
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycolipids from the gut symbiont Bacteroides fragilis are agonists for natural killer T cells and induce their regulatory differentiation†
Garth
Cameron‡
 a,
Tram
Nguyen‡
bc,
Marcin
Ciula
a,
Spencer J.
Williams
*bc and
Dale I.
Godfrey
*a
a,
Tram
Nguyen‡
bc,
Marcin
Ciula
a,
Spencer J.
Williams
*bc and
Dale I.
Godfrey
*a
aDepartment of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Melbourne, VIC 3000, Australia. E-mail: godfrey@unimelb.edu.au
bSchool of Chemistry, University of Melbourne, Parkville, VIC 3010, Australia. E-mail: sjwill@unimelb.edu.au
cBio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, VIC 3010, Australia
First published on 27th May 2023
Abstract
Natural Killer T (NKT) cells are a lipid-antigen reactive T cell subset that is restricted to the antigen presenting molecule CD1d. They possess diverse functional properties that contribute to inflammatory and regulatory immune responses. The most studied lipid antigen target for these T cells is α-galactosylceramide (αGC). The commensal organism Bacteroides fragilis (B. fragilis) produces several forms of αGC, but conflicting information exists about the influence of these lipids on NKT cells. Herein, we report the total synthesis of a major form of αGC from B. fragilis (Bf αGC), and several analogues thereof. We confirm the T cell receptor (TCR)-mediated recognition of these glycolipids by mouse and human NKT cells. Despite the natural structure of Bf αGC containing lipid branching that limits potency, we demonstrate that Bf αGC drives mouse NKT cells to proliferate and differentiate into producers of the immunoregulatory cytokine, interleukin-10 (IL-10). These Bf αGC-experienced NKT cells display regulatory function by inhibiting the expansion of naïve NKT cells upon subsequent exposure to this antigen. Moreover, this regulatory activity impacts more than just NKT cells, as demonstrated by the NKT cell-mediated inhibition of antigen-stimulated mucosal-associated invariant T (MAIT) cells (a T cell subset restricted to a different antigen presenting molecule, MR1). These findings reveal that B. fragilis-derived NKT cell agonists may have broad immunoregulatory activity, providing insight into the mechanisms influencing immune tolerance to commensal bacteria and highlighting a potential means to manipulate NKT cell function for therapeutic benefit.
Introduction
Bacteria of the genus Bacteroides comprise over 25% of the bacterial species within the human colon.1Bacteroides fragilis (B. fragilis) is a normal part of the gut microbiota and typically accounts for 0.5% of human colonic flora, as isolated from the stool.1B. fragilis within the gastrointestinal tract promotes host nutrition and regulatory immune function.2–4 It produces a zwitterionic capsular polysaccharide (polysaccharide A) that can initiate regulatory T cell responses through formation of a polysaccharide-major histocompatibility (MHC) class II complex.3,5–7 In contrast, B. fragilis is also the most commonly isolated anaerobic pathogen1,8 associated with opportunistic infections,9 and expresses a variety of virulence factors.B. fragilis produces a suite of α-galactosylceramide (αGC) glycolipids that bind to CD1d, the antigen-presenting molecule responsible for the selection and activation of natural killer T (NKT) cells.10–13 T cells recognize antigens through a glycoprotein heterodimer termed a T cell receptor (TCR), comprised of either α and β, or γ and δ chains. Type I NKT cells (hereafter referred to as NKT cells), which are the focus of this study, are defined by their recognition of αGC using an αβTCR with limited gene rearrangements in mice and humans comprising of highly evolutionarily-conserved TCR α-chains.14 They also use a limited, but not invariant, range of TCR β-chains, which support recognition of various glycolipid antigens.15,16 Unlike conventional T cells, NKT cells display a memory phenotype from early in their development that includes rapid and potent secretion of cytokines upon primary TCR-mediated activation. Whilst such responses have been reported to contribute both positively and negatively within disease settings,17 NKT cells are also associated with maintenance of immune homeostasis.18,19
Given that B. fragilis is a gut resident, it is an obvious source of antigen for NKT cells, and a key question is therefore how these cells respond to B. fragilis-derived αGC. This is a controversial topic: on the one hand, B. fragilis αGC presented by CD1d has been reported to directly interact with the TCR of NKT cells, initiating their activation.10,12 Conversely, however, it has been suggested that B. fragilis αGC antagonize NKT cell activation by binding to CD1d in a way that blocks the presentation of agonistic endogenous ligands, thereby restricting the inflammatory contribution of NKT cell activation during autoimmune disease.11
To investigate this paradoxical problem, we investigated the impact of B. fragilis glycolipids on mouse and human NKT cells. We synthesized a major naturally-occurring αGC from B. fragilis (hereafter referred to as Bf αGC),10 plus a set of analogues with varied acyl chain compositions. We confirm that Bf αGC, and analogues thereof, are recognized by both mouse and human NKT cells, leading to their activation when presented by CD1d. Further, we demonstrate that following stimulation with Bf αGC, NKT cells develop an immune regulatory phenotype, which manifests in their acquired potential to produce the immunoregulatory cytokine interleukin-10 (IL-10). We show that Bf αGC-experienced NKT cells gain the ability to inhibit the expansion of naïve NKT cells in the presence of this TCR agonist, suggesting an NKT cell-based tolerogenic effect. The immunoregulatory activity of Bf αGC-experienced NKT cells is not limited to other NKT cells: we show that Bf αGC-experienced NKT cells also inhibited the activation of an unrelated T cell population, with the expansion of mucosal-associated invariant T (MAIT) cells also being markedly suppressed in response to their prototypical agonist 5-OP-RU.20 Together, these findings may help explain the apparent discrepancies in earlier studies, suggesting a broad regulatory role for NKT cells following exposure to this commensally derived glycolipid.
Results
Total synthesis of αGC from B. fragilis
B. fragilis produces a series of αGC molecules,10 with branching occurring in both of their lipid tails through incorporation of dietary branched-chain amino acids.21 We chemically synthesized a representative and abundant doubly-branched B. fragilis glycolipid, Bf αGC 1 (Fig. 1A). We developed a convergent approach that involved the individual synthesis of the acyl fragment, the sphinganine chain, and an α-selective galactosyl donor, which were assembled to give the completed glycolipid. The sphinganine fragment 9 was synthesized from 12-methyltridecylbromide 5, which was converted to the corresponding Grignard reagent 6 and coupled with a protected serine-derived Weinreb amide to afford the ketone 7 (Fig. 1B). Stereoselective reduction of 7 using lithium tri(tert-butyloxy)aluminium hydride22 afforded the alcohol 8, which was protected as benzoate 9.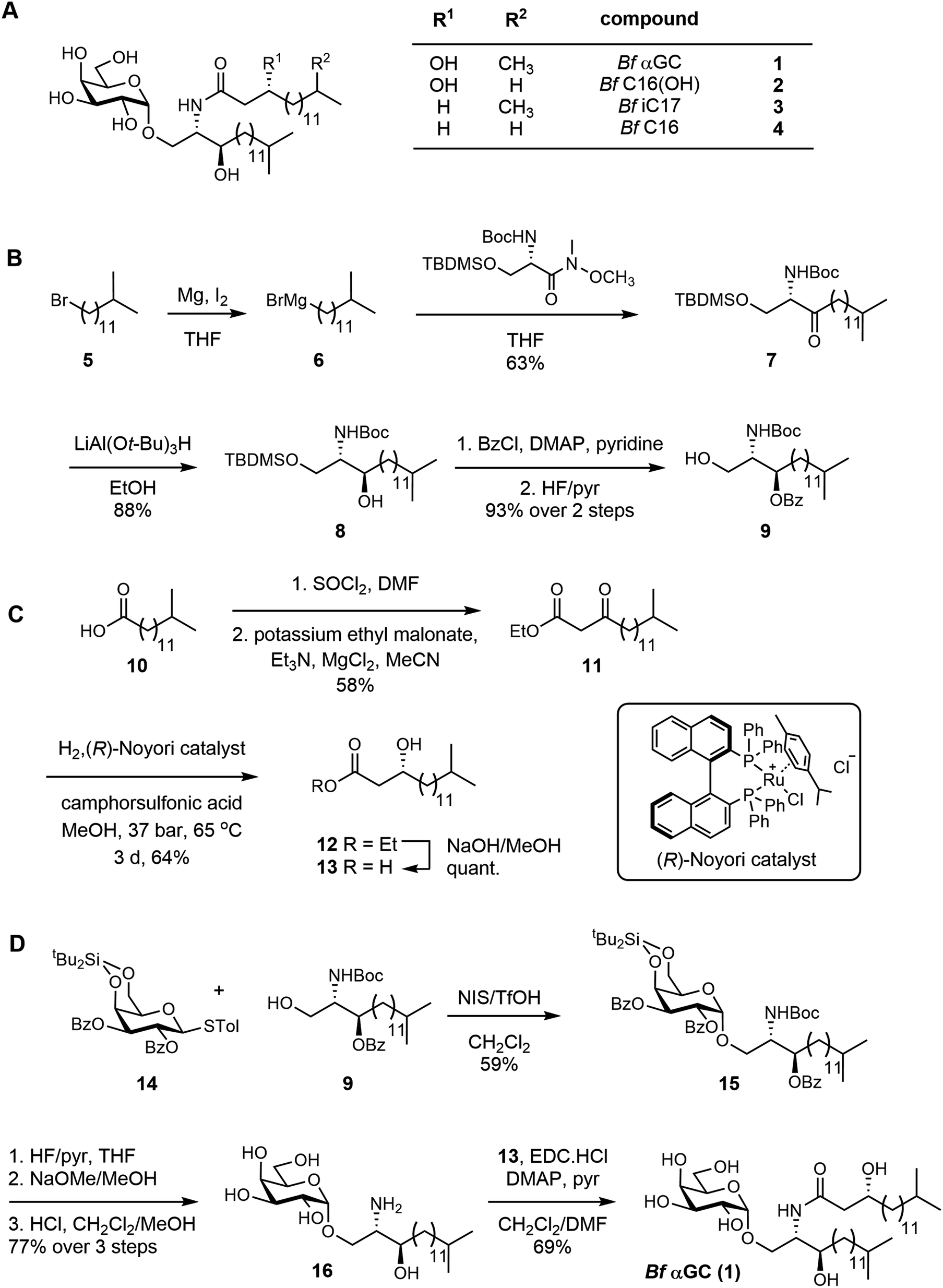 | ||
| Fig. 1 Total synthesis of a representative B. fragilis α-galactosylceramide and acyl chain variants. (A) Structure of Bf αGC (αGalCerBf-716) 1, and analogues 2–4. (B) Synthesis of the protected sphinganine 9. (C) Synthesis of the b-hydroxy acid side-chain. (D) Assembly of Bf αGC. For synthesis of variants, see ESI.† | ||
The acyl fragment was prepared from 13-methyltetradecanoic acid 10, which was converted to the acyl chloride and underwent Claisen condensation with potassium ethyl malonate23 to give ketone 11 (Fig. 1C). Stereoselective reduction of 11 with a p-cymene Noyori catalyst24 afforded R-12 in 95% ee (determined by Mosher ester analysis;25 see Fig. S1–S3†). Saponification afforded the β-hydroxy acid 13. The glycolipid was assembled in three steps from the galactosyl donor 14 (Fig. 1D). Condensation of 14 and 9, promoted by NIS/TfOH, gave the galactoside 15, exclusively as the α-anomer. Stepwise deprotection of 15 using HF/pyr, NaOMe/MeOH and finally HCl in CH2Cl2/MeOH afforded the galactosyl sphinganine 16. Finally, carbodiimide mediated condensation of 16 and β-hydroxy acid 13 in the presence of DMAP and pyridine afforded αGCBf-716 (Bf αGC) (Fig. S4, Table S1†). Using a similar approach, a range of analogues were synthesized to explore the effect of changes in the acyl chain of this compound (Fig. 1A and S5†). Compound 2Bf C16(OH) lacks the isopropyl branch, 3Bf iC17 lacks the acyl 3-OH, and 4Bf C16 lacks both features.
The structure of B. fragilis αGC impacts CD1d-mediated human NKT cell TCR recognition
To examine the recognition of Bf αGC and analogues (Fig. S6†) by human NKT cells we undertook CD1d-tetramer staining experiments on peripheral blood mononuclear cells from healthy human donors. When loaded with an appropriate antigen, CD1d-tetramers enable single-cell detection of T cells that recognize this antigen when bound to the CD1d antigen presenting molecule. Anti-CD36 blockade was used in these experiments to limit TCR-independent CD1d-tetramer staining that can occur when CD1d binds to CD36 and cause higher background staining.26 CD1d-tetramers loaded with Bf αGC weakly stained NKT cells compared to the positive control (αGC/PBS-44, a synthetic glycolipid) (Fig. 2A). However, Bf αGC-loaded CD1d-tetramer staining was consistently higher than vehicle control tetramers. The level of Bf αGC-mediated CD1d-tetramer staining was donor-dependent, with the percentages of NKT cells identified being comparable to the positive control tetramer (PBS-44) in some donors, but lower in others (Fig. 2B). The analogues Bf C16, Bf C16(OH) and Bf iC17 all identified CD1d-tetramer-positive cells to a similar extent as PBS-44 (Fig. 2A and B), suggesting that acyl chain modifications to the natural Bf αGC structure could enhance CD1d-loading and/or affinity for NKT cell TCRs.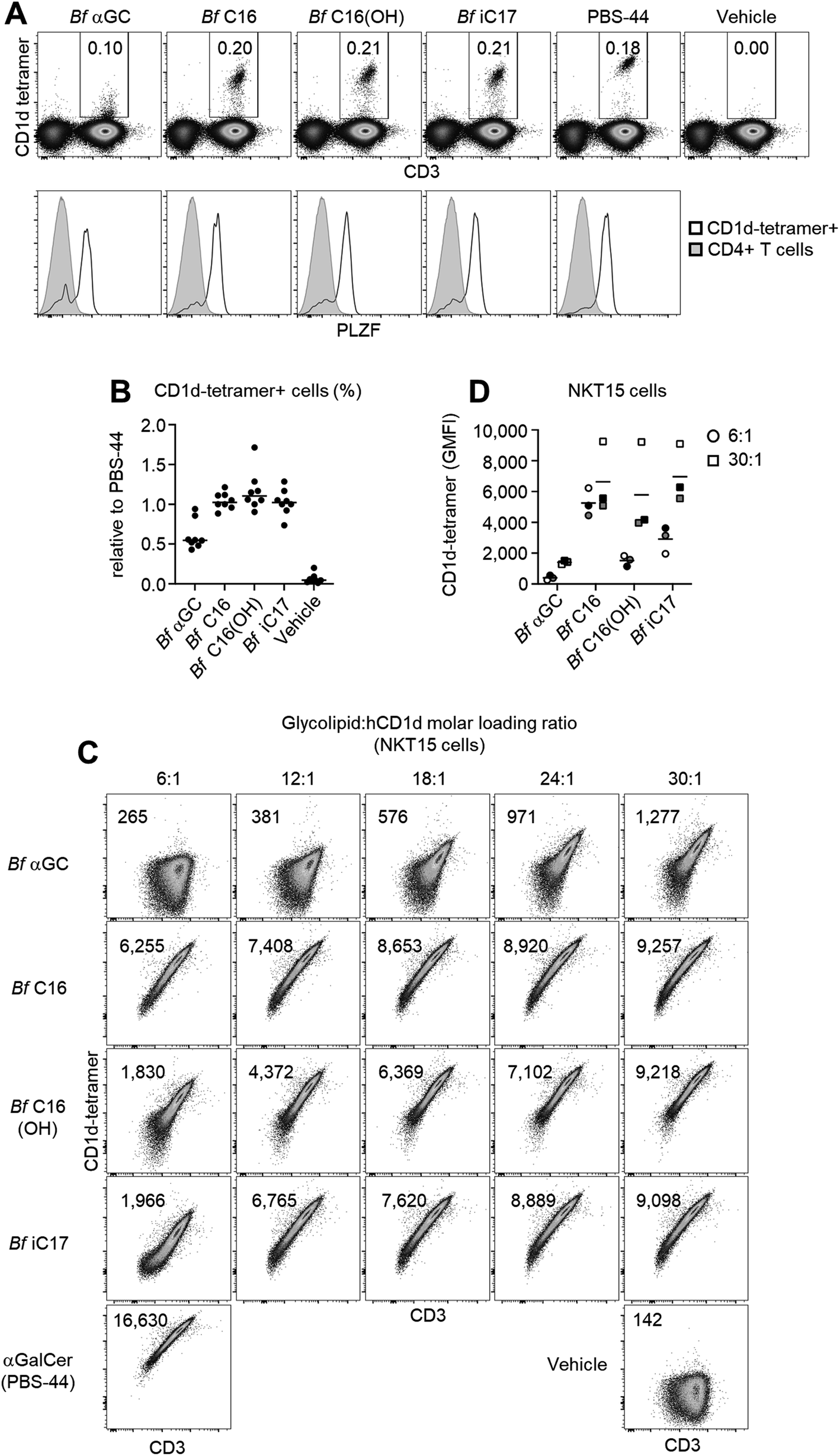 | ||
Fig. 2 Human NKT cells recognize B. fragilis glycolipids presented by CD1d. Human PBMCs were assessed for their reactivity to human CD1d-tetramers loaded with the indicated B. fragilis glycolipid, in comparison to αGalCer (PBS-44) or vehicle control. (A) Representative plots (top) depict the percentage of CD1d-tetramer+ cells amongst viable lymphocytes (excluding CD19+ cells). Plots (bottom) showing degree of PLZF expression amongst gated NKT cells. (B) Pooled data showing the relative percentage of NKT cells identified using B. fragilis analogues in comparison to αGalCer (PBS-44), n = 8 donors. (C and D) Biotinylated CD1d was loaded with increasing molar ratios of the B. fragilis analogues (lipid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD1d) as indicated, then tetramerized using streptavidin–phycoerythrin, and used to stain the NKT15 cell line. (C) Numbers on plots depict the geometric mean of the fluorescence intensity of CD1d-tetramer staining intensity under each condition. Loading ratio of αGalCer (PBS-44) was 6:1, the vehicle (tyloxapol) control was used at an equivalent volume used for the 30:1 B. fragilis αGalCer condition. (D) Data pooled from 3 independent experiments (mean). Colors represent different experiments, shapes represent the indicated molar loading ratios (round = 6:1, square = 30:1). :CD1d) as indicated, then tetramerized using streptavidin–phycoerythrin, and used to stain the NKT15 cell line. (C) Numbers on plots depict the geometric mean of the fluorescence intensity of CD1d-tetramer staining intensity under each condition. Loading ratio of αGalCer (PBS-44) was 6:1, the vehicle (tyloxapol) control was used at an equivalent volume used for the 30:1 B. fragilis αGalCer condition. (D) Data pooled from 3 independent experiments (mean). Colors represent different experiments, shapes represent the indicated molar loading ratios (round = 6:1, square = 30:1). | ||
To determine whether the weak CD1d-tetramer staining observed using Bf αGC was identifying genuine NKT cells, we assessed expression of promyelocytic leukemia zinc finger (PLZF), which is a transcription factor expressed by these cells, but not by most conventional T cells.27 This revealed clear PLZF staining by the bulk of CD1d-tetramer positive T cells when using Bf αGC (Fig. 2A) in comparison to the CD1d-tetramer negative CD4+ T cell population. These data support the concept that there are direct interactions between the naturally occurring version of B. fragilis-derived αGC (Bf aGC) and human NKT cell TCRs when presented by CD1d, and that changes to acyl chain structure modulates the strength of these interactions.
Acyl chain composition of Bf αGC might impact CD1d-loading efficiency in vitro, and thereby CD1d-tetramer staining. Therefore, the molar ratio of each analogue was titrated against biotinylated human CD1d monomers over a range of 6:1 to 30:1 to define the effect of changing loading ratio for each glycolipid. Glycolipid-loaded CD1d monomers were tetramerized using streptavidin-conjugated phycoerythrin (PE, a fluorescent pigment) and used to stain the human NKT-TCR expressing cell line NKT15.28 The CD1d-tetramer staining of Bf αGC correlated with increased loading ratios, with the intensity being clearly above vehicle control tetramer, most notably at the higher molar ratios (Fig. 2C). At the highest molar ratio of 30:1, Bf C16, Bf C16(OH) and Bf iC17 all displayed similar CD1d-tetramer staining intensities. However, at the lowest ratio of 6:1, Bf C16-loaded CD1d-tetramers stained markedly brighter than the other analogues (Fig. 2C and D). These data suggest that the 3-OH present on the acyl chain of Bf C16(OH) and Bf αGC impedes CD1d loading, and that the isopropyl branch contained within the acyl group of Bf iC17 and Bf αGC may also contribute to a reduced in vitro CD1d-loading efficiency.
Bf αGC induces NKT cell proliferation and functional differentiation
The anti-inflammatory influence of B. fragilis colonization has been described in several murine disease settings,29 and is associated with their expression of CD1d ligands.2–4 To better understand the role that Bf αGC has on immune responses, NKT cell-deficient splenocytes from Jα18−/− BALB/c mice were pulsed with our range of analogues, then co-cultured with NKT cell-enriched thymocytes (an immune cell present in the thymus) from wild-type BALB/c mice. Following in vitro culture NKT cell-enriched thymocytes were fluorescently labelled with carboxyfluorescein succinimidyl ester (CFSE) to measure the extent of NKT cell proliferation. At the highest dose of 10 μg ml−1, Bf αGC induced NKT cell expansion and CFSE dilution, to a similar extent as the artificial NKT cell agonist KRN7000 (at the lower concentration of 100 ng ml−1) (Fig. 3A), suggesting that broad recognition of Bf αGC by NKT cells is possible given a sufficient dose. Bf αGC maintained activity at 100 ng ml−1, yet the Bf C16 analogue was the most potent analogue based on the extent of NKT cell proliferation. Interestingly, this correlated with the enhanced CD1d loading efficiency of Bf C16 described above (Fig. 2C and D).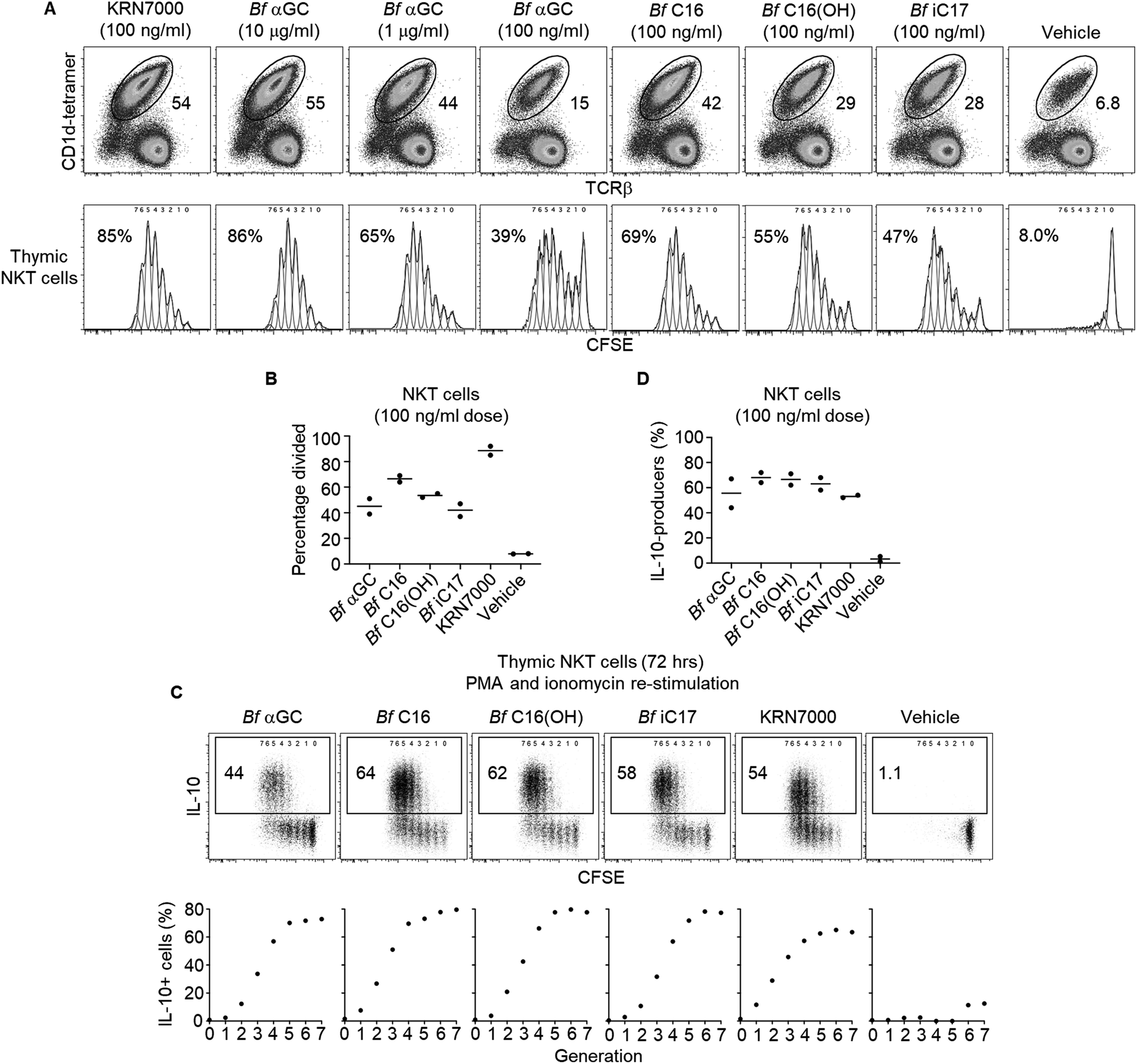 | ||
| Fig. 3 B. fragilis αGalCer analogues are agonists for mouse NKT cells. Jα18−/− (BALB/c) splenocytes were pulsed with the indicated concentration of lipid overnight, then co-cultured with CFSE-labelled, NKT cell-enriched thymocytes (BALB/c) for 3 days. (A) Top rows indicate the percentage of NKT cells detected amongst viable, CD19− lymphocytes. Bottom rows show the CFSE dilution of proliferating NKT cells. Numbers on plots indicate the estimated percentage of dividing NKT cells as gauged by FlowJo proliferation software. (B) Graph shows the division percentage of NKT cells from 2 independent experiments (mean). (C) Cultures described above were re-stimulated for the last 4 h in the presence of PMA and ionomycin in the presence of monensin. Cytokine production was then assessed via intracellular cytokine staining. Representative plots (top) depict the percentages of NKT cells producing IL-10 following culture with the indicated glycolipid. Graphs (bottom) indicate the percentage of IL-10 production from NKT cells within each proliferative generation as gauged by their CFSE dilution. (D) IL-10 production of NKT cells pooled from 2 independent experiments (mean). | ||
As the TCR-mediated activation of NKT cells can induce their capacity to produce the immunoregulatory cytokine interleukin 10 (IL-10),30 we investigated the potential for B. fragilis-derived glycolipids to mediate this functional shift. The co-cultures of Jα18−/− splenocytes and NKT cell-enriched thymocytes described above were activated for the final 4 h with phorbol 12-myristate 13-acetate (PMA) and ionomycin, in the presence of the protein transport inhibitor monensin to prevent cytokine secretion (Fig. 3C and D). Intracellular cytokine staining (ICS) was then performed to measure IL-10 production. This revealed that NKT cells proliferating in response to B. fragilis-derived glycolipids acquired the ability to produce IL-10, a trait that was most evident following multiple rounds of cellular division, and one that was absent among NKT cells not subjected to a TCR agonist (vehicle control).
Bf αGC-experienced NKT cells display regulatory function
Based on the observation that thymic NKT cells acquired IL-10-producing capacity following Bf αGC-induced proliferation (Fig. 3C), we examined whether similar phenotypic changes were detectable from peripheral NKT cells. BALB/c mice derived splenocytes were stimulated in vitro with Bf αGC for 3 days (Fig. 4A) and the cytokine production profiles of NKT cells were assessed following re-stimulation with PMA and ionomycin in the presence of the protein transport inhibitor monensin, as above. Confirming our previous results, production of the immunoregulatory cytokine IL-10 was only evident from NKT cells following exposure to Bf αGC. Relative to vehicle controls, exposure to Bf αGC also increased the proportion of NKT cells producing the Th2-type cytokine IL-13, with reduced levels of the Th1-type cytokine interferon (IFN)-γ (Fig. 4B and C). Whilst a small degree of IFN-γ and IL-10 co-production was observed, expression of these cytokines was largely mutually exclusive. The vast majority of NKT cells produced IL-13 following exposure to Bf αGC; therefore, most IL-10 and IFN-γ producers co-expressed this cytokine. These data indicate how the cytokine profiles of NKT cells are altered following their exposure to the naturally-occurring, bacterially-derived TCR agonist Bf αGC. | ||
| Fig. 4 Bf αGC induces IL-10 differentiation from peripheral NKT cells. Jα18−/− (BALB/c) splenocytes were pulsed with Bf αGC (100 ng ml−1) overnight then co-cultured with CFSE-labelled, NKT cell-enriched thymocytes (BALB/c) for 3 days, the last 4 h in the presence of PMA, ionomycin and monensin. (A) Representative plots of the NKT cell expansion following challenge with Bf αGC or a vehicle control. (B) Cytokine production profiles were then assessed via intracellular cytokine staining. Numbers on plots depict the percentages of NKT cells producing the indicated cytokine. (C) Graph indicates the cytokine production of NKT cells following Bf αGC challenge vs. vehicle control. (C) Data pooled from 4 independent experiments (mean). | ||
To explore if these phenotypic changes, particularly enhanced IL-10 production, might translate to immunoregulatory activity, Bf αGC-experienced splenic NKT cells were sorted after 3 days in vitro culture, alongside CD1d-tetramer− CD4+ CD25− T cells as a control. Both populations were then labelled with CFSE and spiked into a fresh splenocyte culture pulsed with Bf αGC. This enabled the response of previously naïve NKT cells from the secondary splenocyte culture to be assessed separately from the sorted cells (Fig. S2†). This revealed that the addition of Bf αGC-experienced NKT cells markedly inhibited the expansion of naïve NKT cells in response to Bf αGC (Fig. 5A and B), whereas the addition of CD4 T cells from the same Bf αGC-stimulated cultures had no impact.
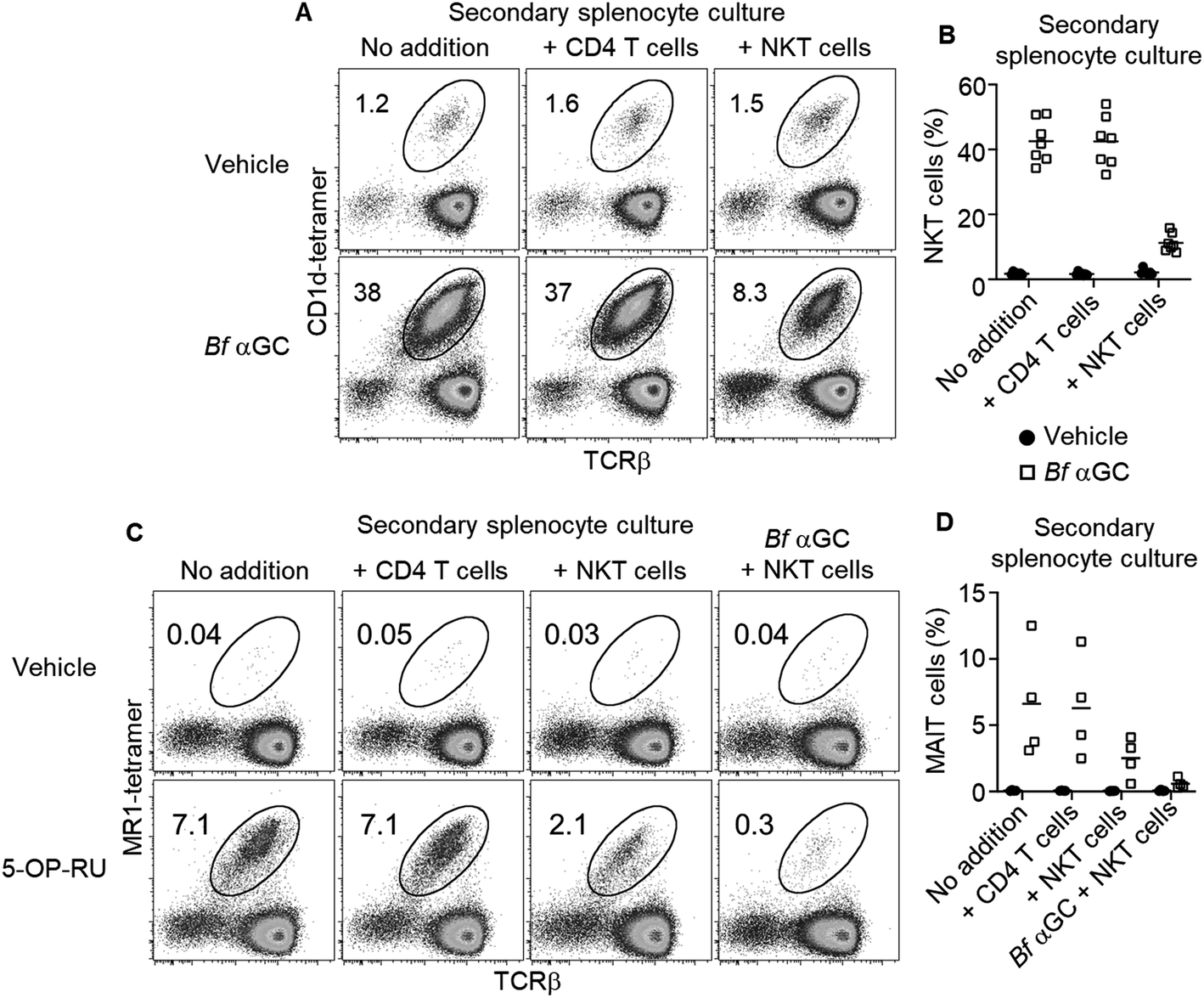 | ||
| Fig. 5 Bf αGC-experienced NKT cells inhibit naïve T cell expansion. Splenic NKT cells (C57BL/6) exposed to Bf αGC (1 μg ml−1) in vitro for 3 days were sorted alongside CD4+ (CD25−) T cells, then labelled with CFSE (1 μM) and added to a secondary splenocyte culture. (A) Representative plots depicting the percentage of NKT cells amongst viable 7-AAD− (7-aminoactinomycin D), CFSE−, CD19− lymphocytes exposed to Bf αGC (1 μg ml−1) or vehicle for 4 days. (B) Graph depicts the percentage of NKT cells detected from 7 independent experiments (mean). (C) Representative plots depicting the percentage of MAIT cells amongst viable 7-AAD−, CFSE−, CD19− lymphocytes exposed to 5-OP-RU (100 nM) or vehicle, with or without Bf αGC (1 μg ml−1) for 4 days. (D) Graph depicts the percentage of MAIT cells detected from 4 independent experiments. | ||
To address whether the observed regulatory function of NKT cells induced following their exposure to Bf αGC might extend beyond the NKT cell lineage, these experiments were repeated using the mucosal-associated invariant T (MAIT) cell agonist 5-OP-RU (Fig. S5†)20 within secondary splenocyte cultures. 5-OP-RU is presented by the distinct antigen presenting molecule MR1 to the TCRs of MAIT cells, causing their activation. The presence of Bf αGC-experienced NKT cells also inhibited the expansion of MAIT cells upon exposure to 5-OP-RU, which was most pronounced when Bf αGC was also present in the secondary culture (Fig. 5C and D). In contrast, the addition of CD4 T cells from the primary culture had no impact on MAIT cell expansion. These data suggest that the regulatory impact of Bf αGC is not limited to CD1d-restricted T cells but extends to other immune-cell subsets.
Taken together, our findings suggest that several structural features of Bf αGC, namely the methyl branches and acyl chain hydroxylation, reduce its potency toward NKT cells, most likely through reduced loading into CD1d. However, Bf αGC tetramer reactive NKT cells are present in donors at levels approaching those reactive to an artificial, high-affinity version of αGC (PBS-44). Despite the relatively lower potency of Bf αGC, NKT cells still proliferate and acquire an IL-10-producing capacity following exposure to this TCR agonist. Moreover, Bf αGC-experienced NKT cells display immunoregulatory function, by inhibiting the expansion of both naïve NKT cells and MAIT cells upon exposure to their cognate antigens.
Discussion
Commensal bacteria contribute to immune homeostasis within the gut,31 but the mechanisms of their action are poorly understood. Here, we investigated the immune activity of a glycolipid identified from the common human commensal bacteria B. fragilis towards lipid-reactive NKT cells. This field of investigation suffers from contradictory studies, with an initial report suggesting that a naturally occurring form of B. fragilis-derived αGC was a CD1d-ligand recognized by NKT cells as an agonist,10 while a subsequent study suggested that B. fragilis αGC plays an antagonistic role by blocking the CD1d-mediated presentation of endogenous NKT cell agonists.Herein, we provide new data that help to explain the existing controversy within the field. We support and expand upon the findings of Wieland Brown et al. that naturally occurring B. fragilis αGC forms agonistic CD1d–glycolipid NKT cell TCR complexes. We also provide a potential mechanism to explain the findings of An et al. that the presence of B. fragilis-derived αGC may limit NKT cell expansion in situ. However, our data suggest that this is not due to these bacterial glycolipids blocking the TCR-mediated activation of NKT cells by endogenous ligands as proposed,11 but due to these commensal lipids inducing a regulatory shift in NKT cell phenotype that develops following their TCR-mediated activation by B. fragilis-derived αGC.
Discrepant reporting of the agonistic nature of such bacterial glycolipids10,11 may be due to the different methodologies used to investigate interactions between CD1d-B. fragilis αGC and NKT cell TCRs. Wieland Brown et al.10 demonstrated activity using the mouse NKT cell lines DN3A4-1.2 and N38-2C12,32 which both express TCRs with the most common variable (V) β segment, Vβ 8.2. An et al. reported antagonism using a different Vβ 8.2 expressing NKT cell clone, DN32.D3 with distinct CDR3β usage,33 and the clone 24.7 (ref. 34) that contains the Vβ6 gene segment, which is a rare configuration within the mouse NKT cell population.35 As the recognition of physiological antigens can be highly dependent on NKT cell TCR β-chain composition,15,16 it is possible that different experimental designs utilized in these studies may have contributed to the contrary findings.
Oh et al. have recently reported that the unique dibranched composition of B. fragilis αGC, which consists of terminal branches within the acyl and sphingosine chains, are incorporated from dietary sources.12 Moreover, they argued that terminal branching within the sphingosine chain of B. fragilis αGC is a critical antigenic determinant. Whilst we were not able to assess the impact of sphingosine branching with our set of analogues, we demonstrate that terminal acyl chain branching reduces the potency of these ligands towards NKT cells. Moreover, we show that the 3′-hydroxyl acyl chain group typically observed within the B. fragilis glycolipids is not just dispensable for agonism,12 but may represent another factor that reduces their potency. This was shown by the enhanced human CD1d loading efficiency of the iC17 analogue in comparison to Bf αGC, which only differs in the absence of this hydroxyl residue.
Despite the native form of Bf αGC10 containing structural elements that appear to diminish its immunogenicity, this lipid clearly maintained activity as a CD1d-restricted NKT cell agonist. Thus, we chose to investigate the regulatory properties of B. fragilis αGC using the naturally occurring form, rather than unnatural analogues exhibiting heightened potency. Interestingly, the B. fragilis analogue (SB2217) used by Oh et al. for their in vivo and structural studies lacked acyl chain branching seen in the natural product, and displayed an affinity for a NKT cell TCR similar to the artificial antigen KRN7000.12 This is important to consider, as the affinity of closely-related αGC variants have been strongly correlated with qualitative and quantitative differences in cytokine responses elicited following NKT cell activation.36–38 Notably, a less potent, lower affinity version of KRN7000, OCH with truncated sphingosine and acyl chains, has been shown to provide enhanced protection against experimental autoimmune encephalomyelitis38 and collagen-induced arthritis.39
Conclusions
This study confirms our previous findings that the TCR-mediated activation of NKT cells by glycolipid agonists can induce their differentiation into IL-10-producers,30 in contrast to another study suggesting that IL-10-producing NKT cells (NKT10 cells) are a functionally distinct lineage.40 Importantly, we show here that this phenotypic shift can be accomplished by a commensal bacterial lipid, which is arguably the most likely source of αGC that human NKT cells will encounter.10 Whilst the precise role of NKT cells in the progress of specific autoimmune diseases remain unclear, our findings suggest that Bf αGC-experienced NKT cells can regulate the activation of CD1d and MR1-restricted T cells in the presence of their respective antigens. Whilst the breadth of this regulatory role is yet to be fully established, these data do suggest a potential explanation for why NKT cell agonism can impede autoimmune progression in a range of disease models.41Our data help to unify the apparently disparate observations previously reported for Bf αGC, highlighting that while it is an NKT cell agonist, it can drive a regulatory phenotype that antagonizes NKT cell responses. We also show that this regulatory phenotype extends to another class of T cells, the MAIT cells. These data indicate how the fine structure of commensal glycolipids can impact their immunogenicity, and how this may impact immune homeostasis within the gut. Finally, these findings highlight the therapeutic potential of naturally occurring NKT cells agonists such as Bf αGC to manipulate T cell function.
Data availability
Data are available within the ESI.†Author contributions
G. C., S. J. W. and D. I. G. designed research; G. C., T. N. and M. C. performed research; G. C., T. N. and M. C. analyzed data; G. C., S. J. W. and D. I. G. wrote the paper.Conflicts of interest
D. I. G. is a member of the scientific advisory board for Avalia Immunotherapies, a company that is developing natural killer T cell-based vaccines.Acknowledgements
This work was supported by grants from the Australian Research Council (DP210100233, DP210100235) and the National Health and Medical Research Council of Australia (NHMRC; 1113293, 1140126). D. I. G. was supported by an NHMRC Senior Principal Research Fellowship (1117766) and subsequently, an NHMRC Investigator Award (2008913). We are grateful to Paul Savage (Brigham Young University, UT, USA) for providing αGC/PBS-44. We thank staff from the flow cytometry facility at the Peter Doherty Institute for Infection and Immunity, The University of Melbourne. David Hay for writing up award to T. Nguyen. 5-OP-RU was kindly provided by David Fairlie (University of Queensland). Jon Clardy (Harvard University) is thanked for providing the proton-NMR spectrum of authentic B. fragilis αGC.References
- H. M. Wexler, Clin. Microbiol. Rev., 2007, 20, 593–621 CrossRef CAS PubMed.
- J. Ochoa-Reparaz, D. W. Mielcarz, Y. Wang, S. Begum-Haque, S. Dasgupta, D. L. Kasper and L. H. Kasper, Mucosal Immunol., 2010, 3, 487–495 CrossRef CAS PubMed.
- C. Ramakrishna, M. Kujawski, H. Chu, L. Li, S. K. Mazmanian and E. M. Cantin, Nat. Commun., 2019, 10, 2153 CrossRef PubMed.
- Y. K. Lee, P. Mehrabian, S. Boyajian, W. L. Wu, J. Selicha, S. Vonderfecht and S. K. Mazmanian, mSphere, 2018, 3, e00587 CAS.
- Y. C. Chang, Y. H. Ching, C. C. Chiu, J. Y. Liu, S. W. Hung, W. C. Huang, Y. T. Huang and H. L. Chuang, PLoS One, 2017, 12, e0180025 CrossRef PubMed.
- S. K. Mazmanian, C. H. Liu, A. O. Tzianabos and D. L. Kasper, Cell, 2005, 122, 107–118 CrossRef CAS PubMed.
- J. Ochoa-Reparaz, D. W. Mielcarz, L. E. Ditrio, A. R. Burroughs, S. Begum-Haque, S. Dasgupta, D. L. Kasper and L. H. Kasper, J. Immunol., 2010, 185, 4101–4108 CrossRef CAS PubMed.
- W. E. Moore, E. P. Cato and L. V. Holdeman, Am. J. Clin. Nutr., 1978, 31, S33–S42 CrossRef CAS PubMed.
- M. Yekani, H. B. Baghi, B. Naghili, S. Z. Vahed, J. Soki and M. Y. Memar, Microb Pathog, 2020, 149, 104506 CrossRef CAS PubMed.
- L. C. Wieland Brown, C. Penaranda, P. C. Kashyap, B. B. Williams, J. Clardy, M. Kronenberg, J. L. Sonnenburg, L. E. Comstock, J. A. Bluestone and M. A. Fischbach, PLoS Biol, 2013, 11, e1001610 CrossRef PubMed.
- D. An, S. F. Oh, T. Olszak, J. F. Neves, F. Y. Avci, D. Erturk-Hasdemir, X. Lu, S. Zeissig, R. S. Blumberg and D. L. Kasper, Cell, 2014, 156, 123–133 CrossRef CAS PubMed.
- S. F. Oh, T. Praveena, H. Song, J. S. Yoo, D. J. Jung, D. Erturk-Hasdemir, Y. S. Hwang, C. C. Lee, J. Le Nours, H. Kim, J. Lee, R. S. Blumberg, J. Rossjohn, S. B. Park and D. L. Kasper, Nature, 2021, 600, 302–307 CrossRef CAS PubMed.
- I. Van Rhijn, D. I. Godfrey, J. Rossjohn and D. B. Moody, Nat. Rev. Immunol., 2015, 15, 643–654 CrossRef CAS PubMed.
- D. I. Godfrey, A. P. Uldrich, J. McCluskey, J. Rossjohn and D. B. Moody, Nat. Immunol., 2015, 16, 1114–1123 CrossRef CAS PubMed.
- G. Cameron, D. G. Pellicci, A. P. Uldrich, G. S. Besra, P. Illarionov, S. J. Williams, N. L. La Gruta, J. Rossjohn and D. I. Godfrey, J. Immunol., 2015, 195, 4604–4614 CrossRef CAS PubMed.
- W. C. Florence, C. Xia, L. E. Gordy, W. Chen, Y. Zhang, J. Scott-Browne, Y. Kinjo, K. O. Yu, S. Keshipeddy, D. G. Pellicci, O. Patel, L. Kjer-Nielsen, J. McCluskey, D. I. Godfrey, J. Rossjohn, S. K. Richardson, S. A. Porcelli, A. R. Howell, K. Hayakawa, L. Gapin, D. M. Zajonc, P. G. Wang and S. Joyce, EMBO J., 2009, 28, 3579–3590 CrossRef CAS PubMed.
- L. Wu and L. Van Kaer, Front. Biosci., Scholar Ed., 2011, 3, 236–251 Search PubMed.
- J. Saez de Guinoa, R. Jimeno, M. Gaya, D. Kipling, M. J. Garzon, D. Dunn-Walters, C. Ubeda and P. Barral, EMBO J., 2018, 37, e97537 CrossRef PubMed.
- C. M. Dowds, R. S. Blumberg and S. Zeissig, Clin. Immunol., 2015, 159, 128–133 CrossRef CAS PubMed.
- L. Kjer-Nielsen, O. Patel, A. J. Corbett, J. Le Nours, B. Meehan, L. Liu, M. Bhati, Z. Chen, L. Kostenko, R. Reantragoon, N. A. Williamson, A. W. Purcell, N. L. Dudek, M. J. McConville, R. A. O'Hair, G. N. Khairallah, D. I. Godfrey, D. P. Fairlie, J. Rossjohn and J. McCluskey, Nature, 2012, 491, 717–723 CrossRef CAS PubMed.
- J. von Gerichten, D. Lamprecht, L. Opalka, D. Soulard, C. Marsching, R. Pilz, V. Sencio, S. Herzer, B. Galy, V. Nordstrom, C. Hopf, H. J. Grone, F. Trottein and R. Sandhoff, J. Lipid Res., 2019, 60, 1892–1904 CrossRef CAS PubMed.
- R. V. Hoffman, N. Maslouh and F. Cervantes-Lee, J. Org. Chem., 2002, 67, 1045–1056 CrossRef CAS PubMed.
- R. J. Clay, T. A. Collom, G. L. Karrick and J. Wemple, Synthesis, 1993, 1993, 290–292 CrossRef.
- K. Mashima, K.-h. Kusano, T. Ohta, R. Noyori and H. Takaya, J. Chem. Soc., Chem. Commun., 1989, 1208–1210, 10.1039/C39890001208.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- N. A. Gherardin, S. J. Redmond, H. E. G. McWilliam, C. F. Almeida, K. H. A. Gourley, R. Seneviratna, S. Li, R. De Rose, F. J. Ross, C. V. Nguyen-Robertson, S. Su, M. E. Ritchie, J. A. Villadangos, D. B. Moody, D. G. Pellicci, A. P. Uldrich and D. I. Godfrey, Sci. Immunol., 2021, 6, eabg4176 CrossRef CAS PubMed.
- A. K. Savage, M. G. Constantinides, J. Han, D. Picard, E. Martin, B. Li, O. Lantz and A. Bendelac, Immunity, 2008, 29, 391–403 CrossRef CAS PubMed.
- J. Le Nours, T. Praveena, D. G. Pellicci, N. A. Gherardin, F. J. Ross, R. T. Lim, G. S. Besra, S. Keshipeddy, S. K. Richardson, A. R. Howell, S. Gras, D. I. Godfrey, J. Rossjohn and A. P. Uldrich, Nat. Commun., 2016, 7, 10570 CrossRef CAS PubMed.
- E. B. Troy and D. L. Kasper, Front. Biosci., Landmark Ed., 2010, 15, 25–34 CrossRef CAS PubMed.
- G. Cameron and D. I. Godfrey, Immunol. Cell Biol., 2018, 96, 759–771 CrossRef CAS PubMed.
- H. J. Wu and E. Wu, Gut Microbes, 2012, 3, 4–14 CrossRef PubMed.
- N. Burdin, L. Brossay, Y. Koezuka, S. T. Smiley, M. J. Grusby, M. Gui, M. Taniguchi, K. Hayakawa and M. Kronenberg, J. Immunol., 1998, 161, 3271–3281 CrossRef CAS.
- O. Lantz and A. Bendelac, J. Exp. Med., 1994, 180, 1097–1106 CrossRef CAS PubMed.
- S. M. Behar, T. A. Podrebarac, C. J. Roy, C. R. Wang and M. B. Brenner, J. Immunol., 1999, 162, 161–167 CrossRef CAS.
- J. L. Matsuda, L. Gapin, N. Fazilleau, K. Warren, O. V. Naidenko and M. Kronenberg, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12636–12641 CrossRef CAS PubMed.
- K. S. Wun, G. Cameron, O. Patel, S. S. Pang, D. G. Pellicci, L. C. Sullivan, S. Keshipeddy, M. H. Young, A. P. Uldrich, M. S. Thakur, S. K. Richardson, A. R. Howell, P. A. Illarionov, A. G. Brooks, G. S. Besra, J. McCluskey, L. Gapin, S. A. Porcelli, D. I. Godfrey and J. Rossjohn, Immunity, 2011, 34, 327–339 CrossRef CAS PubMed.
- S. Oki, A. Chiba, T. Yamamura and S. Miyake, J. Clin. Invest., 2004, 113, 1631–1640 CrossRef CAS.
- K. Miyamoto, S. Miyake and T. Yamamura, Nature, 2001, 413, 531–534 CrossRef CAS PubMed.
- A. Chiba, S. Oki, K. Miyamoto, H. Hashimoto, T. Yamamura and S. Miyake, Arthritis Rheum., 2004, 50, 305–313 CrossRef CAS PubMed.
- D. Sag, P. Krause, C. C. Hedrick, M. Kronenberg and G. Wingender, J. Clin. Invest., 2014, 124, 3725–3740 CrossRef CAS PubMed.
- L. Van Kaer and L. Wu, Front. Immunol., 2018, 9, 519 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02124f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |