
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promoting plasmonic photocatalysis with ligand-induced charge separation under interband excitation†
Ben
Roche‡
,
Tamie
Vo‡
 and
Wei-Shun
Chang
*
and
Wei-Shun
Chang
*
Department of Chemistry and Biochemistry, University of Massachusetts Dartmouth, North Dartmouth, Massachusetts 02747, USA. E-mail: wchang2@umassd.edu
First published on 27th July 2023
Abstract
Plasmonic nanoparticles have been demonstrated to enhance photocatalysis due to their strong photon absorption and efficient hot-carrier generation. However, plasmonic photocatalysts suffer from a short lifetime of plasmon-generated hot carriers that decay through internal relaxation pathways before being harnessed for chemical reactions. Here, we demonstrate the enhanced photocatalytic reduction of gold ions on gold nanorods functionalized with polyvinylpyrrolidone. The catalytic activities of the reaction are quantified by in situ monitoring of the spectral evolution of single nanorods using a dark-field scattering microscope. We observe a 13-fold increase in the reduction rate with the excitation of d-sp interband transition compared to dark conditions, and a negligible increase in the reduction rate when excited with intraband transition. The hole scavenger only plays a minor role in the photocatalytic reduction reaction. We attribute the enhanced photocatalysis to an efficient charge separation at the gold–polyvinylpyrrolidone interface, where photogenerated d-band holes at gold transfer to the HOMO of polyvinylpyrrolidone, leading to the prolonged lifetime of the electrons that subsequently reduce gold ions to gold atoms. These results provide new insight into the design of plasmonic photocatalysts with capping ligands.
Introduction
An ideal photocatalyst requires efficient photon absorption to create highly energetic carriers to enable reactions with a high activation energy.1,2 Plasmonic nanoparticles absorb photons more efficiently at plasmon frequencies than other materials.3 Additionally, the tunability of the plasmon energy by the morphology and composition of nanostructures allows harvesting the photons from UV to IR regions.4–7 Upon photon excitation, plasmons decay non-radiatively into highly energetic electron–hole pairs that can subsequently drive chemical reactions.8–16 Due to these excellent properties, plasmonic nanoparticles have been demonstrated as photocatalysts or photoelectrocatalysts to drive various reactions, such as NH3 decomposition,17,18 CO2 reduction,19–26 hydrogen generation,27–29 and NH3 formation.30–33Photoexcitation of plasmonic nanoparticles through interband and intraband transitions generates hot carriers with various energies.34–37 For gold (Au) nanoparticles, photons with energy higher than the interband threshold (2.4 eV) excite the electrons from the d-band to the sp-band of the metal, creating hot holes and warm electrons populating at the d-band and near the Fermi level, respectively. On the other hand, excitation of surface plasmon resonances promotes electrons near the Fermi level to the unoccupied states within the conduction band, producing hot electrons above the Fermi level and warm holes near the Fermi level34–37 (Fig. S1†). These highly energetic carriers produced by the photoexcitation of plasmonic nanoparticles have been shown to catalyze chemical reactions with high activation barriers.17–33 However, the lifetime of these plasmon-induced hot carriers, limited by electron–hole recombination and electron–electron scattering, is less than 100 fs,34,38–44 which is several orders of magnitude shorter than the chemical reaction time. As a result, extracting the hot carriers before their recombination is critical to the realization of plasmonic photocatalysis.
Several strategies were developed to prolong the hot carrier lifetime by inducing charge separation states at the heterojunction to counteract the ultrafast recombination of electrons and holes in the metal.45 Hybrid nanostructures were created by depositing various materials on Au nanostructures, including semiconductors and molecular adsorbates.46–53 Exciting the surface plasmon resonances of Au/semiconductor nanostructures, the electrons of the metal transfer to the conduction band of the semiconductors while the holes stay in the metal.46–48 In addition, hot holes transfer to the p-GaN with electrons in the Au nanostructures under interband excitation.49 In hybrid nanostructures composed of plasmonic nanoparticles and surface adsorbates, the plasmon excitation directly excites the electrons from the Fermi level of the metal to the LUMO of the molecular adsorbates, leading to charge separation.50–53 An efficient charge separation was also observed in Au nanoparticles with hole scavengers under interband excitation.54–56 Hence, maximizing the charge separation of hybrid nanostructures with the type of excitation will advance our knowledge for the design of plasmonic photocatalysts.
Colloidal nanoparticles are stabilized using capping ligands to avoid aggregation of nanoparticles in the solution. Capping ligands are widely accepted as surface-blocking ligands that inhibit chemical reactions. Recently, Wei has demonstrated the growth of small Au seeds into hexagonal prisms with polyvinylpyrrolidone (PVP) ligands in the presence of hole scavengers under the excitation of the surface plasmon resonances of Au nanoseeds.57,58 The anisotropic growth arises from photochemical reduction of Au3+ ions at the perimeter of the nanoprism, where PVP ligands are specifically adsorbed. After creating the electron–hole pairs by plasmon excitation, the holes were removed by the hole scavenger, methanol, while the hot electrons were stabilized by the positively charged PVP. Therefore, the PVP ligand plays an important role in the plasmon-induced Au3+ reduction reactions rather than serving as a surface-blocking ligand. This strategy of anisotropic growth of nanoseeds into nanoprisms has been utilized to fabricate periodic nanoprisms with various morphologies.59,60 However, the interband and intraband transitions for the Au nanoseeds are spectrally overlapped. It is difficult to distinguish whether interband or intraband transitions promote Au3+ reduction in the presence of PVP ligands. Furthermore, PVP could play a different role in photoinduced growth in addition to stabilizing the hot electrons and AuCl4−. Understanding the underlying mechanism of reduction reactions with PVP is critical to promoting plasmonic catalysis using capping ligands that could potentially induce charge separation.
Here, we utilized PVP-functionalized Au nanorods (AuNRs) as seeds to investigate photoinduced Au3+ reduction by single-particle dark-field scattering (DFS) spectroscopy. AuNRs allow for separating the interband and intraband transitions as the longitudinal plasmon modes are located at an energy lower than 2.0 eV, which is below the interband onset of Au. Single-particle measurements resolve sample heterogeneity to unambiguously quantify the growth rate of individual AuNRs.61 We found that the growth rate of AuNRs in the presence of PVP increases more than ten times under interband transition compared to that in the dark, while intraband excitation does not enhance the growth rate. Hole scavengers only have a minor contribution to the enhancement of the Au3+ reduction reaction mediated by PVP. Additionally, the reduction reaction preferentially occurs at the location of PVP. These results reveal that d-band holes transfer to PVP under interband transition, while the high density of warm electrons in Au reduces Au3+ to promote AuNR growth. Electrons and holes generated by intraband excitation suffer from ultrafast recombination due to the lack of charge separation, prohibiting the reduction of Au3+. Our findings provide insight into the usage of capping ligands to induce charge separation to promote plasmonic catalysis.
