yousf, S. Mane, S. Sharma, M. Lahiri and P. Talukdar, Angew. Chem., Int. Ed., 2020, 59, 7944–7952 CrossRef CAS PubMed.
- M. R. Banghart, M. Volgraf and D. Trauner, Biochemistry, 2006, 45, 15129–15141 CrossRef CAS PubMed.
- R. F. Khairutdinov and J. K. Hurst, Langmuir, 2004, 20, 1781–1785 CrossRef CAS.
- M. Ahmad, S. Metya, A. Das and P. Talukdar, Chem.–Eur. J., 2020, 26, 8703–8708 CrossRef CAS PubMed.
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC.
- Y. R. Choi, G. C. Kim, H.-G. Jeon, J. Park, W. Namkung and K.-S. Jeong, Chem. Commun., 2014, 50, 15305–15308 RSC.
- M. Ahmad, S. Chattopadhayay, D. Mondal, T. Vijayakanth and P. Talukdar, Org. Lett., 2021, 23, 7319–7324 CrossRef CAS PubMed.
- W.-Z. Wang, L.-B. Huang, S.-P. Zheng, E. Moulin, O. Gavat, M. Barboiu and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 15653–15660 CrossRef CAS PubMed.
- Y. Zhou, Y. Chen, P.-P. Zhu, W. Si, J.-L. Hou and Y. Liu, Chem. Commun., 2017, 53, 3681–3684 RSC.
- J.-Y. Chen and J.-L. Hou, Org. Chem. Front., 2018, 5, 1728–1736 RSC.
- C. Wang, S. Wang, H. Yang, Y. Xiang, X. Wang, C. Bao, L. Zhu, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2021, 60, 14836–14840 CrossRef CAS PubMed.
- M. Ahmad, D. Mondal, N. J. Roy, T. Vijayakanth and P. Talukdar, ChemPhotoChem, 2022, 6, e202200002 CrossRef CAS.
- T. G. Johnson, A. Sadeghi-Kelishadi and M. J. Langton, J. Am. Chem. Soc., 2022, 144, 10455–10461 CrossRef CAS PubMed.
- C. Bao, M. Ma, F. Meng, Q. Lin and L. Zhu, New J. Chem., 2015, 39, 6297–6302 RSC.
- L. E. Bickerton and M. J. Langton, Chem. Sci., 2022, 13, 9531–9536 RSC.
- S. K. Choi, M. Verma, J. Silpe, R. E. Moody, K. Tang, J. J. Hanson and J. R. Baker, Bioorg. Med. Chem., 2012, 20, 1281–1290 CrossRef CAS PubMed.
- W. S. Shin, J. Han, R. Kumar, G. G. Lee, J. L. Sessler, J.-H. Kim and J. S. Kim, Sci. Rep., 2016, 6, 29018 CrossRef CAS PubMed.
- N.-C. Fan, F.-Y. Cheng, J.-a. A. Ho and C.-S. Yeh, Angew. Chem., Int. Ed., 2012, 51, 8806–8810 CrossRef PubMed.
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed.
- C.-C. Peng, M.-J. Zhang, X.-X. Sun, X.-J. Cai, Y. Chen and W.-H. Chen, Org. Biomol. Chem., 2016, 14, 8232–8236 RSC.
- X.-H. Yu, C.-C. Peng, X.-X. Sun and W.-H. Chen, Eur. J. Med. Chem., 2018, 152, 115–125 CrossRef CAS PubMed.
- A. Mondal, J. A. Malla, H. Paithankar, S. Sharma, J. Chugh and P. Talukdar, Org. Lett., 2021, 23, 6131–6136 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 1997, 23, 3–25 CrossRef CAS.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
-
http://app.supramolecular.org/bindfit/, accessed July 2017.
- A. Roy, T. Saha, M. L. Gening, D. V. Titov, A. G. Gerbst, Y. E. Tsvetkov, N. E. Nifantiev and P. Talukdar, Chem.–Eur. J., 2015, 21, 17445–17452 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- X. Wu, E. N. W. Howe and P. A. Gale, Acc. Chem. Res., 2018, 51, 1870–1879 CrossRef CAS PubMed.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS PubMed.
- D. Milano, B. Benedetti, M. Boccalon, A. Brugnara, E. Iengo and P. Tecilla, Chem. Commun., 2014, 50, 9157–9160 RSC.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
-
S. O. H. Goto, N. Nakayama and K. Ohta, CONFLEX 8, CONFLEX Corporation, Tokyo, Japan, 2012 Search PubMed.
- H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950–8951 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- J. A. Malla, R. M. Umesh, A. Vijay, A. Mukherjee, M. Lahiri and P. Talukdar, Chem. Sci., 2020, 11, 2420–2428 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01786a |
| ‡ Present address: Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. |
| § Present address: Université libre de Bruxelles (ULB), Ecole polytechnique de Bruxelles, Engineering Molecular NanoSystems, Avenue Franklin Roosevelt 50, 1050 Brussels, Belgium. |
| ¶ Present address: Faculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Zwirkii Wigury 101, Warsaw 02-089, Poland. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Naveen J.
Roy
a,
Naveen J.
Roy