
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA heterologous expression platform in Aspergillus nidulans for the elucidation of cryptic secondary metabolism biosynthetic gene clusters: discovery of the Aspergillus fumigatus sartorypyrone biosynthetic pathway†
Shu-Yi
Lin
 a,
C. Elizabeth
Oakley
b,
Cory B.
Jenkinson
b,
Yi-Ming
Chiang
a,
Ching-Kuo
Lee
c,
Christopher G.
Jones
d,
Paul M.
Seidler
a,
Hosea M.
Nelson
d,
Richard B.
Todd
e,
Clay C. C.
Wang
*af and
Berl R.
Oakley
*b
a,
C. Elizabeth
Oakley
b,
Cory B.
Jenkinson
b,
Yi-Ming
Chiang
a,
Ching-Kuo
Lee
c,
Christopher G.
Jones
d,
Paul M.
Seidler
a,
Hosea M.
Nelson
d,
Richard B.
Todd
e,
Clay C. C.
Wang
*af and
Berl R.
Oakley
*b
aDepartment of Pharmacology and Pharmaceutical Sciences, University of Southern California, Los Angeles, CA 90089, USA. E-mail: clayw@usc.edu
bDepartment of Molecular Biosciences, University of Kansas, 1200 Sunnyside Avenue, Lawrence, KS 66045, USA. E-mail: boakley@ku.edu
cSchool of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei 11031, Taiwan
dThe Arnold and Mabel Beckman Laboratory of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA
eDepartment of Plant Pathology, Kansas State University, Manhattan, KS 66506, USA
fDepartment of Chemistry, University of Southern California, Los Angeles, CA 90089, USA
First published on 26th June 2023
Abstract
Aspergillus fumigatus is a serious human pathogen causing life-threatening Aspergillosis in immunocompromised patients. Secondary metabolites (SMs) play an important role in pathogenesis, but the products of many SM biosynthetic gene clusters (BGCs) remain unknown. In this study, we have developed a heterologous expression platform in Aspergillus nidulans, using a newly created genetic dereplication strain, to express a previously unknown BGC from A. fumigatus and determine its products. The BGC produces sartorypyrones, and we have named it the spy BGC. Analysis of targeted gene deletions by HRESIMS, NMR, and microcrystal electron diffraction (MicroED) enabled us to identify 12 products from the spy BGC. Seven of the compounds have not been isolated previously. We also individually expressed the polyketide synthase (PKS) gene spyA and demonstrated that it produces the polyketide triacetic acid lactone (TAL), a potentially important biorenewable platform chemical. Our data have allowed us to propose a biosynthetic pathway for sartorypyrones and related natural products. This work highlights the potential of using the A. nidulans heterologous expression platform to uncover cryptic BGCs from A. fumigatus and other species, despite the complexity of their secondary metabolomes.
Introduction
Aspergillosis is the spectrum of diseases caused by Aspergillus species that colonize the bronchial trees of patients.1 It is estimated that there are over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of life-threatening Aspergillosis in humans annually, with a mortality rate of 30–80%.2–5 Invasive Aspergillosis (IA) is one of the most common causes of death in severely immunocompromised patients, with mortality rates up to 40% to 50% in patients with acute leukemia and recipients of hematopoietic stem cell transplantations.
000 cases of life-threatening Aspergillosis in humans annually, with a mortality rate of 30–80%.2–5 Invasive Aspergillosis (IA) is one of the most common causes of death in severely immunocompromised patients, with mortality rates up to 40% to 50% in patients with acute leukemia and recipients of hematopoietic stem cell transplantations.
Aspergillus fumigatus is by far the most common species causing IA (70%).5 The World Health Organization “WHO fungal priority pathogens list to guide research, development and public health action 2022” classified A. fumigatus as one of four species in the critical priority group, the most urgent need category.6 Secondary metabolites (SMs) in A. fumigatus contribute to its unique ability to survive in the environment and establish itself in human hosts.7,8 SMs are small molecules produced by fungi, prokaryotes and plants that are not strictly required for growth or reproduction, but they confer a selective advantage to the producing organism. In A. fumigatus, SMs play important roles in iron and copper uptake and homeostasis, for example.8,9A. fumigatus also produces SMs that kill or inhibit competitors and these activities are often important to pathogenesis. Gliotoxin, for one example of several, protects A. fumigatus against amoebae in the environment and against T lymphocytes and the macrophage immune response in pathogenesis. Genetic elimination of gliotoxin production results in attenuation of virulence.10–12
SMs in filamentous fungi are synthesized by enzymes encoded by genes organized in contiguous biosynthetic gene clusters (BGCs).13 The genes of the BGCs are coordinately regulated, and the BGCs often contain genes encoding transcription factors that drive the expression of the other genes of the BGC. SM BGCs vary significantly across species and strains of Aspergillus.14 The two most studied isolates of A. fumigatus are Af293 and CEA10.15,16 Bioinformatics indicate that the A. fumigatus Af293 strain contains 34 predicted SM BGCs, and the CEA10 derivative strain A1163 has 33 predicted SM BGCs. The SM BGCs of the two strains are largely but not completely overlapping, with three BGCs unique to Af293 and two unique to CEA10 making a total of 36 BGCs. The products of only 19 of these BGCs have been determined, leaving, prior to this study, 17 to be determined.17–19
In fungi, many SM BGCs are not expressed under normal laboratory growth conditions, whereas others are expressed at high levels, resulting in a “forest” of SM peaks in LC/MS traces. To determine the SMs produced by cryptic BGCs, one needs to find a way to activate their expression. Once they are expressed, the SMs they produce must be identified and purified from among the forest of metabolites produced by the fungus. Various approaches have been developed in the model fungus Aspergillus nidulans to activate SM production.20–31 In principle, some of these approaches could be transferred to A. fumigatus to elucidate its secondary metabolome, but it would require a great deal of time and effort to develop them. Perhaps more seriously, metabolite profiles from A. fumigatus grown under laboratory conditions are quite complex due to high levels of production of many compounds such as fumigaclavine C, fumiquinazoline, and monomethyl-sulochrin and their pathway intermediates.32–38 Therefore, identifying minor compounds is difficult due to the interference of these major metabolites, and determining the BGC responsible for any minor compound is equally difficult. In A. nidulans, we have developed a strategy we call “genetic dereplication” that facilitates new compound discovery. We have deleted eight BGCs, in their entirety, that produce abundant SMs (>244000 bp deleted), thereby reducing the complexity of SM profiles such that novel compounds are more easily detected.26 Eliminating highly expressed biosynthetic pathways may also increase pools of SM precursors such as acetyl-CoA and malonyl-CoA for pathways expressed at low levels. Because developing the genetic dereplication approach for A. fumigatus would be a lengthy process, we are developing methods to express A. fumigatus BGCs in A. nidulans and determine their products.
In this study, we report the expression of a heretofore cryptic A. fumigatus secondary metabolite biosynthesis pathway in A. nidulans and the determination of the products of the BGC. Using a genetic dereplication strain to reduce the SM background, we refactored the target BGC genes into A. nidulans, placing each gene under the control of an inducible promoter. The heterologously expressed SMs were purified, and the structures were determined by NMR and electron cryo-microscopy (cryoEM) microcrystal electron diffraction (MicroED). We found that the BGC encodes a family of meroterpenoids called sartorypyrones that have never been reported in A. fumigatus. Two of the compounds are novel while two have been found in the closely related nonpathogenic fungus Neosartorya fischeri (KUFC 6344 and FO-5897).39,40 We generated deletant strains that were each missing one gene of the biosynthetic pathway. Some of these strains accumulated chemically stable intermediates and shunt products in sufficient amounts for complete structural characterization, resulting in the identification of a total of eight additional, related compounds. We also individually expressed the non-reducing polyketide synthase (NR-PKS) gene of the BGC and demonstrated that triacetic acid lactone (TAL) is the product of the PKS. TAL is a potentially important biorenewable platform chemical.41 Combining these data with further bioinformatic analysis, we propose a biosynthetic pathway for sartorypyrones. Importantly, the approach reported in this study, and variations thereof, can be used to elucidate other A. fumigatus cryptic secondary metabolism BGCs.
Results
Analysis of a potential gene cluster for production of polyketide diterpenoids (PK-DT) in A. fumigatus
One of the A. fumigatus Af293 SM BGCs with unknown products attracted our interest because it contains a non-reducing polyketide synthase (NR-PKS) gene (Afu8g02350), a prenyltransferase gene (Afu8g02410) and a terpene cyclase (TC) gene (Afu8g02390), suggesting that the SM product of this gene cluster is an unidentified meroterpenoid. As annotated by Inglis et al., this BGC also contains an acetyltransferase gene (Afu8g02360), a flavin-containing monooxygenase (FMO) gene (Afu8g02380), a geranylgeranyl pyrophosphate synthase (GGPPS) gene (Afu8g02400), a hypothetical protein (Afu8g02420), and an alcohol dehydrogenase (Afu8g02430).42 A C-4 methyl sterol oxidase (Afu8g02440) is adjacent. In all, the putative BGC is greater than 24 kbp in length. Synteny analysis in FungiDB reveals that the genes within the putative BGC are conserved and collinear between A. fumigatus isolates Af293 (Afu8g02350–Afu8g02440) and A1163 (AFUB_084240–AFUB_084150).43 Bioinformatic analysis of this BGC using BLASTp showed that genes Afu8g02350–Afu8g02410 are homologous to genes in the sre cluster, which encodes the biosynthetic pathway for sartorypyrone A and D in Aspergillus felis 0260, and the cle cluster, which encodes the biosynthetic pathway for chevalone E in Aspergillus versicolor 0312 (Table 1).44| A. fumigatus spy cluster | A. felis sre cluster | Similarity/identity (%) | A. versicolor cle cluster | Similarity/identity (%) | Putative function |
|---|---|---|---|---|---|
| Afu8g02350 (spyA) | sre6 | 89/82 | cle1 | 59/43 | Polyketide synthase |
| Afu8g02360 (spyB) | sre5 | 86/79 | — | — | Acetyltransferase |
| Afu8g02380 (spyC) | sre4 | 90/83 | cle3 | 73/59 | FAD-dependent monooxygenase |
| Afu8g02390 (spyD) | sre3 | 77/63 | cle7 | 56/37 | Terpene cyclase |
| Afu8g02400 (spyE) | sre2 | 91/87 | cle6 | 81/70 | Geranylgeranyl pyrophosphate synthase |
| Afu8g02410 (spyF) | sre1 | 83/76 | cle5 | 68/57 | Prenyltransferase |
| Afu8g02420 | — | — | — | — | Hypothetical protein |
| Afu8g02430 | — | — | — | — | Alcohol dehydrogenase |
| Afu8g02440 | — | — | — | — | C-4 methyl sterol oxidase |
| No homolog | — | — | cle2 | — | P450 monooxygenase |
| No homolog | — | — | cle4 | — | P450 monooxygenase |
Heterologous expression strategy of the Afu8g02350 BGC in A. nidulans
Our heterologous expression platform has two major elements. One is the use of an updated genetic dereplication strain we have developed, LO11098, and related strains for expression. We have engineered this strain for heterologous SM expression by removing eight of the most highly expressed BGCs which lowers the SM background, making expressed metabolites easier to identify and purify. We have also incorporated seven selectable markers in the strain to facilitate multiple sequential gene transfers. The strain also carries a deletion of the nkuA gene which dramatically reduces non-homologous recombination. The second element is an elaboration of multiplex fusion PCR procedures we have developed45 to rapidly create large linear molecules for transformation with multiple genes under control of inducible promoters.To express our target BGC we used two approaches. In the first approach, we refactored the entire target BGC in A. nidulans, placing each gene under control of the inducible alcA or aldA promoter (Fig. 1A, B and S1†). The alcA and aldA promoters are short (300–400 bp), strongly induced by a variety of alcohols, aldehydes and ketones, and repressed by glucose and certain other carbon sources.46–48 Their shortness and relatively low homology reduce the probability of recombination of these promoters with each other or the native alcA or aldA loci during transformation. Our second approach was to reconstruct the entire BGC intact in A. nidulans and use global regulators of secondary metabolism to activate expression of the BGC. The BGC as predicted by Inglis et al. consisted of Afu8g2350–Afu8g02430.42 We also included Afu8g02440 in case the BGC was larger than predicted (Fig. 1A).
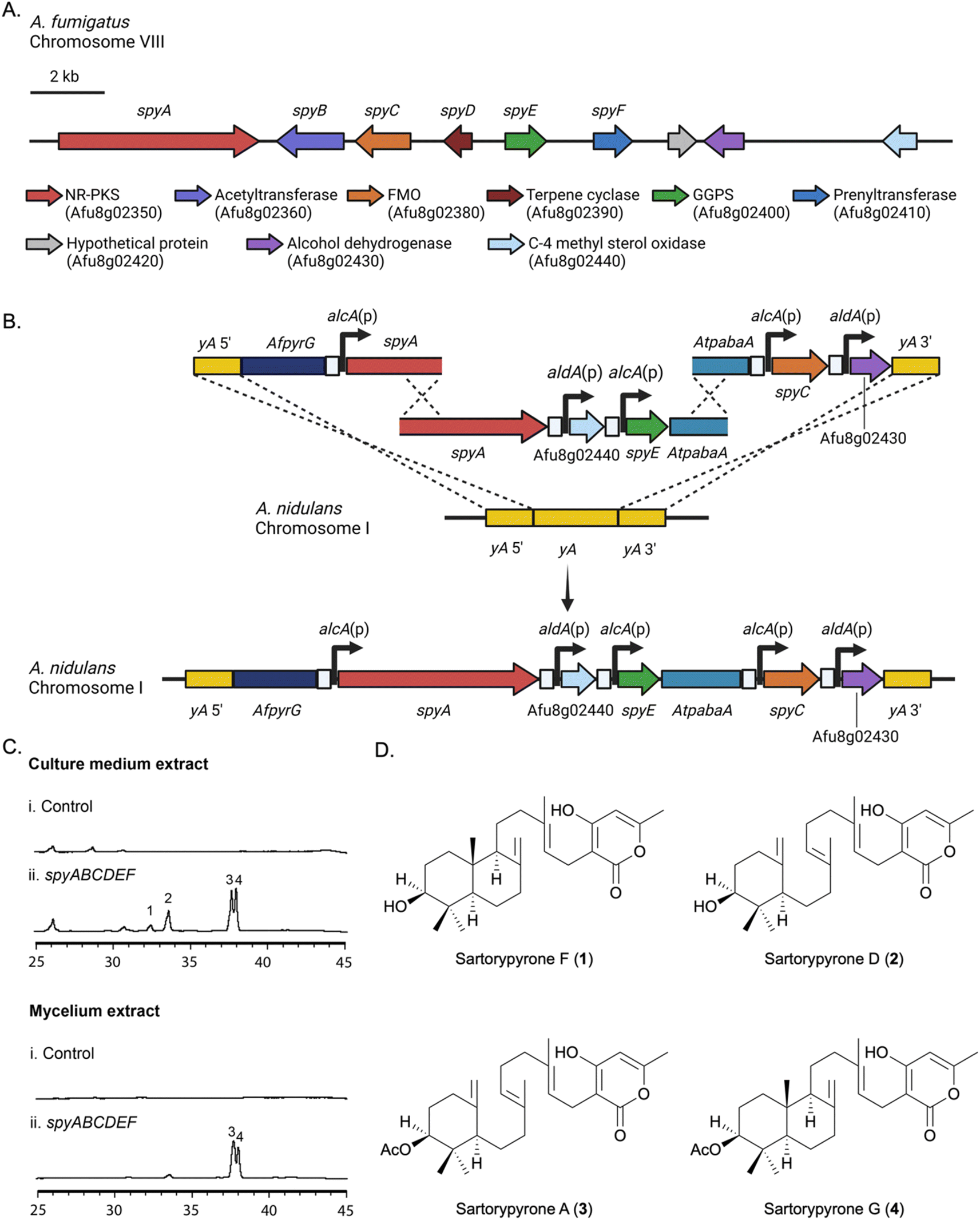 | ||
| Fig. 1 (A) The spy BGC from A. fumigatus. (B) Refactoring the spyA, spyE, spyC, Afu8g02430 and Afu8g02440 genes at the yA locus in A. nidulans. (C) HPLC profiles of culture medium and mycelium extracts from A. nidulans transformants. (D) Structures of compounds 1–4. Afu8g02440 (putative C-4 methyl sterol oxidase) and Afu8g02430 (putative alcohol dehydrogenase) were also refactored at yA as shown, but deletion experiments proved them not to be components of the spy BGC. | ||
Heterologous expression of the Afu8g02350 (spy) BGC in A. nidulans reveals that it produces sartorypyrones
A key in correctly refactoring BGCs is identification of the correct start codon for each gene. BLASTp searches with the amino acid sequence of the putative acetyl transferase, Afu8g02360, as predicted from the FungiDB annotation (https://fungidb.org/fungidb/app),43 suggested that the annotation of this gene might be incorrect, and the correct start site might be 593 base pairs downstream. The second start site also had a better Kozak consensus sequence. We, therefore, refactored this gene in two ways inserting the alcA promoter upstream of the FungiDB annotated start codon (start site 1) and, separately, in front of the downstream putative start codon (start site 2).The putative BGC genes were refactored in two steps. Five genes were refactored at the yA locus (Fig. 1B). Three transforming fragments were first created by fusion PCR. A fragment carrying ∼1 kb of DNA flanking the yA gene as well as the pyrG gene from A. fumigatus (AfpyrG) was fused to the alcA promoter [alcA(p)] and a portion of the Afu8g02350 gene from A. fumigatus. A second fragment constructed by fusion PCR contained an overlapping region of Afu8g02350 as well as two additional genes under control of the aldA promoter [aldA(p)] and alcA(p) and a portion of the Aspergillus terreus pabaA gene (AtpabaA), which was used as a selectable marker. The third fragment contained an overlapping fragment of AtpabaA, two additional genes under control of alcA(p) and aldA(p) and ∼1 kb of yA 3′ flank. Upon transformation with the three fragments, homologous recombination resulted in the replacement of the yA locus with five genes, each under control of alcA(p) or aldA(p). Note that a functional copy of AtpabaA is not formed unless fragments 2 and 3 recombine correctly. Four additional genes were placed under control of alcA or aldA promoters and inserted at the wA locus using essentially the same procedure (Fig. S1†). Using fusion PCR to make transforming fragments, we were, thus, able to refactor the entire >24 kb BGC in two transformations.
Refactored strains were cultivated in lactose minimal medium (LMM), which is non-repressing for the alcA and aldA promoters and induced with methyl-ethyl-ketone (MEK). Extracts from the culture medium and mycelia were analyzed separately by LC/MS for the presence of new metabolites. In comparison with the A. nidulans control strain, which lacked the A. fumigatus BGC genes, compounds 1–4 were detected specifically in the refactored strains (Fig. 1C). The strains in which Afu8g02360 was refactored by fusing alcA(p) at start site 1 (LO11784–LO11793), were not able to produce compounds 3 and 4, however, in comparison with the start site 2 refactored strains (LO11794–LO11803) (Fig. S2†). This indicated to us that the FungiDB annotation of the acetyl transferase is incorrect. Refactoring Afu8g02360 at start site 2 results in a functional acetyl transferase and production of 3 and 4, the acetylated final products of the BGC. Data with deletion strains (below) support this conclusion.
Yields of compounds from the initial strains were inadequate to allow determination of the structures of 1–4. To increase yields, we deleted the A. nidulans agsB gene. agsB encodes α-1,3-glucan synthase and agsB deletants cause hyphae to disperse in liquid culture.49 We hypothesized that deleting agsB would allow denser growth and increased metabolite yields, and this proved to be the case. An agsBΔ, Afu8g02360 start site 2 refactored strain (LO11839) was subjected to large-scale cultivation and compounds 1–4 were isolated by flash chromatography and semi-preparative HPLC. Spectroscopic analysis (NMR, MS, and UV-Vis data) allowed us to determine that compounds 2 and 3 are sartorypyrone D and sartorypyrone A, respectively, which were previously identified in Neosartorya fischeri strains KUFC-6344 and FO-5897 (Fig. 1D and Tables S3 and S4†).39,40 (Note: N. fischeri is a homotypic synonym of Aspergillus fischeri and both designations are used in the scientific literature.) Sartorypyrone D has also been produced synthetically in Aspergillus oryzae by expressing chevalone E BGC genes from A. versicolor along with a terpene cyclase from A. felis.44 Because it produces sartorypyrones we have designated the Afu8g02350 SM BGC as the spy BGC. Compounds 1 and 4 are new to science and were structurally elucidated by 1D and 2D NMR spectra (Fig. 1D, for detailed structural elucidation, see ESI† – detailed structural characterizations of new compounds). Furthermore, the relative configuration was confirmed by MicroED analysis on compound 1 (Fig. 2, S53 and Table S13†). Taking the above spectroscopic analyses together, compounds 1 and 4 are new bicyclic meroditerpenoids, which we have named sartorypyrone F and sartorypyrone G, respectively.
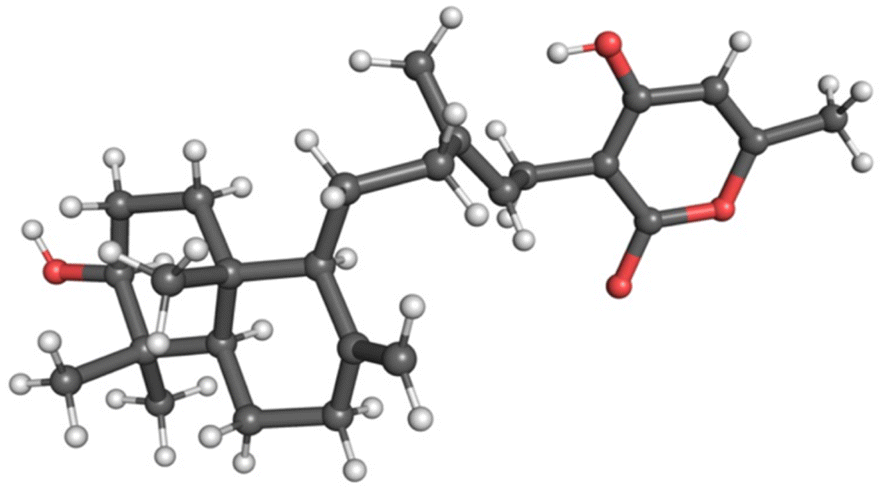 | ||
| Fig. 2 MicroED structure of compound 1. The relative stereochemistry of compound 1 was confirmed by MicroED. | ||
In our second heterologous expression approach, we reconstructed the entire target BGC in A. nidulans and attempted to use global regulators of secondary metabolism to activate its expression. We amplified portions of the target BGC and used PCR to fuse them to sequences flanking the yA (yellow spore color) gene and to selectable markers (A. terreus pyrG [AtpyrG] and A. fumigatus pyroA [AfpyroA]) (Fig. S3†). This created two fragments carrying a total of six genes. Afu8g02420 was on both fragments. Upon co-transformation (Fig. S3A–D†), the two fragments recombined homologously with the yA flanking sequences and the Afu8g02420 sequence on the two fragments recombined homologously with each other. The result is a replacement of the yA coding sequence with six genes from the target cluster. A second transformation with two additional fragments (Fig. S3A and E–G†) resulted in the reconstruction of the entire target BGC in A. nidulans. The second transformation also removes the AfpyroA selectable marker. Including primers, the entire sequence transferred into A. nidulans was 25832 bp.
To potentially upregulate expression of the BGC in the reconstructed BCG strain, we separately deleted mcrA, a negative regulator of secondary metabolism,29 and we placed two positive regulators of secondary metabolism, laeA and llmG under control of the inducible alcA promoter [alcA(p)laeA and alcA(p)llmG].50,51 We also created pairwise combinations of the mcrA deletion (mcrAΔ), alcA(p)laeA and alcA(p)llmG. Initial metabolite profiles (not shown) revealed that while deletion of mcrA upregulated secondary metabolite production, it created a sufficiently high background that identification of products of the target BGC wasn't feasible. Overexpression of laeA or llmG, however, did not create an excessive background, nor did overexpression of these two positive regulators in tandem. Co-overexpression of laeA and llmG did not yield strong new peaks that could easily be ascribed to the reconstructed BGC. However, knowing the masses of the sartorypyrones produced by the refactored cluster allowed us to unambiguously detect sartorypyrone A (3) and sartorypyrone G (4) in the laeA, llmG overexpressing strain by mass spectrometry in extracted ion chromatogram (EIC) mode (Fig. S3B†), while they were not detectable in control strains. This result provides a useful confirmation that 3 and 4 are the final products of the BGC.
Identification, purification, and structural elucidation of the intermediates and shunt products from single-gene-deleted mutant strains
In order to determine the roles of the individual genes in the BGC, we generated a series of single gene deletion strains (Table S1†) from the completely refactored strain LO11911. LO11911 is LO11839 engineered to give even higher yields by replacing the promoter of the alcR gene with the strong constitutive gpdA promoter.52 The deletant strains were verified by multiple diagnostic PCR reactions. They were cultivated under the same conditions as were used to produce sartorypyrones in the refactored strain (i.e., under inducing conditions) and their SM profiles were examined by LC/MS. The results showed that the final products, 3 and 4, were not produced in the spyA–F single gene deletion strains (Fig. 3). Detectable intermediates or shunt products accumulated, however, in spyB–E single gene deletion strains (Fig. 3). The metabolite profiles of mutants missing Afu8g02420, Afu8g02430, or Afu8g02440 did not show significant differences from their parent strain indicating that they were not involved in the biosynthesis of sartorypyrones (Fig. S4†). These data are consistent with transcription data available at FungiDB indicating that these three genes are regulated independently of the spy genes.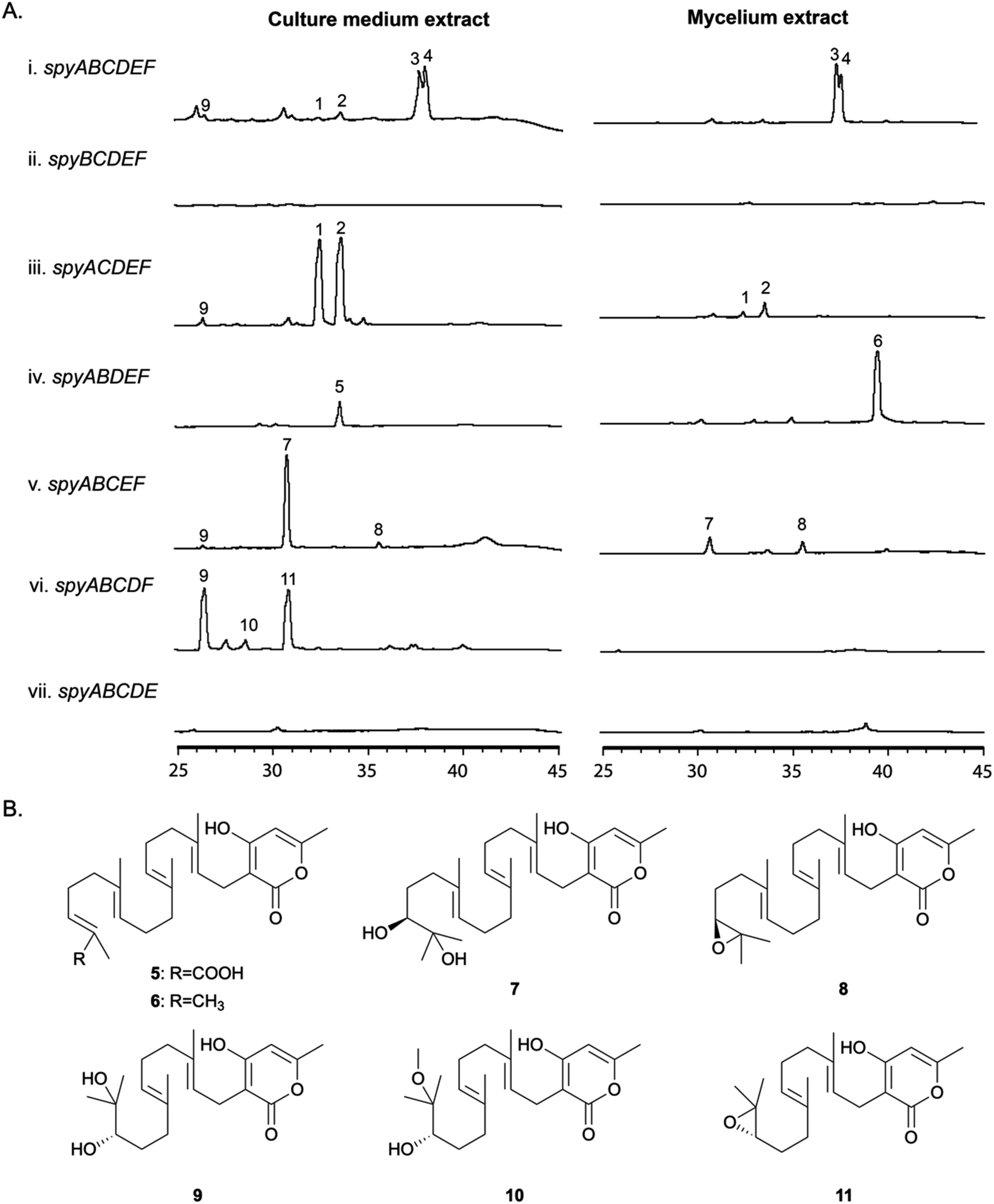 | ||
| Fig. 3 (A) HPLC profiles of culture medium and mycelium extracts from A. nidulans transformants, which carry individual gene deletions from the completely refactored spy strain LO11911 (Afu8g02420, Afu8g02430, Afu8g02440 were included). (B) Structures of compounds 5–11. | ||
Deleting the PKS gene, spyA (Afu8g02350), which is expected to catalyze the first metabolic step in the pathway, dramatically reduced production of the products of the pathway. However, compounds 3 and 4 were produced in very small amounts (more than a 100-fold reduction relative to the strain with spyA present, as determined by EIC), suggesting that an alternative mechanism exists in A. nidulans for the production of small amounts of the polyketide produced by SpyA (Fig. S5†). Based on their domain structures, two A. nidulans PKS genes were candidates for producing the same compound as SpyA, AN6448, the PKS in the cichorine BGC and AN12440, the product of which is unknown.53 However, deletion of these genes along with spyA left a very small residuum of 3 and 4. These PKSs, therefore are not responsible for the production of trace amounts of 3 and 4 in the absence of spyA (Fig. S5†).
The spyB (Afu8g02360) gene is predicted to encode an acetyltransferase, and the mutant missing the spyB gene was unable to produce the acetylated compounds 3 and 4. Instead, the unacetylated derivatives, compounds 2 and 1, respectively, accumulated in the culture medium and, at lower concentrations, in mycelia. We detected substantially more of compounds 1 and 2 than in the parental strain with the intact refactored BGC (more than a 100-fold increase as determined by EIC). These data confirm that the protein encoded by spyB is an acetyl transferase and, as indicated above, that start site 2 is correct.
In the mutant missing the spyC (Afu8g02380) gene, we isolated two prenylated polyketides 5 and 6, suggesting that the spyC gene codes for an FAD-dependent monooxygenase as the terminal olefin in the diterpene moiety was incapable of epoxidation. The analysis of 1H, 13C, and 2D NMR confirm compound 6 is a known intermediate, geranylgeranyl-triacetate lactone (Table S7†),44 while compound 5 is a new carboxylate shunt product of 6 (Fig. 3B, for detailed structural elucidation see ESI†).
The spyD (Afu8g02390) gene is predicted to encode a terpene cyclase. In the mutant lacking the spyD gene, we detected two uncyclized prenylated polyketides 7 and 8. The 1H, 13C, and 2D NMR analysis confirmed compound 7 is sartorypyrone E, which has been previously characterized in the literature (Table S8†),54 while compound 8 is the epoxide-containing intermediate in the biosynthetic pathway of sartorypyrones (Table S9 and Fig. S33–S37†). Although other studies have proposed the hypothetical biosynthetic intermediate structure of compound 8,44,55 our study provides the first evidence of its existence as a metabolite of the producing fungus. We named this new compound epoxygeranylgeranyl-triacetate lactone. The epoxide in compound 8 is relatively unstable and thus significantly more of the hydroxylated shunt product compound 7 is generated.
Compound 9 was present at low levels in the culture medium of the strain with the full refactored BGC and in the spyB and spyD deletants, but it accumulated at much higher levels in the spyE (Afu8g02400) deletant. 10 and 11 also accumulated in the spyE deletant but were not detected by LC/MS in other strains. Large scale isolation and characterization via NMR spectroscopy showed that metabolites 9–11 were produced by utilizing farnesyl pyrophosphate (FPP) instead of geranylgeranyl pyrophosphate (GGPP) (Table S10–S12 and Fig. S38–S52†), consistent with spyE encoding a GGPS that converts FPP to GGPP. The production of these compounds suggests that the prenyltransferase and FMO have a broad substrate tolerance and can utilize both GGPP and FPP as substrates. Furthermore, the relative configurations of these compounds were deduced to be identical to their analogs with the GGPP moiety because of their biosynthetic relationships. We named these new compounds dihydroxyfarnesyl-triacetate lactone (9), 17-methoxy-16-hydroxyfarnesyl-triacetate lactone (10) and epoxyfarnesyl-triacetate lactone (11).
In the mutant missing the spyF (Afu8g02410) gene, which encodes a putative prenyltransferase, all the peaks were abolished, as expected if SpyF is required for prenylation of triacetic acid lactone (TAL) for production of compounds 6 and 11, but we did not detect the polyketide products. Matsuda et al. showed that co-expression in A. oryzae of the A. felis sre3 terpene cyclase with the A. versicolor chevalone E BGC genes encoding the PKS, prenyl transferase, geranylgeranyl pyrophosphate synthase and FAD-dependent monooxygenase produced sartorypyrone D. The proposed biosynthetic pathway suggested that the polyketide TAL was the likely product of the chevalone E BGC PKS. TAL was not detected, however, when the chevalone E BGC NR-PKS Cle1 was expressed in A. oryzae.44 Similarly, we deduced that TAL is the likely polyketide product of the SpyA NR-PKS, because compound 6 is a geranylgeranylated derivative of TAL. Our proposed biosynthetic pathway predicts that TAL should accumulate in the spyF deletion strain, but we did not initially detect TAL in this deletant. We therefore constructed an A. nidulans strain (LO12091) in which the SpyA NR-PKS, alone, is expressed under control of the inducible alcA promoter. Cultivation and direct analysis of the induced culture medium of the spyA-expressing strain compared with that of a control strain lacking spyA did not initially reveal any polyketide products of SpyA. In order to extract acidic phenolic polyketide compounds, we lowered the pH value of the culture medium filtrate and subsequently extracted the acidified medium by ethyl acetate. Acidification of the culture medium from the sypA-expressing strain, but not the control strain lacking spyA, revealed a new peak, and we were able to confirm that the peak is TAL (12) by comparing it to a TAL standard (Fig. 4). Therefore, SpyA is a TAL synthase. Armed with this information, we acidified culture medium from a spyF deletant and were able to detect 12. The deletant strains in addition to the complete refactored strain allowed us to isolate, and characterize by NMR, all the intermediates in the sartorypyrone biosynthetic pathway (Fig. 5).
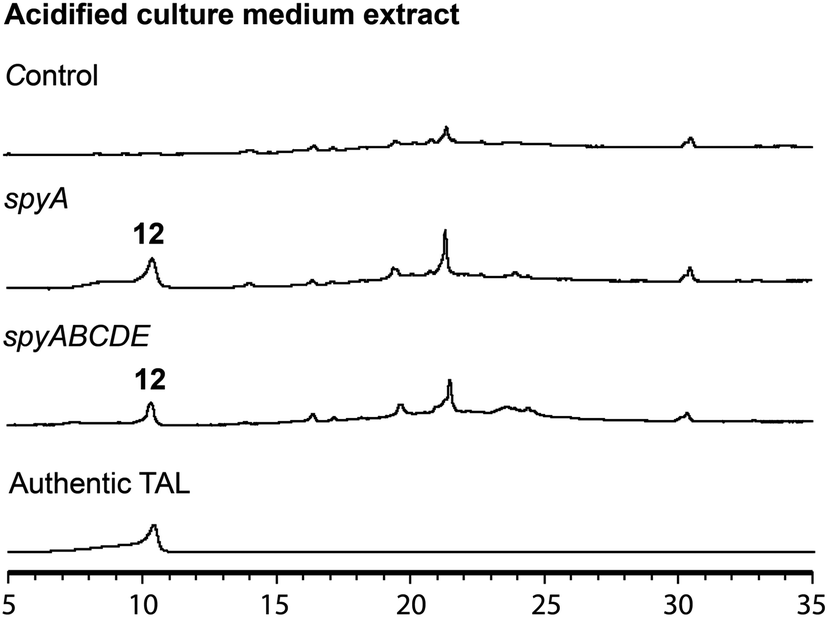 | ||
| Fig. 4 HPLC profiles of acidified induced culture medium extracts from an A. nidulans strain lacking spyA, and transformants which carry inducible spyA alone or carry the entire inducible spy BGC with spyF deleted (spyABCDE). An authentic TAL standard HPLC profile was included for comparison. | ||
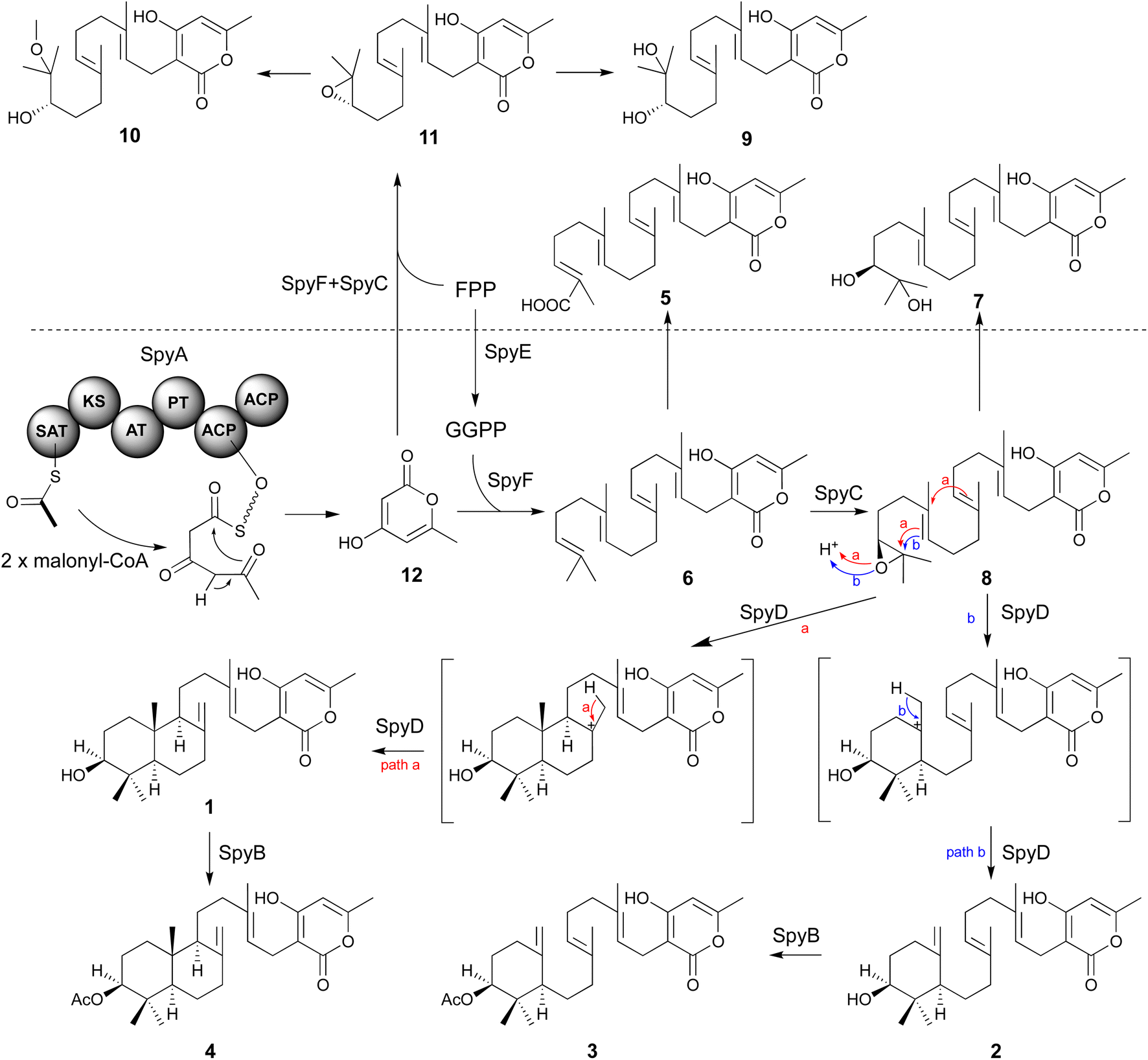 | ||
| Fig. 5 Proposed biosynthetic pathway for 3, 4, and related shunt products. The pathway bifurcates to 3–4 and related shunt products, which are separated by a dashed line. Brackets indicate hypothetical parts of the pathway. The following domains in the NR-PKS SpyA are indicated: SAT: starter unit-ACP transacylase; KS: ketosynthase; AT: acyl transferase; PT: product template; ACP: acyl carrier protein. SpyA is shown with acetyl-CoA conjugated to the SAT domain. | ||
Discussion
Secondary metabolites contribute to the reproductive success and pathogenicity of fungi, but many of the secondary metabolites produced by the BGCs of the serious fungal pathogen A. fumigatus are unknown. We have used a combination of heterologous expression and natural products chemistry to determine the products of a heretofore cryptic A. fumigatus meroterpenoid BGC, which we have named the spy BGC. The spy BGC harbors homologs of all the genes in the sartorypyrone A (sre) BGC in A. felis. Several sartorypyrones have been isolated from Aspergillus species, but this study is, to our knowledge, the first to show the biosynthesis of this class of compounds from an A. fumigatus BGC. The isolation of compounds 1–4 from the refactored strain and the cluster reconstructed strain suggested that a single terpene cyclase in the pathway is able to produce both trans-decalin meroterpenoids and monocyclic meroterpenoids. Promiscuous behaviors of terpene cyclases have been reported in other studies.56,57We created a set of targeted gene deletions that have allowed us to elucidate roles of the spy genes in the biosynthesis of compounds 3 and 4. Cultivation of deletant strains allowed us to identify and fully characterize twelve compounds from the pathway, seven of which have not been reported previously. Based on the intermediates and shunt products isolated from the mutant strains, we were able to propose a biosynthetic pathway for the sartorypyrones produced by the spy BGC (Fig. 5). First, compound 12 is generated by the NR-PKS SpyA using one molecule of acetyl-CoA and two molecules of malonyl-CoA. As SpyA lacks a thioesterase (TE) domain, 12 is likely generated through self-release from SpyA by spontaneous lactonization. The prenyltransferase SpyF then conjugates GGPP to 12 to form compound 6, for which the pathway-specific GGPS SpyE is required to provide GGPP. Subsequently, compound 6 is epoxidized at the terminal olefin by the FMO SpyC, followed by cyclization of the terpenoid component catalyzed by the terpene cyclase SpyD. SpyD exhibits promiscuous activity, resulting in the formation of bicyclic sartorypyrone F (1) and monocyclic sartorypyrone D (2). While both compounds are generated by direct terminating deprotonation, the degrees of polyene cyclization are different (path a and b). Finally, the last step of the biosynthesis involves the acetylation of the meroterpenoids by the acetyltransferase SpyB to produce compound 3 and compound 4. In the absence of GGPP (i.e. in the mutant lacking spyE), SpyF is able to catalyze addition of FPP to 12 to produce shunt products 9–11. If spyE is intact, GGPP is produced, and we hypothesize that SpyF favors the addition of GGPP over addition of FPP such that little of the shunt products accumulate (although a small amount of 9 was detected). In addition, by driving conversion of FPP to GGPP, SpyE may reduce the available levels of FPP, thereby further reducing production of 9–11. As to why 9 is detected in strains with SpyE but 10 and 11 are not, 11 is likely to be somewhat unstable which may reduce its levels and 11 appears to break down to 9 in preference to 10 such that little 10 is produced even in spyE deletants. A very small amount of 3 and 4 are produced in the spyA deletant, which indicates that 12 is also produced by an unknown minor mechanism. Note, however, that 12 is one of four products produced when the A. terreus PKS gene ATEG_00145.1 is expressed in A. nidulans.58 The major product of ATEG_00145.1 is a pentaketide, but some triketide (12) and tetraketide is produced. It is likely that the PT domain in ATEG_00145.1 that controls product length is not stringent, allowing release of shorter products in addition to the main product. It is possible that an endogenous A. nidulans PKS produces a small amount of 12 as an early release product similar to ATEG_00145.1.
Several additional points are worth making. First, we were able to refactor the entire A. fumigatus BGC as annotated by Inglis et al.,42 and an additional gene, in two transformations using transforming fragments created by fusion PCR, and we were easily able to detect metabolites produced by the BGC in our reduced SM background A. nidulans genetic dereplication strain. This demonstrates the efficiency and practicality of our heterologous expression system for elucidating the products of cryptic BGCs from A. fumigatus and potentially from other fungi. Second, in the native BGC, promoters are likely of different strengths, optimized by evolution to produce the final products of the biosynthetic pathway. In our refactored BGC, all promoters are strong and this likely results in the accumulation of intermediate compounds such as 1 and 2. This can be advantageous in elucidating the biosynthetic pathway. Furthermore, we demonstrated the feasibility of MicroED for elucidating structures of SMs produced in our heterologous expression system. Our strategy of reconstructing the target BGC in A. nidulans and up-regulating by manipulating global regulators of secondary metabolism, while very feasible, was of minor value with the sartorypyrone BGC, only providing a confirmation of the final products of the BGC. However, nearly all of the remaining cryptic BGCs of A. fumigatus harbor putative transcription factors that are predicted to drive expression of the genes of the BGCs. Reconstructing A. fumigatus BGCs in A. nidulans and upregulating the transcription factors of the BGCs could prove to be a very efficient approach toward elucidating the remaining A. fumigatus BGCs.
Finally, TAL is a potentially very valuable biorenewable platform chemical, and efforts are underway to produce it efficiently by a number of routes. The most well-known route is via type III PKSs, such as 2-pyrone synthase (2-PS), which catalyze decarboxylative Claisen condensation with one acetyl-CoA molecule as the initial unit and two malonyl-CoA molecules as extension units, followed by spontaneous lactonization to generate TAL.59,60 Some research has employed heterologous expression of 2-PS in various organisms, such as Escherichia coli, Yarrowia lipolytica, and Saccharomyces cerevisiae to achieve a higher production yield.61–64 Alternatively, polyketoacyl-CoA thiolases (PKTs) were shown to be capable of producing TAL using acetyl-CoA as both initial and extension units.65 In this study, we have now demonstrated that the NR-PKS SpyA is also able to form TAL. Although our yields of TAL are not large, our system has not been optimized for TAL production, and there are many routes to much higher yields. Unambiguous identification of a type I NR-PKS that makes TAL herein potentially opens the way to more efficient and commercially viable TAL production.
Conclusions
In summary, we have deciphered an unknown SM BGC in the human pathogen A. fumigatus using a heterologous expression approach. The spy BGC consists of six contiguous genes involved in the biosynthesis of the sartorypyrones. By integrating data obtained from bioinformatic analysis and intermediates or shunt products isolated from the individual gene deletion mutants, we were able to propose a biosynthetic pathway for this family of compounds. Our approach of refactoring the entire gene cluster in the dereplicated A. nidulans host system provides us with a straightforward way to dissect the biosynthetic pathway. This work provides an appealing demonstration that the A. nidulans heterologous expression platform can be used for the elucidation of cryptic BGCs in A. fumigatus and other species.Data availability
All experimental procedures and characterization data are available in the ESI.† The NMR spectra data are available in the Natural Products Magnetic Resonance Database (NP-MRD).Author contributions
C. C. C. W. and B. R. O. supervised the project. S. L. and C. E. O. performed all the experiments. S. L. completed data analysis. C. B. J. and R. B. T. contributed to the design of the experiments. S. L., Y. C.and C. L. contributed to the elucidation of the structures. The MicroED data collection and processing was performed by C. G. J. and P. M. S. with the help of H. M. N.; B. R. O. and R. B. T. contributed to data interpretation. The initial manuscript was written by S. L. with the help of R. B. T., C. C. C. W. and B. R. O. and all authors participated in reviewing and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
S. L. acknowledges the financial support from National Defense Medical Center, Taiwan. B. R. O. and C. C. C. W. are supported by a grant R21AI156320 from the National Institute for Allergies and Infectious Diseases. B. R. O. is supported by the Irving S. Johnson Fund of the University of Kansas Endowment. Contribution number 23-271-J from the Kansas Agricultural Experiment Station.References
- D. A. Stevens, R. B. Moss, V. P. Kurup, A. P. Knutsen, P. Greenberger, M. A. Judson, D. W. Denning, R. Crameri, A. S. Brody, M. Light, M. Skov, W. Maish and G. Mastella, and participants in the Cystic Fibrosis Foundation Consensus Conference, Clin. Infect. Dis., 2003, 37, S225–S264 CrossRef PubMed.
- K. Pianalto and J. Alspaugh, J. Fungi, 2016, 2, 26 CrossRef PubMed.
- F. Bongomin, S. Gago, R. Oladele and D. Denning, J. Fungi, 2017, 3, 57 CrossRef PubMed.
- T. R. T. Dagenais and N. P. Keller, Clin. Microbiol. Rev., 2009, 22, 447–465 CrossRef CAS PubMed.
- J.-P. Latgé and G. Chamilos, Clin. Microbiol. Rev., 2019, 33, e00140 CrossRef.
- WHO fungal priority pathogens list to guide research, development and public health action, https://www.who.int/publications/i/item/9789240060241, accessed June 27, 2023 Search PubMed.
- J. L. Steenwyk, M. E. Mead, S. L. Knowles, H. A. Raja, C. D. Roberts, O. Bader, J. Houbraken, G. H. Goldman, N. H. Oberlies and A. Rokas, Genetics, 2020, 216, 481–497 CrossRef CAS PubMed.
- N. Raffa and N. P. Keller, PLoS Pathog., 2019, 15, e1007606 CrossRef CAS PubMed.
- S. M. Leal, S. Roy, C. Vareechon, S. deJesus Carrion, H. Clark, M. S. Lopez-Berges, A. diPietro, M. Schrettl, N. Beckmann, B. Redl, H. Haas and E. Pearlman, PLoS Pathog., 2013, 9, e1003436 CrossRef CAS PubMed.
- J. A. Sugui, J. Pardo, Y. C. Chang, K. A. Zarember, G. Nardone, E. M. Galvez, A. Müllbacher, J. I. Gallin, M. M. Simon and K. J. Kwon-Chung, Eukaryotic Cell, 2007, 6, 1562–1569 CrossRef CAS.
- S. Spikes, R. Xu, C. K. Nguyen, G. Chamilos, D. P. Kontoyiannis, R. H. Jacobson, D. E. Ejzykowicz, L. Y. Chiang, S. G. Filler and G. S. May, J. Infect. Dis., 2008, 197, 479–486 CrossRef CAS PubMed.
- R. A. Cramer, M. P. Gamcsik, R. M. Brooking, L. K. Najvar, W. R. Kirkpatrick, T. F. Patterson, C. J. Balibar, J. R. Graybill, J. R. Perfect, S. N. Abraham and W. J. Steinbach, Eukaryotic Cell, 2006, 5, 972–980 CrossRef CAS PubMed.
- N. P. Keller, G. Turner and J. W. Bennett, Nat. Rev. Microbiol., 2005, 3, 937–947 CrossRef CAS PubMed.
- A. E. Barber, T. Sae-Ong, K. Kang, B. Seelbinder, J. Li, G. Walther, G. Panagiotou and O. Kurzai, Nat. Microbiol., 2021, 6, 1526–1536 CrossRef CAS PubMed.
- W. C. Nierman, A. Pain, M. J. Anderson, J. R. Wortman, H. S. Kim, J. Arroyo, M. Berriman, K. Abe, D. B. Archer, C. Bermejo, J. Bennett, P. Bowyer, D. Chen, M. Collins, R. Coulsen, R. Davies, P. S. Dyer, M. Farman, N. Fedorova, N. Fedorova, T. V. Feldblyum, R. Fischer, N. Fosker, A. Fraser, J. L. García, M. J. García, A. Goble, G. H. Goldman, K. Gomi, S. Griffith-Jones, R. Gwilliam, B. Haas, H. Haas, D. Harris, H. Horiuchi, J. Huang, S. Humphray, J. Jiménez, N. Keller, H. Khouri, K. Kitamoto, T. Kobayashi, S. Konzack, R. Kulkarni, T. Kumagai, A. Lafton, J.-P. Latgé, W. Li, A. Lord, C. Lu, W. H. Majoros, G. S. May, B. L. Miller, Y. Mohamoud, M. Molina, M. Monod, I. Mouyna, S. Mulligan, L. Murphy, S. O'Neil, I. Paulsen, M. A. Peñalva, M. Pertea, C. Price, B. L. Pritchard, M. A. Quail, E. Rabbinowitsch, N. Rawlins, M.-A. Rajandream, U. Reichard, H. Renauld, G. D. Robson, S. R. de Córdoba, J. M. Rodríguez-Peña, C. M. Ronning, S. Rutter, S. L. Salzberg, M. Sanchez, J. C. Sánchez-Ferrero, D. Saunders, K. Seeger, R. Squares, S. Squares, M. Takeuchi, F. Tekaia, G. Turner, C. R. V. de Aldana, J. Weidman, O. White, J. Woodward, J.-H. Yu, C. Fraser, J. E. Galagan, K. Asai, M. Machida, N. Hall, B. Barrell and D. W. Denning, Nature, 2005, 438, 1151–1156 CrossRef CAS PubMed.
- N. D. Fedorova, N. Khaldi, V. S. Joardar, R. Maiti, P. Amedeo, M. J. Anderson, J. Crabtree, J. C. Silva, J. H. Badger, A. Albarraq, S. Angiuoli, H. Bussey, P. Bowyer, P. J. Cotty, P. S. Dyer, A. Egan, K. Galens, C. M. Fraser-Liggett, B. J. Haas, J. M. Inman, R. Kent, S. Lemieux, I. Malavazi, J. Orvis, T. Roemer, C. M. Ronning, J. P. Sundaram, G. Sutton, G. Turner, J. C. Venter, O. R. White, B. R. Whitty, P. Youngman, K. H. Wolfe, G. H. Goldman, J. R. Wortman, B. Jiang, D. W. Denning and W. C. Nierman, PLoS Genet., 2008, 4, e1000046 CrossRef.
- J. Romsdahl and C. C. C. Wang, Med. Chem. Commun., 2019, 10, 840–866 RSC.
- M. C. Stroe, T. Netzker, K. Scherlach, T. Krüger, C. Hertweck, V. Valiante and A. A. Brakhage, eLife, 2020, 9, e52541 CrossRef CAS PubMed.
- D. Kalb, T. Heinekamp, G. Lackner, D. H. Scharf, H.-M. Dahse, A. A. Brakhage and D. Hoffmeister, Appl. Environ. Microbiol., 2015, 81, 1594–1600 CrossRef PubMed.
- J. W. Bok, Y.-M. Chiang, E. Szewczyk, Y. Reyes-Dominguez, A. D. Davidson, J. F. Sanchez, H.-C. Lo, K. Watanabe, J. Strauss, B. R. Oakley, C. C. C. Wang and N. P. Keller, Nat. Chem. Biol., 2009, 5, 462–464 CrossRef CAS PubMed.
- A. A. Brakhage and V. Schroeckh, Fungal Genet. Biol., 2011, 48, 15–22 CrossRef CAS.
- Y.-M. Chiang, S.-L. Chang, B. R. Oakley and C. C. Wang, Curr. Opin. Chem. Biol., 2011, 15, 137–143 CrossRef CAS PubMed.
- M. Ahuja, Y.-M. Chiang, S.-L. Chang, M. B. Praseuth, R. Entwistle, J. F. Sanchez, H.-C. Lo, H.-H. Yeh, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 8212–8221 CrossRef CAS PubMed.
- A. A. Soukup, Y.-M. Chiang, J. W. Bok, Y. Reyes-Dominguez, B. R. Oakley, C. C. C. Wang, J. Strauss and N. P. Keller, Mol. Microbiol., 2012, 86, 314–330 CrossRef CAS PubMed.
- J. Yaegashi, B. R. Oakley and C. C. C. Wang, J. Ind. Microbiol. Biotechnol., 2014, 41, 433–442 CrossRef CAS.
- Y.-M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, C. Zutz, C. C. C. Wang and B. R. Oakley, Angew. Chem., Int. Ed., 2016, 55, 1662–1665 CrossRef CAS PubMed.
- H.-H. Yeh, M. Ahuja, Y.-M. Chiang, C. E. Oakley, S. Moore, O. Yoon, H. Hajovsky, J.-W. Bok, N. P. Keller, C. C. C. Wang and B. R. Oakley, ACS Chem. Biol., 2016, 11, 2275–2284 CrossRef CAS PubMed.
- J. Macheleidt, D. J. Mattern, J. Fischer, T. Netzker, J. Weber, V. Schroeckh, V. Valiante and A. A. Brakhage, Annu. Rev. Genet., 2016, 50, 371–392 CrossRef CAS PubMed.
- C. E. Oakley, M. Ahuja, W.-W. Sun, R. Entwistle, T. Akashi, J. Yaegashi, C.-J. Guo, G. C. Cerqueira, J. Russo Wortman, C. C. C. Wang, Y.-M. Chiang and B. R. Oakley, Mol. Microbiol., 2017, 103, 347–365 CrossRef CAS PubMed.
- M. F. Grau, R. Entwistle, Y.-M. Chiang, M. Ahuja, C. E. Oakley, T. Akashi, C. C. C. Wang, R. B. Todd and B. R. Oakley, ACS Chem. Biol., 2018, 13, 3193–3205 CrossRef CAS PubMed.
- L. K. Caesar, N. L. Kelleher and N. P. Keller, Fungal Genet. Biol., 2020, 144, 103477 CrossRef CAS PubMed.
- K. A. O'Hanlon, L. Gallagher, M. Schrettl, C. Jöchl, K. Kavanagh, T. O. Larsen and S. Doyle, Appl. Environ. Microbiol., 2012, 78, 3166–3176 CrossRef PubMed.
- I. A. Unsöld and S.-M. Li, ChemBioChem, 2006, 7, 158–164 CrossRef PubMed.
- P. Wiemann, B. E. Lechner, J. A. Baccile, T. A. Velk, W.-B. Yin, J. W. Bok, S. Pakala, L. Losada, W. C. Nierman, F. C. Schroeder, H. Haas and N. P. Keller, Front. Microbiol., 2014, 5, 530 Search PubMed.
- B. D. Ames, X. Liu and C. T. Walsh, Biochemistry, 2010, 49, 8564–8576 CrossRef CAS PubMed.
- B. D. Ames and C. T. Walsh, Biochemistry, 2010, 49, 3351–3365 CrossRef CAS PubMed.
- K. Throckmorton, F. Y. Lim, D. P. Kontoyiannis, W. Zheng and N. P. Keller, Environ. Microbiol., 2016, 18, 246–259 CrossRef CAS PubMed.
- A. Blachowicz, N. Raffa, J. W. Bok, T. Choera, B. Knox, F. Y. Lim, A. Huttenlocher, C. C. C. Wang, K. Venkateswaran and N. P. Keller, mBio, 2020, 11, e03415–e03419 CrossRef CAS PubMed.
- A. Eamvijarn, N. M. Gomes, T. Dethoup, J. Buaruang, L. Manoch, A. Silva, M. Pedro, I. Marini, V. Roussis and A. Kijjoa, Tetrahedron, 2013, 69, 8583–8591 CrossRef CAS.
- S. Kaifuchi, M. Mori, K. Nonaka, R. Masuma, S. Ōmura and K. Shiomi, J. Antibiot., 2015, 68, 403–405 CrossRef CAS PubMed.
- M. Chia, T. J. Schwartz, B. H. Shanks and J. A. Dumesic, Green Chem., 2012, 14, 1850–1853 RSC.
- D. O. Inglis, J. Binkley, M. S. Skrzypek, M. B. Arnaud, G. C. Cerqueira, P. Shah, F. Wymore, J. R. Wortman and G. Sherlock, BMC Microbiol., 2013, 13, 91 CrossRef CAS PubMed.
- B. Amos, C. Aurrecoechea, M. Barba, A. Barreto, E. Y. Basenko, W. Bażant, R. Belnap, A. S. Blevins, U. Böhme, J. Brestelli, B. P. Brunk, M. Caddick, D. Callan, L. Campbell, M. B. Christensen, G. K. Christophides, K. Crouch, K. Davis, J. DeBarry, R. Doherty, Y. Duan, M. Dunn, D. Falke, S. Fisher, P. Flicek, B. Fox, B. Gajria, G. I. Giraldo-Calderón, O. S. Harb, E. Harper, C. Hertz-Fowler, M. J. Hickman, C. Howington, S. Hu, J. Humphrey, J. Iodice, A. Jones, J. Judkins, S. A. Kelly, J. C. Kissinger, D. K. Kwon, K. Lamoureux, D. Lawson, W. Li, K. Lies, D. Lodha, J. Long, R. M. MacCallum, G. Maslen, M. A. McDowell, J. Nabrzyski, D. S. Roos, S. S. C. Rund, S. W. Schulman, A. Shanmugasundram, V. Sitnik, D. Spruill, D. Starns, C. J. Stoeckert, S. S. Tomko, H. Wang, S. Warrenfeltz, R. Wieck, P. A. Wilkinson, L. Xu and J. Zheng, Nucleic Acids Res., 2022, 50, D898–D911 CrossRef CAS PubMed.
- W.-G. Wang, L.-Q. Du, S.-L. Sheng, A. Li, Y.-P. Li, G.-G. Cheng, G.-P. Li, G. Sun, Q.-F. Hu and Y. Matsuda, Org. Chem. Front., 2019, 6, 571–578 RSC.
- E. Szewczyk, T. Nayak, C. E. Oakley, H. Edgerton, Y. Xiong, N. Taheri-Talesh, S. A. Osmani and B. R. Oakley, Nat. Protoc., 2006, 1, 3111–3120 CrossRef CAS.
- M. Flipphi, M. Mathieu, I. Cirpus, C. Panozzo and B. Felenbok, J. Biol. Chem., 2001, 276, 6950–6958 CrossRef CAS.
- R. B. Waring, G. S. May and N. R. Morris, Gene, 1989, 79, 119–130 CrossRef CAS PubMed.
- B. Felenbok, J. Biotechnol., 1991, 17, 11–17 CrossRef CAS.
- A. Yoshimi, M. Sano, A. Inaba, Y. Kokubun, T. Fujioka, O. Mizutani, D. Hagiwara, T. Fujikawa, M. Nishimura, S. Yano, S. Kasahara, K. Shimizu, M. Yamaguchi, K. Kawakami and K. Abe, PLoS One, 2013, 8, e54893 CrossRef CAS PubMed.
- N. Keller, J. Bok, D. Chung, R. M. Perrin and E. Keats Shwab, Med. Mycol., 2006, 44, 83–85 CrossRef PubMed.
- M. F. Grau, R. Entwistle, C. E. Oakley, C. C. C. Wang and B. R. Oakley, ACS Chem. Biol., 2019, 14, 1643–1651 CrossRef CAS PubMed.
- P. J. Punt, N. D. Zegers, M. Busscher, P. H. Pouwels and C. A. M. J. J. van den Hondel, J. Biotechnol., 1991, 17, 19–33 CrossRef CAS PubMed.
- J. F. Sanchez, R. Entwistle, D. Corcoran, B. R. Oakley and C. C. C. Wang, Med. Chem. Commun., 2012, 3, 997–1002 RSC.
- S. Bang, J. H. Song, D. Lee, C. Lee, S. Kim, K. S. Kang, J. H. Lee and S. H. Shim, J. Agric. Food Chem., 2019, 67, 1831–1838 CrossRef CAS PubMed.
- K. Kanokmedhakul, S. Kanokmedhakul, R. Suwannatrai, K. Soytong, S. Prabpai and P. Kongsaeree, Tetrahedron, 2011, 67, 5461–5468 CrossRef CAS.
- M.-C. Tang, H.-C. Lin, D. Li, Y. Zou, J. Li, W. Xu, R. A. Cacho, M. E. Hillenmeyer, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2015, 137, 13724–13727 CrossRef CAS PubMed.
- L. Barra and I. Abe, Nat. Prod. Rep., 2021, 38, 566–585 RSC.
- Y.-M. Chiang, C. E. Oakley, M. Ahuja, R. Entwistle, A. Schultz, S.-L. Chang, C. T. Sung, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2013, 135, 7720–7731 CrossRef CAS.
- S. Eckermann, G. Schröder, J. Schmidt, D. Strack, R. A. Edrada, Y. Helariutta, P. Elomaa, M. Kotilainen, I. Kilpeläinen, P. Proksch, T. H. Teeri and J. Schröder, Nature, 1998, 396, 387–390 CrossRef CAS.
- L. Zhu, M. Pietiäinen, J. Kontturi, A. Turkkelin, P. Elomaa and T. H. Teeri, New Phytol., 2022, 236, 296–308 CrossRef CAS PubMed.
- Y. Li, S. Qian, R. Dunn and P. C. Cirino, J. Ind. Microbiol. Biotechnol., 2018, 45, 789–793 CrossRef CAS PubMed.
- J. Yu, J. Landberg, F. Shavarebi, V. Bilanchone, A. Okerlund, U. Wanninayake, L. Zhao, G. Kraus and S. Sandmeyer, Biotechnol. Bioeng., 2018, 115, 2383–2388 CrossRef CAS PubMed.
- K. A. Markham, C. M. Palmer, M. Chwatko, J. M. Wagner, C. Murray, S. Vazquez, A. Swaminathan, I. Chakravarty, N. A. Lynd and H. S. Alper, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2096–2101 CrossRef CAS PubMed.
- J. Cardenas and N. A. Da Silva, Metab. Eng., 2014, 25, 194–203 CrossRef CAS PubMed.
- Z. Tan, J. M. Clomburg, S. Cheong, S. Qian and R. Gonzalez, Nat. Catal., 2020, 3, 593–603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2259814. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02226a |
| This journal is © The Royal Society of Chemistry 2023 |