Results and discussion
The plasmon-catalyzed Au3+ reduction reaction was performed using PVP-functionalized AuNRs (21.6 ± 3.1 × 53.5 ± 5.9 nm, Fig. S2†) as seeds, and the reaction kinetics was probed by single-particle DFS spectroscopy. AuNRs with an aspect ratio of 2.5 ± 0.4 exhibit the longitudinal and transverse modes at 700 and 520 nm, respectively, in an aqueous solution (Fig. S3†). As a result, it is possible to separately excite the intraband and interband transitions by photons with wavelengths longer than 600 nm and shorter than 500 nm, respectively. Additionally, the peak energy of AuNRs blueshifts with a smaller aspect ratio and the intensity scales with their size. One can measure the spectral evolution of AuNRs by single-particle DFS spectroscopy to probe their morphology change without using high-resolution electron microscopy. Furthermore, single-particle measurements enable acquiring the spectra of hundreds of AuNRs during the reaction (see the Method section in the ESI† and the discussion below) to obtain the statistical information, which is inaccessible by electron microscopy. Finally, quantifying the photocatalytic efficiency of Au3+ reduction as a function of the aspect ratio of AuNRs can be achieved by single-particle spectroscopy with “one” measurement, as AuNRs exhibit a heterogeneous distribution of the aspect ratio (Fig. S2†).To perform the photocatalytic reduction reaction, PVP functionalized AuNRs were spin-cast on a silanized coverslip that was assembled into a flow cell with another glass slide spaced with a parafilm (Method section in the ESI†). The flow cell was mounted on a DFS microscope with hyperspectral imaging capacity. Hyperspectral imaging allows one to acquire the scattering spectra of hundreds of single AuNRs in a few minutes (Fig. 1A). The recorded spectrum (blue symbol in Fig. 1B) was fit with a Lorentzian function (red line in Fig. 1B) to obtain the peak energy (Eres), full width at half maximum (Γ), maximum intensity (Imax), and amplitude 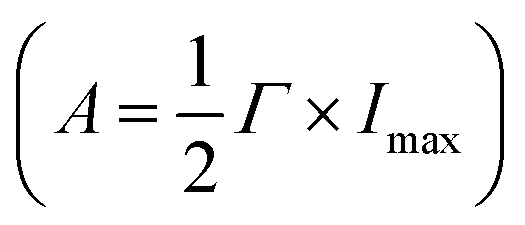 . A series of hyperspectral images of the same AuNRs were acquired at various reaction times (t) with and without photoexcitation using the experimental sequence shown in Fig. 1C and S4† (see the section of the Method in the ESI† for the detailed procedure). A reagent containing 3 μM HAuCl4, 15 μM PVP, and 2.9 M ethanol was introduced into the cell and incubated for 10 minutes to allow the reduction of Au3+ (peak of the blue line). Subsequently, the reaction was stopped by flowing DI water to the cell to completely remove the reagent (valley of the blue line), and the hyperspectral images of AuNRs were acquired (color bars). We defined the reaction time as the total length of the incubation time in the reagent. The spectral evolution of AuNRs was obtained at t = 0–50 min without light illumination and t = 60–110 min under photoexcitation (golden line). The excitations of interband and intraband transitions were realized by placing a blue filter (500 nm short-pass filter) and a red filter (610 nm long-pass filter) after the lamp, respectively. Two IR filters were also inserted in the excitation path to avoid heating the aqueous solution by absorbing IR photons.
. A series of hyperspectral images of the same AuNRs were acquired at various reaction times (t) with and without photoexcitation using the experimental sequence shown in Fig. 1C and S4† (see the section of the Method in the ESI† for the detailed procedure). A reagent containing 3 μM HAuCl4, 15 μM PVP, and 2.9 M ethanol was introduced into the cell and incubated for 10 minutes to allow the reduction of Au3+ (peak of the blue line). Subsequently, the reaction was stopped by flowing DI water to the cell to completely remove the reagent (valley of the blue line), and the hyperspectral images of AuNRs were acquired (color bars). We defined the reaction time as the total length of the incubation time in the reagent. The spectral evolution of AuNRs was obtained at t = 0–50 min without light illumination and t = 60–110 min under photoexcitation (golden line). The excitations of interband and intraband transitions were realized by placing a blue filter (500 nm short-pass filter) and a red filter (610 nm long-pass filter) after the lamp, respectively. Two IR filters were also inserted in the excitation path to avoid heating the aqueous solution by absorbing IR photons.
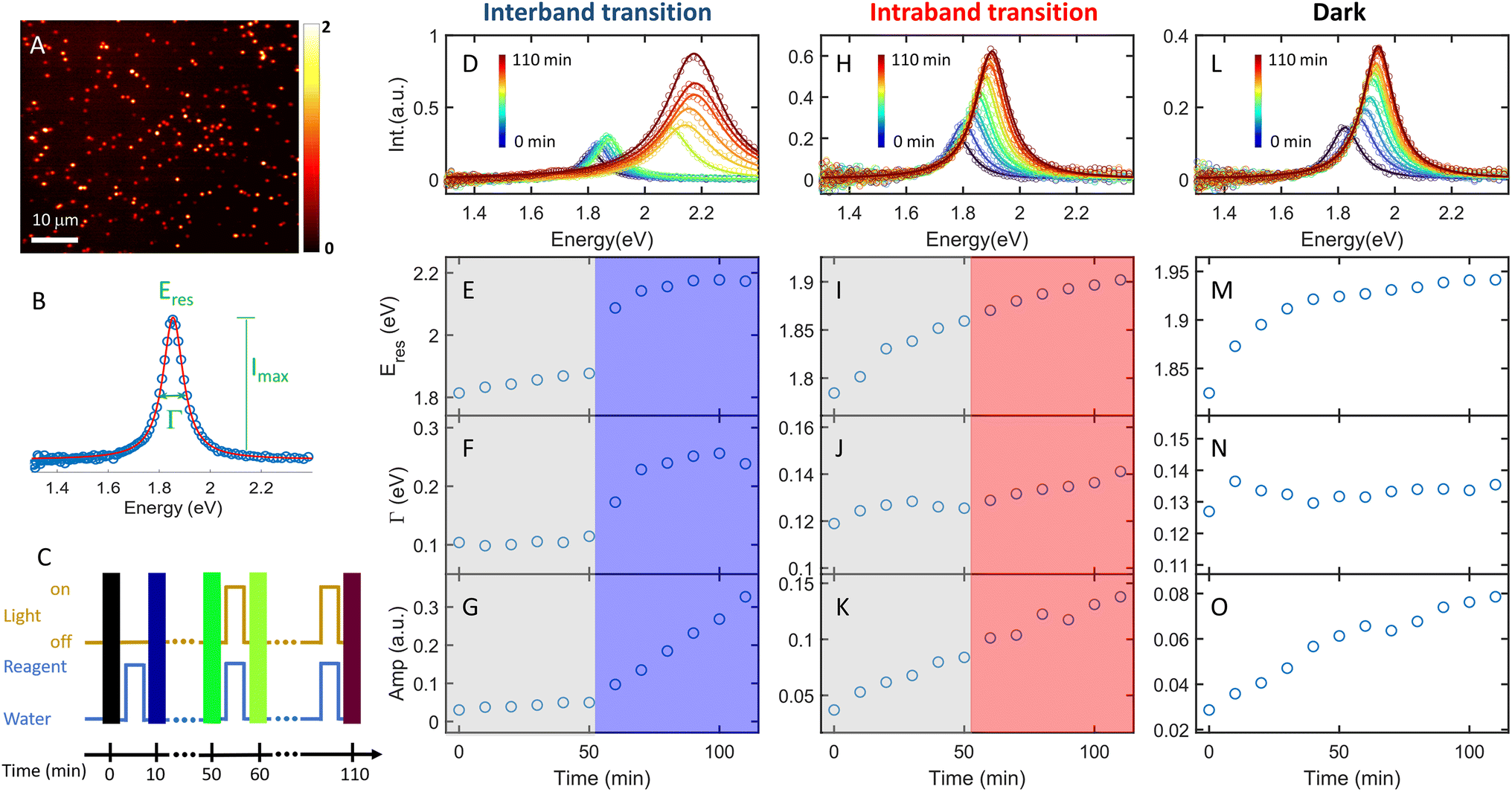 | ||
| Fig. 1 In situ monitoring of single AuNR growth under photoexcitation and in the dark. (A) Hyperspectral images of AuNRs in water. The scale bar is 10 μm. (B) Scattering spectrum (blue symbols) of a single AuNR fit to a Lorentzian function (red) to obtain plasmon energy (Eres), full width at half maximum (Γ), and peak intensity (Imax). (C) Sequence of the experiment. The blue line represents the injection of water (valley) and the reagent (peak) into the cell. The golden line indicates the light on (peak) and off (valley). The color bars are the sequence of hyperspectral images. The time duration of the peak for the blue and golden lines is 10 minutes. (D) Spectral evolution of a representative AuNR during the Au3+ reduction reaction with interband excitation. (E–G) Temporal evolution of Eres (E), Γ (F), and the amplitude (G) of the scattering spectra shown in (D). The grey and blue shadows indicate the blue light off and on, respectively. (H) Spectral evolution of a representative AuNR during the Au3+ reduction reaction with intraband excitation. (I–K) Temporal evolution of Eres (I), Γ (J), and the amplitude (K) of the scattering spectra shown in (H). The grey and red shadows indicate the red light off and on, respectively. (L) Spectral evolution of a representative AuNR during the Au3+ reduction reaction in the dark. (M–O) Temporal evolution of Eres (M), Γ (N), and the amplitude (O) of the scattering spectra shown in (L). | ||
The AuNRs exhibit fast growth under interband excitation, while the intraband transition has a minimal contribution. Fig. 1D shows the spectral evolution of a representative AuNR with the interband excitation. The time-dependent Eres, Γ, and amplitude are shown in Fig. 1E, F, and G, respectively. At t = 0–50 min, the spectra show a small blueshift in Eres (Fig. 1E) and a slight increase in the amplitude (Fig. 1G), suggesting a minor growth of the AuNR into a smaller aspect ratio in the dark. In stark contrast, the spectra blueshift to ∼2.2 eV with a momentous increase in the amplitude at t = 60–110 min, indicating an efficient growth of AuNRs under the interband excitation. The AuNR was reshaped into a large isotropic nanoparticle (Fig. S5†). The spectral evolution of a AuNR excited by intraband transition shows a gradual shift to higher energy and a small increase in the amplitude (Fig. 1H–K). Compared to a control experiment where the reaction was carried out in the dark at t = 0–110 min, the spectral evolution of the AuNR in the dark (Fig. 1L–O) is similar to that with intraband excitation. This result indicates a negligible contribution of the intraband transition to AuNR growth in the presence of the PVP ligands.
We have measured 353, 251, and 187 AuNRs excited by interband and intraband transitions and in the dark, respectively. The subensemble of spectral evolution of these AuNRs exhibits a behavior similar to that of the representative AuNRs shown in Fig. 1. Fig. 2A shows the correlation of Eres at t = 0 min and t = 110 min with interband (blue) and intraband (red) transitions and without illumination (black). The Eres of AuNRs at t = 0 min ranging from 1.5–2 eV suggests a heterogeneous distribution of the aspect ratio of AuNRs consistent with the results obtained with a scanning electron microscope (SEM, Fig. S2†). Such sample heterogeneity is beneficial for single-particle measurements because one can access the growth mechanism of AuNRs with a variety of initial AuNR morphology in one measurement.62 At t = 110 min, the Eres of all AuNRs shifts to ∼2.2 eV under interband excitation (blue dots), indicating that AuNRs transform into isotropic nanoparticles regardless of their initial aspect ratio, supported by the SEM image of the nanoparticles (Fig. S6†). Exciting intraband transition results in a blueshift of ∼50 meV for all AuNRs (red dots), suggesting a slight reshaping of AuNRs into a smaller aspect ratio. The degrees of the peak shift of AuNRs for the intraband transition and in the dark (black dots) are almost identical, indicating that the intraband transition does not provide additional energy to transform AuNRs. To obtain the statistics of the amplitudes for different AuNRs during the reaction, the time-dependent amplitude of each AuNR was normalized by its amplitude at t = 0 min to eliminate the variation in the scattering intensity of AuNRs due to different illumination powers in the DFS measurements between cells. Fig. 2B displays the normalized amplitude as a function of the reaction time with interband (blue) and intraband (red) excitations and without illumination (black). The AuNRs grow slightly in the dark as the averaged amplitudes at t = 50 min increase by a factor of 2 for these three samples (grey area). After photon illumination, the subensemble average of the normalized amplitude exhibits a stark difference under interband and intraband transitions. The normalized amplitude increases more than 13 times at t = 110 min by the interband excitation. In contrast, the time-dependent normalized amplitude behaves similarly for the intraband transition and in the dark, where the averaged amplitudes increase by a factor of 3 for both cases at t = 110 min. Combining evidence in the change of Eres, normalized amplitude, and SEM images, we can conclude that the AuNRs transform into large isotropic nanoparticles under the interband transition regardless of their initial aspect ratio. No significant reshaping was observed for AuNRs in the area without illumination (Fig. S7†). The intraband transition has a minimal impact on the Au3+ reduction reaction because the Eres shift and amplitude change are similar between the intraband excitation and in the dark. In both cases, the AuNRs slightly grow into a smaller aspect ratio with a slight increase in volume.
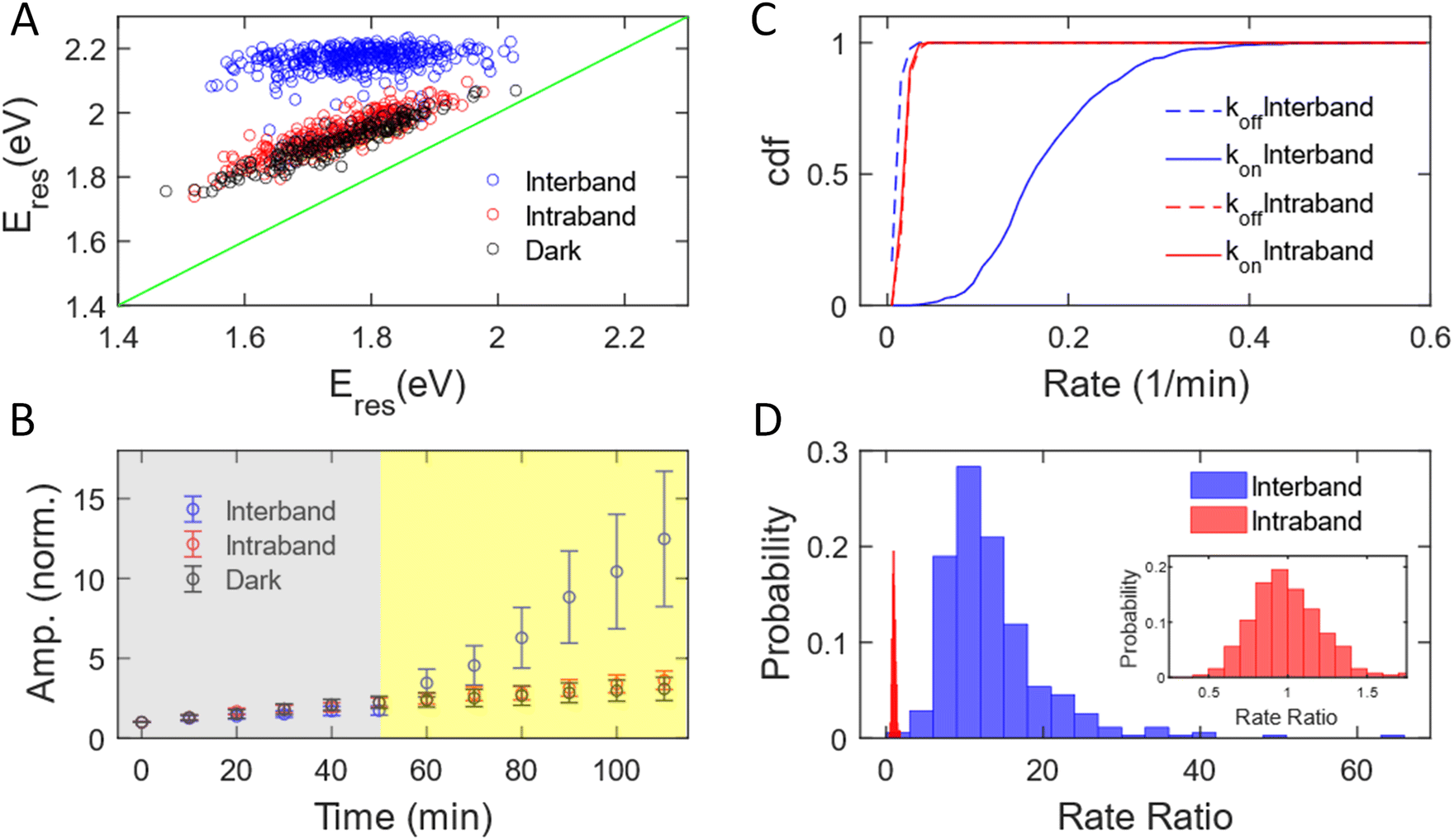 | ||
| Fig. 2 Growth of AuNRs with PVP ligands under interband and intraband transition. (A) Correlation between Eres of individual AuNRs at t = 0 (x-axis) and 110 min (y-axis) under interband (blue) and intraband (red) transitions and in the dark (black). The blue, red, and black symbols include data from 353, 251, and 187 AuNRs, respectively. (B) Subensemble average of the normalized amplitude of AuNRs during the growth reaction under interband (blue) and intraband (red) excitations and in the dark (black). The symbols and error bars represent the mean and standard deviation of the normalized amplitudes of AuNRs at a specific reaction time. (C) Cumulative density function of kon (solid lines) and koff (dashed lines) under interband (blue) and intraband (red) transitions. (D) Histogram of the rate ratio for individual AuNRs excited by interband (blue) and intraband (red) transitions. (Inset) Histogram of the rate ratio for AuNRs by intraband excitation with the rate ratio at 0–2. | ||
The growth rate of AuNRs excited by the interband transition is 13 times higher than that under intraband transition and in the dark. We observed that the time-dependent normalized amplitude is linear to the reaction time both in the dark and with light illumination (Fig. S8†). As a result, the time-dependent amplitude was fit to a linear equation, y = kt + a, at t = 0–50 min and t = 50–110 min to quantify the growth rate in the dark (koff) and under illumination (kon). We define the measured growth rate as an apparent growth rate because it is quantified using the increased spectral amplitude rather than the increased volume. The fitting was performed on the normalized amplitude as a function of the reaction time for all AuNRs with both interband transition (353 AuNRs) and intraband transition (251 AuNRs). Fig. 2C shows the cumulative density function of koff (dashed) and kon (solid) under interband (blue) and intraband (red) transitions. The cumulative density function for koff (0.023 ± 0.006 min−1) overlaps with that for kon (0.023 ± 0.006 min−1) with red-light illumination (red). The identical koff and kon suggest that intraband excitation does not enhance the photocatalytic activity of the Au3+ reduction. In contrast, kon is 0.180 ± 0.072 min−1, while koff is 0.015 ± 0.006 min−1 under interband excitation. The increase in the growth rate by more than 10-fold suggests a much more efficient photocatalytic reduction of Au3+ with the interband transition. Additionally, single-particle measurements provide enriched information on the morphology-dependent growth rate. Fig. S9† shows koff as a function of Eres at t = 0 min, representing the aspect ratio of AuNRs before the reaction. The anti-correlation in Fig. S9† demonstrates a higher koff for AuNRs with a larger aspect ratio, probably because AuNRs with a higher aspect ratio possess more surface area for chemical reactions. Furthermore, we can also correlate the kon and koff of the same AuNRs, as shown in Fig. S10.† Under interband transition, there is no correlation between kon and koff, suggesting that AuNRs lose their memory with blue-light illumination. Indeed, all AuNRs transformed into isotropic shapes within 10 minutes of interband excitation, regardless of the initial aspect ratio of AuNRs, as evidenced by the fact that Eres shifted to 2.0 eV for most of the AuNRs after illumination with blue light for 10 minutes, as shown in Fig. S11.† On the other hand, koff and kon show a positive correlation with the intraband transition, meaning that AuNRs with a higher growth rate in the dark also grow faster with red-light illumination. These results further demonstrate the benefit of single-particle spectroscopy to take advantage of sample heterogeneity to obtain more information in “one” measurement.
We further quantified the enhanced growth rate under illumination for each AuNR by defining the rate ratio as kon/koff. Fig. 2D shows the histogram of the rate ratio for single AuNRs excited by the interband (blue) and intraband (red) transitions. The large and broad distribution of the rate ratio (13.46 ± 6.94) demonstrates an enhanced growth rate of more than one order of magnitude with a significant variation between AuNRs under interband transition. In contrast, the enhanced growth rate with the intraband transition was calculated to be 0.99 ± 0.22, indicating no increased photocatalytic activity of the Au3+ reduction. Based on these observations, we conclude that the excitation of the interband transition is responsible for the photoreduction of Au3+ in the presence of PVP.
The adsorption of PVP on AuNRs is critical to enhancing the photocatalytic reduction of Au3+ under interband excitation. To investigate the role of PVP, we performed experiments on AuNR growth by illuminating blue light with three different growth solutions containing (1) 3 μM HAuCl4, 15 μM PVP, and 2.9 M ethanol, (2) 3 μM HAuCl4 and 15 μM PVP, and (3) 3 μM HAuCl4 and 2.9 M ethanol. Fig. 3A and B show the subensemble average of the change of Eres (ΔEres(t) = Eres(t) − Eres(0)) and normalized amplitude as a function of reaction time, respectively. In the dark, the ΔEres and normalized amplitude of AuNRs increase slightly with the reaction time, indicating the growth of AuNRs into smaller aspect ratios in the three reagents. The small variation (mean values within error bars) between these three cases suggests that AuNRs grow similarly in the solutions regardless of the presence of PVP and ethanol. When illuminated with blue light, AuNRs in growth solution 3 (without PVP) only show a small increase in ΔEres and the normalized amplitude (magenta symbols). Another control experiment using AuNRs functionalized with cetyltrimethylammonium bromide (CTAB) in growth solution 3 also exhibits a slight increase in the normalized amplitude with interband excitation (Fig. S12†). These results suggest no significant growth of AuNRs in the absence of PVP under interband transition. In contrast, Eres blueshifts by 0.4 eV, and the normalized amplitude increases by a factor of 10 in growth solution 2 (without ethanol) at t = 110 min (green symbols). Such a large blueshift in Eres and increase in the amplitude are comparable to the results in growth solution 1 (blue symbols), indicating that all AuNRs reshape into large and isotropic nanoparticles. Based on these observations, PVP is the key to promoting the photocatalytic reduction of Au3+, regardless of the presence of ethanol, despite a slightly less increase in the amplitude without ethanol. Ethanol is utilized as a hole scavenger in plasmonic catalysis to remove holes and prolong the lifetime of hot electrons.55,57 Our results suggest that PVP ligands could serve as hole scavengers.
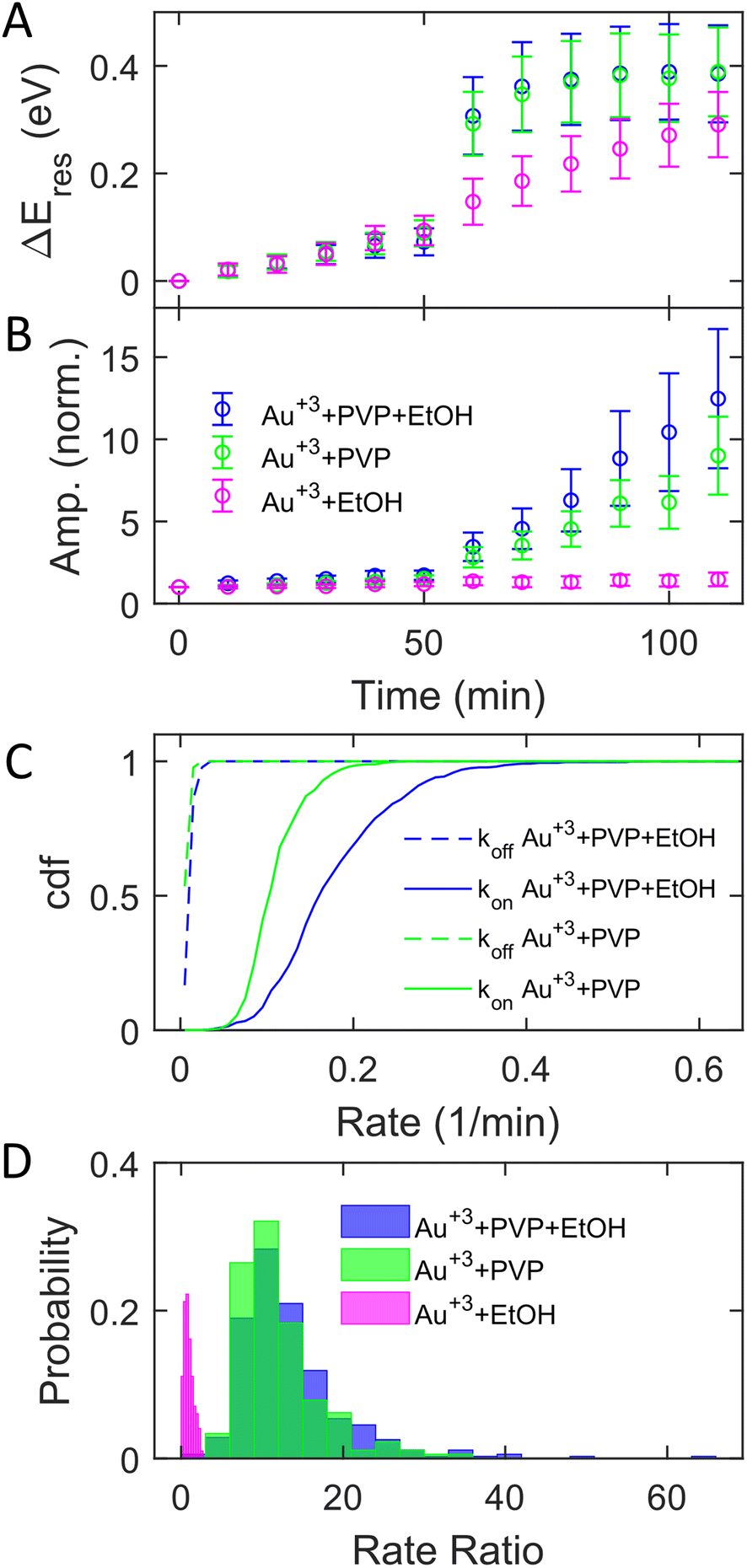 | ||
| Fig. 3 The role of PVP in the photoinduced growth of AuNRs under interband excitation. (A and B) Evolution of ΔEres (A) and the normalized amplitude (B) in a growth solution containing Au3+, PVP, and ethanol (blue), Au3+ and PVP (green), and Au3+ and ethanol (magenta). The symbols and error bars represent the mean and standard deviation of the values for AuNRs, respectively. (C) Cumulative density function of kon (solid) and koff (dashed) with a growth solution containing Au3+, PVP, and ethanol (blue) and Au3+ and PVP (green). (D) Histogram of the rate ratio for individual AuNRs with growth solutions containing Au3+, PVP, and ethanol (blue), Au3+ and PVP (green), and Au3+ and ethanol (magenta). | ||
Further analysis of the rate ratio confirms that the enhanced growth rates with and without ethanol are comparable under interband excitation in the presence of PVP. Fig. 3C shows koff (dashed) and kon (solid) in growth solutions 1 (blue) and 2 (green). Averaging from 353 (blue) and 355 (green) AuNRs, koff = 0.015 ± 0.006 min−1 and kon = 0.180 ± 0.072 min−1 with solution 1, while koff = 0.011 ± 0.004 min−1 and kon = 0.113 ± 0.034 min−1 with solution 2. In solutions 1 and 2, it shows that the averaged kon increased more than 10 times compared to koff. The rate ratios of each AuNR were also calculated. Fig. 3D shows the histogram of the rate ratio with solution 1 (blue), solution 2 (green), and solution 3 (magenta). The average rate ratios are 13.46 ± 6.94 and 12.08 ± 5.17 for solutions 1 and 2, respectively, demonstrating that the growth rates increase more than 12 times under interband excitation with PVP regardless of the presence of the hole scavenger. No enhanced growth rate is observed when excited by interband transition without PVP as the rate ratio for solution 3 is 0.95 ± 0.57. As the hole scavenger separates the electron–hole pair and promotes the hot-electron lifetime, we proposed that PVP rather than ethanol induces electron–hole separation. Indeed, the HOMO of PVP is −5.93 eV,63 which is higher than the onset of the Au d-band (∼−7.6 eV) and lower than the Fermi level of Au (−5.2 eV). The interband excitation creates electrons and holes populating near the Fermi level and d-band, respectively. The d-band holes subsequently transfer to the HOMO of PVPs, leading to the charge separation at the Au–PVP interface and enhancing the lifetime of the electron–hole pairs. This process is efficient because PVP ligands are adsorbed on the Au surface. The long-lived electrons on the Au surface reduce Au3+ to deposit Au atoms on AuNRs for growth. Therefore, efficient charge separation by PVP is responsible for the enhanced growth rate. The holes in PVP ligands further oxidize ethanol or water to prevent electron–hole recombination. This is evidenced by a slightly higher enhanced growth rate with ethanol, which more effectively removes holes on PVPs to inhibit the recombination of holes and electrons. Furthermore, no enhanced growth was observed in the presence of ethanol but without PVP because of the lack of hole quenching, as ethanol needs to diffuse to the Au surface to remove holes before electron–hole recombination.
Next, we controlled the adsorption position of PVP on AuNRs to identify the location of electrons after the interband excitation. The Liz-Marzán group has shown that PVP ligands preferentially bind to the {111} facet, which is located at the tip of AuNRs, compared to {110} and {100}.64 Therefore, PVP ligands are adsorbed at the tip of AuNRs with a low PVP concentration, while uniformly covering AuNRs at a high PVP concentration. If the electrons localize near PVP after charge separation, the Au3+ reduction should occur at the tip of AuNRs, leading to a redshift of Eres due to a larger aspect ratio. In contrast, if the electrons delocalize in AuNRs after the holes transfer to PVP, a blueshift of Eres is expected because Au3+ is reduced at a random position, causing an isotropic growth. To prove this concept, we performed experiments with the growth solution containing 3 μM HAuCl4 and 2.9 M ethanol with various concentrations of PVP. The reduction reaction was carried out in the dark at t = 0–30 min, and then under the illumination of blue light at t = 40–60 min. Fig. 4 shows 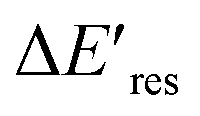 versus Eres at t = 0 min, where
versus Eres at t = 0 min, where 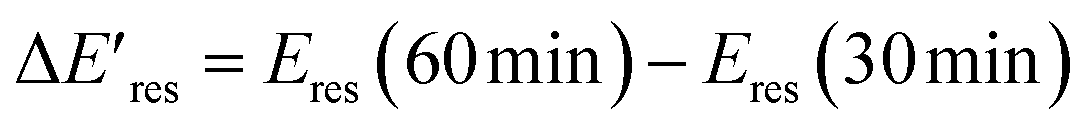 representing the difference in peak energy after illuminating blue light for 30 min. For the PVP concentration of 15 μM (blue),
representing the difference in peak energy after illuminating blue light for 30 min. For the PVP concentration of 15 μM (blue), 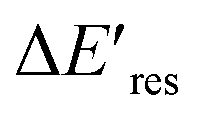 is positive for all AuNRs, indicating a blueshift of Eres with interband excitation. Interestingly, approximately 63% of AuNRs show a negative
is positive for all AuNRs, indicating a blueshift of Eres with interband excitation. Interestingly, approximately 63% of AuNRs show a negative 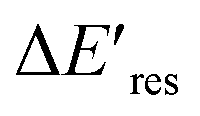 (redshift) when changing the PVP concentration to 1.5 μM (red). Further reducing the concentration to 1.0 μM leads to more redshift in Eres (green), with 88% of AuNRs showing a negative
(redshift) when changing the PVP concentration to 1.5 μM (red). Further reducing the concentration to 1.0 μM leads to more redshift in Eres (green), with 88% of AuNRs showing a negative 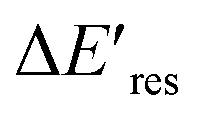 . Because PVP ligands preferentially adsorb at the tip of AuNRs at low PVP concentration, the redshift of Eres arises from the deposition of Au atoms at the tip of AuNRs, implying the localized electrons at the Au–PVP interface after electron–hole separation. With a high concentration of PVP, the AuNRs are covered with PVP ligands, and the electrons are evenly distributed on the AuNR surface after charge separation, causing isotropic growth, which leads to a blueshift of Eres. A schematic illustration of this mechanism is depicted in Fig. S13.† All these results strongly indicate that the electrons are localized at the Au–PVP interface after charge separation.
. Because PVP ligands preferentially adsorb at the tip of AuNRs at low PVP concentration, the redshift of Eres arises from the deposition of Au atoms at the tip of AuNRs, implying the localized electrons at the Au–PVP interface after electron–hole separation. With a high concentration of PVP, the AuNRs are covered with PVP ligands, and the electrons are evenly distributed on the AuNR surface after charge separation, causing isotropic growth, which leads to a blueshift of Eres. A schematic illustration of this mechanism is depicted in Fig. S13.† All these results strongly indicate that the electrons are localized at the Au–PVP interface after charge separation.
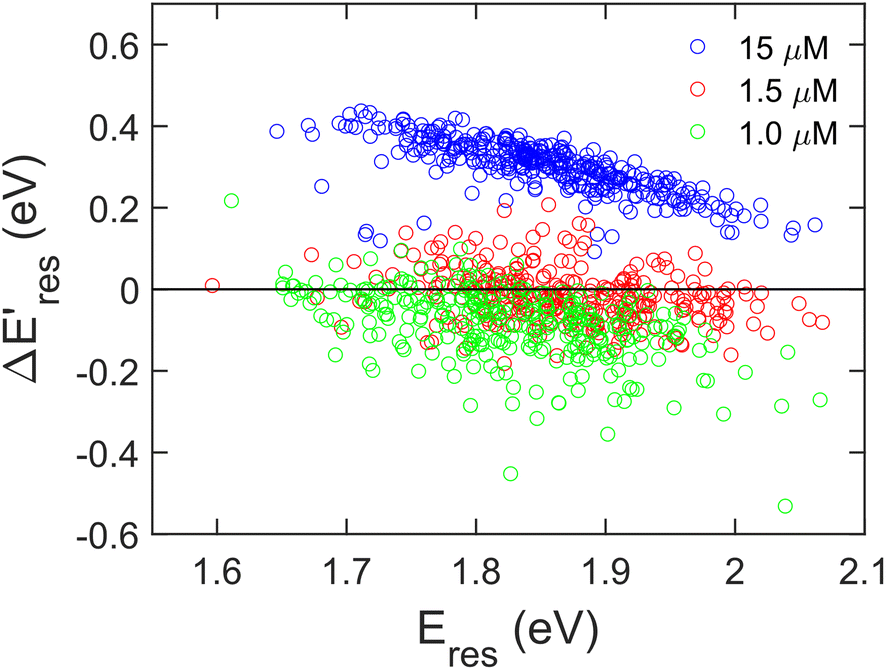 | ||
Fig. 4 The change of Eres (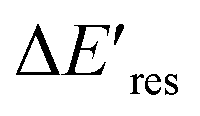 ) as a function of Eres at t = 0 min with 15 μM (blue), 1.5 μM (red), and 1.0 μM (green) PVP in the growth solution. The black line indicates ) as a function of Eres at t = 0 min with 15 μM (blue), 1.5 μM (red), and 1.0 μM (green) PVP in the growth solution. The black line indicates 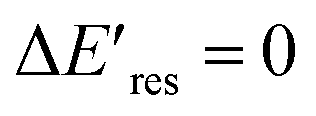 . . | ||
Based on these results, we propose the mechanisms of the photocatalytic reduction of Au3+ on AuNRs with PVP, as depicted in Fig. 5. When excited with an interband transition, it creates d-band holes and sp-band electrons, which populate near the Fermi level. The d-band holes at −7.6 eV subsequently transfer to the HOMO of PVP at −5.93 eV as the holes move upward in energy spontaneously, leading to charge separation to prolong the lifetime of the hot carriers. The electrons at the Au–PVP interface reduce Au3+ to induce AuNR growth, while the holes in PVP undergo an ethanol or water oxidation reaction. On the other hand, the intraband excitation generates electrons populating at the states with energy equivalent to the photon energy above the Fermi level and the holes near the Fermi level. The energy of holes is higher than that of the PVP HOMO. Additionally, the LUMO of PVP locates at −2.01 eV, which is ∼3.2 eV higher than the Fermi level of Au. As the energy of longitudinal plasmons is less than 2 eV, the intraband excitation could not promote the electron population from the Fermi level of Au to the LUMO of PVP. Therefore charge separation is forbidden under the excitation of intraband transition. Then electron–hole recombination occurs before the electrons reduce Au3+. As a result, no photocatalytic reduction was observed under the intraband transition. It is worth noting that the activities of plasmonic photocatalysts behave linearly with excitation power under interband and intraband transitions.54,55 The growth rate of AuNRs is expected to increase linearly with the excitation power excited by the interband transition. However, increasing the illumination power for the intraband transition should have a minimum impact on the growth rate due to ultrafast electron–hole recombination. Photoexcitation of plasmonic nanoparticles elevates the temperature on the nanoparticle surface and enhances the catalytic activities. Given the lower excitation power in our experiment, the temperature only increases less than 1 °C on the nanoparticle surface, and therefore the increased temperature due to the photoexcitation has no impact on the growth rate, as observed in the inset of Fig. 2D.
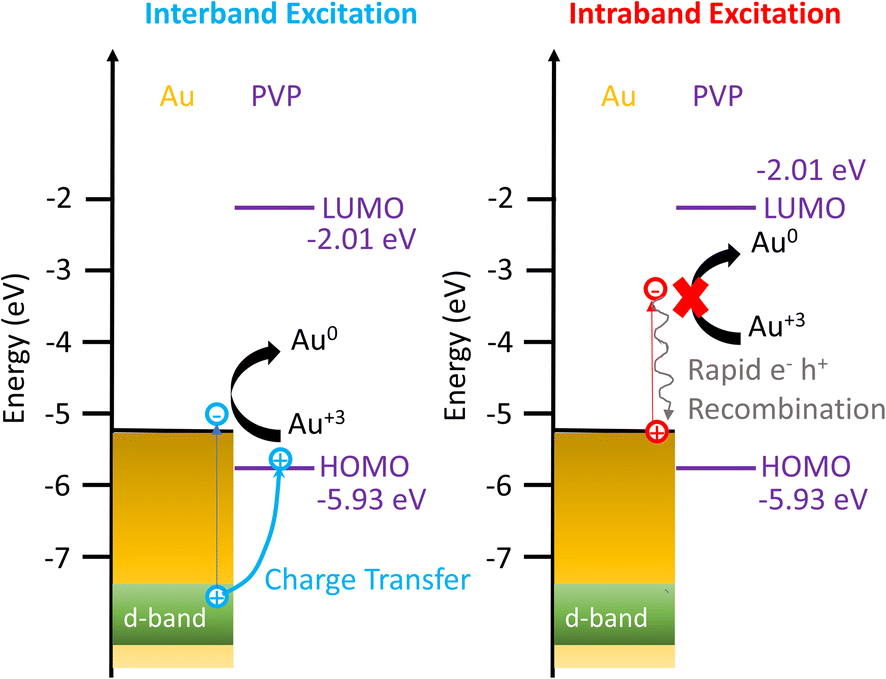 | ||
| Fig. 5 Energy diagram showing the mechanism of enhanced Au3+ reduction driven by interband excitation and no enhancement of Au3+ reduction excited by the intraband transition. | ||
The results of this study can be expanded to plasmonic nanoparticles with various materials and morphologies. The HOMO of PVP is located at −5.93 eV, which is higher than the d-band of silver (Ag) and copper (Cu), located at approximately −8.2 eV and −6.6 eV, respectively.36 Charge separation occurs at the metal–PVP interface upon photoexciting the interband transition for Au, Ag, and Cu. Additionally, the interband transition is determined by the bulk materials and is independent of the morphology of nanoparticles. As a result, our findings can be applied to noble nanoparticles of any shape.
Conclusion
In summary, using single-particle DFS spectroscopy, we have demonstrated PVP-induced charge separation to enhance the catalytic activity of the Au3+ reduction reaction under interband excitation. Probing the spectral evolution of single AuNRs in the presence of PVP ligands confirms that the AuNRs transform into large and isotropic nanoparticles excited by the interband transition. The growth rate increases 13 times under interband excitation compared to without illumination. The intraband transition does not contribute to the Au3+ reduction. PVP ligands rather than hole scavengers are critical to enhancing the reduction reaction. These results elucidate that PVP induces electron–hole separation with interband excitation to prolong the lifetime of sp-band electrons, which subsequently reduce Au3+. The hot carriers generated by the intraband excitation suffer from ultrafast electron–hole recombination, prohibiting the reduction reaction. This study provides an alternative method to utilize capping ligands to promote the photocatalysis of plasmonic nanoparticles.Data availability
Materials, protocols for sample preparation and single-particle measurements, SEM images of AuNRs and Au nanoparticles, the extinction spectrum of AuNR solution, color images of AuNRs during the reduction reaction, results of a control experiment using CTAB-functionalized AuNRs, and detailed data analysis for single-particle measurements can be found in the ESI.Author contributions
W. S. C. and B. R. conceived the idea and designed the experiments. B. R. and T. V. performed experiments and analyzed the data. W. S. C. wrote the manuscript with contributions from all authors. All authors have given the approval of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the ACS Petroleum Research Fund (62122-DNI6) and the National Science Foundation (CHE 2203612, CMI program) with partial co-funding from the CAT, MSN, and CSDM-A programs. The electron microscopy images in this work were obtained using a scanning electron microscope supported by the National Science Foundation under Grant no. 1726239. The authors also thank Mr. Paul Sousa for fabricating sample holders for the optical microscope.References
- Y. Sun and Z. Tang, MRS Bull., 2020, 45, 20–25 CrossRef CAS.
- L. Zhou, M. Lou, J. L. Bao, C. Zhang, J. G. Liu, J. M. P. Martirez, S. Tian, L. Yuan, D. F. Swearer, H. Robatjazi, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022109118 CrossRef CAS.
- S. Li, H. Huang, L. Shao and J. Wang, ACS Nano, 2021, 15, 10759–10768 CrossRef CAS PubMed.
- E. A. Coronado, E. R. Encina and F. D. Stefani, Nanoscale, 2011, 3, 4042–4059 RSC.
- M. W. Knight, L. Liu, Y. Wang, L. Brown, S. Mukherjee, N. S. King, H. O. Everitt, P. Nordlander and N. J. Halas, Nano Lett., 2012, 12, 6000–6004 CrossRef CAS PubMed.
- L. S. Slaughter, W.-S. Chang, P. Swanglap, A. Tcherniak, B. P. Khanal, E. R. Zubarev and S. Link, J. Phys. Chem. C, 2010, 114, 4934–4938 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- S. Wu, Y. Chen and S. Gao, Phys. Rev. Lett., 2022, 129, 086801 CrossRef CAS PubMed.
- J. U. Salmón-Gamboa, M. Romero-Gómez, D. J. Roth, A. V. Krasavin, P. Wang, W. Dickson and A. V. Zayats, Nanoscale Adv., 2021, 3, 767–780 RSC.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- P. Wang, A. V. Krasavin, M. E. Nasir, W. Dickson and A. V. Zayats, Nat. Nanotechnol., 2018, 13, 159–164 CrossRef CAS.
- X. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS PubMed.
- Y. Zhang, T. Nelson, S. Tretiak, H. Guo and G. C. Schatz, ACS Nano, 2018, 12, 8415–8422 CrossRef CAS PubMed.
- Y. Yuan, L. Zhou, H. Robatjazi, J. L. Bao, J. Zhou, A. Bayles, L. Yuan, M. Lou, M. Lou, S. Khatiwada, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2022, 378, 889–893 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- J. S. DuChene, G. Tagliabue, A. J. Welch, W.-H. Cheng and H. A. Atwater, Nano Lett., 2018, 18, 2545–2550 CrossRef CAS.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- E. B. Creel, E. R. Corson, J. Eichhorn, R. Kostecki, J. J. Urban and B. D. McCloskey, ACS Energy Lett., 2019, 4, 1098–1105 CrossRef CAS.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- D. Devasia, A. J. Wilson, J. Heo, V. Mohan and P. K. Jain, Nat. Commun., 2021, 12, 2612 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Low, Y.-W. Yang, H. Li, D. Wu, S. Chen, J. Jin, H. Li, H. Ju, C.-H. Wang, Z. Lu, R. Long, L. Song and Y. Xiong, Nat. Commun., 2023, 14, 221 CrossRef CAS PubMed.
- R. Li, W.-H. Cheng, M. H. Richter, J. S. DuChene, W. Tian, C. Li and H. A. Atwater, ACS Energy Lett., 2021, 6, 1849–1856 CrossRef CAS.
- H. Lai, W. Xiao, Y. Wang, T. Song, B. Long, S. Yin, A. Ali and G.-J. Deng, Chem. Eng. J., 2021, 417, 129295 CrossRef CAS.
- Y. Wang, Y. Wang, I. Aravind, Z. Cai, L. Shen, B. Zhang, B. Wang, J. Chen, B. Zhao, H. Shi, J. M. Dawlaty and S. B. Cronin, J. Am. Chem. Soc., 2022, 144, 3517–3526 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS.
- G. Joshi, A. Saha, A. Dutta and S. Khatua, ACS Appl. Mater. Interfaces, 2022, 14, 38815–38823 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 9802–9805 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- E. Contreras, R. Nixon, C. Litts, W. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS.
- M. Bernardi, J. Mustafa, J. B. Neaton and S. G. Louie, Nat. Commun., 2015, 6, 7044 CrossRef CAS PubMed.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard III and H. A. Atwater, Nat. Commun., 2014, 5, 5788 CrossRef CAS.
- A. O. Govorov, H. Zhang and Y. K. Gun'ko, J. Phys. Chem. C, 2013, 117, 16616–16631 CrossRef CAS.
- A. M. Brown, R. Sundararaman, P. Narang, W. A. Goddard III and H. A. Atwater, ACS Nano, 2016, 10, 957–966 CrossRef CAS PubMed.
- G. V. Hartland, L. V. Besteiro, P. Johns and A. O. Govorov, ACS Energy Lett., 2017, 2, 1641–1653 CrossRef CAS.
- S. A. Lee, C. T. Kuhs, E. K. Searles, H. O. Everitt, C. F. Landes and S. Link, Nano Lett., 2023, 23, 3501–3506 CrossRef CAS.
- W. R. Jeffries, K. Park, R. A. Vaia and K. L. Knappenberger Jr, Nano Lett., 2020, 20, 7722–7727 CrossRef CAS PubMed.
- C. Voisin, N. Del Fatti, D. Christofilos and F. Vallée, J. Phys. Chem. B, 2001, 105, 2264–2280 CrossRef CAS.
- H. Harutyunyan, A. B. F. Martinson, D. Rosenmann, L. K. Khorashad, L. V. Besteiro, A. O. Govorov and G. P. Wiederrecht, Nat. Nanotechnol., 2015, 10, 770–774 CrossRef CAS PubMed.
- N. Hogan, S. Wu and M. Sheldon, J. Phys. Chem. C, 2020, 124, 4931–4945 CrossRef CAS.
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, ACS Nano, 2020, 14, 16202–16219 CrossRef PubMed.
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702–704 CrossRef CAS PubMed.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632–635 CrossRef CAS.
- B. Foerster, M. Hartelt, S. S. E. Collins, M. Aeschlimann, S. Link and C. Sönnichsen, Nano Lett., 2020, 20, 3338–3343 CrossRef CAS PubMed.
- G. Tagliabue, J. S. DuChene, M. Abdellah, A. Habib, D. J. Gosztola, Y. Hattori, W.-H. Cheng, K. Zheng, S. E. Canton, R. Sundararaman, J. Sá and H. A. Atwater, Nat. Mater., 2020, 19, 1312–1318 CrossRef CAS PubMed.
- B. Foerster, A. Joplin, K. Kaefer, S. Celiksoy, S. Link and C. Sönnichsen, ACS Nano, 2017, 11, 2886–2893 CrossRef CAS PubMed.
- B. Foerster, V. A. Spata, E. A. Carter, C. Sönnichsen and S. Link, Sci. Adv., 2019, 5, eaav0704 CrossRef CAS.
- S. Y. Lee, P. V. Tsalu, G. W. Kim, M. J. Seo, J. W. Hong and J. W. Ha, Nano Lett., 2019, 19, 2568–2574 CrossRef CAS PubMed.
- B. Xiang, Y. Li, C. H. Pham, F. Paesani and W. Xiong, Sci. Adv., 2017, 3, e1701508 CrossRef PubMed.
- J. Zhao, S. C. Nguyen, R. Ye, B. Ye, H. Weller, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, ACS Cent. Sci., 2017, 3, 482–488 CrossRef CAS PubMed.
- Y. Kim, J. G. Smith and P. K. Jain, Nat. Chem., 2018, 10, 763–769 CrossRef CAS.
- Z. Mao, R. Espinoza, A. Garcia, A. Enwright, H. Vang and S. C. Nguyen, ACS Nano, 2020, 14, 7038–7045 CrossRef CAS PubMed.
- Y. Zhai, J. S. DuChene, Y.-C. Wang, J. Qiu, A. C. Johnston-Peck, B. You, W. Guo, B. DiCiaccio, K. Qian, E. W. Zhao, F. Ooi, D. Hu, D. Su, E. A. Stach, Z. Zhu and W. D. Wei, Nat. Mater., 2016, 15, 889–895 CrossRef CAS PubMed.
- W. Guo, A. C. Johnston-Peck, Y. Zhang, Y. Hu, J. Huang and W. D. Wei, J. Am. Chem. Soc., 2020, 142, 10921–10925 CrossRef CAS.
- S. D. Golze, S. Porcu, C. Zhu, E. Sutter, P. C. Ricci, E. C. Kinzel, R. A. Hughes and S. Neretina, Nano Lett., 2021, 21, 2919–2925 CrossRef CAS.
- S. D. Golze, R. A. Hughes, S. Rouvimov, R. D. Neal, T. B. Demille and S. Neretina, Nano Lett., 2019, 19, 5653–5660 CrossRef CAS PubMed.
- B. S. Hoener, S. R. Kirchner, T. S. Heiderscheit, S. S. E. Collins, W.-S. Chang, S. Link and C. F. Landes, Chem, 2018, 4, 1560–1585 CAS.
- S. S. E. Collins, E. K. Searles, L. J. Tauzin, M. Lou, L. Bursi, Y. Liu, J. Song, C. Flatebo, R. Baiyasi, Y.-Y. Cai, B. Foerster, T. Lian, P. Nordlander, S. Link and C. F. Landes, ACS Nano, 2021, 15, 9522–9530 CrossRef CAS PubMed.
- F. Meng, S. Liu, Y. Wang, C. Tao, P. Xu, W. Guo, L. Shen, X. Zhang and S. Ruan, J. Mater. Chem., 2012, 22, 22382–22386 RSC.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 679–688 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02167j |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |