
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencepH-tunable membrane-active polymers, NCMNP2a-x, and their potential membrane protein applications†
Thi Kim Hoang
Trinh‡
 ab,
Andres Jorge
Cabezas‡
cd,
Soumil
Joshi‡
e,
Claudio
Catalano
ab,
Abu Bakkar
Siddique
ab,
Weihua
Qiu
ab,
Sanket
Deshmukh
e,
Amedee
des Georges
cdf and
Youzhong
Guo
*ab
ab,
Andres Jorge
Cabezas‡
cd,
Soumil
Joshi‡
e,
Claudio
Catalano
ab,
Abu Bakkar
Siddique
ab,
Weihua
Qiu
ab,
Sanket
Deshmukh
e,
Amedee
des Georges
cdf and
Youzhong
Guo
*ab
aDepartment of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23298, USA. E-mail: yguo4@vcu.edu
bInstitute for Structural Biology, Drug Discovery and Development, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA
cStructural Biology Initiative, CUNY Advanced Science Research Center, City University of New York, New York, New York 10017, USA
dPhD Program in Biochemistry, The Graduate Center of the City University of New York, New York, New York 10017, USA
eDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA2 4060, USA
fDepartment of Chemistry & Biochemistry, City College of New York, New York, New York 10017, USA
First published on 9th June 2023
Abstract
Accurate 3D structures of membrane proteins are essential for comprehending their mechanisms of action and designing specific ligands to modulate their activities. However, these structures are still uncommon due to the involvement of detergents in the sample preparation. Recently, membrane-active polymers have emerged as an alternative to detergents, but their incompatibility with low pH and divalent cations has hindered their efficacy. Herein, we describe the design, synthesis, characterization, and application of a new class of pH-tunable membrane-active polymers, NCMNP2a-x. The results demonstrated that NCMNP2a-x could be used for high-resolution single-particle cryo-EM structural analysis of AcrB in various pH conditions and can effectively solubilize BcTSPO with the function preserved. Molecular dynamic simulation is consistent with experimental data that shed great insights into the working mechanism of this class of polymers. These results demonstrated that NCMNP2a-x might have broad applications in membrane protein research.
Introduction
Membrane proteins are involved in numerous vital biological processes, such as chemotaxis in bacteria, photosynthesis in plants, vision, hearing, and heart rhythm in animals, as well as more complex cognitive processes in humans.1,2 Due to their necessity, membrane proteins are also among the primary drug targets on the market.3 Precise three-dimensional atomic structures are in high demand to comprehend the active mechanisms of membrane proteins. Before 2013, most membrane protein 3D structures were determined using X-ray crystallography; nevertheless, the recent resolution revolution in single-particle cryo-transmission electron microscopy (cryo-EM) has garnered tremendous attention. It can circumvent significant limitations of crystallography, such as difficulties in obtaining diffracting membrane protein crystals. Cryo-EM is particularly useful for visualizing the conformation of a target protein in a frozen-hydrated state and requires only a minute amount of protein. Membrane proteins are typically extracted from the cell membrane using detergents for structural analysis. Unfortunately, these detergents can wreak havoc on the natural bilayers of cell membranes, resulting in instability, loss of function, and structural alterations of membrane proteins.4 In cryo-EM studies, they can cause a decrease in water surface tension, which makes it challenging to control ice thickness and protein distribution in cryo-EM grids. Their micelles degrade the background of cryo-EM images.5,6 Therefore, several modern methods (e.g., liposome,7 nanodiscs,8 saposin-derived lipid nanoparticle,9 and amphipol,10etc.) have been applied to reconstitute membrane protein into more stable single particles. Although these strategies improve the success of cryo-EM structural analysis, they necessitate an initial detergent solubilization step prior to reconstitution, which in some cases still faces the same challenges as previously described.Recently, detergent-free techniques, such as styrene–maleic acid lipid particle (SMALP) and native cell membrane nanoparticle (NCMN) systems, have emerged as promising alternatives to detergent-based techniques.11,12 Membrane-active polymers, such as styrene–maleic acid (SMA) copolymers and derivatives, are the most important element of detergent-free systems. In an ideal situation, membrane-active polymers will preserve the native macromolecular complex in nanoparticles, including membrane proteins in their native state and their native lipid bilayer belt surrounding the transmembrane domain. In this scenario, native protein–protein and protein–lipid interactions are preserved, and consequently, the membrane protein retains the same potential functionality as its counterpart on the cell membrane. SMA copolymers are the most prevalent membrane-active polymers capable of advancing structure determination by various techniques, such as solid-state NMR,13 X-ray crystallography,14 and cryo-EM.6 However, they still have significant limitations. SMA copolymers are not always capable of preserving membrane proteins in their native state15 and they are incompatible with divalent cations and low pH conditions.16,17 Moreover, buffer conditions with a pH higher than 7 are necessary for the successful application of SMA copolymers, in which the carboxyl groups of polymer serve as the only water-soluble functional groups.18
Numerous efforts have been made to produce SMA derivatives and other alternative polymers to circumvent these constraints. Four broad strategies have been employed to date: (1) advances in synthetic methods,19,20 (2) diversification of styrene: maleic acid ratio,19,21 (3) modification of the hydrophobic domain, and (4) modification of the hydrophilic groups.22,23 Efforts in substituting hydrophobic styrene units afford several novel backbones (e.g., stilbene–maleic acid,24 diisobutylene–maleic acid,25 and cycloalkane–maleic acid,26etc.). Changing the hydrophilic carboxylic groups is the most common way to improve pH and compatibility with divalent cations. Most attempts rely on the ring-opening of the inactive styrene–maleic anhydride (SMAnh) copolymer by primary amines. Thus, the carboxylic groups are replaced with other chemical groups. Various replacement head groups are available, including but not limited to alcohol,27 amine,22 or phosphobetaine21 can alter the inherent properties of SMA in part or whole. Alternatively, the dehydration method that yields carboxylic-free polymers with positively charged maleimide subunits offers SMA-QA,28 SMA-AA,22 and SMI29 exhibit excellent tolerance towards divalent cations at concentrations over 100 mM. However, they showed a peculiar/unusual pH behavior, where precipitation occurs out of pH 2.5–10 for SMA-QA, pH < 6 for SMA-AA, and pH < 7.8 for SMI. Variations in the intrinsic pKa values of their positive head groups are the underlying cause of the differences. Although several disc-forming polymers have been developed, their use in biophysical characterization and high-resolution cryo-EM structure determination of membrane proteins has never been reported.
The primary objective of this research is to develop NCMN polymers compatible with a wide pH range and suitable for functional studies and high-resolution membrane protein structure determination. We have developed a new class of membrane-active polymers by ring-opening industrial SMAnh with a Brønsted base, tris(hydroxymethyl) aminomethane (Tris). These polymers are designated as NCMNP2a-x, where “x” represents the desired level of Tris grafting. We investigated the modified SMA to determine the effect of Tris content on applicability range (e.g., pH-active range and divalent cation resistance), membrane-active performance (e.g., solubilization efficiency and NCMN particle characteristics), and the effect of neutral side groups on lipid selectivity. The applicability of these novel polymers was evaluated on two membrane protein model systems, the E. coli multidrug efflux pump AcrB and Bacillus cereus TSPO (BcTSPO). Depending on the level of Tris-grafting, the NCMNP2a-x are found to enhance their pH stability slightly or significantly, but they are still susceptible to Ca2+ concentrations of a few millimolar. We determined the high-resolution structure of AcrB and performed enzyme assays of functional BcTSPO under varying pH conditions to test the ability of these novel polymers to stabilize membrane proteins for functional and structural studies. To comprehend the working mechanism of membrane-active polymers, a coarse-grained (CG) molecular dynamics (MD) simulation was conducted to investigate the polymer–lipid bilayer interactions. Having proved that the MARTINI CG force field (FF)30,31 is useful for SMA copolymers with styrene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :maleic anhydride = 2:1,32,33 it was applied in this study. We are convinced that NCMNP2a-x membrane-active polymers could serve as new substitutes for SMA copolymers and may be appropriate for functional and structural studies of various membrane proteins when pH conditions must be altered.
:maleic anhydride = 2:1,32,33 it was applied in this study. We are convinced that NCMNP2a-x membrane-active polymers could serve as new substitutes for SMA copolymers and may be appropriate for functional and structural studies of various membrane proteins when pH conditions must be altered.
Results and discussion
Synthesis and characterization of NCMNP2a-x
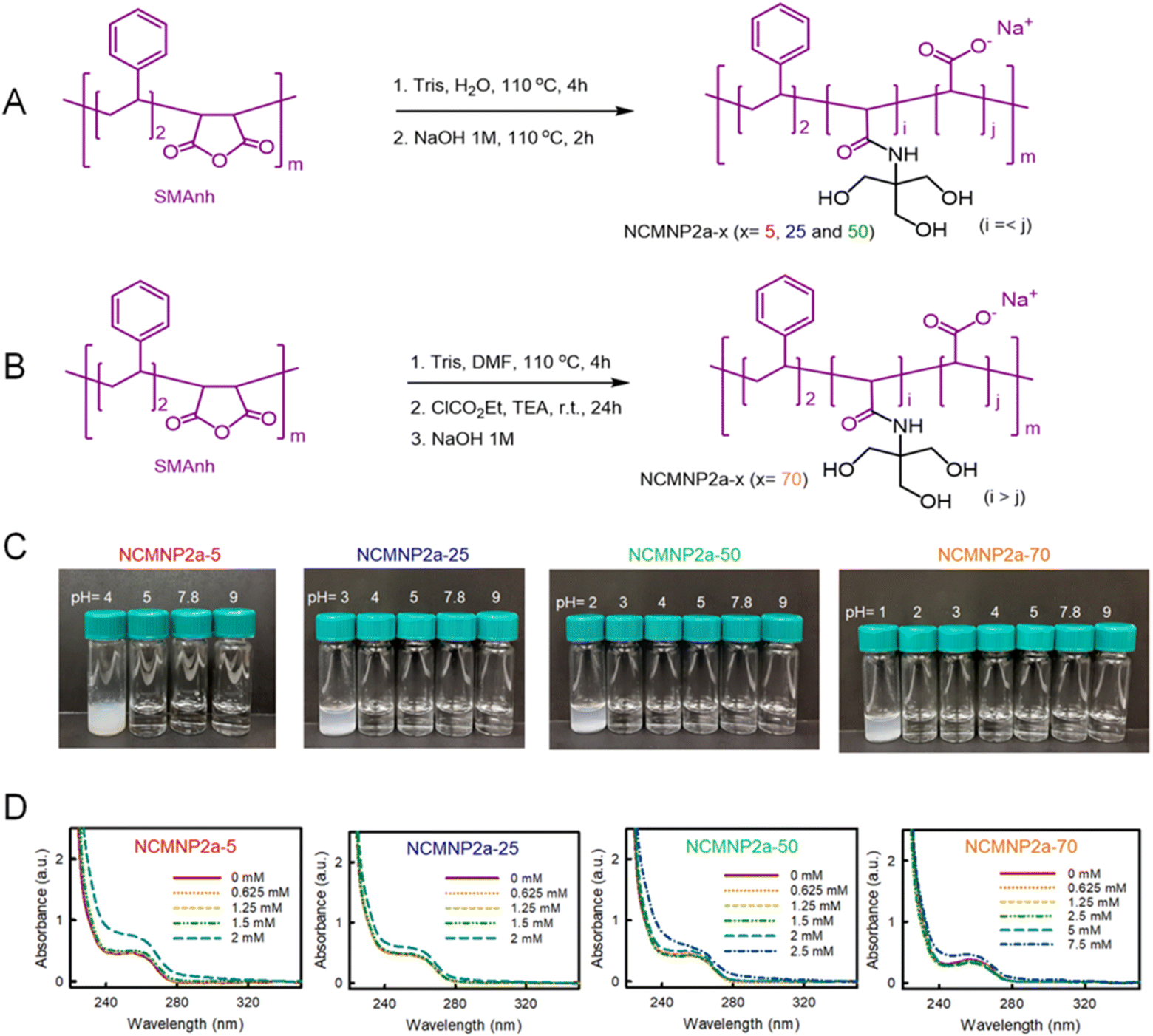 | ||
| Fig. 1 Synthesis routine and physical characterizations of NCMNP2a-x polymers. (A) General schematic reaction for producing low Tris-grafting polymers (x = 5, 25 and 50). (B) General schematic reaction for producing high Tris-grafting polymer (x = 70). (C) Influence of Tris incorporation on the solubility of NCMNP2a-x (0.05% w/v) in the standard buffers containing 100 mM NaCl at various pH values. (D) A comparison of various NCMNP2a-x (0.05% w/v) in response to increasing quantities of divalent cation Ca2+. Absorbance increase corresponds to polymer aggregates because of inter- and intermolecular binding of polymer chains to Ca2+. | ||
In support of the 1H NMR characterization, a quantitative measurement has been performed to validate the grafting of Tris using FTIR (Fig. S1B and C†). The peak for vibrational C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of amide obtained by the reaction of Tris with maleic anhydride and CO bond of unreacted carboxylic acid were identified at 1645.1 cm−1 and 1697.2 cm−1. However, because of their proximity it was hard to quantify by normal analysis. To date several approaches has been explored such as absorbance, integral area and the occupied volume to quantify the functionality using FTIR data.34–36 These days determination of peak integral area by deconvolution curve fitting has become popular because it offers detail qualitative and quantitative analysis of highly overlap or hidden absorbance peak which inspired the spectroscopic analysis of complex protein structure.35,37,38 The spectra were deconvoluted considering two target Gaussian center 1645.1 cm−1 and 1697.2 cm−1 for amide and carboxylic CO bond. The deconvolution gives the occupied area of respective peaks. The grafting ratio was then calculated using this integrated area. We observed an incremental order for grafting functionalities as 24.46%, 50.33%, and 61.999% for NCMNP2a-25, NCMNP2a-50 and NCMNP2a-70 respectively which supports methods and 1H NMR data analysis (Table S10†). However, the amide signal for NCMNP2a-5 inclined significantly and converged to the next broad peak which made it difficult to fit for functionality calculation.
O bond of amide obtained by the reaction of Tris with maleic anhydride and CO bond of unreacted carboxylic acid were identified at 1645.1 cm−1 and 1697.2 cm−1. However, because of their proximity it was hard to quantify by normal analysis. To date several approaches has been explored such as absorbance, integral area and the occupied volume to quantify the functionality using FTIR data.34–36 These days determination of peak integral area by deconvolution curve fitting has become popular because it offers detail qualitative and quantitative analysis of highly overlap or hidden absorbance peak which inspired the spectroscopic analysis of complex protein structure.35,37,38 The spectra were deconvoluted considering two target Gaussian center 1645.1 cm−1 and 1697.2 cm−1 for amide and carboxylic CO bond. The deconvolution gives the occupied area of respective peaks. The grafting ratio was then calculated using this integrated area. We observed an incremental order for grafting functionalities as 24.46%, 50.33%, and 61.999% for NCMNP2a-25, NCMNP2a-50 and NCMNP2a-70 respectively which supports methods and 1H NMR data analysis (Table S10†). However, the amide signal for NCMNP2a-5 inclined significantly and converged to the next broad peak which made it difficult to fit for functionality calculation.
Applications in membrane protein studies
The previous results demonstrate that their physical properties have improved. Consequently, this section examines whether our modified polymers can directly assemble NCMN particles from the native membrane for structural and functional studies. | ||
| Fig. 2 Effect of Tris-grafting degree on membrane protein solubilization and size of AcrB–NCMN particles therefrom. (A) The typical procedure applied in the solubilization and purification of membrane proteins in this work. (B) Ni–NTA affinity chromatography profiles of purified AcrB in different NCMN polymers. (C) Analysis of AcrB–NCMN particles on SDS-PAGE along with protein marker (M). Lanes 1, 2, 3, and 4 correspond to purified AcrB extracted by NCMNP2a-5, NCMNP2a-25, NCMNP2a-50, and NCMNP2a-70, respectively. (D) Negative stain analysis of AcrB particles extracted by NCMNP2a-x polymers (x = 5, 25, 50, and 70). The bottom-left scale bar represents 50 nm. (E) Compositions of lipid species extracted from membrane fraction and purified AcrB determined by ESI-MS. Abbreviations included are cardiolipin (CL), cholesterol ester (ChE), coenzyme (Co), diglyceride (DG), phosphatidylcholines (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylserine (PS), sphingomyelin (SPH) and triglyceride (TG). | ||
All NCMNP2a-x solubilized cell membrane samples were purified using the His-tag to eliminate other soluble fractions. The chromatograph of supernatants proceeded with the low Tris-grafting polymers, displaying single and sharp peaks at ∼5 mL (Fig. 2B), demonstrating that the NCMN particles are present in eluent fractions. In contrast, no elution peak was observed after treatment with NCMNP2a-70, suggesting that no or very few NCMN particles were obtained. The collected peak fractions were additionally characterized by SDS-PAGE gel to detect contaminants, which revealed a single band with a molecular mass of approximately 110 kDa (Fig. 2C, lanes 1–3). This corresponds to the monomeric state of AcrB protein,50 and demonstrates the success in the purification.
In contrast, the yields of NCMN particles decreased in inverse proportion to the number of reduced carboxylic acid groups on NCMNP2a-x side chains. NCMNP2a-5, which retained the most carboxylic acid side groups, produced the highest yield of NCMN particles, highly comparable to SMA2000 (Fig. S4A†), highlighting the significance of carboxylic acid in solubilization efficiency. Scheidelaar et al. proposed that membrane solubilization's “insight mechanism” for non-modified SMA51 primarily occurs via the hydrophobic effect, wherein planar phenyl rings can insert between lipid acyl chains. Consequently, pendant carboxylic acid groups may also contribute to this effect. Due to their chemical nature, these acid groups can modulate the electrostatic interaction between polymer chains and lipid bilayers. The small size of the hydrophilic moieties provides a small cross-sectional area suitable for deeply penetrating the polymer into membranes, permitting the passage of solutes with a low molecular mass. Compared to SMA, NCMNP2a-x has uncharged and larger side groups, which eliminates electrostatic interaction and results in a larger cross-sectional area. Because of these chemical properties, the extraction efficiency of NCMNP2a-x is reduced.
We performed additional solubilization experiments with NCMNP2a-70 to confirm the formation of protein–lipid patches. Considering the presence of high molar mass fractions, the supernatant was collected using a centrifuge force of 64000×g instead of 200000×g as in previous tests. Predictably, A higher level of protein was obtained. The detectable chromatographic peak in the elution profile shifted to a lower retention volume than before. Our experience suggests that the cell membrane has been solubilized into larger NCMN particles that may not interact strongly with the Ni–NTA resin. (Fig. 2B and C (lane 4) and S4B†). The elution fraction displayed multiple bands on the SDS-PAGE gel, including the AcrB monomer. Therefore, it was assumed that these larger nanodiscs contained few AcrB molecules, with the majority of their mass coming from a significant contribution of surrounding lipids and other molecules with low molar mass.
In contrast, the smaller and more homogeneous AcrB–NCMN particles should be ideal for resolving 3D structures with high resolution using single-particle cryo-EM.
However, the composition of the associated lipids in SMALPs differs from that of native membranes depending on membrane proteins and regions of cell membranes. As isolated by SMA, the major lipid components of S. cerevisiae mitochondria, where CytcO was expressed, did not change.59 In contrast, Ayub et al. reported that CD81-SMALP isolated from Drosophila S2 appeared to be enriched in negatively charged lipids.57 The disparity in lipid content is primarily the result of interactions between membrane proteins and lipids that surround them.56 To identify potential lipid component alterations, we compared the extracted lipids of AcrB–NCMN particles derived from NCMNP2a-50 and the E. coli cell membrane. As depicted in Fig. 2E, the overall lipid profiles identified were comparably similar. Cholesterol ester (ChE), diglyceride (DG), and triglyceride (TG) concentrations exhibited the most notable variations (TG). We used a similar lipid extraction procedure,58 but identified more lipid species instead of detecting only phosphatidylglycerol (PG). Phosphatidylethanolamine (PE), phosphoglycerol (PG), and cardiolipin (CL) were the major lipid components in both cases, which is consistent with the lipid components previously identified in the inner membrane of E. coli (55% PE, 15% PG, and 5% CL).60,61 These promising findings suggest that our NCMN particles are capable of resolving the AcrB structure in a nearly native state.
None of our refined structures caused notable changes in the internal structure compared to the SMA2000-derived structure and each other (root-mean-square deviation values in a range of 0.425–0.897 Å, see Table S4†). The SMA2000 structure, however, lacked one-helix (Iα), whereas all 36 transmembrane helices (TMs) were identified in our three structures (Fig. 3A and S6†). It is tempting to hypothesize that, in comparison to SMA2000, those unique polymers better retain native lipids and, as a result, better stabilize the structure of the more flexible regions, like the outer-helix (Iα).64 Despite varying Tris concentrations, all NCMN polymers retain endogenous lipids within the lipid cavity of AcrB (Fig. 3B). Liposomes and CyclAPols, on the other hand, could not achieve this.7,63 In contrast to outcomes reported with detergent systems, the tight packing of these lipids in the inner leaflet can prevent the collapse of TM domains (Fig. S7†) and support efflux activity.65 According to the aforementioned lipid analysis, it shows that the new hydroxyl groups may preserve native lipids as well as SMA2000, or even better, making them appropriate for cryo-EM research.
 | ||
| Fig. 3 Cryo-EM structures of AcrB–NCMN particles. (A) Ribbon diagrams of the entire AcrB complex warped by different polymers at various pH. These structures were obtained through manual fitting of the 6BAJ structure into the maps using Coot software. (B) Expanding views of blue box regions featuring the lipid bilayer located in the central cavity. The lipid bilayer was confidentially assigned with good quality of the EM map. (C) Configuration of protomer L of AcrB in different NCMN polymers. Left panel: NCMNP2a-5, pH = 7.8, middle panel: NCMNP2a-50, pH = 7.8 and right panel: NCMNP2a-50, pH = 5. (D) Configuration of protomer T of AcrB in different NCMN polymers. Left panel: NCMNP2a-5, pH = 7.8, middle panel: NCMNP2a-50, pH = 7.8 and right panel: NCMNP2a-50, pH = 5. (E) Configuration of protomer O of AcrB in different NCMN polymers with dotted lines (yellow) depicting the possible hydrogen bonds between residues. Left panel: NCMNP2a-5, pH = 7.8, middle panel: NCMNP2a-50, pH = 7.8 and right panel: NCMNP2a-50, pH = 5. | ||
Three subunits comprise the asymmetric trimer-like AcrB structures, and each subunit has a unique conformation corresponding to one of the three substrate/proton transportation phases (L state, T state, and O state).62 As previously documented, specific residues such as D407, D408, K940, N941, and T978 are important components of the proton-relay network that undergoes rearrangements triggered by D408 protonation/deprotonation.66,67 Although this conformational change is a protonation-driven mechanism, no experiment has yet demonstrated the effect of solution pH on it. In this study, the enhanced pH resistance of NCMNP2a-50 enables us to investigate their potential structural change at pH = 5. However, the residue-level structural analysis revealed no conformational changes in the proposed proton transport mechanism (Fig. 3C–E). K940 continues to form stable salt bridges with either D407 or D408 in protomers L and T, and it transfers from D408 to T978 in protomer O. In the latter instance, K940 maintains its ionic connection with D407, which is sustained by the formation of new hydrogen bonds between K940 and T978, and N941. At such an acidic pH, the protonation of D407 and D408, whose pKas are lower than 5 by at least one pH unit, is assumed to be challenging.66,67 These are consistent with previous structures (2HRT,686BAJ62) and the computational work of Yue et al.,66 where the significant pH-dependent shift relative to basic pH was only found at pH values less than 5.
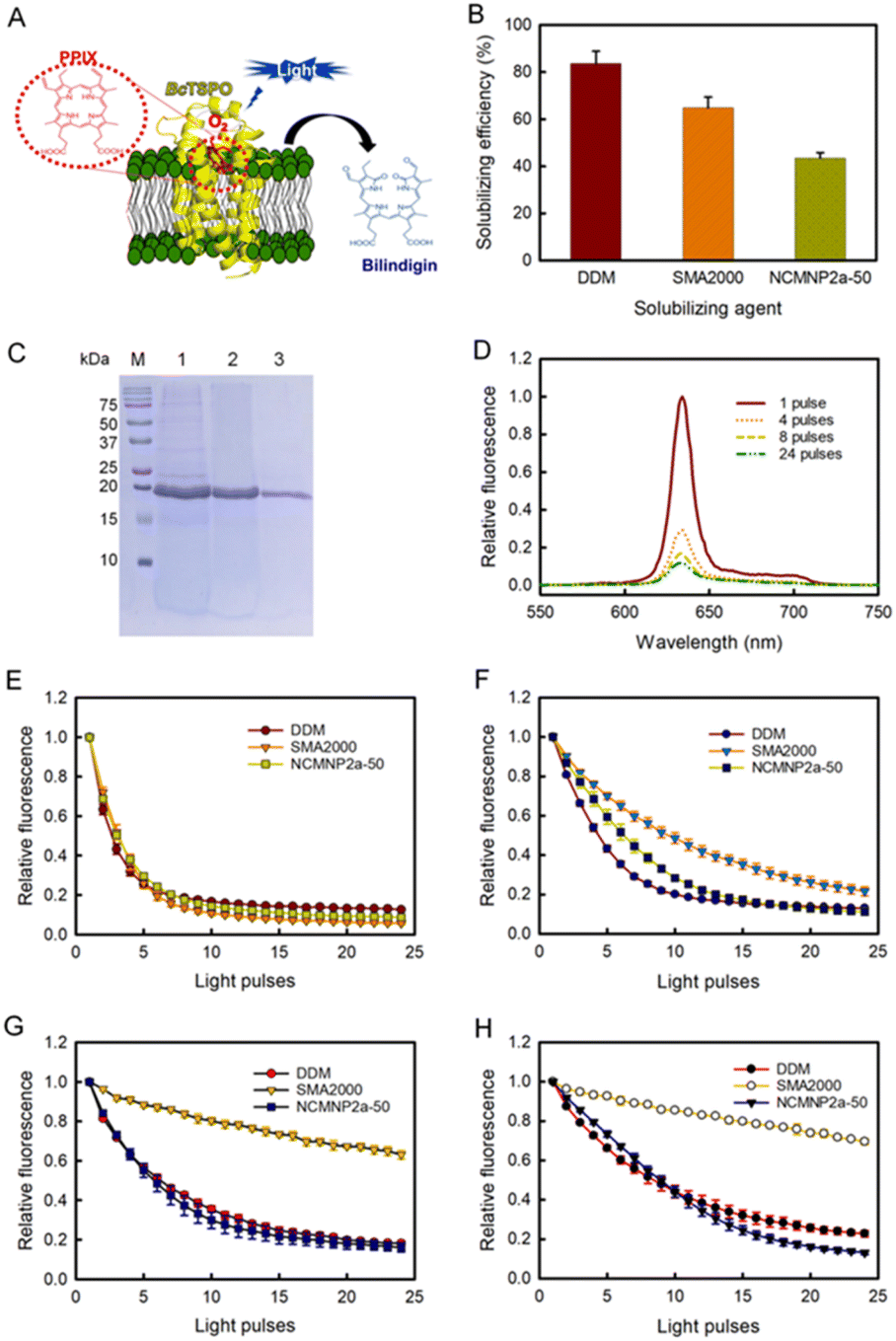 | ||
| Fig. 4 Comparative effectiveness of differing solubilizing agents toward BcTSPO solubilization and catalytic activity of corresponding BcTSPO therefrom in the photodegradation of PpIX. (A) Schematic BcTSPO induced photodegradation of PpIX into bilindigin. (B) Solubilization effectiveness of recombinant protein BcTPSO in DDM, SMA2000 and NCMNP2a-50. (C) Visualization of purified BcTSPO particles on Coomassie-stained SDS-PAGE compared to protein ladder (M). Lane 1, 2 and 3 correspond to BcTSPO particles extracted by DDM, SMA2000 and NCMNP2a-50, respectively. (D) Representative fluorescent emission spectra of PpIX associated with BcTSPO-NCMN particles upon excitation at 410 nm. (E)–(H) Photodegradation of PpIX in the presence of BcTSPO at various pH conditions. Decays of PpIX at 632 nm as a function of the light pulse at pH = 7.8 (E). Decays of PpIX at 632 nm as a function of the light pulse at pH = 5 (F). Decays of PpIX at 632 nm as a function of the light pulse at pH = 4 (G). Decays of PpIX at 632 nm as a function of the light pulse at pH = 3 (H). | ||
For the enzyme activity assay, all investigations utilized an established methodology based on the fluorescence decay of PpIX.71 In the absence of BcTSPO, exposure of PpIX to light with a wavelength of 405 nm did not result in PpIX degradation (Fig. S9A†). Significant fluorescence degradation was seen when PpIX was exposed to 405 nm wavelength light in the presence of BcTSPO–NCMN particles (Fig. 4D). This indicates that BcTSPO–NCMN particles have catalytic properties. At pH = 7.8, the intrinsic tryptophan fluorescence quenching profiles of BcTSPO treated with NCMNP2a-5 and NCMNP2a-50 were remarkably similar to those of detergent-purified BcTSPO and SMA2000-purified BcTPSO samples (Fig. S10† and 4E). This demonstrates that even with additional functional side chains, there may only be a polymer–lipid interaction and no protein binding, keeping the protein intact and functional.72,73 These findings indicate that BcTSPO remained active, independent of the amount of Tris-grafting.
Next, we incubated BcTSPO particles solubilized by DDM, SMA2000, and NCMNP2a-50 with PpIX at pH = 5, 4, and 3 to determine the effects of acidic pH on the degradation of PpIX catalyzed by BcTSPO. All resulted in the degradation of PpIX upon excitation (Fig. 4F–H), with fluorescence decay rates depending on the individual BcTSPO construct and the acidity of the reactive system. BcTSPO particles derived from DDM and NCMNP2a-50 degrade at similar rates at the same pH settings but significantly more slowly than at pH = 7.8. In contrast, the activity of SMA2000-purified BcTSPO is dramatically reduced at pH = 4 and pH = 3, while it is still effective at pH = 5. The latter observation may be related to the aggregation of BcTPSO particles produced by a long-standing problem with SMA2000 belts, in which their carboxylic side groups undergo protonation at pH 5, resulting in electrostatic interaction between protein particles. Consequently, the creation of protein clusters likely hinders PpIX from associating with the active side of BcTPSO.
At an acidic pH, the partial aggregation of PpIX (Fig. S9B†) and its protonation state reduce the catalytic activity.74,75 Low pH could result in protonation of the propionic moiety on PpIX, a decrease in the solubility of PpIX in the reaction system, and a significant drop in its binding affinity to receptors. In conclusion, these results demonstrate the pH sensitivity of protoporphyrin IX breakdown by BcTSPO. In addition, using NCMNP2a-50 with improved pH compatibility can circumvent the limitations of the present SMA2000 when speculating about the functional pH dependence of possible membrane proteins.
 | ||
| Fig. 5 Averaged RDF between (A) polymer–water (inset: zoomed-in first peak), (B) polymer–polymer, and (C) polymer–lipids (inset: zoomed-in first peak). (D) Simulation snapshots for all NCMNP2a-x systems studied. (E) Schematic showing the different angles calculated, (F) values for mean and standard deviations of angle 1 calculated for all monomers in the polymer chain, (G) values for mean and standard deviations of angle 2 calculated for all monomers in the polymer chain. | ||
To understand the chemical basis of the experimentally observed behavior of nanoparticle formation, we performed CG MD simulations of the self-assembly of lipid–polymer nanoparticles. All simulations resulted in the formation of well-defined nanoparticles as shown in Fig. 5D, with the average diameters of most self-assembled nanoparticles between 8 and 10 nm, which agrees with our experimentally observed values as well as published literature.32,33 At higher percentages of Tris substitution, nanoparticle interactions were found to be stronger with separate but connected nanoparticles observed in the case of NCMNP2a-50 and a solitary coalesced nanoparticle observed in the case of NCMNP2a-70. This was also reflected in the polymer–lipid RDFs (Fig. 5C) and was consistent with the experimentally observed behavior of high Tris-substituted polymer nanoparticles. To investigate the role of bulkier Tris groups in nanoparticle aggregation, we analyzed the angles formed by styrene and backbone beads across the maleic acid groups (Fig. 5E). As seen in Fig. 5F and G, The average values of the observed angles increased as the amount of Tris substitution increased. The standard deviation values also indicated that these angles fluctuated more in the presence of increased Tris groups, providing the polymer chains greater conformational flexibility. Furthermore, calculating the solvent-accessible surface area (SASA) values for all styrene groups in the polymer chains showed an increase in styrene accessibility with an increase in Tris groups, which can be attributed to the conformational flexibility of these highly-substituted polymer chains. This increase in SASA, and thus, the accessibility of styrene, may result in increased lipid–polymer interactions in NCMNP2a-70, resulting in an aggregated mass opposite to individual nanoparticles in cases of lower Tris substitution.
Conclusions
We have designed, synthesized, and characterized a new class of membrane-active polymers, NCMNP2a-x. We demonstrated that this new class of polymers is suitable for high-resolution structure determination of the model membrane protein, AcrB, at different pH conditions. We also showed that NCMNP2a-x could be used to analyze the enzyme activity of BcTSPO at different pH conditions. Variation of the grafting ratio of Tris on the SMA2000 backbone determines the pH sensitivity of the NCMNP2a-x polymers and also governs the cell membrane solubilization efficiency and subsequent size of NCMN particles. When solubilizing the cell membrane using NCMNP2a-70, very few AcrB–NCMN particles were obtained, and the size of such particles was much larger than that obtained using NCMNP2a-5 and NCMNP2a-50. Non-targeted membrane proteins were also encased as contaminants in such lipid bilayer patches. However, NCMNP2a-70 may be particularly useful when studying large membrane protein complexes. For example, these large and enriched cell membrane patches may be suitable for cryo-EM tomography analysis.In contrast, the low Tris-grafted NCMNP2a-x polymers (≤50% of Tris) favored the solubilization of AcrB and its endogenous lipids into single particles suitable for single-particle cryo-EM analysis and can produce atomic-level membrane protein structures. Notably, Iα helix (TM) residues at the outer of helical bundles were fully resolved using NCMNP2a-x. In contrast, one of the Iα helix (TM) within the three subunits gave inferior EM density when using SMA2000. The lipid bilayer plugs were well preserved in all three cryo-EM structures at different pH conditions.
Molecular dynamic simulation analyses, including conformations of single polymer chains in water, interactions between multiple polymer chains in water and their self-organization into nanoparticles, and the formation of NCMN particles through polymer–lipid bilayer interaction, faithfully mimic the behaviors of various polymer systems as they are observed in experiments. This gives us a wealth of knowledge on the mechanics behind the interactions between polymers and lipid bilayers and paves the path for the future computer-aided rational design of membrane-active polymers that will enlarge the NCMN polymer library.
NCMNP2a-x are a new class of pH-tunable membrane-active polymers that retain native membrane protein function and are suitable for high-resolution structure determination with single-particle cryo-EM. Therefore, they may have a wide range of applications in future membrane protein research.
Methods
Materials
Tris[hydroxymethyl]aminomethane (Tris), sodium dodecyl sulfate (SDS), ammonium persulfate (APS), 30% acrylamide/bisacrylamide solution, 37.5:1 (2.7% crosslinker), N,N,N′,N′-tetramethylethylenediamine (TEMED) and tris(2-carboxyethyl)phosphine (TCEP) were purchased from Bio-Rad. On the other hand, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES, ≥99%), glycerol (≥99.5%), nickel(II) sulfate hexahydrate (NiSO4, >98%), imidazole (≥99%), hydrochloric acid (HCl, 36.5–38%), acetic acid (glacial, ≥99.7% w/w), methanol (MeOH, 99.8%), phenolphthalein (≥100% w/v), chloroform (≥99.8%, stabilized by ethanol) and sodium chloride (NaCl, ≥99%) with BioReagent or ACS grade were received from Fisher Chemical. DL-Dithiothreitol (DTT, ≥98% (HPLC), ≥99% (titration)), sodium hydroxide (pellets, ≥98%, reagent grade), sodium acetate (anhydrous, ≥99%, for molecular biology), protoporphyrin IX (≥95%) and triethylamine (TEA, ≥99%) were obtained from Sigma-Aldrich. n-Dodecyl-β-D-maltoside (DDM, Anagrade) was received from Anatrace. Ethyl chloroformate (ClO2Et, 99%), deuterium oxide (D2O, 99.9 atom% D), and N,N-dimethylformamide (DMF, anhydrous, 99.8%) were ordered from Acros that were stored in a dark brown glass bottle with a self-sealing septum. Poly(styrene-co-maleic anhydride) with a 2:1 ratio of styrene to MAnh (SMAnh, acid number: 355 mg KOH per g) were sold by Cray Valley. Before preparing different buffer solutions, water was double-deionized by a Millipore Milli-Q system to produce 18 MΩ deionized water (DI H2O).
Synthesis of NCMN polymers
Structural characterization of NCMNP2a-x
Stability characterization of NCMNP2a-x
Changes in absorbance in the range of 220–350 nm of all above samples were monitored by a NanoDrop™ 2000 spectrophotometer. Each of the experiments was repeated three times.
Expression, solubilization and purification of AcrB and BcTSPO
The expression and cell lysis of AcrB and BcTSPO membrane were carried out based on our previous protocol.71,76 Following a typical solubilization procedure, 1 g membrane fraction was suspended in 10 mL NCMN Buffer A and then homogenized by using a Dounce homogenizer. Consequently, the suspended membrane was transferred to a 50 mL polypropylene tube and mixed with NCMNP2a-x for a final concentration of 2.5% w/v. After shaking the sample for 2 h at 20 °C, the insoluble species were centrifuged at 64000×g (for NCMNP2a-70 media) and 200000×g (for all other media) for 1 h 20 °C. The collected supernatant was loaded onto a 5 mL Ni–NTA column (GE Healthcare) pre-equilibrated with NCMN Buffer A at a flow rate of 0.5 ml ml−1. Then, the column was washed with 30 mL of NCMN Buffer B and 30 mL of NCMN Buffer C before the protein was eluted with a mixture buffer of the NCMN Buffer C and NCMN Buffer D (1:1 v/v).
All the buffers were filtered with 0.22 μm MCE Membrane (MF-MilliporeTM) before use and their compositions are listed below:
- NCMN Buffer A: 50 mM HEPES, pH 8.4, 500 mM NaCl, 5% glycerol, 20 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer B: 25 mM HEPES, pH 7.8, 500 mM NaCl, 40 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer C: 25 mM HEPES, pH 7.8, 500 mM NaCl, 75 mM imidazole, 0.1 mM TCEP.
- NCMN Buffer D: 25 mM HEPES, pH 7.8, 500 mM NaCl, 500 mM imidazole, 0.1 mM TCEP.
AcrB–NCMN particles characterizations
000× magnification on a 4k × 4k CCD camera.
000×g for 10 min, the organic layer was rinsed three times with 2 volumes of cold DI H2O. Afterward, the organic phase was characterized by electrospray ionization mass spectrometry (ESI-MS). Another lipid extraction was performed with a native cell membrane using a similar method to compare lipid composition.
000 with a data-dependent MS2 triggered for any analyte reaching 3e6 or above signal. Data-dependent MS2 was collected at 30000 resolutions. Data were analyzed using Thermo Scientific's Lipid Search 4.2 software.
Activity assay of BcTSPO
Before performing the activity assays, different reaction solutions were prepared following the procedures below:• PpIX stock solution was prepared as follows: adding 1 μL of saturated PpIX in DMSO to 199 μL of Buffer E (pH 7.8) and then taking 50 μL of this mixture and diluting 4× with Buffer E (pH 7.8).
• Buffer E contains 40 mM HEPES, 100 mM NaCl, 0.1 mM TCEP, and 0.05% w/v DDM. The pH value was adjusted to 7.8, 5, 4, or 3, depending on the target pH of the experiments. The solution was filtered with 0.22 μm MCE membrane before use.
The enzyme activity tests were conducted following our previous protocol with some modifications.71 Typically, 100 μL of BcTSPO ([BcTSPO] ∼ 1 mg ml−1) was mixed with 129 μL of Buffer E (pH 7.8, pH 5, pH 4 or pH 3) and 1 μL of PpIX stock solution. This media was transferred into a 5 mm square quartz cuvette and then incubated in the dark for 5 min. The reaction was initiated by opening the shutters and exciting the mixture at 405 nm. The fluorescence emission was monitored from 550 to 750 nm.
All experiments were conducted on a Shimadzu RF-53301PC fluorescence spectrometer. The samples were illuminated at 405 nm with excitation bandwidth at 1.5 nm. On the other hand, the emission spectra were recorded with emission bandwidth at 3 nm.
Determination of repeating units
In general, SMAnh's chain end contains cumene because, as known, it was industrially synthesized via radical polymerization with dicumyl peroxide as a thermal initiator. Unfortunately, the methyl protons (Hb and Hi) signals at δ = 1.25–1.27 ppm, which could be used as reference peaks to characterize the structure of SMAnh, were disturbed by other signals. Therefore, we used other data such as molecular weight (approximately 3 kDa) and St:MAnh (2:1) of SMAnh to determine the repeating units, x, and m. By combining them, a set of the following equations is obtained:
Molecular weight:
| Mn = MCH3 + (MCH2 + MCH + MC6H5)x + MC4H2O3m + MC6H5 + 2MCH3 |
| 3000 = 15.03 + (14.03 + 13.02 + 77.10)x + 98.06m + 77.10 + 30.06 |
| 2877.81 = 104.15x + 98.06m | (1) |
| Relationship of repeating unit: x = 2m | (2) |
| From (1) and (2): x = 18.6 and m = 9.3 |
That gives x = 18.6 and m = 9.3, respectively. Secondly, to validate accuracy, we recheck the calculation by integrating chemical shifts in the 1H-NMR spectrum of SMAnh. However, it is impossible due to an overlap of aromatic protons (δ = 5.8–8.0 ppm) from St and protons from solvent (δ = 7.26 ppm). Take note that, as ignoring –COOH protons, the SMA has the same number of aromatic and aliphatic protons as its mother, SMAnh. Besides, the chemical shift of D2O solvent at δ = 4.79 ppm does not perturb any signals of SMA. Thus, SMA was selected to testify the calculation instead of SMAnh. With x = 18.6, the total aromatic protons were set at 98 protons, including 93 protons from St in repeating units and 5 protons from cumene chain end. As a result, the total aliphatic protons observed were 81.02 protons (3IHb + (2IHc + IHd) × x + IHe × y + IHg × z + 6IHi = 81.02). Given that relationship between y and z is y + z = 16.2. Because SMA is obtained from the hydrolysis of SMAnh, the m value, in this case, is equivalent to y = z = (y + z)/2 = 8.1, which is relatively lower than the initial calculation. However, when applying these new values (x = 18.6 and m = 8.1) to recalculate Mn, it was found that Mn ∼ 2900 g mol−1 is highly comparable to SEC data documented by the provider. On the other hand, setting the number of aliphatic protons of SMAnh to 81.02 protons gives 100.21 protons in the range of 5.7–8.5 ppm. That probably combines 98 aromatic protons from SMAnh and ∼2 protons solvent. These results are relevant and consistent. Thereby, x = 18.6 and y + z = 16.2 were used as references to estimate the number of protons of structures shown in Fig. S2† and the degree of carboxylic acid modified by Tris in a later section.
Determination of Tris grafting degree
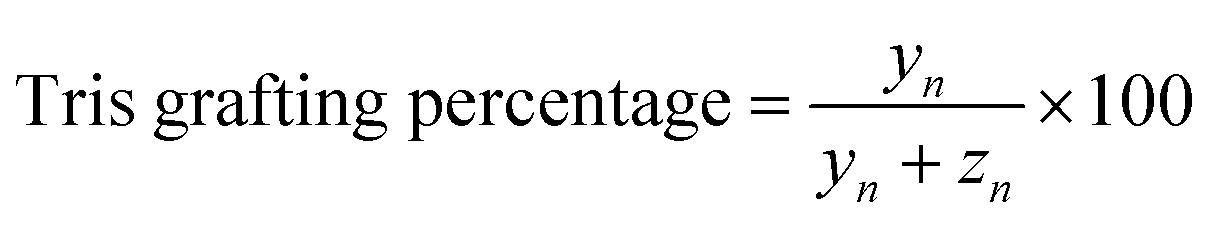 | (3) |
Repeating units:
| xn = 18.6 | (4) |
| yn + zn = 16.2 | (5) |
Total integration of aliphatic protons:
| 3IHb + (2IHc + IHd) × xn + (IHe + 6IHf) × yn + IHg × zn + 6IHi = ∑aliphatic protons |
| 3xn + 7yn + zn = ∑aliphatic protons − 9 | (6) |
From (3)–(6), we calculated the Tris degrees of NCMNP2a-x and data are summarized in Table S1.†
Molecular dynamic simulation
:1 ratio. Here, the COO− group was modeled using the negatively charged SQ5n bead to appropriately mimic lower pH conditions. The residue SMT consisted of a single COO− (SQ5n bead) replaced with the Tris group while the residue TRI consisted of both COO− beads replaced with Tris groups. As COO− groups were replaced with the larger Tris groups, the total charge on the residues decreased as these substituted groups could not dissociate in low pH conditions. Thus, these residues differed significantly in their bulkiness as well as total charge in lower pH conditions, replicating experimentally. Using different arrangements of these building residues, polymers with 0%, 5%, 25%, 50%, and 70% Tris substitution were generated, representing the experimentally studied NCMNP2a-x systems as shown in Fig. S12A.† Polymer chains were created with 23 repeating units to maintain coherence with the aforementioned CG MD studies on SMALP formation and are shown in Fig. S12B.‡32,33
In the case of the single and multi-polymer simulations, polymer chains representing NCMNP2a-x systems were randomly dispersed in a box of water along with neutralizing sodium ions and water beads. Polymer chains with varying initial configurations and arrangement of residues were simulated to incorporate a factor of disorder that may be observed in experiments. In order to study the self-assembly of lipids and polymers into nanoparticles, systems containing 6, 12, and 18 polymer chains were created with fixed numbers of lipids, randomly dispersed into a cubic box with size 25 nm × 25 nm × 25 nm, which was solvated with ∼40000 water beads (∼164000 all-atom water molecules). To appropriately reproduce the distribution of lipid components as observed in the experimentally isolated nanoparticles, we used PE (75%) and PG (25%) lipids in our CG MD simulations. Particularly, the smaller 1,2-dilauroyl-sn-glycero-3-phosphoethanolamine (DLPE) (580 molecules) and 1,2-dilauroyl-sn-glycero-3-phosphoglycerol (DLPG) (200 molecules) were utilized to facilitate faster self-assembly.32 Sodium ions were added for system neutralization. A representative snapshot of the system can be found in Fig. S12C.† Furthermore, uncharged simulations were also carried out for all three cases mentioned below to ascertain the impact charged beads had on polymer conformations, interactions, and nanoparticle formation.
Table S5† provides detailed information regarding all the systems and simulation run times. Simulations were performed for over 2 μs at 310 K with the NPT ensemble using most of the hyperparameters defined in the aforementioned studies. The NAMD package was used for this purpose and the system was considered to be periodic in all directions.79 A timestep of 20 fs was used with a cutoff of 12 Å and dielectric value of 15, consistent with the MARTINI 3.0 FF. Polymer chains were visualized using the VMD package and analyzed for their general conformations using Radius of Gyration (Rg) calculations as well as their structural correlations with other system components from Radial Distribution Functions (RDFs).80 All analysis was carried out using CPPTRAJ and in-house TCL and Python scripts for the last 1 μs of the simulation after the systems were observed to have been equilibrated.81
Next, we analyzed the structure of the solvent around the different polymer chains by plotting several RDFs between polymer components and water. To identify the origin of this behavior, we further analyzed the RDFs between water and the important individual components of every polymer chain as presented in Fig. S8.† It was observed that, while all individual components of the polymer showed dehydration as the percentage of Tris in the polymers increased, the difference was particularly large for the dehydration of the backbone (TC1 beads). The TC1 beads attached to charged COO− groups showed significantly taller peaks when these beads were present in high numbers resulting in an ordered structure of water around them. However, as the percentage of Tris groups increased in polymer chains, the structure of water appeared considerably disordered, as a result of the steric effect from bulkier groups. The structure of water around all hydrophilic beads (i.e. SQ5n, P2, and TN4a) showed slight dehydration with an increase in Tris content but did not affect the overall ordering of the water beads. RDFs were also calculated between the uncharged polymer chain components and water and followed the same general trend as the charged systems hinting that the presence of charges did not affect the bead's structural correlation with water.
To study the effect of Tris groups on the accessibility of hydrophobic backbone and styrene beads, we investigated the solvent-accessible surface areas (SASA) for the TC1 and TC5 beads as shown in Table S8.† The SASA for these hydrophobic beads can be imagined as the direct result of polymer conformations attained to stabilize these chains in their respective simulations. It was observed that in the case of backbone TC1 beads, SASA decreased as the percentage of Tris increased. However, this trend was exactly the opposite for the styrene TC5 beads where SASA increased with Tris substitution. This is especially interesting as it indicates that attributing the changes in SASA values to the steric hindrance of bulkier groups may not be adequate. Having previously observed that the presence of charges hampered the polymer backbone conformations, we also studied the SASA for the uncharged polymer systems as shown in Table S8.† It was observed that the SASA values for backbone TC1 beads were significantly lower for polymer chains with under 50% Tris substitution, while the SASA values for 50% and 70% substituted polymers were comparable to their charged counterparts. This, in addition to the increased backbone Rg for lower substituted polymer chains indicated that when the number of charged groups in polymers was high, to reduce their repulsions and assist their stabilization through solvent and ionic interactions, the charged groups had to be directed outwards, inevitably making the backbone beads more accessible to the solvent. This is also supported by the observation that the RDFs between TC1 beads attached to maleic acid groups were much more hydrated as compared to the TC1 beads attached to styrene groups at low percentages of Tris substitution (Fig. S13†). As the Tris substitution increased, there were reduced repulsions within the polymer and most of the backbone beads formed a central hydrophobic core, decreasing their accessibility. This highlights the importance of electrostatic interactions on polymer stabilization. The increase in SASA for the styrene TC5 beads. However, remained the same in the case of charged or uncharged simulations indicating that this was most likely due to the steric effects of the larger substituted groups.
To probe this further, the polymer–polymer RDF was plotted as shown in Fig. 5B, along with the RDFs between all individual components in the system shown in Fig. S14.† Contrary to those between polymer chains and water, polymer–polymer RDFs showed the highest structural correlations as the Tris content in polymers increased. Furthermore, all polymer systems with Tris groups displayed a predominant peak at ∼3 Å indicating some amount of polymer–polymer interactions. This peak was missing in the RDF for SMA polymer chains as they never interacted with each other. However, when the same RDFs for uncharged simulations were observed, there did appear a peak for the SMA polymer chains indicating that the absence of charge improved polymer–polymer interactions, albeit only in case of the otherwise entirely charged polymer. Similar trends, of greater structural correlation with increasing Tris substitution, were observed for individual polymer component RDFs including TC1–TC1 and TC5–TC5 indicating that polymer chains that instantaneously interacted with each other did so through their hydrophobic backbone and styrene groups. Of these, the structural correlation between styrene groups of interacting chains was found to be much more dominant where clear ordering was observed in these RDFs (Fig. S14B†). Other RDFs including SQ5n–SQ5n, P2–P2, and TN4a–TN4a also displayed the same trend as shown by the overall polymer–polymer RDFs. Thus, the new polymer chains developed and investigated in this study showed much stronger inter-polymeric interactions than the conventionally studied SMA polymers. These increased interactions between polymers at higher Tris substitutions are hypothesized to result in nanoparticle aggregation that was observed experimentally.
To identify the reasons for this particular behavior, we studied the RDFs between the polymer chains and lipid molecules as shown in Fig. 5C. Here, complementary to the differences in simulation trajectories, it was observed that the structural correlation between polymer chains and lipids molecules was quite similar for SMA, NCMNP2a-5, and NCMNP2a-25, all of which displayed similar numbers of nanoparticles in quantity and size. In the case of NCMNP2a-50 and NCMNP2a-70, the first RDF peaks were found to be higher than the previous systems with NCMNP2a-70 polymers having the highest structural correlation with lipid molecules indicating the highest interactions with lipids. The same was also observed for the uncharged simulations indicating that charge had little role to play in the aggregation of polymers and lipids with over 50% Tris substitution (Fig. S17†). Similar behavior was observed in most individual component analyses between polymer beads and lipid head, glycerol, and tail groups (Fig. S15 and S16†). While most of this appeared to be resulting from increased correlations of the Tris component beads with lipid groups, the RDF between styrene TC5 beads and lipid tails also showed a significant increase in first peak height for NCMNP2a-70 (Fig. S15B†). This indicated that polymer chains with higher Tris substitution possibly interact strongly with lipids, and other polymers as previously mentioned, resulting in the aggregation of multiple nanoparticles. A detailed analysis of simulation trajectories suggested that styrene groups from polymers initiate the formation of lipid nanoparticles by interacting with the hydrophobic tails of lipid molecules. Note, similar observations have been reported in previous studies.10,11
Next, to probe the effect of bulkier groups on the conformation of styrenes, if any, we studied the angles created by styrene and backbone beads, across the maleic acid groups. These were calculated for each monomeric unit, in the last 1 μs of the simulation trajectories. It was clearly observed that the mean angle formed between the two benzene rings constituting N1–N2–N3 and N4–N5–N6 beads increased from ∼60° to ∼70°. As also seen from the standard deviations for these angles, the angles fluctuated more in the presence of Tris groups, providing the polymer chains greater conformational flexibility. The mean angles formed by individual styrene rings with the backbone beads also showed similar trends, where systems with higher Tris groups showed angles about ∼20–25° larger, also resulting from larger fluctuations in these angles. This suggests that the presence of bulkier Tris groups certainly affected the conformations of styrene groups resulting in higher polymer–lipid interactions. Hence, to identify how these angles affected the accessibility of each benzene ring, we calculated the solvent-accessible surface area (SASA) values for these rings, as shown in Table S9.† In both, single polymer, and self-assembly simulations, it was found that the SASA increased for the systems containing 70% Tris substitution. However, this increase was tremendously more significant in the case of the benzene rings constituting N4–N5–N6 beads, in both charged and uncharged simulations. Furthermore, it was observed that as the Tris substitution increased, the benzenes comprising of N1–N2–N3 and N4–N5–N6 showcased similar SASA values as compared to SMA and NCMNP2a-5 polymer chains where this difference was as large as 70 Å2.
Data availability
The datasets of EM maps are provided in ESI Tables S2 and S3.† The experimental structure coordinates can be downloaded from RCSB PDB and EM maps can be downloaded from EMDB.Author contributions
Y. G. conceived the project. Y. G., W. Q., and T. K. H. T. designed the experiments. T. K. H. T. synthesized and analysed all the polymers and performed protein purifications, cryo-EM structure determinations, and enzyme assays. A. J. C. and A. G. collected and processed the cryo-EM data. S. J., S. D., and Y. G. designed the molecular dynamic experiment, S. J. performed the computational molecular dynamic analysis. C. C. participated in solving the cryo-EM structures of AcrB. A. B. S. performed quantitative FTIR analysis. W. Q. performed molecular cloning and initial optimization of AcrB–NCMN particles. T. K. H. T. and Y. G. drafted the manuscript. All authors contributed to the revision of the manuscript. Y. G. supervised the project.Conflicts of interest
Y. G. and T. K. H. T. are inventors of NCMNP2a-x (patent pending).Acknowledgements
We would like to thank Abu Bakkar Siddique for his review and suggestions on the manuscript. Y. G. was supported by the VCU School of Pharmacy and Department of Medicinal Chemistry through startup funds, the Institute for Structural Biology, Drug Discovery, and Development through laboratory space and facilities. This work was supported by NIH grant R01GM132329 to Y. G. and R35GM133598 to A. G. A. J. C. was supported by a G-RISE PhD traineeship from the National Institutes of Health (T32GM136499). The lipidomic analyses were performed at the VCU Lipidomics/Metabolomics Core with the NIH-NCI Cancer Center Support Grant P30 CA016059 to the VCU Massey Cancer Center and shared resource grant (S10RR031535) from the National Institutes of Health. Cryo-EM data were collected at the Imaging Facility of CUNY Advanced Science Research Center with help from Tong Wang for instrument use and scientific and technical assistance. The funders had no role in study design, data collection, analysis, decision to publish, or manuscript preparation. The content is solely the authors' responsibility and does not necessarily represent the National Institutes of Health or other funding organizations' official views.References
- G. H. Wadhams and J. P. Armitage, Nat. Rev. Mol. Cell Biol., 2004, 5, 1024–1037 CrossRef CAS PubMed.
- M. Farhadi, E. Razmara, M. Balali, Y. Hajabbas Farshchi and M. Falah, J. Cell. Mol. Med., 2021, 25, 5869–5883 CrossRef PubMed.
- H. Stahlberg, D. Fotiadis, S. Scheuring, H. Rémigy, T. Braun, K. Mitsuoka, Y. Fujiyoshi and A. Engel, FEBS Lett., 2001, 504, 166–172 CrossRef CAS PubMed.
- Y. Guo, Crystals, 2020, 10, 86 CrossRef CAS PubMed.
- H. E. Autzen, D. Julius and Y. Cheng, Curr. Opin. Struct. Biol., 2019, 58, 259–268 CrossRef CAS PubMed.
- K. S. Simon, N. L. Pollock and S. C. Lee, Biochem. Soc. Trans., 2018, 46, 1495–1504 CrossRef CAS PubMed.
- X. Yao, X. Fan and N. Yan, Proc. Natl. Acad. Sci., 2020, 117, 18497–18503 CrossRef CAS PubMed.
- W. Mi, Y. Li, S. H. Yoon, R. K. Ernst, T. Walz and M. Liao, Nature, 2017, 549, 233–237 CrossRef CAS PubMed.
- J. Frauenfeld, R. Löving, J.-P. Armache, A. F. P. Sonnen, F. Guettou, P. Moberg, L. Zhu, C. Jegerschöld, A. Flayhan, J. A. G. Briggs, H. Garoff, C. Löw, Y. Cheng and P. Nordlund, Nat. Methods, 2016, 13, 345–351 CrossRef PubMed.
- M. Liao, E. Cao, D. Julius and Y. Cheng, Nature, 2013, 504, 107–112 CrossRef CAS PubMed.
- C. J. Brown, C. Trieber and M. Overduin, Curr. Opin. Struct. Biol., 2021, 69, 70–77 CrossRef CAS PubMed.
- Y. Guo, Biochem. Soc. Trans., 2021, 49, 1361–1374 CrossRef CAS PubMed.
- J. Radoicic, S. H. Park and S. J. Opella, Biophys. J., 2018, 115, 22–25 CrossRef CAS PubMed.
- J. Broecker, B. T. Eger and O. P. Ernst, Structure, 2017, 25, 384–392 CrossRef CAS PubMed.
- C. Catalano, D. Ben-Hail, W. Qiu, P. Blount, A. des Georges and Y. Guo, Membranes, 2021, 11, 849 CrossRef CAS PubMed.
- S. C. Lee, T. J. Knowles, V. L. G. Postis, M. Jamshad, R. A. Parslow, Y.-p. Lin, A. Goldman, P. Sridhar, M. Overduin, S. P. Muench and T. R. Dafforn, Nat. Protoc., 2016, 11, 1149–1162 CrossRef CAS PubMed.
- L. Unger, A. Ronco-Campaña, P. Kitchen, R. M. Bill and A. J. Rothnie, Biochem. Soc. Trans., 2021, 49, 1349–1359 CrossRef CAS PubMed.
- J. M. Dörr, S. Scheidelaar, M. C. Koorengevel, J. J. Dominguez, M. Schäfer, C. A. van Walree and J. A. Killian, Eur. Biophys. J., 2016, 45, 3–21 CrossRef PubMed.
- A. A. A. Smith, H. E. Autzen, T. Laursen, V. Wu, M. Yen, A. Hall, S. D. Hansen, Y. Cheng and T. Xu, Biomacromolecules, 2017, 18, 3706–3713 CrossRef CAS PubMed.
- B. D. Harding, G. Dixit, K. M. Burridge, I. D. Sahu, C. Dabney-Smith, R. E. Edelmann, D. Konkolewicz and G. A. Lorigan, Chem. Phys. Lipids, 2019, 218, 65–72 CrossRef CAS PubMed.
- M. C. Fiori, W. Zheng, E. Kamilar, G. Simiyu, G. A. Altenberg and H. Liang, Sci. Rep., 2020, 10, 9940 CrossRef CAS PubMed.
- T. Ravula, N. Z. Hardin, S. K. Ramadugu and A. Ramamoorthy, Langmuir, 2017, 33, 10655–10662 CrossRef CAS PubMed.
- S. Lindhoud, V. Carvalho, J. W. Pronk and M.-E. Aubin-Tam, Biomacromolecules, 2016, 17, 1516–1522 CrossRef CAS PubMed.
- M. Esmaili and M. Overduin, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 257–263 CrossRef CAS PubMed.
- A. O. Oluwole, B. Danielczak, A. Meister, J. O. Babalola, C. Vargas and S. Keller, Angew. Chem., Int. Ed., 2017, 56, 1919–1924 CrossRef CAS PubMed.
- A. Marconnet, B. Michon, C. Le Bon, F. Giusti, C. Tribet and M. Zoonens, Biomacromolecules, 2020, 21, 3459–3467 CrossRef CAS PubMed.
- A. F. Craig, E. E. Clark, I. D. Sahu, R. Zhang, N. D. Frantz, M. S. Al-Abdul-Wahid, C. Dabney-Smith, D. Konkolewicz and G. A. Lorigan, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2931–2939 CrossRef CAS PubMed.
- T. Ravula, N. Z. Hardin, S. K. Ramadugu, S. J. Cox and A. Ramamoorthy, Angew. Chem., Int. Ed., 2018, 57, 1342–1345 CrossRef CAS PubMed.
- S. C. L. Hall, C. Tognoloni, J. Charlton, É. C. Bragginton, A. J. Rothnie, P. Sridhar, M. Wheatley, T. J. Knowles, T. Arnold, K. J. Edler and T. R. Dafforn, Nanoscale, 2018, 10, 10609–10619 RSC.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- X. Periole and S.-J. Marrink, in Biomolecular Simulations: Methods and Protocols, ed. L. Monticelli and E. Salonen, Humana Press, Totowa, NJ, 2013, pp. 533–565, DOI:10.1007/978-1-62703-017-5_20.
- M. Xue, L. Cheng, I. Faustino, W. Guo and S. J. Marrink, Biophys. J., 2018, 115, 494–502 CrossRef CAS PubMed.
- P. S. Orekhov, M. E. Bozdaganyan, N. Voskoboynikova, A. Y. Mulkidjanian, H.-J. Steinhoff and K. V. Shaitan, Langmuir, 2019, 35, 3748–3758 CrossRef CAS PubMed.
- M. Tazreiter, P. Christian, R. Schennach, T. Grießer and A. M. Coclite, Anal. Methods, 2017, 9, 5266–5273 RSC.
- P. Laurson, P. Raudsepp, H. Kaldmäe, A. Kikas and U. Mäeorg, AIP Adv., 2020, 10, 085214 CrossRef CAS.
- R. Mao, M. B. Huglin and T. P. Davis, Eur. Polym. J., 1993, 29, 475–481 CrossRef CAS.
- A. Sadat and I. J. Joye, Appl. Sci., 2020, 10, 5918 CrossRef CAS.
- A. Brangule and K. A. Gross, IOP Conf. Ser.: Mater. Sci. Eng., 2015, 77, 012027 Search PubMed.
- T. Ravula, S. K. Ramadugu, G. Di Mauro and A. Ramamoorthy, Angew. Chem., 2017, 129, 11624–11628 CrossRef.
- W. J. Cook, U. Thewalt and C. E. Bugg, Biochem. Biophys. Res. Commun., 1976, 68, 143–148 CrossRef CAS PubMed.
- Y. Lu, G. Deng, F. Miao and Z. Li, J. Inorg. Biochem., 2003, 96, 487–492 CrossRef CAS PubMed.
- W. J. Cook and C. E. Bugg, J. Am. Chem. Soc., 1973, 95, 6442–6446 CrossRef CAS PubMed.
- S. J. Angyal, in Carbohydrate Chemistry–VI, ed. W. M. Doane, Butterworth-Heinemann, 1973, pp. 131–146 Search PubMed.
- K. Poole, Curr. Opin. Microbiol., 2001, 4, 500–508 CrossRef CAS PubMed.
- D. Ma, D. N. Cook, J. E. Hearst and H. Nikaido, Trends Microbiol., 1994, 2, 489–493 CrossRef CAS PubMed.
- S. Scheidelaar, M. C. Koorengevel, C. A. van Walree, J. J. Dominguez, J. M. Dörr and J. A. Killian, Biophys. J., 2016, 111, 1974–1986 CrossRef CAS PubMed.
- A. O. Oluwole, J. Klingler, B. Danielczak, J. O. Babalola, C. Vargas, G. Pabst and S. Keller, Langmuir, 2017, 33, 14378–14388 CrossRef CAS PubMed.
- M. C. Fiori, Y. Jiang, G. A. Altenberg and H. Liang, Sci. Rep., 2017, 7, 7432 CrossRef PubMed.
- A. H. Kopf, J. M. Dörr, M. C. Koorengevel, F. Antoniciello, H. Jahn and J. A. Killian, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183125 CrossRef CAS PubMed.
- V. Postis, S. Rawson, J. K. Mitchell, S. C. Lee, R. A. Parslow, T. R. Dafforn, S. A. Baldwin and S. P. Muench, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 496–501 CrossRef CAS PubMed.
- S. Scheidelaar, M. C. Koorengevel, J. D. Pardo, J. D. Meeldijk, E. Breukink and J. A. Killian, Biophys. J., 2015, 108, 279–290 CrossRef CAS PubMed.
- S. Murakami, R. Nakashima, E. Yamashita and A. Yamaguchi, Nature, 2002, 419, 587–593 CrossRef CAS PubMed.
- D. Du, Z. Wang, N. R. James, J. E. Voss, E. Klimont, T. Ohene-Agyei, H. Venter, W. Chiu and B. F. Luisi, Nature, 2014, 509, 512–515 CrossRef CAS PubMed.
- M. Jamshad, V. Grimard, I. Idini, T. J. Knowles, M. R. Dowle, N. Schofield, P. Sridhar, Y. Lin, R. Finka, M. Wheatley, O. R. T. Thomas, R. E. Palmer, M. Overduin, C. Govaerts, J.-M. Ruysschaert, K. J. Edler and T. R. Dafforn, Nano Res., 2015, 8, 774–789 CrossRef CAS PubMed.
- J. M. Dörr, M. C. Koorengevel, M. Schäfer, A. V. Prokofyev, S. Scheidelaar, E. A. W. van der Cruijsen, T. R. Dafforn, M. Baldus and J. A. Killian, Proc. Natl. Acad. Sci., 2014, 111, 18607–18612 CrossRef PubMed.
- A. C. K. Teo, S. C. Lee, N. L. Pollock, Z. Stroud, S. Hall, A. Thakker, A. R. Pitt, T. R. Dafforn, C. M. Spickett and D. I. Roper, Sci. Rep., 2019, 9, 1813 CrossRef PubMed.
- H. Ayub, M. Clare, I. Milic, N. P. Chmel, H. Böning, A. Devitt, T. Krey, R. M. Bill and A. J. Rothnie, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183419 CrossRef CAS PubMed.
- S. J. Hesketh, D. P. Klebl, A. J. Higgins, M. Thomsen, I. B. Pickles, F. Sobott, A. Sivaprasadarao, V. L. G. Postis and S. P. Muench, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183192 CrossRef CAS PubMed.
- I. A. Smirnova, D. Sjöstrand, F. Li, M. Björck, J. Schäfer, H. Östbye, M. Högbom, C. von Ballmoos, G. C. Lander, P. Ädelroth and P. Brzezinski, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2984–2992 CrossRef CAS PubMed.
- I. Prabudiansyah, I. Kusters, A. Caforio and A. J. M. Driessen, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2050–2056 CrossRef CAS PubMed.
- C. R. Raetz, Microbiol. Rev., 1978, 42, 614–659 CrossRef CAS PubMed.
- W. Qiu, Z. Fu, G. G. Xu, R. A. Grassucci, Y. Zhang, J. Frank, W. A. Hendrickson and Y. Guo, Proc. Natl. Acad. Sci., 2018, 115, 12985–12990 CrossRef CAS PubMed.
- A. J. Higgins, A. J. Flynn, A. Marconnet, L. J. Musgrove, V. L. G. Postis, J. D. Lippiat, C.-w. Chung, T. Ceska, M. Zoonens, F. Sobott and S. P. Muench, Commun. Biol., 2021, 4, 1–9 CrossRef PubMed.
- C. Catalano, D. Ben-Hail, W. Qiu, P. Blount, A. des Georges and Y. Guo, Membranes, 2021, 11, 849, DOI:10.3390/membranes11110849..
- E. W. Yu, J. R. Aires and H. Nikaido, J. Bacteriol., 2003, 185, 5657–5664 CrossRef CAS PubMed.
- Z. Yue, W. Chen, H. I. Zgurskaya and J. Shen, J. Chem. Theory Comput., 2017, 13, 6405–6414 CrossRef CAS PubMed.
- T. Yamane, S. Murakami and M. Ikeguchi, Biochemistry, 2013, 52, 7648–7658 CrossRef CAS PubMed.
- M. A. Seeger, A. Schiefner, T. Eicher, F. Verrey, K. Diederichs and K. M. Pos, Science, 2006, 313, 1295–1298 CrossRef CAS PubMed.
- Y. Guo, R. C. Kalathur, Q. Liu, B. Kloss, R. Bruni, C. Ginter, E. Kloppmann, B. Rost and W. A. Hendrickson, Science, 2015, 347, 551–555 CrossRef CAS PubMed.
- C. Ginter, I. Kiburu and O. Boudker, Biochemistry, 2013, 52, 3609–3611 CrossRef CAS PubMed.
- Y. Guo, R. C. Kalathur, Q. Liu, B. Kloss, R. Bruni, C. Ginter, E. Kloppmann, B. Rost and W. A. Hendrickson, Science, 2015, 347, 551–555 CrossRef CAS PubMed.
- S. Gulati, M. Jamshad, T. J. Knowles, K. A. Morrison, R. Downing, N. Cant, R. Collins, J. B. Koenderink, R. C. Ford, M. Overduin, I. D. Kerr, T. R. Dafforn and A. J. Rothnie, Biochem. J., 2014, 461, 269–278 CrossRef CAS PubMed.
- D. J. K. Swainsbury, S. Scheidelaar, R. van Grondelle, J. A. Killian and M. R. Jones, Angew. Chem., 2014, 126, 11997–12001 CrossRef.
- D. Maitra, B. M. Pinsky, A. Soherawardy, H. Zheng, R. Banerjee and M. B. Omary, bioRxiv, 2021, preprint, DOI:10.1101/2021.01.11.426224.
- L. M. Scolaro, M. Castriciano, A. Romeo, S. Patanè, E. Cefalì and M. Allegrini, J. Phys. Chem. B, 2002, 106, 2453–2459 CrossRef CAS.
- K. G. Kroeck, W. Qiu, C. Catalano, T. K. H. Trinh and Y. Guo, JoVE, 2020, e61298, DOI:10.3791/61298.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domański, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, X. Periole, D. P. Tieleman, A. H. de Vries and S. J. Marrink, Nat. Methods, 2021, 18, 382–388 CrossRef CAS PubMed.
- R. Alessandri, J. Barnoud, A. S. Gertsen, I. Patmanidis, A. H. de Vries, P. C. T. Souza and S. J. Marrink, Adv. Theory Simul., 2022, 5, 2100391 CrossRef CAS.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01890c |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |