
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoluminescence control by atomically precise surface metallization of C-centered hexagold(I) clusters using N-heterocyclic carbenes†
Zhen
Lei‡
 a,
Pei
Zhao‡
b,
Xiao-Li
Pei
a,
Hitoshi
Ube
a,
Masahiro
Ehara
*b and
Mitsuhiko
Shionoya
*a
a,
Pei
Zhao‡
b,
Xiao-Li
Pei
a,
Hitoshi
Ube
a,
Masahiro
Ehara
*b and
Mitsuhiko
Shionoya
*a
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shionoya@chem.s.u-tokyo.ac.jp
bResearch Center for Computational Science, Institute for Molecular Science, Myodaiji, Okazaki, Aichi 444-8585, Japan. E-mail: ehara@ims.ac.jp
First published on 11th May 2023
Abstract
The properties of metal clusters are highly dependent on their molecular surface structure. The aim of this study is to precisely metallize and rationally control the photoluminescence properties of a carbon(C)-centered hexagold(I) cluster (CAuI6) using N-heterocyclic carbene (NHC) ligands with one pyridyl, or one or two picolyl pendants and a specific number of silver(I) ions at the cluster surface. The results suggest that the photoluminescence of the clusters depends highly on both the rigidity and coverage of the surface structure. In other words, the loss of structural rigidity significantly reduces the quantum yield (QY). The QY in CH2Cl2 is 0.04 for [(C)(AuI-BIPc)6AgI3(CH3CN)3](BF4)5 (BIPc = N-isopropyl-N′-2-picolylbenzimidazolylidene), a significant decrease from 0.86 for [(C)(AuI-BIPy)6AgI2](BF4)4 (BIPy = N-isopropyl-N′-2-pyridylbenzimidazolylidene). This is due to the lower structural rigidity of the ligand BIPc because it contains a methylene linker. Increasing the number of capping AgI ions, i.e., the coverage of the surface structure, increases the phosphorescence efficiency. The QY for [(C)(AuI-BIPc2)6AgI4(CH3CN)2](BF4)6 (BIPc2 = N,N′-di(2-pyridyl)benzimidazolylidene) recovers to 0.40, 10-times that of the cluster with BIPc. Further theoretical calculations confirm the roles of AgI and NHC in the electronic structures. This study reveals the atomic-level surface structure–property relationships of heterometallic clusters.
Introduction
Gold clusters have attracted a lot of attention in recent years due to their molecular structures based on aurophilicity between gold atoms and their optical properties such as structure-specific photoluminescence.1,2 Gold clusters with such properties have been applied to a variety of applications such as OLEDs and bioimaging etc.3 To realize on-demand synthesis of cluster-based phosphorescent materials, great efforts have been made over the past decades to correlate the structure of gold clusters with their photoluminescence.4–6 In the meantime, several general strategies were developed to enhance or tune the photoluminescence. Examples include changes in electronic structure due to alloying of metal kernels,7–10 surface hardening by additives to suppress non-radiative relaxation,11–14 supramolecular networks due to self-assembly of clusters,15–18 and chemical modification of supporting ligands by electronic donating/withdrawing groups and use of steric hindrance.19On the other hand, surface molecular design of metal clusters is a promising strategy for developing chemical and physical properties specific to metal arrays. Compared to the widely used phosphine ligands, NHC ligands are similarly stable monodentate ligands with flexible electronic properties such as electron-donating properties and high designability of chemical structures.20–27 Therefore, we have been working on photoluminescence control of C-centered AuI and AuI–AgI clusters by chemical modification with N-heterocyclic carbene (NHC) ligands.28–32 We found that the introduction of different types of ligands on the surface of AuI and AuI–AgI clusters dramatically changed their photoluminescence behavior, including excitation and emission energies, quantum yields (QYs, φ) and phosphorescence lifetimes (τ). For example, the homometallic [(C)(AuI-BIiPr)6](BF4)2 (1, BIiPr = N,N′-diisopropylbenzimidazolylidene) and the classic [(C)(AuI-PPh3)6](BF4)2 (ref. 33–36) are structurally similar but exhibit very different emission properties in the solid state.30 More surprisingly, the heterometallic [(C)(AuI-BIPy)6AgI2](BF4)4 (2, BIPy = N-isopropyl-N′-2-pyridylbenzimidazolylidene) shows blue-shifted emission and 3 times higher QY in solution than [(C)(AuI-dppy)6AgI2](BF4)4 (dppy = 2-pyridyldiphenylphosphine).32,37
Previously, Wang and co-workers reported a series of carbon-centered, phosphine-protected AuI–AgI clusters.37–44 The second layer of AgI ions on the cluster surface fully protects the core portion, resulting in long life and highly efficient emission.43,44 In our recent work, we found that NHC ligands may further enhance the emission of heterometallic clusters.32 These results suggest that, the silver(I) adatoms and NHC ligands synergistically contribute to the photoluminescence properties of heterometallic clusters.
In this study, we aimed to investigate this synergistic effects of NHC ligands and the silver(I) adatoms on the surface of heterometallic clusters. By taking advantage of pre-designed benzimidazolylidene-type NHC ligands with one or two picolyl pendant(s), we have achieved atomically precise metallization to the CAuI6 cluster site, and established correlations among the ligand structure, metal adatom number, and photoluminescence behavior (Scheme 1). We emphasize that the results of this work are not a mirror universe of phosphine-protected clusters, since (a) ligand design here is more elaborate; (b) the heterometallic clusters obtained show distinctly different molecular geometries; and (c) the photoluminescence of the clusters does not simply follow the “full protection” rule.
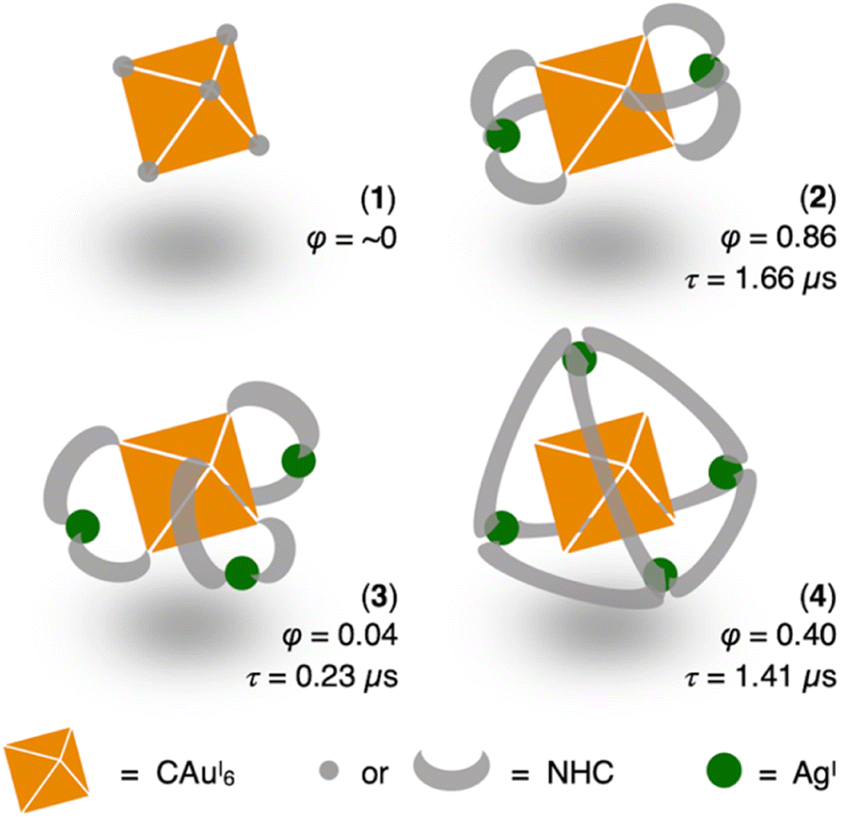 | ||
| Scheme 1 Phosphorescence properties of CAuI6 clusters surrounded by different NHC or NHC–AgI shells in CH2Cl2 at room temperature (φ represents QY, τ represents lifetime). | ||
We first added a methylene linker between the benzimidazole and pyridyl groups of BIPy (Fig. 1). When acting as a bidentate ligand as in the case of BIPy, the methylene group seemed to give some structural flexibility to the resulting ligand N-isopropyl-N′-2-picolylbenzimidazolylidene (BIPc). Furthermore, since the pyridyl groups of BIPy mainly affect the electronic structure of the hexagold(I) cluster (Scheme S1†),32 the fundamental motivation for this design was the thought that the introduced methylene linker could separate the two heterocyclic moieties, thereby breaking the electronic perturbation from the directly connected imidazole and benzimidazole groups. By using this BIPc, a moderately phosphorescent cluster [(C)(AuI-BIPc)6AgI3(CH3CN)3](BF4)5 (3) was constructed, in which the octahedral CAuI6 core was capped by three (BIPc)2Ag moieties in a triangular arrangement (Scheme 1). Note that the tricapped octahedral structure has not been achieved yet in neither the NHC- nor the phosphine-protected CAuI6 cluster family (Scheme S2†). The newly synthesized CAuI6AgI3-type cluster 3 showed a red-shifted emission, but the QY was significantly lower (φ = 0.04) and the lifetime was shorter (0.23 μs) compared to the excellent photophysical properties of bicapped octahedral, CAuI6AgI2-type cluster 2. The use of the picolyl group resulted in the coordination of another silver ion around the octahedral CAuI6 structure, causing a red-shift in the emission. On the other hand, the introduction of a methylene linker to the picolyl group resulted in a much looser structure of the coordination sphere, which was found to be responsible for the low QY and short lifetime.
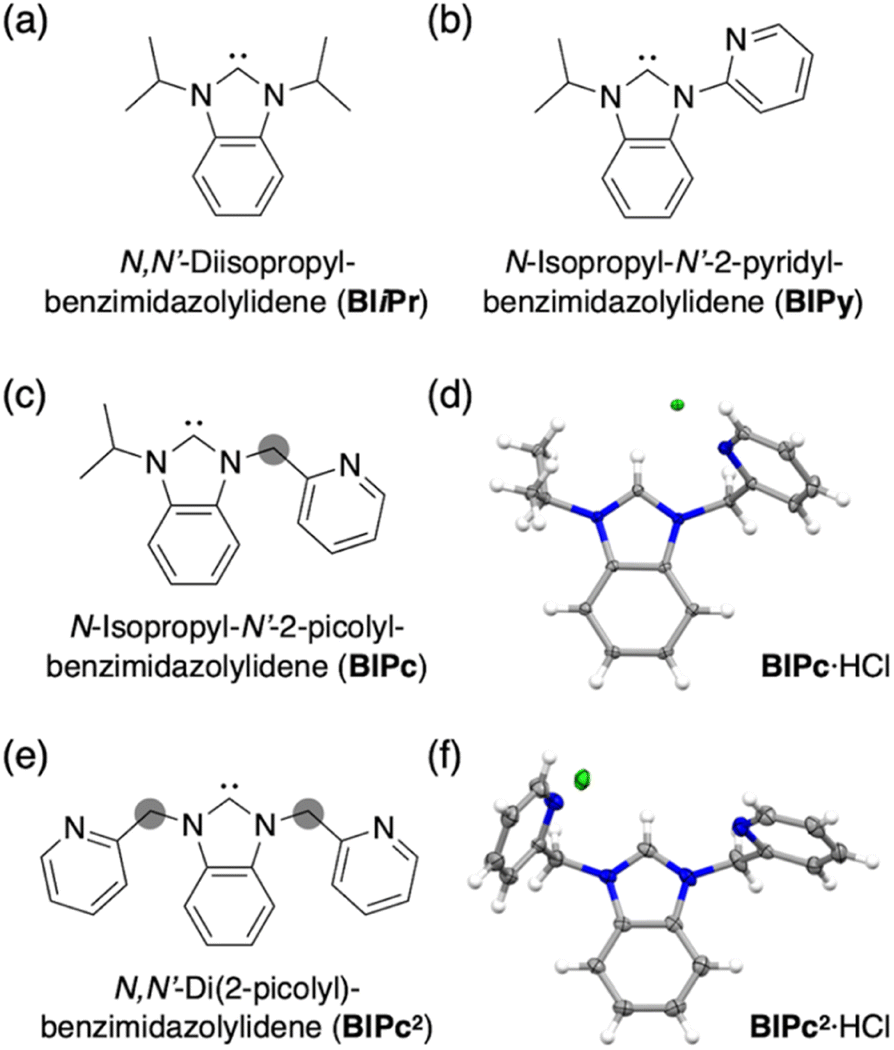 | ||
| Fig. 1 (a) Structure of ligand BIiPr. (b) Structure of ligand BIPy. (c) Structure 2 of ligand BIPc. (d) Molecular structure of BIPc·HCl. (e) Structure of ligand BIPc. (f) Molecular structure of BIPc2·HCl. | ||
Next, the carbene ligand N,N′-di(2-picolyl)benzimidazolylidene (BIPc2), which has a higher symmetry with two picolyl groups, was used to make the structure of the coordination sphere more rigid (Fig. 1).45–47 As a result, a tetrahedral form of [(C)(AuI-BIPc2)6AgI4(CH3CN)2](BF4)6 (4), a CAuI6AgI4-type cluster, was obtained in which the outer shell part composed of (BIPc2)6AgI4 covered the octahedral CAuI6 cluster (Scheme 1). By introducing an additional ligand in the wingtip group of the carbene ligand and increasing the number of AgI ions bound to CAuI6, strong red phosphorescence with a QY of 0.40 and a lifetime of 1.41 μs was observed in CH2Cl2, both of which were significantly improved compared to cluster 3. The CAuI6AgI4 tetrahedron has edges of 5.544–5.947 Å, which is comparable to the Au10 nanocluster,48,49 one of the long pursued tetrahedral gold nanoclusters. This result indicates that not only the coverage of the cluster surface by additional metal ions but also the rigidity of the coordination structure is important for the strong and long-lived phosphorescent properties of metal clusters. The structure will also provide a useful new idea for rational synthesis of tetrahedral Au10 and Au20.48–54
Results and discussion
Synthesis
Structures of BIiPr, BIPy, BIPc and BIPc2 are shown in Fig. 1. The ligands BIPc and BIPc2 were synthesized as hydrochloride salts by a modified procedure of the literature (Fig. 1 and S1†). The CAuI6 clusters [(C)(AuI-BIPc)6](BF4)2 (5) and [(C)(AuI-BIPc2)6](BF4)2 (6) as precursors were prepared in 46% and 43% overall yields based on the amount of gold, respectively (Fig. 2a, S2–S22 and Tables S1–S3†).§ CAuI6AgI2-type cluster 2 was synthesized according to our previously reported literature.32Next, heterometallic clusters were synthesized from clusters 5 and 6. The CAuI6 cluster of 5 or 6 was mixed with AgBF4 in CH2Cl2/CH3CN (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 9:1) and the reaction was monitored by ESI-MS spectrometry. When AgBF4 was added to cluster 5, signals indicating the formation of heterometallic species of [(C)(AuI-BIPc)6AgI](BF4)2+, [(C)(AuI-BIPc)6AgI2](BF4)22+, [(C)AuI5AgI(BIPc)4](BF4)+, and [(C)AuI5AgI2(BIPc)4](BF4)2+ appeared (Fig. S23†), which were found to be similar to the pattern of peaks seen in the BIPy-protected cluster.32 This complexation process in solution was also monitored by UV-vis absorption, fluorescence and 1H NMR spectroscopies (Fig. S24–S26†). In the UV-vis spectra, the intensity of the peak around 340 nm decreased significantly with the addition of silver ions, and a new series of peaks and shoulders appeared. The emission spectra showed an increase in the intensity of the broad peak in the lower energy region. In fact, when the reaction mixture was irradiated with a UV lamp, a bright orange-yellow emission was observed. The decrease and increase in absorbance at 343 and 440 nm, respectively, and the increase in emission intensity at 609 nm were all strongly suggestive of the incorporation of three AgI ions into cluster 5. The 1H NMR spectra showed that the sharp signals became unstructured with the addition of AgI ions, suggesting a flexible coordination structure of the product.
:v = 9:1) and the reaction was monitored by ESI-MS spectrometry. When AgBF4 was added to cluster 5, signals indicating the formation of heterometallic species of [(C)(AuI-BIPc)6AgI](BF4)2+, [(C)(AuI-BIPc)6AgI2](BF4)22+, [(C)AuI5AgI(BIPc)4](BF4)+, and [(C)AuI5AgI2(BIPc)4](BF4)2+ appeared (Fig. S23†), which were found to be similar to the pattern of peaks seen in the BIPy-protected cluster.32 This complexation process in solution was also monitored by UV-vis absorption, fluorescence and 1H NMR spectroscopies (Fig. S24–S26†). In the UV-vis spectra, the intensity of the peak around 340 nm decreased significantly with the addition of silver ions, and a new series of peaks and shoulders appeared. The emission spectra showed an increase in the intensity of the broad peak in the lower energy region. In fact, when the reaction mixture was irradiated with a UV lamp, a bright orange-yellow emission was observed. The decrease and increase in absorbance at 343 and 440 nm, respectively, and the increase in emission intensity at 609 nm were all strongly suggestive of the incorporation of three AgI ions into cluster 5. The 1H NMR spectra showed that the sharp signals became unstructured with the addition of AgI ions, suggesting a flexible coordination structure of the product.
The complexation between cluster 6 and AgBF4 in CH2Cl2/CH3CN was first monitored by 1H NMR spectroscopy. The heterometallic species formed in situ exhibited a single set of relatively sharp signals, distinct from the complexation of cluster 5 and AgBF4 (Fig. S26 and 27†). ESI-MS spectroscopic analysis revealed the gradual formation of heterometallic species ([(C)(AuI-BIPc2)6AgIn](BF4)n2+ (n = 1–4)) containing one to four AgI ions (Fig. S28†). In the UV-vis absorption and emission spectra, a plot of absorbance/emission intensity against the amount of AgI added to cluster 6 indicates that three to four AgI ions were incorporated into cluster 6 (Fig. S29 and 30†). These data strongly suggest that a tetra-capped heterometallic structure was formed.
Molecular structures and characterizations
Single crystals of heterometallic clusters were obtained by layering dry Et2O on a CH2Cl2/CH3CN solution of products in tubes. Single-crystal X-ray diffraction (ScXRD) analysis determined the structures of the tricapped CAuI6AgI3-type 3 and the tetrahedral CAuI6AgI4-type 4.§ The molecular structure and simplified scheme of the cationic part of cluster 3 are shown in Fig. 2b and c, respectively. The structure of 3 is clearly different from that of CAuI6AgI2-type cluster 2, as the three AgI atoms, supported by the ligand BIPc, cap the three octahedral faces of the CAuI6 cluster, forming a triangulated monorectified tetrahedron (triangular frustum) structure. Specifically, two BIPc ligands adjacent via pyridyl groups and one additional acetonitrile ligand are coordinated to each AgI ion in cluster 3, so the overall structure can be viewed as a CAuI6 center supported by three (BIPc)2Ag motifs. The reason why the synthesis in CH2Cl2/CH3OH did not work (Fig. S31 and S32, see ESI† for details) can be explained by the co-stabilization with acetonitrile. The AuI–AgI distances in cluster 3 (2.8268(14)–2.9754(14) Å, Table S4†) were found slightly longer than that in cluster 2 (2.8467(17) Å). The ligand BIPc has one more methylene group than BIPy, making it relatively flexible. Notably, when cluster 3 was formed, the 1H NMR spectra showed that the singlet signal of the methylene group of CAuI6-type cluster 5 (δ = 5.77 ppm in CD2Cl2) was split into two doublet signals (δ = 6.10, 6.66 ppm in CD2Cl2) (Fig. S4 and S33†). This is because, when the pyridyl group coordinates to AgI, the two hydrogen atoms of the methylene group become chemically inequivalent. One hydrogen atom is oriented toward the metal kernel to form a hydrogen-gold bond, while the other is oriented away from it. Indeed, in the structure of cluster 3, the estimated intramolecular C(sp3)–H⋯Au bond between the hydrogen atom and the gold atom was very short55–60 (Fig. S37,† 2.677 Å). The results of the 1H NMR measurements of cluster 3 were found to be affected from the deuterated solvent in the solvent used. In fact, very broadened signals were observed when measured in CD2Cl2/CD3CN (v:v = 9:1, Fig. S38†), reproducing the results of the complexation between cluster 5 and AgBF4 (Fig. S26†). This result suggests that the presence of coordinating acetonitrile molecules makes the coordination of ligand BIPc in cluster 3 slightly more fluxional. UV-vis absorption spectroscopy and ESI-MS were also employed to characterize cluster 3 (Fig. S39 and S40†). The results were similar to the spectra of heterometallic species formed in situ (Fig. S23 and S24†).
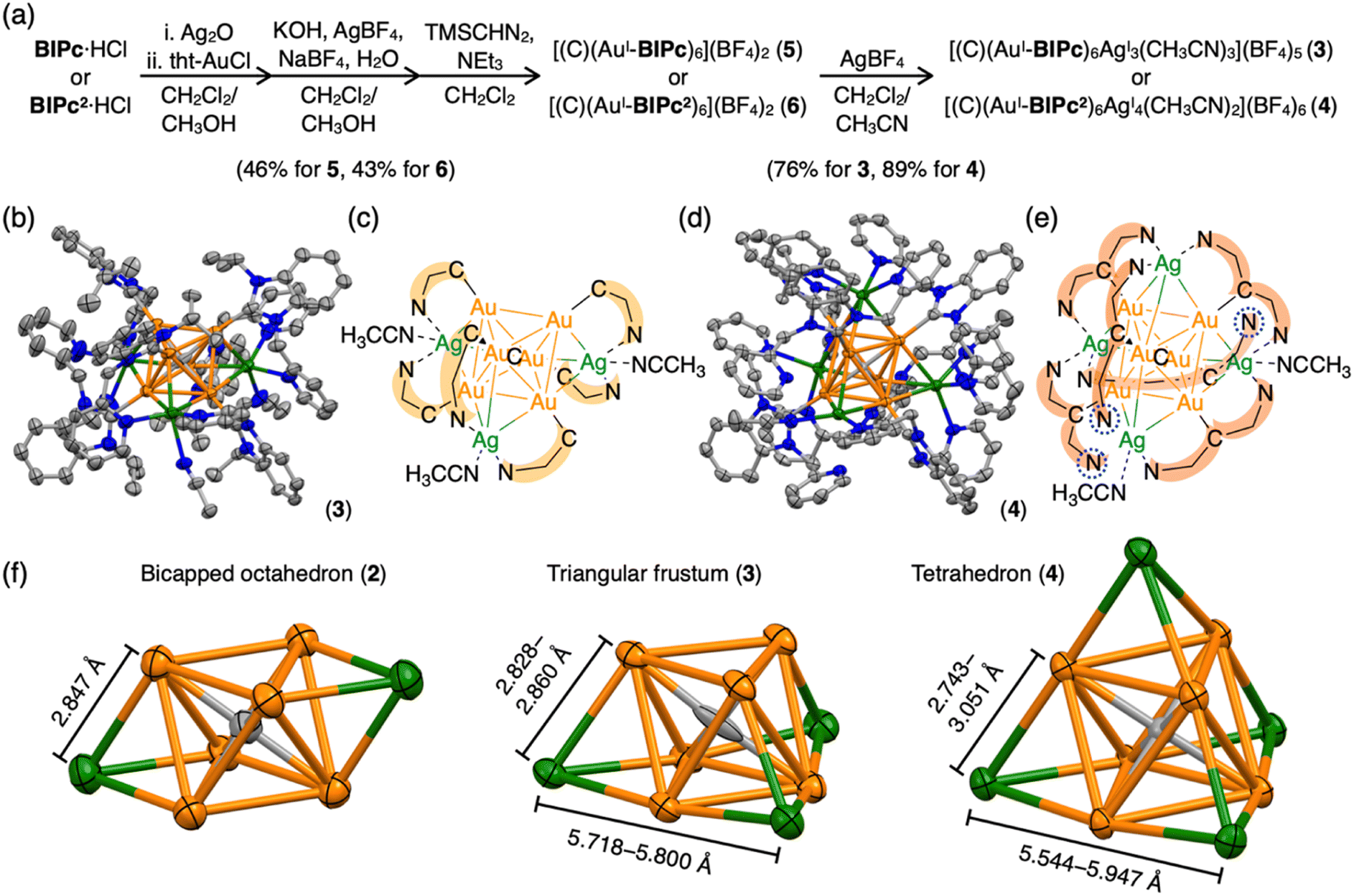 | ||
| Fig. 2 (a) Synthetic schemes of the homometallic clusters [(C)(AuI-BIPc)6](BF4)2 (5) and [(C)(AuI-BIPc2)6](BF4)2 (6), and heterometallic clusters [(C)(AuI-BIPc)6AgI3(CH3CN)3](BF4)5 (3) and [(C)(AuI-BIPc2)6Ag4(CH3CN)2](BF4)6 (4). (b) Molecular structure of cluster 3. (c) A simplified scheme showing the structure of cluster 3. C^N represents ligand BIPc. (d) Molecular structure of cluster 4. (e) A simplified scheme showing the structure of cluster 4. N^C^N represents ligand BIPc2. Uncoordinated pyridyl groups are highlighted by dashed blue cycles. (f) Comparison of structures of bicapped octahedral CAuI6AgI2 in cluster 2, triangular frustum-shaped CAuI6AgI3 in cluster 3, and tetrahedral CAuI6AgI4 in cluster 4. The length of the edge of each structure is shown. | ||
More interestingly, CAuI6AgI4-type cluster 4 has a tetrahedral structure (Fig. 2d and e). Tetrahedral gold clusters (e.g. Au4, Au10, Au20) have long been explored because they can be regarded as fragments of bulk gold and have been found or predicted to exhibit unique structural properties such as wide energy gap and spherical aromaticity.48–54,61–63 Despite the fact that Au4 has actually been reported, structure information on the latter two clusters is not yet available. As shown in the simplified CAuI6AgI4 structure in Fig. 2e, four AgI atoms cap the four faces of the octahedral CAuI6 in the central of cluster 4. The AuI–AgI distances for cluster 4 are in the range of 2.7425(7) to 3.0507(7) Å, slightly off distances in 2 and 3. Another way to look at it is that the central CAuI6 is encapsulated in a tetrahedral (BIPc2)6Ag4 cage. Three of the twelve pyridyl groups were found not to be involved in metal coordination in the crystalline state, as indicated by the blue dashed circles. The coordinating acetonitrile was thought to contribute to the stabilization. Similarly, in the case of cluster 3, two pyridyl groups and one acetonitrile are considered to be uniquely coordinated to the three AgI ions to stabilize the complex in solution. In the case of cluster 4, on the other hand, the ligand exchange reaction in solution is considered to be more complex because there can be multiple combinations of pyridyl groups and acetonitrile as ligands to the four AgI ions. The mismatched structure of the ligand BIPc2 probably prevents the formation of a perfect tetrahedral structure.
An important structural point is that the composition of 4 is similar to previously reported [(C)(AuI-PPhPy2)AgI4](BF4)6 (PPhPy2 = bis(2-pyridyl)phenylphosphine), but the arrangement of four AgI ions on the octahedral CAuI6 surface is distinctly different from each other.43 The latter cluster has a butterfly-like molecular structure, that is, two opposite pairs of neighboring triangular faces are capped; whereas the four AgI ions in 4 are in a tetrahedral arrangement as far apart from each other as possible (Scheme S2†). This difference may be attributed to the different geometries of the NHC and phosphine ligands. In PPhPy2, the two coordinative pyridyl groups are bonded directly connected to the same phosphorous atom, so the angle between these two pyridyl groups should be approximately 120°, and the distance between the two N donors are relatively short. These two features are in geometrical contrast to BIPc2, in which the two picolyl groups are on two sides of the benzimidazolylidene. The introduction of a methylene linker at the picolyl group further increased the distance between the two N donors in BIPc2, making the coordination mode more flexible. These results demonstrate the designability of the NHC ligands and their superiority.
For heterometallic cluster 4, we first performed a detailed evaluation of the solution structure by 1H NMR spectroscopy (Fig. S41–S44†) and observed only one set of signals in the spectrum. Second, all peaks in the 1H and 13C NMR spectra were sharp and showed no apparent fluxing, despite the use of a deuterated mixed solvent containing coordinating acetonitrile. This suggests that cluster 4 exists in a fairly symmetric structure in solution. A splitting of the methylene signal was also observed in the 1H NMR spectrum of 4, as in cluster 3. The sharp singlet peak at 5.63 ppm in CAuI6 cluster 6 split into two doublet peaks at 5.38 and 6.06 ppm when forming cluster 4 (Fig. S8 and S41†). This result suggests that the hydrogen atom on one side of the methylene chain of cluster 4 forms an intramolecular C(sp3)–H⋯Au bond and the pyridyl group is fixed in solution, as shown in Fig. S37.† Cluster 4 was further characterized by ESI-MS and UV-vis spectroscopy (Fig. S45 and S46†). As a result, we were able to reproduce most of the data for the heterometallic products formed at the site (Fig. S28 and S29†). This indicates that cluster 4 is the main product of the complexation of CAuI6 cluster 6 with AgBF4.
Correlation between phosphorescence and structure
Clusters 2–4 have a common CAuI6 octahedral core structure and exhibit properties characteristic of different external structures. In clusters 2–4, the two, three, and four faces of the central octahedral CAuI6 were capped by AgI ions, forming bicapped octahedral, triangular frustum-shaped (monorectified tetrahedral), and tetrahedral metal kernels, respectively (Fig. 2f). In cluster 2, the AuI–AgI distance is 2.847 Å. In cluster 3, the side edges of the frustum (AuI–AgI) ranged from 2.828 to 2.860 Å and the lower bottom (AgI–AgI) side edges ranged from 5.718 to 5.800 Å. Tetrahedral cluster 4 was slightly distorted, with all edges in the 5.544–5.947 Å range. In the case of NHC ligands, on the other hand, the rigidity of the entire molecule depends on the presence or absence of a methylene linker in the ligand. As a result, it is inferred that clusters 2–4 exhibit structure-specific emission.In the solid state, clusters 3 and 4 show orange and red emission with maxima at 640 and 661 nm, respectively, both significantly red-shifted compared to cluster 2 (573 nm; Fig. 3a, S47 and S48†). The emission maxima of clusters 2–4 also showed a similar red-shift in the CH2Cl2 solution (Fig. 3b, S49 and S50†). Both clusters 3 and 4 are very stable under ambient conditions. The absorption spectra of CH2Cl2 solutions of 3 and 4 (c = 3.3 × 10−5 mol L−1) remain virtually unchanged after one week in sunlight at room temperature (Fig. S51†). Surprisingly, 4 remains robust in CH2Cl2/CH3CN (v:v = 9:1, c = 3.3 × 10−5 mol L−1) at 4 °C for about 18 months (Fig. S52†). The wingtip group of ligand BIPc in cluster 3 is one methylene chain longer than that of BIPy in cluster 2, so that one more AgI ion could be accommodated on the central hexagold(I) cluster. In the case of cluster 4, the number of pyridyl groups increased to twelve, creating new chances to bind further one more adatom. As a result, more red-shifted emission was observed from the clusters with more metal atoms, and the stability of cluster 4 was also improved by introducing a fourth silver atom.
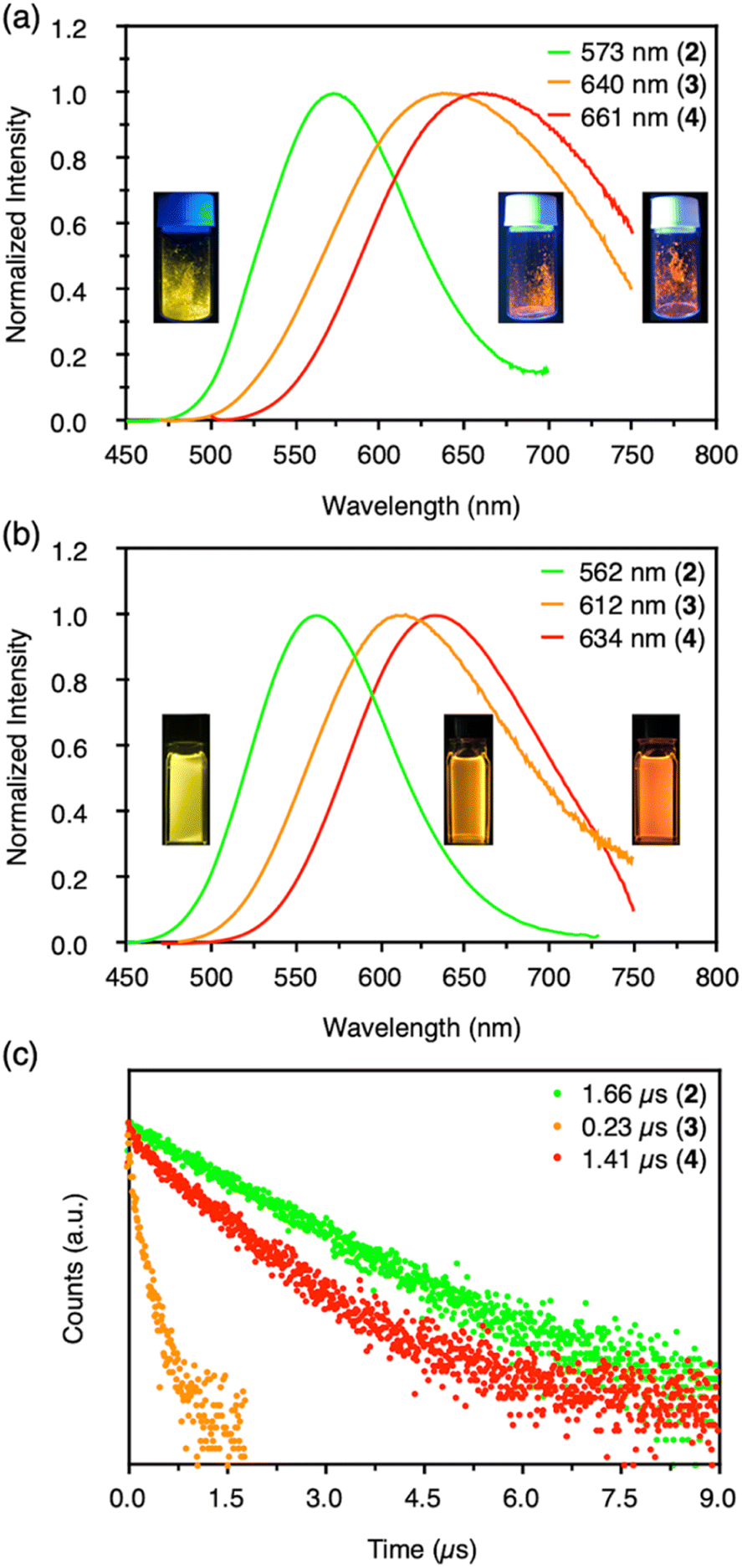 | ||
| Fig. 3 (a) Emission spectra of CAuI6AgIn-type clusters 2–4 (n = 2, 3, 4) in the solid state at room temperature and corresponding emission photographs of 365 nm excitation (from left to right). (b) Emission spectra of clusters 2–4 in CH2Cl2 at 20 °C and corresponding emission photographs of 365 nm excitation (from left to right). (c) Emission decays of clusters 2–4 in CH2Cl2 at room temperature. | ||
More importantly, the photoluminescence QYs and lifetimes of clusters 2–4 were very different from each other. First, the QY and lifetime of cluster 3 were measured to be 0.04 and 0.23 μs, respectively, about an order of magnitude smaller than those of cluster 2 (0.86 and 1.66 μs, respectively). However, these values for cluster 4 were 0.40 and 1.41 μs, respectively, almost recovering to the level of the values for cluster 2. The high QY, microsecond-scale lifetime, and large Stokes shift indicate that the emissions of clusters 2–4 are phosphorescence.
The radiative and non-radiative rate constants (kr and knr, respectively) of each cluster were then calculated from the QY and lifetime values (Table 1). Because all of these clusters are protected by benzimidazolylidene-type ligands, we were able to correlate two factors with respect to the emissions of clusters: the wingtip groups of the carbene ligand, and number of AgI atoms in the cluster.
| Cluster | φ | τ (μs) | k r (×105 s−1) | k nr (×105 s−1) |
|---|---|---|---|---|
| 2 | 0.86 | 1.66 | 5.18 | 0.84 |
| 3 | 0.04 | 0.23 | 1.74 | 41.7 |
| 4 | 0.40 | 1.41 | 2.84 | 4.26 |
First, the values of kr and knr for CAuI6AgI3-type cluster 3 were changed by a factor of 1/3 and about 50 times, respectively, compared to those of cluster 2. These marked differences are likely due to a significant decrease in the stability of the bond between the methylene group of the BIPc ligand of cluster 3 and the AgI ion (Scheme S2†). This increase in the non-radiation deactivation process of 3 is due to the introduction of a flexible structure, which may have had a significant impact on the emission efficiency. Furthermore, the knr value of CAuI6AgI4-type cluster 4 decreased by about a factor of 10, indicating that non-radiation deactivation process was significantly suppressed. In cluster 3, the six ligands have one picolyl ligand each, with two picolyl nitrogens and one acetonitrile coordinating to the three AgI ions. In cluster 4, six ligands have two picolyl groups each, and most or all of them can interact with the four AgI atoms, as shown by ScXRD and NMR data. Considering that the bond formation between the picolyl group and AgI has a small effect on the bond lengths and bond angles of the methylene-bridged N–C–C bonds in the ligand moiety of clusters 3 and 4, it is estimated that the incorporation of the fourth AgI atom into the CAuI6 core of cluster 4 serves to further enhance QY by efficiently suppressing the non-radiation process, and that the immobilizing effect of the picolyl groups, although not negligible, is not a decisive factor.
Theoretical calculations
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were carried out to disclose electronic structures of the clusters and the origin of the phosphorescence. First, the Bader and NPA (natural population analysis) charges of the central C, Au and Ag atoms in CAuI6AgI3-type cluster 3 and CAuI6AgI4-type cluster 4 were analyzed (Table S5†). Compared to CAuI6-type clusters 5 and 6, the negative charges of the central C atoms in clusters 2, 3 and 4 are reduced and the positive charges of the Au atoms are reduced. These results indicate that Ag ions covering the CAuI6 kernel affect the charge distribution of the metal clusters. Furthermore, the larger the number of Ag ions, the greater the effect on the charge distribution.Next, we simulated the UV-vis absorption spectra of 3 and 4. As shown in Fig. 4a and b, the appearance of shoulder bands in the lower energy region (∼410 nm for cluster 3, 405 nm for cluster 4) was well reproduced. The first peak at 403 nm of cluster 3 is mainly due to the transition from HOMO−2 (highest occupied molecular orbital) to LUMO (lowest unoccupied molecular orbital), as shown in Fig. 4c, S54 and Table S6.† The strongest peak at 377 nm is mostly derived from transitions between three occupied molecular orbitals (HOMO, HOMO−1, and HOMO−2) and two unoccupied molecular orbitals (LUMO and LUMO+1). The occupied MOs are localized in the Au kernel, and a large contribution from Ag atoms is observed in the unoccupied MOs. In the case of 4 with four Ag atoms, an onset peak appears around 416 nm, with the strongest peak located around 359 nm. As summarized in Table S7 and Fig. S55,† these absorption peaks originate from transitions among MOs of Au and Ag, and the distribution of orbitals involved is similar to that of 3. Orbital composition analysis by Mulliken partition was then performed for 2–6 (Table S8†). In all clusters, the central C and Au atoms show large contributions (∼70%) to the occupied molecular orbitals (HOMO, HOMO−1, and HOMO−2). For the virtual molecular orbitals (LUMO+2, LUMO+1, and LUMO), the ligands contribute significantly (∼60%) in homometallic clusters 5 and 6, while the silver atoms contribute mainly in LUMO (∼30–40%) in heterometallic clusters 2–4.
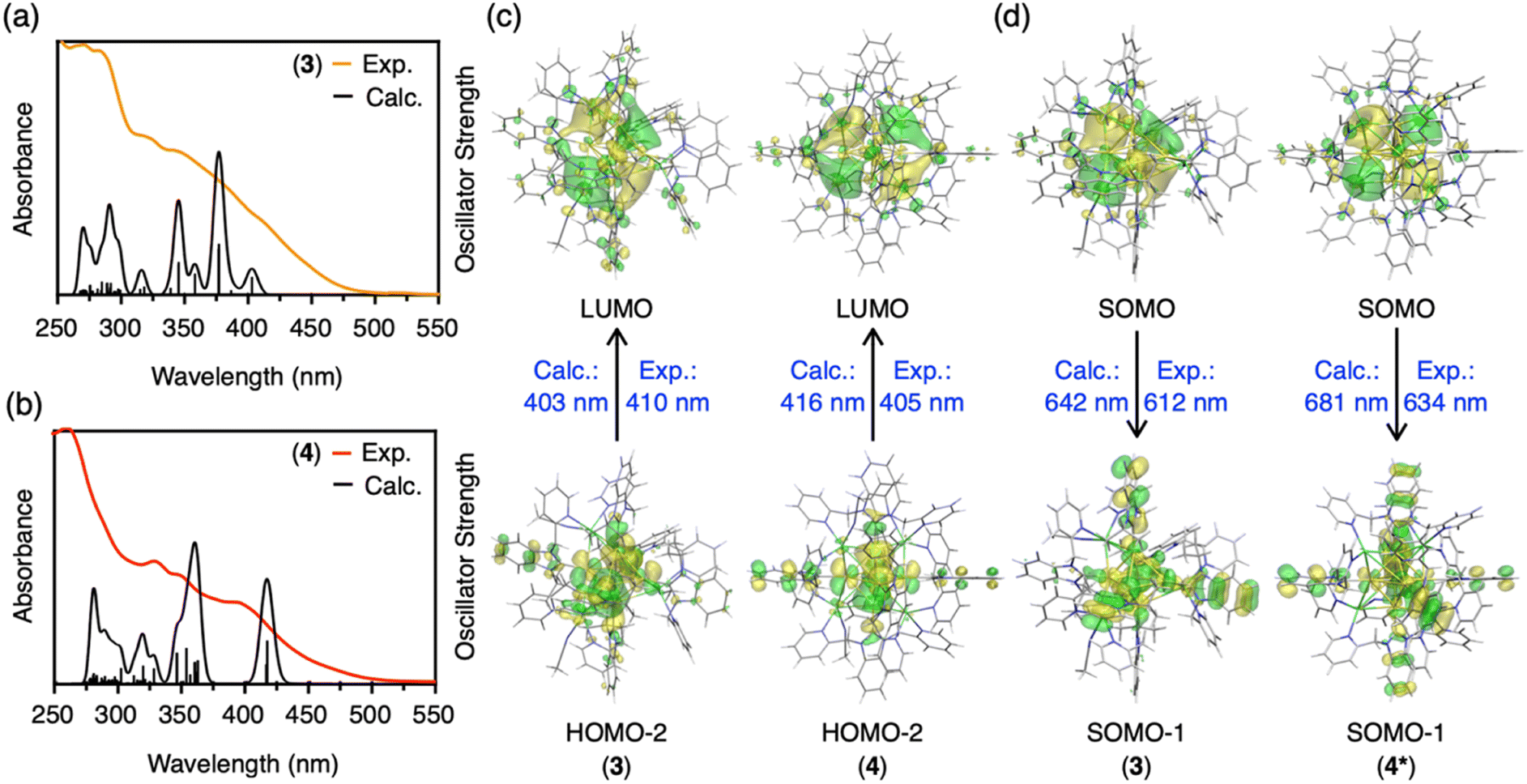 | ||
| Fig. 4 (a) Calculated and experimental UV-vis absorption spectra of cluster 3. (b) Calculated and experimental UV-vis absorption spectra of cluster 4. (c) Calculated HOMO and LUMO, and comparison of calculated and experimental excitation energies of clusters 3 and 4. (d) Calculated SOMOs (singly occupied molecular orbitals, in the triplet state), and comparisons of calculated and experimental emission energies of clusters 3 and 4*. | ||
Finally, we calculated the lowest triplet excited state to estimate the phosphorescence energies of CAuI6AgI3-type cluster 3 and CAuI6AgI4-type cluster 4, which were determined as 1.94 (642 nm) and 1.97 eV (629 nm), respectively (Fig. 4d, S56 and Table S9†). However, these results were in conflict with the experimental results, in which clusters 2–4 clearly showed a trend of red-shift. Considering that cluster 4 maintains a fairly symmetric structure in solution, as indicated by the NMR results, we removed the two acetonitrile molecules from cluster 4 and constructed a highly symmetric structure with a T point group, namely [(C)(AuI-BIPc2)6AgI4](BF4)6 (4*, Table S10 and Fig. S57, S58†). The phosphorescence energy of 4* was calculated to be 1.82 eV (681 nm), supporting a red-shift compared to cluster 3. As shown in Fig. 4d, the SOMOs of clusters 3 and 4* were mainly located in the silver atoms of the clusters, similar to the LUMOs of clusters 3 and 4. On the other hand, a large contribution from the benzimidazolylidene motif of the ligand was observed in the SOMO−1 orbitals of clusters 3 and 4*. These are very different from the occupied MOs of clusters 3 and 4, because the central CAuI6 cores are more involved in HOMO−2 etc. Thus, the calculations indicate that the contribution of the outer shells (ligands and silver atoms) in the phosphorescent relaxation process is significant. Theoretical evaluation of the phosphorescence lifetimes and radiative rate constants of 3 and 4* lead to similar conclusions that the outer shells are significantly involved in each state, which changed the coupling of the spin–orbit states of each cluster (Tables S11 and S12, for the details see ESI†).
Conclusions
By using pre-designed NHC ligands with one pyridyl, or one or two picolyl pendants, we have achieved atomically precise surface metallization of C-centered AuI6 clusters with a specific number of silver(I) atoms. It was confirmed experimentally and theoretically that the NHC–AgI shells contribute significantly to the photoluminescence properties of the resulting heterometallic clusters. Specifically, reducing rigidity will favor non-radiative pathways more, while increasing coverage and protection will efficiently suppress those pathways. This study has precisely elucidated the relationship between heterometallic cluster structure and phosphorescent properties at the atomic level. Advancements in basic design methods for highly photoluminescent metal assemblies confined in organic and organometallic shells are expected.Data availability
All the data are shown in the ESI.†Author contributions
Z. L. performed the synthesis, characterizations and analysed the data. P. Z. performed the theoretical calculations. X.-L. P. assisted in the synthesis and characterizations. H. U., M. E. and M. S. directed the study. All authors prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP21H05022 (M. S.), JP20H02718 (M. E.), JP22H05133 (M. E.), and JP22K14691 (X. P.), and MEXT KAKENHI grant number JP16H06509 (M. S.), and JST, CREST grant number JPMJCR22B2 (M. S.), Japan.Notes and references
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- N. Mirzadeh, S. H. Privér, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Chem. Rev., 2020, 120, 7551–7591 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- Q. Li, M. Zhou, W. Y. So, J. Huang, M. Li, D. R. Kauffman, M. Cotlet, T. Higaki, L. A. Peteanu, Z. Shao and R. Jin, J. Am. Chem. Soc., 2019, 141, 5314–5325 CrossRef CAS PubMed.
- K. L. D. M. Weerawardene and C. M. Aikens, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS PubMed.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and S. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS PubMed.
- X.-Y. Xie, P. Xiao, X. Cao, W.-H. Fang, G. Cui and M. Dolg, Angew. Chem., Int. Ed., 2018, 57, 9965–9969 CrossRef CAS PubMed.
- Z. Lu, Y.-J. Yang, W.-X. Ni, M. Li, Y. Zhao, Y.-L. Huang, D. Luo, X. Wang, M. A. Omary and D. Li, Chem. Sci., 2021, 12, 702–708 RSC.
- J.-S. Yang, Z. Han, X.-Y. Dong, P. Luo, H.-L. Mo and S.-Q. Zang, Angew. Chem., Int. Ed., 2020, 59, 11898–11902 CrossRef CAS PubMed.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 9934–9939 CrossRef CAS PubMed.
- C. Yao, C.-Q. Xu, I.-H. Park, M. Zhao, Z. Zhu, J. Li, X. Hai, H. Fang, Y. Zhang, G. Macam, J. Teng, L. Li, Q.-H. Xu, F.-C. Chuang, J. Lu, C. Su, J. Li and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 8270–8276 CrossRef CAS PubMed.
- B. Santiago-Gonzalez, A. Monguzzi, J. M. Azpiroz, M. Prato, S. Erratico, M. Campione, R. Lorenzi, J. Pedrini, C. Santambrogio, Y. Torrente, F. De Angelis, F. Meinardi and S. Brovelli, Science, 2016, 353, 571–575 CrossRef CAS PubMed.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- Z. Wu, Y. Du, J. Liu, Q. Yao, T. Chen, Y. Cao, H. Zhang and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 8139–8144 CrossRef CAS PubMed.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS PubMed.
- Y. Lin, P. Charchar, A. J. Christofferson, M. R. Thomas, N. Todorava, M. M. Mazo, Q. Chen, J. Doutch, R. Richardson, I. Yarovsky and M. M. Stevens, J. Am. Chem. Soc., 2018, 140, 18217–18226 CrossRef CAS PubMed.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed.
- A. M. Polgar, F. Weigend, A. Zhang, M. J. Stillman and J. F. Corrigan, J. Am. Chem. Soc., 2017, 139, 14045–14048 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- J. L. Peltier, M. Soleilhavoup, D. Martin, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2020, 142, 16479–16485 CrossRef CAS PubMed.
- H. Shen, Z. Xu, M. S. A. Hazer, Q. Wu, J. Peng, R. Qin, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 3752–3758 CrossRef CAS PubMed.
- J. Wei, J.-F. Halet, S. Kahlal, J.-Y. Saillard and A. Muñoz-Castro, Inorg. Chem., 2020, 59, 15240–15249 CrossRef CAS PubMed.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, J. Am. Chem. Soc., 2022, 144, 9000–9006 CrossRef CAS PubMed.
- H. Shen, Q. Wu, M. S. A. Hazer, X. Tang, Y.-Z. Han, R. Qin, C. Ma, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Chem, 2022, 8, 2380–2392 CAS.
- Z. Lei, X.-L. Pei, H. Ube and M. Shionoya, Bull. Chem. Soc. Jpn., 2021, 94, 1324–1330 CrossRef CAS.
- H. Ube, Q. Zhang and M. Shionoya, Organometallics, 2018, 37, 2007–2009 CrossRef CAS.
- Z. Lei, K. Nagata, H. Ube and M. Shionoya, J. Organomet. Chem., 2020, 917, 121271 CrossRef CAS.
- X.-L. Pei, P. Zhao, H. Ube, Z. Lei, K. Nagata, M. Ehara and M. Shionoya, J. Am. Chem. Soc., 2022, 144, 2156–2163 CrossRef CAS PubMed.
- Z. Lei, M. Endo, H. Ube, T. Shiraogawa, P. Zhao, K. Nagata, X.-L. Pei, T. Eguchi, T. Kamachi, M. Ehara, T. Ozawa and M. Shionoya, Nat. Commun., 2022, 13, 4288 CrossRef CAS PubMed.
- F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1988, 27, 1544–1546 CrossRef.
- H. Schmidbaur, B. Brachthäuser and O. Steigelmann, Angew. Chem., Int. Ed. Engl., 1991, 30, 1488–1490 CrossRef.
- O. D. Häberlen, H. Schmidbaur and N. Rösch, J. Am. Chem. Soc., 1994, 116, 8241–8248 CrossRef.
- F. P. Gabbaï, A. Schier, J. Riede and H. Schmidbaur, Chem. Ber., 1997, 130, 111–113 CrossRef.
- J.-H. Jia and Q.-M. Wang, J. Am. Chem. Soc., 2009, 131, 16634–16635 CrossRef CAS PubMed.
- Z. Lei and Q.-M. Wang, Coord. Chem. Rev., 2019, 378, 382–394 CrossRef CAS.
- J.-H. Jia, J.-X. Liang, Z. Lei, Z.-X. Cao and Q.-M. Wang, Chem. Commun., 2011, 47, 4739–4741 RSC.
- Y. Yang, X.-L. Pei and Q.-M. Wang, J. Am. Chem. Soc., 2013, 135, 16184–16191 CrossRef CAS PubMed.
- X.-Y. Liu, Y. Yang, Z. Lei, Z.-J. Guan and Q.-M. Wang, Chem. Commun., 2016, 52, 8022–8025 RSC.
- Z. Lei, X.-L. Pei, Z.-G. Jiang and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 12771–12775 CrossRef CAS PubMed.
- Z. Lei, X.-L. Pei, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 7117–7120 CrossRef CAS PubMed.
- J.-J. Li, C.-Y. Liu, Z.-J. Guan, Z. Lei and Q.-M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201549 CAS.
- P. Ai, M. Mauro, L. De Cola, A. A. Danopoulos and P. Braunstein, Angew. Chem., Int. Ed., 2016, 55, 3338–3341 CrossRef CAS PubMed.
- P. Ai, M. Mauro, C. Gourlaouen, S. Carrara, L. De Cola, Y. Tobon, U. Giovanella, C. Botta, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2016, 55, 8527–8542 CrossRef CAS PubMed.
- P. Ai, K. Y. Monakhov, J. van Leusen, P. Kögerler, C. Gourlaouen, M. Tromp, R. Welter, A. A. Danopoulos and P. Braunstein, Chem. - Eur. J., 2018, 24, 8787–8796 CrossRef CAS PubMed.
- A. A. A. Attia, A. M. V. Brânzanic, A. Muñoz-Castro, A. Lupan and R. B. King, Phys. Chem. Chem. Phys., 2019, 21, 17779–17785 RSC.
- P. M. Petrar, M. B. Sárosi and R. B. King, J. Phys. Chem. Lett., 2012, 3, 3335–3337 CrossRef CAS.
- Q.-F. Zhang, X. Chen and L.-S. Wang, Acc. Chem. Res., 2018, 51, 2159–2168 CrossRef CAS PubMed.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- Z. Li, H.-Y. T. Chen, K. Schouteden, T. Picot, T.-W. Liao, A. Seliverstov, C. V. Haesendonck, G. Pacchioni, E. Janssens and P. Lievens, Sci. Adv., 2020, 6, eaay4289 CrossRef CAS PubMed.
- H.-F. Zhang, M. Stender, R. Zhang, C. Wang, J. Li and L.-S. Wang, J. Phys. Chem. B, 2004, 108, 12259–12263 CrossRef CAS.
- J. Li, X. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzańska, Chem. Soc. Rev., 2014, 43, 345–380 RSC.
- H. Schmidbaur, Angew. Chem., Int. Ed., 2019, 58, 5806–5809 CrossRef CAS PubMed.
- M. Rigoulet, S. Massou, E. D. S. Carrizo, S. Mallet-Ladeira, A. Amgoune, K. Miqueu and D. Bourissou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 46–51 CrossRef CAS PubMed.
- F. Rekhroukh, L. Estevez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414–3418 CrossRef CAS PubMed.
- M. Straka, E. Andris, J. Vícha, A. Ružička, J. Roithová and L. Rulíšek, Angew. Chem., Int. Ed., 2019, 58, 2011–2016 CrossRef CAS PubMed.
- M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8, 576 CrossRef PubMed.
- O. Crespo, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, Angew. Chem., Int. Ed. Engl., 1997, 36, 993–995 CrossRef CAS.
- E. Zeller, H. Beruda and H. Schmidbaur, Inorg. Chem., 1993, 32, 3203–3204 CrossRef CAS.
- D.-e. Jiang and M. Walter, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 193402 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, characterizations, spectroscopic analysis, and crystallographic data. CCDC 2153516–2153521. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01976d |
| ‡ These authors contributed equally to this work. |
| § Crystallographic data for BIPc·HCl: C16H18N3Cl, a = 9.26934(12), b = 14.15286(15), c = 12.30478(15) Å, β = 111.3536(14)°, V = 1503.42(3) Å3, monoclinic space group P21/c, Z = 4, T = 93(2) K, 9271 reflections measured, 2931 unique (Rint = 0.0357), final R1 = 0.0366, wR2 = 0.0974 for 2797 observed reflections [I > 2σ(I)]. For BIPc2·HCl: C19H17N4Cl·H2O, a = 13.3991(2), b = 9.82400(10), c = 13.8269(2) Å, β = 105.9170(10)°, V = 1750.29(4) Å3, monoclinic space group P21/n, Z = 4, T = 93(2) K, 8598 reflections measured, 3359 unique (Rint = 0.0176), final R1 = 0.0393, wR2 = 0.1039 for 3247 observed reflections [I > 2σ(I)]. For [(C)(AuI-BIPc)6Ag3(CH3CN)3](BF4)5 (3): C103H111B5N21F20Ag3Au6, a = 28.2746(8), b = 33.2019(8), c = 29.4805(10) Å, β = 106.484(3)°, V = 26537.9(14) Å3, monoclinic space group I2/a, Z = 8, T = 93(2) K, 62923 reflections measured, 25505 unique (Rint = 0.0481), final R1 = 0.0970, wR2 = 0.2906 for 15540 observed reflections [I > 2σ(I)]. For [(C)(AuI-BIPc2)6Ag4(CH3CN)2](BF4)6 (4): C119H102B6N26F24Ag4Au6·2CH2Cl2·CH3CN, a = 35.5410(3), b = 15.07664(17), c = 50.5683(5) Å, β = 93.2056(9)°, V = 27054.0(5) Å3, monoclinic space group I2/a, Z = 8, T = 93(2) K, 74304 reflections measured, 26513 unique (Rint = 0.0522), final R1 = 0.0605, wR2 = 0.1686 for 22778 observed reflections [I > 2σ(I)]. For [(C)(AuI-BIPc)6](BF4)2 (5): C97H102B2N18F8Au6·CH2Cl2, a = 13.57251(16), b = 22.5778(2), c = 17.07448(17) Å, β = 99.9220(11)°, V = 5153.99(10) Å3, monoclinic space group P21/n, Z = 2, T = 93(2) K, 31050 reflections measured, 10136 unique (Rint = 0.0363), final R1 = 0.0496, wR2 = 0.1311 for 8499 observed reflections [I > 2σ(I)]. For [(C)(AuI-BIPc2)6](BF4)2 (6): C115H96B2N24F8Au6·CH2Cl2, a = 14.44142(13), b = 43.0804(4), c = 17.47381(12) Å, β = 92.7197(7)°, V = 10858.95(16) Å3, monoclinic space group P21/c, Z = 4, T = 93(2) K, 59653 reflections measured, 21354 unique (Rint = 0.0392), final R1 = 0.0379, wR2 = 0.0949 for 18478 observed reflections [I > 2σ(I)]. All crystallographic data have been deposited at the CCDC under deposition numbers CCDC 2153516 (for BIPc·HCl), 2153517 (for BIPc2·HCl), 2153518 (for 3), 2153519 (for 4), 2153520 (for 5), and 2153521 (for 6). |
| This journal is © The Royal Society of Chemistry 2023 |