
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCluster-selective 57Fe labeling of a Twitch-domain-containing radical SAM enzyme†
Gil
Namkoong
 and
Daniel L. M.
Suess
*
and
Daniel L. M.
Suess
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: suess@mit.edu
First published on 2nd June 2023
Abstract
57Fe-specific techniques such as Mössbauer spectroscopy are invaluable tools in mechanistic studies of Fe–S proteins. However, they remain underutilized for proteins that bind multiple Fe–S clusters because such proteins are typically uniformly enriched with 57Fe. As a result, it can be unclear which spectroscopic responses derive from which cluster, and this in turn obscures the chemistry that takes place at each cluster. Herein, we report a facile method for cluster-selective 57Fe enrichment based on exchange between the protein's Fe–S clusters and exogenous Fe ions. Through a combination of inductively coupled plasma mass spectrometric and 57Fe Mössbauer spectroscopic analysis, we show that, of the two [Fe4S4] clusters in BtrN (a Twitch-domain-containing radical S-adenosyl-L-methionine (SAM) enzyme), the Fe ions in the SAM-binding cluster undergo faster exchange with exogenous Fe2+; the auxiliary cluster is essentially inert under the reaction conditions. Exploiting this rate difference allows for either of the two [Fe4S4] clusters to be selectively labeled: the SAM-binding cluster can be labeled by exchanging unlabeled BtrN with 57Fe2+, or the auxiliary cluster can be labeled by exchanging fully labeled BtrN with natural abundance Fe2+. The labeling selectivity likely originates primarily from differences in the clusters' accessibility to small molecules, with secondary contributions from the different redox properties of the clusters. This method for cluster-selective isotopic labeling could in principle be applied to any protein that binds multiple Fe–S clusters so long as the clusters undergo exchange with exogenous Fe ions at sufficiently different rates.
Introduction
Radical S-adenosyl-L-methionine (SAM) enzymes constitute the largest superfamily of proteins and are found in all domains of life.1–3 They catalyze a variety of radical-mediated reactions, including those involved in cofactor biosynthesis, DNA repair, antibiotic biosynthesis, and nucleotide modification.1 Almost every such reaction is initiated by the generation of the 5′-deoxyadenosyl radical, which is a product of the one-electron reductive cleavage of SAM. In this reaction, the electron is supplied by an [Fe4S4] cluster bound to the highly conserved CX3CX2C motif (the “RS” cluster).1,4–11 Additionally, some radical SAM enzymes bind one or more auxiliary clusters (“Aux” clusters)3 that are in many cases catalytically essential. Although the mechanistic roles of the Aux clusters are often unknown, some Aux clusters have been shown to coordinate substrate, to donate or accept electrons, and/or to supply S atoms (Fig. 1A).12–17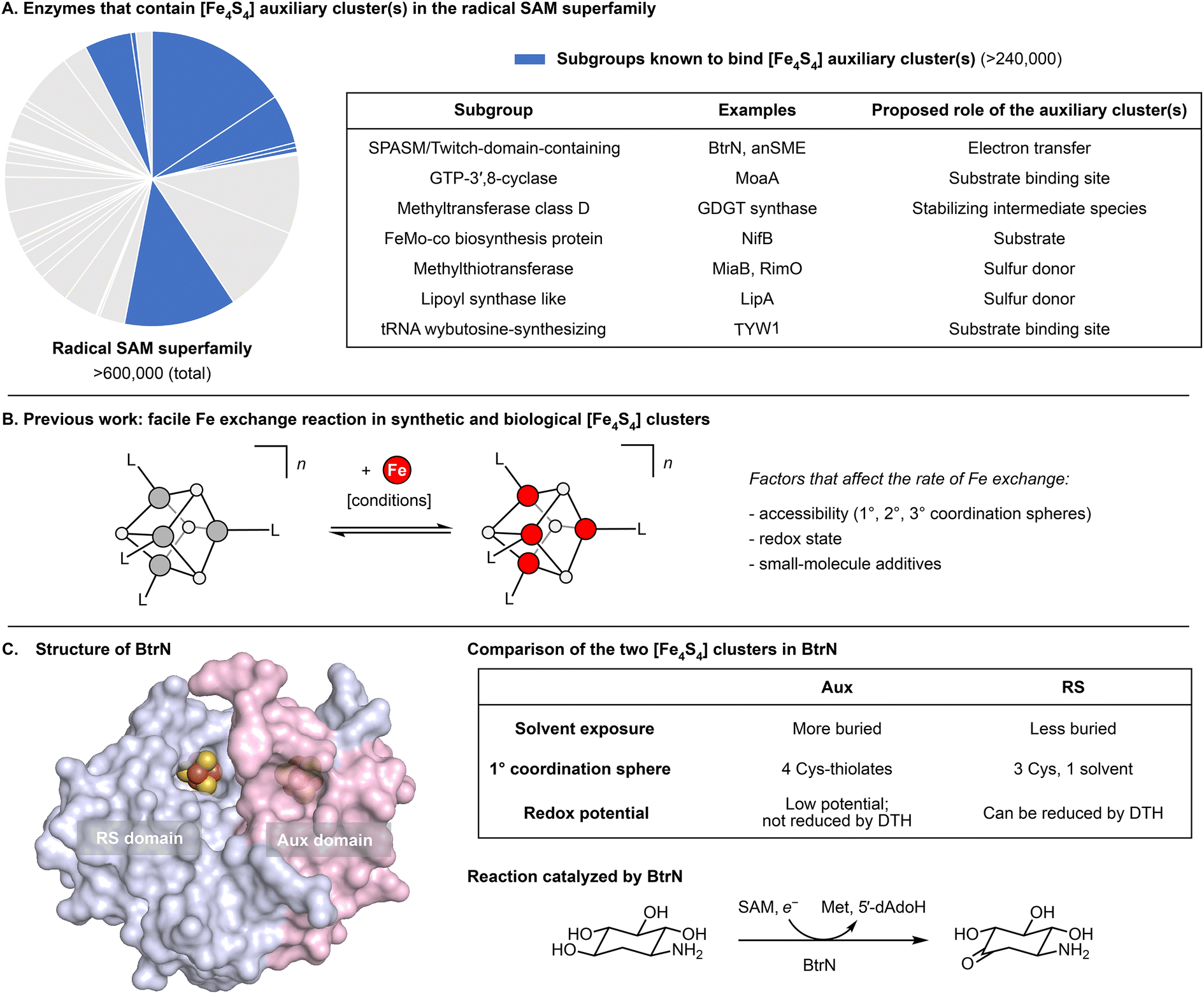 | ||
| Fig. 1 Functions and properties of selected [Fe4S4] clusters. (A) Examples of enzymes in the radical SAM superfamily that contain auxiliary [Fe4S4] cluster(s) and proposed roles of their auxiliary cluster(s).12–17 (B) Our previous work28 revealed some factors that govern the rate of exchange between [Fe4S4] clusters and exogenous Fe ions. (C) Structure and properties of BtrN, a radical SAM dehydrogenase. The surface representation was generated from PDB 4M7S.30 | ||
A substantial barrier for understanding the functions of Aux clusters in catalysis is distinguishing the spectroscopic responses arising from the Aux cluster(s) from those arising from the RS cluster. For 57Fe-based techniques (e.g., Mössbauer and electron-nuclear double resonance (ENDOR) spectroscopy), the large number of Fe sites in each cluster results in severe spectral overlap and ambiguity about spectroscopic assignments. These challenges are compounded by the presence of multiple clusters, and, for mixtures of intermediates, by the presence of multiple states of each cluster. As a consequence, 57Fe-specific spectroscopies have remained underutilized in mechanistic studies of proteins that bind multiple FeS clusters (with notable exceptions, examples of which can be found in ref. 18–23). Such complications arise because 57Fe is incorporated in uniform isotopic abundance across all clusters during protein overexpression and/or cluster reconstitution. If feasible, cluster-selective labeling—the incorporation of spectroscopically active isotopes (here, 57Fe) into a single cluster—would ameliorate these problems.
A few examples of cluster-selective labeling have been reported, including the catalytic cofactors in the [FeFe] hydrogenase24,25 and the Mo-dependent nitrogenase.26,27 In these cases, the labeling selectivity was achieved by (i) overproducing and purifying ‘partially’ apo-forms of the enzymes that are absent of a single cofactor, (ii) separately (bio)synthesizing the labeled (or unlabeled) cofactor, and (iii) inserting the cofactor into the unlabeled (or labeled) apo-enzyme. However, such approaches cannot be applied to most Fe–S proteins, which are often unstable in their apo-forms and for which there are no general mechanisms for inserting clusters into specific binding sites with high selectivity (especially when the clusters are of the same composition (e.g., two [Fe4S4] clusters)).
Herein, we report a method for cluster-selective labeling that relies on neither the isolation of apo-protein nor the insertion of preassembled cofactors, and thus could in principle apply to any Fe–S protein that binds multiple clusters. Specifically, our protocol entails exchanging cluster Fe ions with exogenous Fe, where the labeling selectivity derives from differences in exchange rates between the various clusters. This builds on a recent study28 in which we observed that the Fe exchange reaction into cuboidal Fe–S clusters is generally facile and that its rate is dependent on several factors, including the core oxidation state and the steric environment of the cluster (Fig. 1B). In the current work, we show that, even within a single protein, two clusters can exhibit dramatically different rates of Fe exchange. These insights enable the cluster-selective 57Fe labeling of BtrN, a radical SAM enzyme with a Twitch domain that harbors a single Aux cluster in addition to the conserved RS cluster,29,30 and point to general strategy for spectroscopic/mechanistic analysis of Fe–S proteins bearing multiple clusters.
Materials and methods
Materials
All reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. 57FeCl2 powder31 and 57FeCl3 (aq)32 were prepared according to literature procedures, using 57Fe powder purchased from Trace Science (95.5% 57Fe, 3.6% 56Fe).Overproduction, purification, and chemical reconstitution of BtrN
All procedures were adapted from reported protocols29,32 with minor modifications. See ESI† for experimental details.Preparation of BtrN-RS(57Fe4) and BtrN-Aux(57Fe4) samples
All procedures were carried out in an MBraun UNIlab glovebox (<5 ppm O2). To a 2 mL solution of 0.25 mM BtrN (natural isotopic abundance Fe) in 100 mM HEPES, pH 7.5, 10% glycerol, 500 mM NaCl were added 50 μL of 500 mM sodium dithionite (DTH; 50 equiv.) in the same buffer, 100 μL of 100 mM 57FeCl2 (20 equiv.) in the same buffer, and 100 μL of 500 mM dithiothretiol (DTT; 100 equiv.) in the same buffer, and incubated at room temperature for 15 min, before the resulting mixture was concentrated to approximately 0.6 mL using 10 kDa AMICON centrifugal filters. The concentrate was diluted with 1.4 mL of 100 mM HEPES, pH 7.5, 10% glycerol, 500 mM NaCl, before the same amounts of DTH, 57FeCl2, and DTT were added again. This procedure was repeated three additional times, and the final mixture was gel-filtered twice through PD-10 columns (GE Healthcare) equilibrated with 100 mM HEPES, pH 7.5, 10% glycerol, 500 mM NaCl, 2 mM DTH. The resulting eluate was concentrated to approximately 900 μL. Aliquots were taken to prepare samples for UV/vis, EPR, inductively coupled plasma mass spectrometry (ICP-MS), and Bradford and ferene assays. The remaining solution was used to prepare the Mössbauer sample. The BtrN-Aux(57Fe4) sample was prepared by following the same procedure for preparing BtrN-RS(57Fe4) sample, except that BtrN(57Fe8) and FeCl2 (natural isotopic abundance) were used.Time-course ICP-MS experiment for monitoring 57Fe incorporation
All procedures were carried out in an MBraun UNIlab glovebox (<5 ppm O2). To a solution of BtrN (100 mM HEPES, pH 7.5, 10% glycerol, 500 mM NaCl) were added 20 equiv. 57FeCl2 from a 100 mM stock solution in the same buffer and 100 equiv. DTT from a 500 mM stock solution in the same buffer. For the experiment in the DTH-reduced state, 50 equiv. DTH from a 500 mM stock solution in the same buffer was added to the protein solution and incubated at room temperature for 10 min, prior to the addition of 57FeCl2 and DTT. The resulting mixture was incubated at room temperature for 15 min, 30 min, 1 h, and 2 h before 200 μL aliquots were taken and gel-filtered through NAP-10 columns (GE Healthcare), equilibrated with 100 mM HEPES, pH 7.5, 10% glycerol, 500 mM NaCl. The resulting eluates were analyzed by ICP-MS for the 56Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :57Fe ratio.
:57Fe ratio.
Calculating the label incorporation
The label incorporation (I) of a sample obtained from an isotope exchange reaction is defined as the proportion of Fe ions in the protein substituted by exogenously added Fe. It can be calculated from the following equations using the 56Fe:57Fe ratio (r) obtained by ICP-MS (see ESI† for details).
(i) Exchange between natural abundance protein and exogenous 57Fe
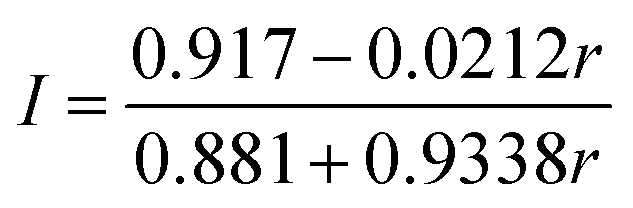 | (1) |
(ii) Exchange between BtrN(57Fe8) (57Fe enrichment = 85.2%) and natural abundance Fe
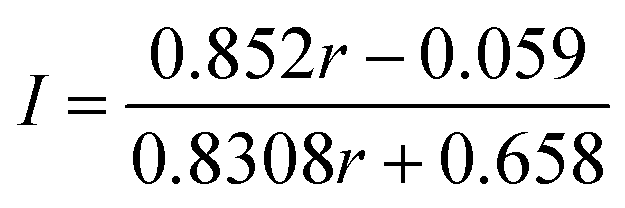 | (2) |
Mössbauer spectroscopy
57Fe Mössbauer spectra were recorded on a spectrometer from SEE Co. (formerly WEB Research Co.) operating in the constant-acceleration mode in a transmission geometry, equipped with a closed cycle He gas refrigerator cryostat from Janis (Wilmington, MA). The quoted isomer shifts are relative to the centroid of the spectrum of a metallic foil of α-Fe at room temperature. Data were collected on frozen solutions. Samples were transferred to a Delrin sample cup in a 50 mL conical tube in an anaerobic glovebox (<5 ppm O2) and frozen in liquid N2 outside the glovebox. Each sample contains 800–900 μL of 0.2–0.4 mM BtrN. Data analysis was performed using version 4 of the program WMOSS (https://www.wmoss.org)33 or using the linear least-squared fitting function (lsqcurvefit) in version R2020b of MATLAB.34,35 Data were fitted using Lorentzian lineshapes (see ESI† for simulation details).ICP-MS28,34,35
ICP-MS data were recorded on an Agilent 7900 ICP-MS instrument. Samples were prepared by first digesting the concentrated sample in 70% nitric acid (TraceMetal Grade, Fischer) at 60 °C, and then diluting it with Milli-Q water such that the final concentration of nitric acid is 2%. Standards for 56Fe were prepared from 1000 ppm Fe standard solution (SPEX Certiprep). Standards for 57Fe were prepared by dissolving 57Fe powder (Trace Science) in concentrated nitric acid. The concentrations of 56Fe and 57Fe in the standard solutions were based on the natural abundance of each isotope in the unenriched standard (91.7% 56Fe, 2.12% 57Fe) and the isotope enrichment in 57Fe powder (95.5% 57Fe, 3.6% 56Fe). All samples and standards contained 1 ppb Tb (final concentration) as an internal standard.Results and discussion
BtrN catalyzes the oxidation of 2-deoxy-scyllo-inosamine to a ketone in butirosin biosynthesis (Fig. 1C).36,37 In addition to a canonical RS cluster, BtrN harbors a catalytically essential [Fe4S4] Aux cluster (Fig. 1C) that has been proposed to play a role in electron transfer.29,30,38 Whereas the Aux cluster has Cys4 ligation, the RS cluster is ligated by only three cysteinyl groups, leaving its ‘unique’ Fe to bind SAM.30 The fact that the RS cluster binds SAM suggests that it may be more accessible to small molecules than the Aux cluster. Additionally, the RS cluster can be selectively reduced to the [Fe4S4]+ state, leaving the Aux cluster in the [Fe4S4]2+ state.29,38 On the basis of our previous work,28 we hypothesized that both factors—the differences in the accessibility and in the redox properties of the two clusters—would result in faster exchange between the RS cluster and added Fe2+, and that this could be exploited for cluster-selective labeling.We began by preparing two samples with uniform isotopic enrichment: unlabeled BtrN and fully labeled BtrN (BtrN(57Fe8)), each generated following reported methods29,32 for protein overexpression and cluster reconstitution using natural-abundance or 57Fe-enriched Fe sources, respectively (Fig. 2). We then incubated the unlabeled BtrN sample with 20 equiv. 57FeCl2, 100 equiv. DTT, and 50 equiv. DTH for 15 min. As shown below, these conditions result in exchange between the natural-abundance Fe in BtrN and the 57Fe2+ in solution. The small molecules—including the liberated natural-abundance Fe2+—were partly removed by concentrating the mixture four-fold using spin filters with a 10 kDa cutoff. The solution was then diluted to the original volume, and 57FeCl2, DTT, and DTH were re-introduced (see Materials and methods for details). This procedure was performed a total of four times, resulting in iterative 57Fe exchange into BtrN. Based on evidence discussed below, we found that exogenous 57Fe washed into only the RS cluster, giving the selectively labeled sample, BtrN-RS(57Fe4).
 | ||
| Fig. 2 Cluster-selective (un)labeling of BtrN and each cluster's 57Fe content obtained from ICP-MS and Mössbauer analysis. BtrN in natural isotopic abundance or 57Fe-enriched (BtrN(57Fe8)) were prepared by reported procedures.29,32 BtrN-RS(57Fe4) and BtrN-Aux(57Fe4) samples were prepared as described in the text. Red and gray balls are 57Fe and natural-abundance Fe, respectively (note that although we represent the RS cluster in the BtrN-Aux(57Fe4) sample with gray balls, these sites contain some residual 57Fe). The 57Fe content of each cluster in BtrN-RS(57Fe4) and BtrN-Aux(57Fe4) samples is calculated from the 56Fe:57Fe ratio assuming 100% exchange selectivity for the RS cluster. | ||
A sample with 57Fe in only the Aux cluster (BtrN-Aux(57Fe4)) was generated by an identical procedure except beginning with BtrN(57Fe8) and replacing 57FeCl2 with natural-abundance FeCl2; here, the natural-abundance exogenous FeCl2 effectively ‘unlabels’ the RS cluster, leaving the Aux cluster enriched. Note that, consistent with our previous study,28 these Fe exchange procedures did not alter the total cluster content as evidenced by the similar UV/vis spectra (ESI Fig. S1†), EPR signal intensities (ESI Fig. S2†), and total Fe content (ESI Table S1†) of the pre- vs. post-exchange samples.
ICP-MS analysis of both samples, combined with Mössbauer spectroscopic analysis (vide infra), is consistent with only a single cluster being exchanged with the added mononuclear Fe. The BtrN-RS(57Fe4) sample has a 56Fe:57Fe ratio of 59:41, which corresponds to a label incorporation of 40% (eqn (1)), meaning that 40% of the total Fe sites in the protein has exchanged with the exogenous pool of Fe. Likewise, the 56Fe:57Fe ratio for the BtrN-Aux(57Fe4) sample (40:60) corresponds to a label incorporation of 42% (eqn (2)). Although these values do not directly inform on the cluster-selectivity of the exchange—the modest yield could instead be the result of unselective, low-yielding exchange—the observation that both values do not exceed 50% is at least consistent with exchange at only one cluster.
For the Mössbauer spectroscopic analysis, the three samples—BtrN(57Fe8), BtrN-RS(57Fe4), and BtrN-Aux(57Fe4)—were reduced with DTH to poise the RS cluster in the [Fe4S4]+ state while leaving the Aux cluster in the [Fe4S4]2+ state;29,38 as shown below, the difference in the core charges of the two clusters affords sufficient resolution to determine the selectivity of the label incorporation by Mössbauer spectroscopy. Additionally, all Mössbauer spectra were recorded under identical conditions (80 K, zero field) and simultaneously simulated34,35 using the 56Fe:57Fe ratios determined from ICP-MS (see further discussion below, Table 1, and the ESI† for the simulation details).
| Component | δ (mm s−1) | |ΔEQ| (mm s−1) | Γ (mm s−1) | Area ratio | |
|---|---|---|---|---|---|
|
a Simulations were performed by fitting a common set of parameters (δ, |ΔEQ|, Γ, and area ratio) for all three samples. For the BtrN-RS(57Fe4) and BtrN-Aux(57Fe4) samples, the labeling selectivity was fit using the 56Fe:57Fe ratios obtained by ICP-MS analysis. See ESI for simulation details.
|
|||||
| RS [Fe4S4]+ | Fe2+ | 0.61 | 1.68 | 0.35 | 1 |
| Fe2+ | 0.63 | 1.02 | 0.30 | 1 | |
| Fe2.5+ | 0.45 | 1.00 | 0.31 | 2 | |
| Aux [Fe4S4]2+ | Fe2.5+ | 0.45 | 0.79 | 0.30 | 1 |
| Fe2.5+ | 0.43 | 1.22 | 0.30 | 3 | |
We begin our analysis by describing the qualitative features of the spectra. The Mössbauer spectrum of the BtrN(57Fe8) sample has been previously reported with both clusters poised in the [Fe4S4]2+ state.29 For our DTH-reduced sample, the spectrum (Fig. 3, “1”) was fit in the global simulation to five quadrupole doublets representing eight Fe sites that arise from the RS cluster in the [Fe4S4]+ state (1:1:2 ratio of individual components) and the Aux cluster in the [Fe4S4]2+ state (1:3 ratio of individual components, consistent with previous simulations29); the parameters are provided in Table 1 and are typical for [Fe4S4] enzymes.1,39 The shoulder at ∼1.4 mm s−1 arises from one of the Fe2+ sites of the [Fe4S4]+ RS cluster, and this shoulder is strikingly more pronounced in the BtrN-RS(57Fe4) spectrum (Fig. 3A, “2”), suggesting that the [Fe4S4]+ RS cluster in the BtrN-RS(57Fe4) sample is enriched with 57Fe relative to the Aux cluster. Indeed, subtracting the BtrN-RS(57Fe4) spectrum from the BtrN(57Fe8) spectrum (with the total area of the two spectra normalized to be equal) reveals that the BtrN-RS(57Fe4) spectrum has a higher average isomer shift, consistent with the [Fe4S4]+ RS cluster being selectively enriched (Fig. 3A, “1–2”). In contrast, in the Mössbauer spectrum of BtrN-Aux(57Fe4), the shoulder at ∼1.4 mm s−1 resulting from the Fe2+ site in the reduced RS cluster is diminished in intensity (Fig. 3B, “3”), qualitatively suggesting that the 57Fe now mostly resides in the [Fe4S4]2+ Aux cluster.
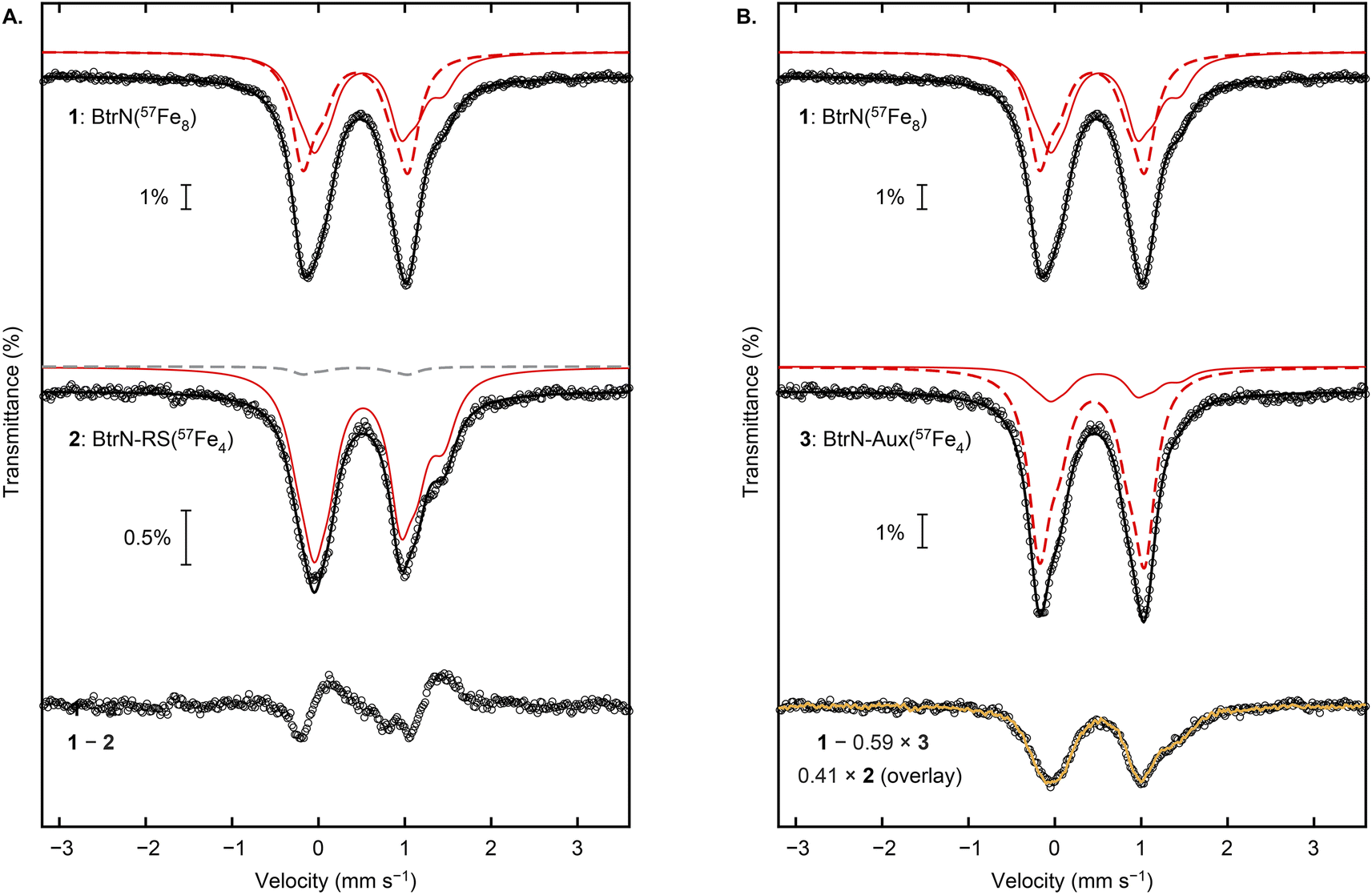 | ||
| Fig. 3 Mössbauer spectroscopic analysis of BtrN(57Fe8), BtrN-RS(57Fe4), and BtrN-Aux(57Fe4) at zero-field, 80 K. (A) Comparison of BtrN(57Fe8) and BtrN-RS(57Fe4) spectra and the difference spectrum. (B) Comparison of BtrN(57Fe8) and BtrN-Aux(57Fe4) spectra and the difference spectrum, overlayed with the BtrN-RS(57Fe4) spectrum (normalized as shown in the figure and as described in the text). Experimental data (black circles), total simulations (solid black line), simulation for the RS [Fe4S4]+ cluster enriched with 57Fe (solid red line), simulation for the Aux [Fe4S4]2+ cluster enriched with 57Fe (dashed red line), simulation for the Aux [Fe4S4]2+ cluster in natural isotopic abundance (dashed gray line), overlay of BtrN-RS(57Fe4) experimental data (yellow trace). Small contributions from adventitious Fe2+ were subtracted from spectra 1 and 2 for clarity (raw spectra are presented in ESI Fig. S3–S5†). | ||
Further evidence for high labeling selectivity is presented in Fig. 3B. If 100% exchange selectivity for the RS cluster is assumed, then, based on the ICP-MS numbers, the RS cluster in the BtrN-Aux(57Fe4) sample has lost 82% of its 57Fe via exchange with exogenous, natural-abundance Fe2+. Therefore, subtraction of the BtrN-Aux(57Fe4) spectrum from the BtrN(57Fe8) spectrum, where the area of the former is normalized to 59% of the latter (50% contribution from the Aux cluster plus 9% contribution from the RS cluster ((100 − 82) ÷ 2%)), should give a difference spectrum with a spectroscopic response from only the RS cluster. Indeed, the difference spectrum (black circles, Fig. 3B, “1 − 0.59 × 3”) is indistinguishable from the BtrN-RS(57Fe4) spectrum (yellow trace, Fig. 3B, “0.41 × 2 (overlay)”), further supporting the high selectivity of our labeling protocol.
In the global simulation, we fixed the label incorporation to the values determined by ICP-MS analysis (vide supra) and fit the cluster selectivity for (un)labeling the two clusters (see ESI† for the simulation details). The outcome revealed 99% selectivity for incorporating 57Fe into the RS cluster in the conversion of BtrN to BtrN-RS(57Fe4), and 97% selectivity for incorporating natural abundance Fe into the RS cluster in the conversion of BtrN-(57Fe8) to BtrN-Aux(57Fe4). Based on this analysis, as well as the qualitative observations of the Mössbauer spectra described above, we conclude that exogenous Fe2+ exchanges essentially exclusively with the RS cluster.
To gain additional insights into the factors that control the selectivity of cluster labeling, we tested whether the redox state of the RS cluster affects the rate of exchange with exogenous Fe. Specifically, we mixed BtrN with 20 equiv. 57FeCl2 and 100 equiv. DTT in the presence or absence of 50 equiv. DTH. We then took aliquots at various time points, removed the small molecules by gel-filtration, and studied the 56Fe:57Fe ratio by ICP-MS. Note that the reaction conditions and the redox state of the RS cluster ([Fe4S4]2+) in the –DTH sample are identical to those used for labeling RlmN in our previous study.28
For the –DTH BtrN sample (Fig. 4, circles with dashed line), we found the label incorporation to be 23% after 2 h (over eight Fe sites, which is equivalent to 46% over four sites if 100% selectivity for the RS cluster is assumed). A somewhat higher label incorporation of 32% (equivalent to 64% over four sites) was observed after 2 h for the +DTH sample (Fig. 4, crosses with solid line), consistent with our findings in synthetic Fe–S clusters28 that reducing Fe–S clusters accelerates the rate of Fe exchange. Nevertheless, the finding that exchange with the RS cluster still occurs to an appreciable extent in the –DTH sample (i.e., when the RS cluster is oxidized) suggests that the difference in the Fe exchange rate between the two clusters—and therefore the origins of cluster-selective labeling—results primarily from differences in the degree of cluster accessibility, and that the selective reduction of the RS cluster is a secondary effect that accelerates the exchange process. The slower exchange for the RS cluster in BtrN compared with RlmN (for which, under identical conditions, we observed ca. 80% incorporation over four sites after 3 h incubation with the cluster in the [Fe4S4]2+ state)28 indirectly lends further credence to this proposal because RlmN likely has a more accessible active site owing to its substrate being a macromolecule40–43 compared with BtrN, whose substrate is a small molecule.
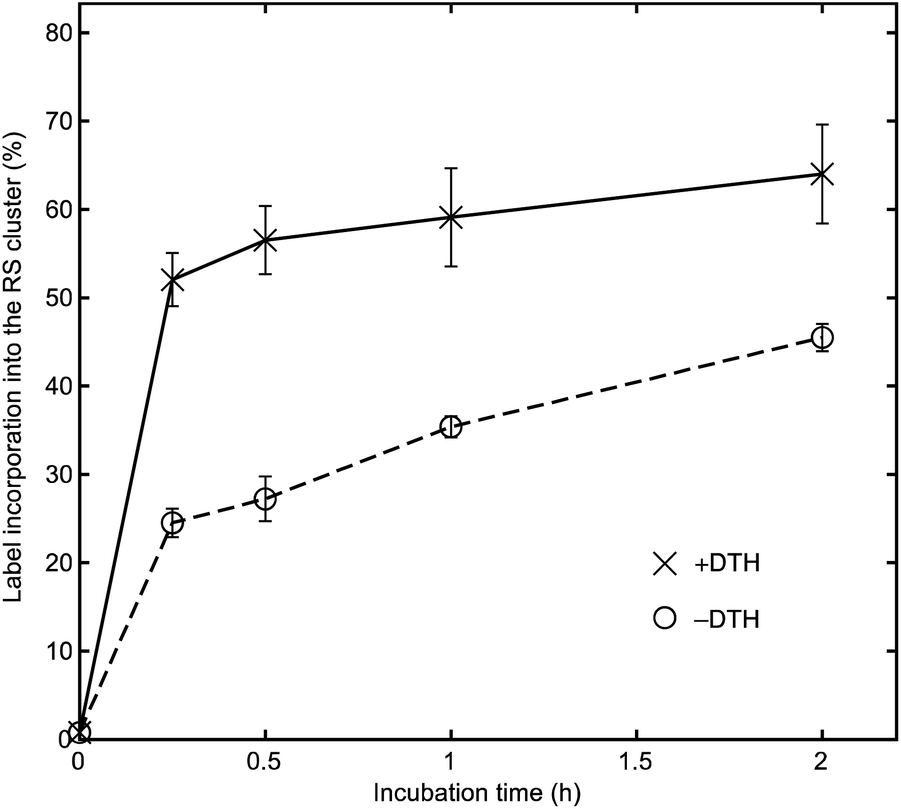 | ||
| Fig. 4 Monitoring incorporation of 57Fe into the RS cluster of BtrN (assuming 100% cluster selectivity) in the presence of 20 equiv. 57FeCl2 and 100 equiv. DTT, in the presence (crosses with solid line) or absence (circles with dashed line) of 50 equiv. DTH. Error bars represent uncertainty propagated from the ICP-MS measurements. | ||
Conclusion
Using BtrN as test case, we have demonstrated that Fe ion exchange occurs at different rates for different clusters within a protein scaffold and that this property can be exploited to enable cluster-selective 57Fe labeling for proteins that contain multiple Fe–S clusters. The quantitative selectivity for (un)labeling the RS cluster can be attributed to differences in the local environments of the clusters, including their accessibility, site-differentiation, and, to a lesser extent, redox properties. We anticipate this method will be applicable to spectroscopic and mechanistic studies of any Fe–S protein bearing multiple clusters (so long as the clusters' rates of Fe ion exchange are sufficiently different), including many of the ∼40% of the members of the radical SAM superfamily that are thought to contain [Fe4S4] Aux cluster(s) in addition to the canonical RS cluster (Fig. 1A).2,3Author contributions
G. N. and D. L. M. S. designed the research; G. N. conducted the experiments; G. N. and D. L. M. S. analyzed the data; and G. N. and D. L. M. S. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Squire Booker and Matthew Radle (Pennsylvania State University) for providing the plasmids used in this work. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM141203). Support for the ICP-MS instrument was provided by a core center grant P30-ES002109 from the National Institute of Environmental Health Sciences of the National Institutes of Health.Notes and references
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Radical S-Adenosylmethionine Enzymes, Chem. Rev., 2014, 114(8), 4229–4317, DOI:10.1021/cr4004709.
- G. L. Holliday, E. Akiva, E. C. Meng, S. D. Brown, S. Calhoun, U. Pieper, A. Sali, S. J. Booker and P. C. Babbitt, Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain, Methods Enzymol., 2018, 606, 1–71, DOI:10.1016/bs.mie.2018.06.004.
- N. Oberg, T. W. Precord, D. A. Mitchell and J. A. Gerlt, RadicalSAM.Org: A Resource to Interpret Sequence-Function Space and Discover New Radical SAM Enzyme Chemistry, ACS Bio Med Chem Au, 2022, 2(1), 22–35, DOI:10.1021/acsbiomedchemau.1c00048.
- C. J. Walsby, D. Ortillo, W. E. Broderick, J. B. Broderick and B. M. Hoffman, An Anchoring Role for FeS Clusters: Chelation of the Amino Acid Moiety of S-Adenosylmethionine to the Unique Iron Site of the [4Fe−4S] Cluster of Pyruvate Formate-Lyase Activating Enzyme, J. Am. Chem. Soc., 2002, 124(38), 11270–11271, DOI:10.1021/ja027078v.
- C. Krebs, W. E. Broderick, T. F. Henshaw, J. B. Broderick and B. H. Huynh, Coordination of Adenosylmethionine to a Unique Iron Site of the [4Fe-4S] of Pyruvate Formate-Lyase Activating Enzyme: A Mössbauer Spectroscopic Study, J. Am. Chem. Soc., 2002, 124(6), 912–913, DOI:10.1021/ja017562i.
- G. Layer, J. Moser, D. W. Heinz, D. Jahn and W.-D. Schubert, Crystal Structure of Coproporphyrinogen III Oxidase Reveals Cofactor Geometry of Radical SAM Enzymes, EMBO J., 2003, 22(23), 6214–6224, DOI:10.1093/emboj/cdg598.
- M. Horitani, K. Shisler, W. E. Broderick, R. U. Hutcheson, K. S. Duschene, A. R. Marts, B. M. Hoffman and J. B. Broderick, Radical SAM Catalysis via an Organometallic Intermediate with an Fe–[5′-C]-Deoxyadenosyl Bond, Science, 2016, 352(6287), 822–825, DOI:10.1126/science.aaf5327.
- A. S. Byer, H. Yang, E. C. McDaniel, V. Kathiresan, S. Impano, A. Pagnier, H. Watts, C. Denler, A. L. Vagstad, J. Piel, K. S. Duschene, E. M. Shepard, T. P. Shields, L. G. Scott, E. A. Lilla, K. Yokoyama, W. E. Broderick, B. M. Hoffman and J. B. Broderick, Paradigm Shift for Radical S-Adenosyl-I-Methionine Reactions: The Organometallic Intermediate Ω Is Central to Catalysis, J. Am. Chem. Soc., 2018, 140(28), 8634–8638, DOI:10.1021/jacs.8b04061.
- W. E. Broderick, B. M. Hoffman and J. B. Broderick, Mechanism of Radical Initiation in the Radical S-Adenosyl-I-Methionine Superfamily, Acc. Chem. Res., 2018, 51(11), 2611–2619, DOI:10.1021/acs.accounts.8b00356.
- R. I. Sayler, T. A. Stich, S. Joshi, N. Cooper, J. T. Shaw, T. P. Begley, D. J. Tantillo and R. D. Britt, Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate, ACS Cent. Sci., 2019, 5(11), 1777–1785, DOI:10.1021/acscentsci.9b00706.
- M. N. Lundahl, R. Sarksian, H. Yang, R. J. Jodts, A. Pagnier, D. F. Smith, M. A. Mosquera, W. A. van der Donk, B. M. Hoffman, W. E. Broderick and J. B. Broderick, Mechanism of Radical S-Adenosyl-I-Methionine Adenosylation: Radical Intermediates and the Catalytic Competence of the 5′-Deoxyadenosyl Radical, J. Am. Chem. Soc., 2022, 144(11), 5087–5098, DOI:10.1021/jacs.1c13706.
- N. D. Lanz and S. J. Booker, Identification and Function of Auxiliary Iron–Sulfur Clusters in Radical SAM Enzymes, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824(11), 1196–1212, DOI:10.1016/j.bbapap.2012.07.009.
- N. D. Lanz and S. J. Booker, Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853(6), 1316–1334, DOI:10.1016/j.bbamcr.2015.01.002.
- T. A. J. Grell, P. J. Goldman and C. L. Drennan, SPASM and Twitch Domains in S-Adenosylmethionine (SAM) Radical Enzymes, J. Biol. Chem., 2015, 290(7), 3964–3971, DOI:10.1074/jbc.R114.581249.
- E. Mulliez, V. Duarte, S. Arragain, M. Fontecave and M. Atta, On the Role of Additional [4Fe-4S] Clusters with a Free Coordination Site in Radical-SAM Enzymes, Front. Chem., 2017, 5 DOI:10.3389/fchem.2017.00017.
- A. R. Balo, L. Tao and R. D. Britt, Characterizing SPASM/Twitch Domain-Containing Radical SAM Enzymes by EPR Spectroscopy, Appl. Magn. Reson., 2022, 53(3–5), 809–820, DOI:10.1007/s00723-021-01406-2.
- C. T. Lloyd, D. F. Iwig, B. Wang, M. Cossu, W. W. Metcalf, A. K. Boal and S. J. Booker, Discovery, Structure and Mechanism of a Tetraether Lipid Synthase, Nature, 2022, 609(7925), 197–203, DOI:10.1038/s41586-022-05120-2.
- N. D. Lanz, M.-E. Pandelia, E. S. Kakar, K.-H. Lee, C. Krebs and S. J. Booker, Evidence for a Catalytically and Kinetically Competent Enzyme–Substrate Cross-Linked Intermediate in Catalysis by Lipoyl Synthase, Biochemistry, 2014, 53(28), 4557–4572, DOI:10.1021/bi500432r.
- B. Zhang, A. J. Arcinas, M. I. Radle, A. Silakov, S. J. Booker and C. Krebs, First Step in Catalysis of the Radical S-Adenosylmethionine Methylthiotransferase MiaB Yields an Intermediate with a [3Fe-4S]0-Like Auxiliary Cluster, J. Am. Chem. Soc., 2020, 142(4), 1911–1924, DOI:10.1021/jacs.9b11093.
- N. B. Ugulava, K. K. Surerus and J. T. Jarrett, Evidence from Mössbauer Spectroscopy for Distinct [2Fe-2S]2+ and [4Fe-4S]2+ Cluster Binding Sites in Biotin Synthase from Escherichia coli, J. Am. Chem. Soc., 2002, 124(31), 9050–9051, DOI:10.1021/ja027004j.
- M. M. Cosper, G. N. L. Jameson, H. L. Hernández, C. Krebs, B. H. Huynh and M. K. Johnson, Characterization of the Cofactor Composition of Escherichia coli Biotin Synthase, Biochemistry, 2004, 43(7), 2007–2021, DOI:10.1021/bi0356653.
- M.-E. Pandelia, D. Bykov, R. Izsak, P. Infossi, M.-T. Giudici-Orticoni, E. Bill, F. Neese and W. Lubitz, Electronic Structure of the Unique [4Fe-3S] Cluster in O2-Tolerant Hydrogenases Characterized by 57Fe Mossbauer and EPR Spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(2), 483–488, DOI:10.1073/pnas.1202575110.
- V. Kathirvelu, P. Perche-Letuvée, J.-M. Latour, M. Atta, F. Forouhar, S. Gambarelli and R. Garcia-Serres, Spectroscopic Evidence for Cofactor–Substrate Interaction in the Radical-SAM Enzyme TYW1, Dalton Trans., 2017, 46(39), 13211–13219, 10.1039/C7DT00736A.
- J. M. Kuchenreuther, Y. Guo, H. Wang, W. K. Myers, S. J. George, C. A. Boyke, Y. Yoda, E. E. Alp, J. Zhao, R. D. Britt, J. R. Swartz and S. P. Cramer, Nuclear Resonance Vibrational Spectroscopy and Electron Paramagnetic Resonance Spectroscopy of 57Fe-Enriched [FeFe] Hydrogenase Indicate Stepwise Assembly of the H-Cluster, Biochemistry, 2013, 52(5), 818–826, DOI:10.1021/bi301336r.
- R. Gilbert-Wilson, J. F. Siebel, A. Adamska-Venkatesh, C. C. Pham, E. Reijerse, H. Wang, S. P. Cramer, W. Lubitz and T. B. Rauchfuss, Spectroscopic Investigations of [FeFe] Hydrogenase Maturated with [57Fe2(Adt)(CN)2(CO)4]2−, J. Am. Chem. Soc., 2015, 137(28), 8998–9005, DOI:10.1021/jacs.5b03270.
- P. A. McLean, V. Papaefthymiou, W. H. Orme-Johnson and E. Münck, Isotopic Hybrids of Nitrogenase. Mössbauer Study of MoFe Protein with Selective 57Fe Enrichment of the P-Cluster, J. Biol. Chem., 1987, 262(27), 12900–12903, DOI:10.1016/S0021-9258(18)45141-1.
- S. J. Yoo, H. C. Angove, V. Papaefthymiou, B. K. Burgess and E. Münck, Mössbauer Study of the MoFe Protein of Nitrogenase from Azotobacter vinelandii Using Selective 57Fe Enrichment of the M-Centers, J. Am. Chem. Soc., 2000, 122(20), 4926–4936, DOI:10.1021/ja000254k.
- N. B. Thompson, G. Namkoong, B. A. Skeel and D. L. M. Suess, Facile and Dynamic Cleavage of Every Iron–Sulfide Bond in Cuboidal Iron–Sulfur Clusters, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(6), e2210528120, DOI:10.1073/pnas.2210528120.
- T. L. Grove, J. H. Ahlum, P. Sharma, C. Krebs and S. J. Booker, A Consensus Mechanism for Radical SAM-Dependent Dehydrogenation? BtrN Contains Two [4Fe-4S] Clusters, Biochemistry, 2010, 49(18), 3783–3785, DOI:10.1021/bi9022126.
- P. J. Goldman, T. L. Grove, S. J. Booker and C. L. Drennan, X-Ray Analysis of Butirosin Biosynthetic Enzyme BtrN Redefines Structural Motifs for AdoMet Radical Chemistry, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(40), 15949–15954, DOI:10.1073/pnas.1312228110.
- T. C. Berto, M. B. Hoffman, Y. Murata, K. B. Landenberger, E. E. Alp, J. Zhao and N. Lehnert, Structural and Electronic Characterization of Non-Heme Fe(II)–Nitrosyls as Biomimetic Models of the FeB Center of Bacterial Nitric Oxide Reductase, J. Am. Chem. Soc., 2011, 133(42), 16714–16717, DOI:10.1021/ja111693f.
- N. D. Lanz, T. L. Grove, C. B. Gogonea, K.-H. Lee, C. Krebs and S. J. Booker, RlmN and AtsB as Models for the Overproduction and Characterization of Radical SAM Proteins, Methods Enzymol., 2012, 516, 125–152, DOI:10.1016/B978-0-12-394291-3.00030-7.
- Prisecaru, I., WMOSS4 Mössbauer Spectral Analysis Software, 2009-2016, http://www.wmoss.org.
- S. Srisantitham, E. D. Badding and D. L. M. Suess, Postbiosynthetic Modification of a Precursor to the Nitrogenase Iron–Molybdenum Cofactor, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(11), e2015361118, DOI:10.1073/pnas.2015361118.
- E. D. Badding, S. Srisantitham, D. A. Lukoyanov, B. M. Hoffman and D. L. M. Suess, Connecting the Geometric and Electronic Structures of the Nitrogenase Iron–Molybdenum Cofactor through Site-Selective 57Fe Labelling, Nat. Chem., 2023, 15(5), 658–665, DOI:10.1038/s41557-023-01154-9.
- K. Yokoyama, M. Numakura, F. Kudo, D. Ohmori and T. Eguchi, Characterization and Mechanistic Study of a Radical SAM Dehydrogenase in the Biosynthesis of Butirosin, J. Am. Chem. Soc., 2007, 129(49), 15147–15155, DOI:10.1021/ja072481t.
- K. Yokoyama, D. Ohmori, F. Kudo and T. Eguchi, Mechanistic Study on the Reaction of a Radical SAM Dehydrogenase BtrN by Electron Paramagnetic Resonance Spectroscopy, Biochemistry, 2008, 47(34), 8950–8960, DOI:10.1021/bi800509x.
- S. J. Maiocco, T. L. Grove, S. J. Booker and S. J. Elliott, Electrochemical Resolution of the [4Fe-4S] Centers of the AdoMet Radical Enzyme BtrN: Evidence of Proton Coupling and an Unusual, Low-Potential Auxiliary Cluster, J. Am. Chem. Soc., 2015, 137(27), 8664–8667, DOI:10.1021/jacs.5b03384.
- M.-E. Pandelia, N. D. Lanz, S. J. Booker and C. Krebs, Mössbauer Spectroscopy of Fe/S Proteins, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853(6), 1395–1405, DOI:10.1016/j.bbamcr.2014.12.005.
- S.-M. Toh, L. Xiong, T. Bae and A. S. Mankin, The Methyltransferase YfgB/RlmN Is Responsible for Modification of Adenosine 2503 in 23S rRNA, RNA, 2007, 14(1), 98–106, DOI:10.1261/rna.814408.
- A. K. Boal, T. L. Grove, M. I. McLaughlin, N. H. Yennawar, S. J. Booker and A. C. Rosenzweig, Structural Basis for Methyl Transfer by a Radical SAM Enzyme, Science, 2011, 332(6033), 1089–1092, DOI:10.1126/science.1205358.
- A. Benitez-Paez, M. Villarroya and M. E. Armengod, The Escherichia coli RlmN Methyltransferase Is a Dual-Specificity Enzyme That Modifies Both rRNA and tRNA and Controls Translational Accuracy, RNA, 2012, 18(10), 1783–1795, DOI:10.1261/rna.033266.112.
- E. L. Schwalm, T. L. Grove, S. J. Booker and A. K. Boal, Crystallographic Capture of a Radical S-Adenosylmethionine Enzyme in the Act of Modifying tRNA, Science, 2016, 352(6283), 309–312, DOI:10.1126/science.aad5367.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02016a |
| This journal is © The Royal Society of Chemistry 2023 |