
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ oxidative polymerization of platinum(IV) prodrugs in pore-confined spaces of CaCO3 nanoparticles for cancer chemoimmunotherapy†
Fangmian
Wei
a,
Libing
Ke
a,
Siyuan
Gao
a,
Johannes
Karges
 b,
Jinquan
Wang
c,
Yu
Chen
a,
Liangnian
Ji
a and
Hui
Chao
*ad
b,
Jinquan
Wang
c,
Yu
Chen
a,
Liangnian
Ji
a and
Hui
Chao
*ad
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Guangdong Provincial Key Laboratory of Digestive Cancer Research, The Seventh Affiliated Hospital, Sun Yat-Sen University, Guangzhou, 510006, P. R. China. E-mail: ceschh@mail.sysu.edu.cn
bFaculty of Chemistry and Biochemistry, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany
cGuangdong Provincial Key Laboratory of Biotechnology Drug Candidate, Guangdong Pharmaceutical University, Guangzhou, 510006, China
dMOE Key Laboratory of Theoretical Organic Chemistry and Functional Molecule, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan, 400201, P. R. China
First published on 2nd June 2023
Abstract
Drug resistance and metastases are the leading causes of death in clinics. To overcome this limitation, there is an urgent need for new therapeutic agents and drug formulations that are able to therapeutically intervene by non-traditional mechanisms. Herein, the physical adsorption and oxidative polymerization of Pt(IV) prodrugs in pore-confined spaces of CaCO3 nanoparticles is presented, and the nanomaterial surface was coated with DSPE-PEG2000-Biotin to improve aqueous solubility and tumor targeting. While the nanoparticle scaffold remained stable in an aqueous solution, it quickly degraded into Ca2+ in the presence of acid and into cisplatin in the presence of GSH. The nanoparticles were found to interact in cisplatin-resistant non-small lung cancer cells by a multimodal mechanism of action involving mitochondrial Ca2+ overload, dual depletion of GSH, nuclear DNA platination, and amplification of ROS and lipid peroxide generation, resulting in triggering cell death by a combination of apoptosis, ferroptosis and immunogenic cell death in vitro and in vivo. This study could present a novel strategy for the treatment of drug-resistant and metastatic tumors and therefore overcome the limitations of currently used therapeutic agents in the clinics.
Introduction
Pt(II) complexes are widely used chemotherapeutic drugs for various types of tumours.1 Despite their clinical success, these compounds are associated with severe side effects (i.e., nerve/kidney damage, nausea, vomiting, and bone marrow suppression).2 In addition, an increasing number of (cisplatin) drug-resistant tumors are reported in the clinics, limiting the clinical use of these compounds.3 While cancer cells have developed different mechanisms to prevent the therapeutic effect of these metal complexes, low cellular uptake, efficient excretion, and detoxification of the Pt(II)-based drugs are considered the leading causes.4 In particular, high levels of glutathione (GSH) in the tumor microenvironment (TME) can generate GSH–Pt adducts, which are subsequently excreted from the cancer cells through GSH S-conjugate export pumps (GS-X pump) before entering the cell nucleus.5 To overcome this resistance mechanism, studies have indicated that providing pathways to target and consume GSH and restoring cisplatin sensitivity in tumor cells is a very effective new strategy for overcoming tumor resistance.6Besides the excretion of GSH–Pt adducts, high levels of GSH are also able to limit the ability of treated cancer cells to trigger programmed cell death through apoptosis.7 To circumvent this drawback, researchers have focused on discovering novel cell death mechanisms.8,9 Among the clinically most promising cell death mechanisms for anticancer therapy, ferroptosis has been highlighted.10 Ferroptosis is an iron-dependent non-apoptotic form of cell death. This cell death mechanism is characterized by the accumulation of lipid peroxides which are formed through the irreversible peroxidation of membrane lipids by free radicals such as hydroxy radials or the misregulation of specific enzymes in the cells.11 Recent studies have indicated that cancer cells are highly susceptible to the depletion of intracellular GSH levels, causing reactive oxygen species-mediated ferroptosis through the inactivation of glutathione peroxidase 4 (GPX4).12 Capitalizing on this, the combination of apoptosis and ferroptosis cell death pathways presents a promising anticancer strategy. To reduce potential side effects, much interest has been devoted to Pt(IV) prodrugs which remain stable and inactive in healthy human cells but are reduced to Pt(II) and therefore activated in the reducing TME.13 Importantly, Pt(IV) complexes have been demonstrated to strongly bind and deplete intracellular GSH.14 To enhance the therapeutic properties, Pt(II) and Pt(IV) complexes could be encapsulated into nanoparticles with other anticancer agents to combat drug resistance7,15 or cancer-targeting moieties for tumor-selective delivery.14,16
Despite their promising properties, the therapeutic Pt complexes were found to leak from the nanomaterial,17,18 limiting their application in a clinical setting.
Besides chemotherapy, cancer immunotherapy is frequently used in clinics.19 Despite its promising therapeutic outcome, the immunosuppressive microenvironment of cancer cells strongly limits the treatment's therapeutic efficiency. Capitalizing on this, there is a high demand for compounds that induce an immunogenic response inside the targeted cancer cells.20,21 Among other strategies, chemotherapy-induced immunogenic cell death (ICD) may offer a general strategy to kill the tumor cells by eliciting broad antitumor immunity.22 Previous studies23–29 have demonstrated that Pt(II) and Pt(IV) complexes can cause ICD. Interestingly, while cisplatin was found not to induce ICD, the structurally related metal complex oxaliplatin showed a strong ICD effect in (drug-resistant) cancer cells.30 Recent studies have indicated that ferroptosis-inducing compounds can enhance the immunogenic effect through the activation of cytotoxic T lymphocytes (CTLs).31 Reciprocally, activated CD8+ T cells can secrete high levels of interferon γ (IFN-γ) to trigger ferroptosis-specific lipid peroxidation in cancer cells.32 Capitalizing on these preliminary studies, developing therapeutic agents that can trigger cell death by a combination of ferroptosis and ICD is of high interest.
Herein, a novel strategy for the selective delivery and multimodal treatment of drug-resistant cancer tumors is presented. Thiol functionalized Pt(IV) complexes were physically loaded into CaCO3 nanoparticles and then covalently linked by in situ oxidative polymerization to form Pt(IV)SS NPs-2 within the supramolecular CaCO3 nanoparticle scaffold (Pt(IV)SS@CaCO3). This prevents drug leakage of the molecular agent and allows for efficient and selective delivery to the tumorous tissue. To enhance aqueous solubility as well as improve tumor targeting, the nanomaterial Pt(IV)SS@CaCO3@Biotin was coated on the surface with DSPE-PEG2000-Biotin.33 While the nanomaterial was found to be stable in an aqueous solution, the particles quickly degraded into Ca2+ ions and soluble therapeutic Pt complexes in the presence of acid upon depletion of intracellular glutathione. Ca2+ levels and storage in the mitochondria are crucial for cell survival and proliferation.34 Upon misregulation of the Ca2+ balance, cytochrome C in the mitochondria is released, and caspase-3 activated, resulting in apoptosis.35 In this study, we report on nanoparticles that are able to therapeutically intervene in cisplatin-resistant non-small lung cancer cells by a multimodal mechanism of action involving Ca2+ overloading of the mitochondria, the depletion of intracellular level glutathione, binding to nuclear DNA, and amplification of the generation of reactive oxygen species and lipid peroxides, resulting in triggering cell death by a combination of apoptosis, ferroptosis, and ICD (Scheme 1). This combination of cell death mechanisms is associated with high clinical relevance. Strikingly, the nanomaterial was found to show a strong anticancer effect towards cisplatin-resistant non-small lung cancer tumors in a mouse model without any indication of side effects such as those observed for the parent drug cisplatin (i.e., hematopoietic toxicity, hepatotoxicity, or nephrotoxicity), therefore indicating a high potential to overcome (cisplatin) drug-resistant tumors. Importantly, the ability to induce ICD inside immunocompetent C57BL/6J mice was demonstrated to nearly fully eradicate a metastatic syngeneic tumor. These findings suggest that the here presented promising therapeutic strategy for cancer chemoimmunotherapy could overcome drug-resistant and metastatic tumors, which are considered the leading cause of cancer-associated deaths in clinics.
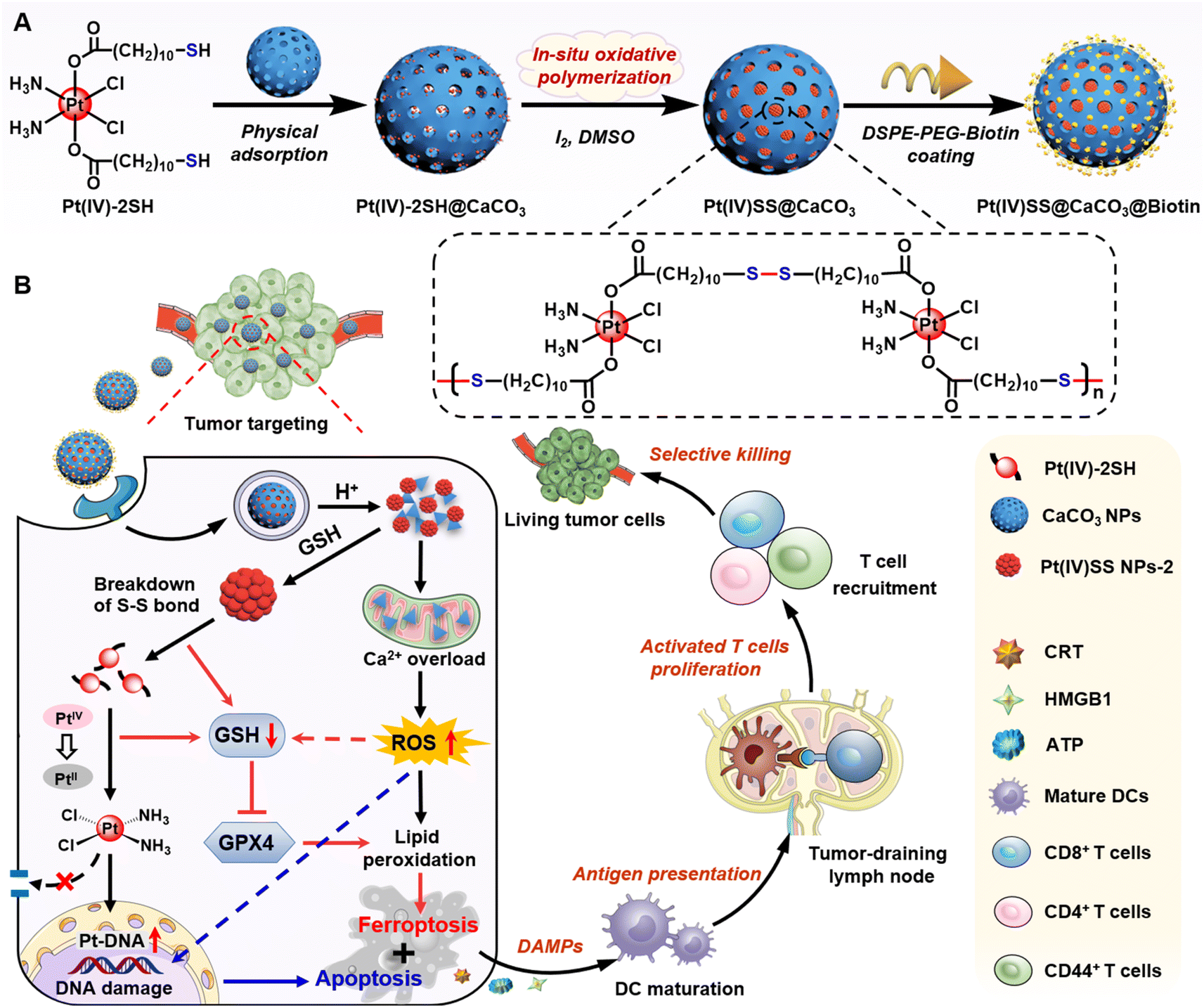 | ||
| Scheme 1 Schematics of physical adsorption and oxidative polymerization of a Pt(IV) prodrug inside of supramolecular calcium carbonate nanoparticle (Pt(IV)SS@CaCO3@Biotin)-mediated efficient cancer immunotherapy by releasing cisplatin, generating ROS and dual GSH depletion to trigger the combination of apoptosis and ferroptosis. | ||
Results and discussion
Synthesis and characterization
The Pt(IV) prodrug (Pt(IV)-2SH) was synthesized by the oxidation of cisplatin and subsequent functionalization with 11-mercaptoundecanoic acid in the axial positions (Scheme S1†). The identity of the metal complex was verified by high-resolution mass spectrometry, 1H NMR, 13C NMR, 195Pt NMR spectroscopy, and Fourier transform infrared spectrometry (FTIR) (Fig. S1–S4†). As no significant changes were observed upon storage of Pt(IV)-2SH for one week (Fig. S5†), the stability of the metal complex is indicated. CaCO3 nanoparticles were prepared from (NH4)HCO3 and CaCl2 (Fig. S7†).36Pt(IV)-2SH was adsorbed into CaCO3 NPs to form Pt(IV)-2SH@CaCO3. Transmission electron microscopy (TEM) images showed a change in the morphology of the particles by the deposition of metal complexes inside the pores of the CaCO3 nanoparticles (Fig. 1A). The terminal thiol groups of the metal complex were covalently linked by confined in situ oxidative polymerization with iodine and DMSO to form Pt(IV)SS@CaCO3. The (elemental mapping) TEM images confirmed that Ca, O, Pt, and S are homogeneously distributed in both nanoparticles (Fig. 1D and S8†) and their hydrodynamic diameter with a size of ∼100 nm is slightly higher than that of CaCO3 (Fig. S8B†). Next, the chemical structure of Pt(IV)SS@CaCO3 was studied by recording the FTIR spectrum in the process of the physical adsorption and oxidative polymerization of a Pt(IV) prodrug inside of CaCO3 (Fig. S6†). Particularly, for obtaining a self-polymerizing Pt(IV) nano-prodrug (Pt(IV)SS NPs-2) in pore-confined spaces of CaCO3 nanoparticles, Pt(IV)SS@CaCO3 was degraded by acid (pH 5.0) and purified using centrifugation, washing with dichloromethane, and dialysis, resulting in the presence of less than 3.56% free thiol groups as determined by Ellman's method.37 The as-obtained Pt(IV)SS NPs-2 was observed to be uniformly spherical at ∼5.1 nm by using TEM, and with a hydrodynamic diameter of ∼7.5 nm (Fig. 1A and S8B†). Inversely, Pt(IV)-2SH self-assembled to form a Pt(IV) nano-prodrug (Pt(IV)SS NPs-1) by oxidative polymerization in the presence of iodine and DMSO. The particle size of Pt(IV)SS NPs-1 was ∼405 nm (Fig. S9†). This result indicated that our new strategy could successfully prepare Pt(IV)SS NPs-2. Interestingly, Pt(IV)SS NPs-2 was dissolved in tetrahydrofuran (1.0 mg mL−1) and studied by gel permeation chromatography, which revealed the broad chromatographic peaks of the polymer (Fig. 1E). However, no peaks of the small molecule Pt complexes were observed with high-resolution mass spectrometry (Fig. S10†). Upon polymerization, the appearance of a stretching frequency at 473 cm−1 in the FTIR spectrum was observed (Fig. S6B†). Previous studies have indicated that this frequency is attributed to the formation of a S–S bond.38 Notably, the average pore size and specific surface area of Pt(IV)SS@CaCO3 were determined to be 11.9 nm and 14.7 m2 g−1via the Brunauer–Emmett–Teller (BET) measurements, which were slightly smaller than 17.3 nm and 44.5 m2 g−1 for bare CaCO3, respectively (Fig. S11†). What's more, the X-ray diffraction pattern (Fig. S12†) matches the standard card of CaCO3 (PDF #33-0268), further verifying the basic structure of CaCO3, whereas Pt(IV)SS NPs-2 exhibits an amorphous structure. Collectively, the above results demonstrate the successful synthesis of the Pt(IV) prodrug inside CaCO3 by confined in situ oxidative polymerization.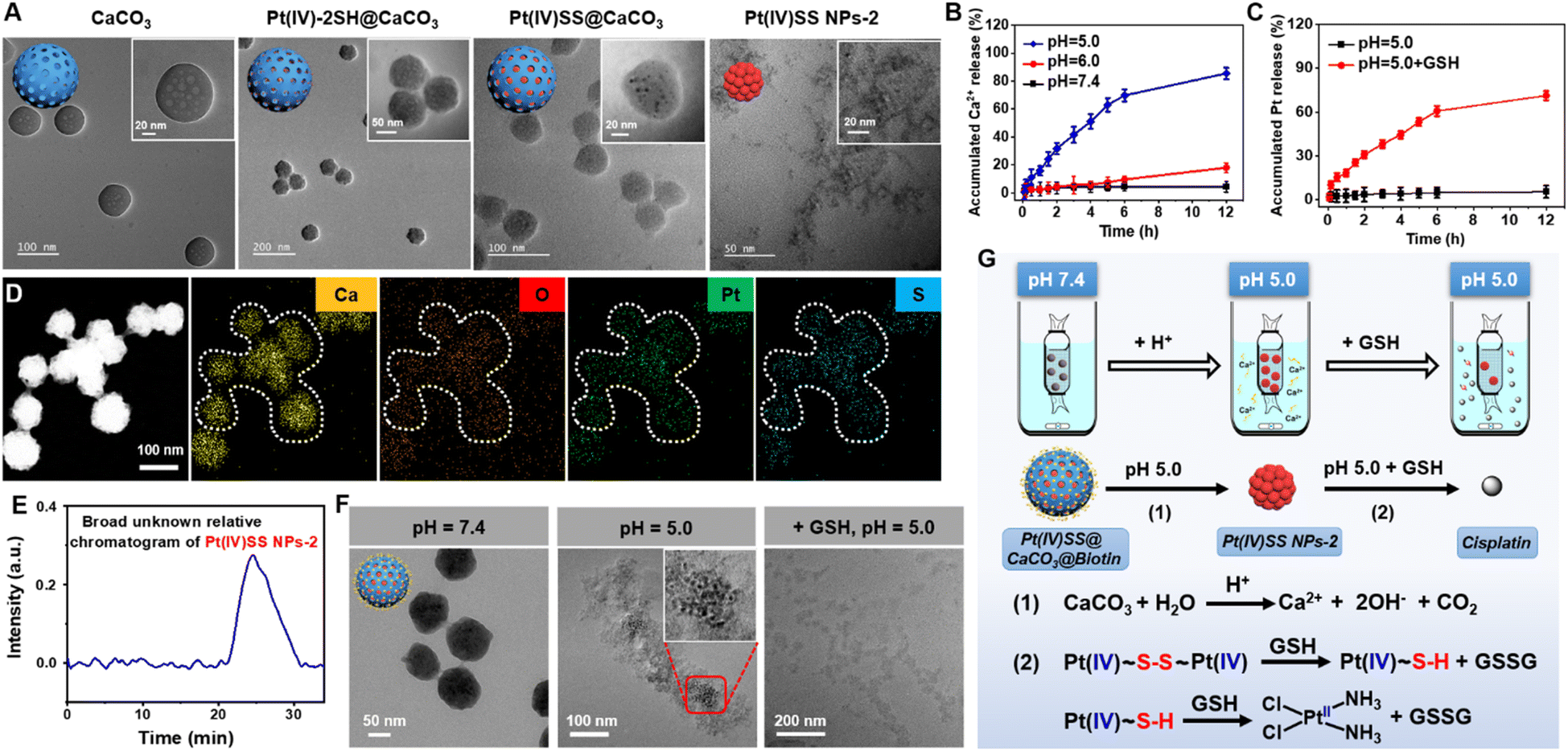 | ||
| Fig. 1 Characterization, pH response, and depletion of GSH of Pt(IV)SS@CaCO3@Biotin. (A) TEM image of CaCO3, Pt(IV)-2SH@CaCO3, Pt(IV)SS@CaCO3 and Pt(IV)SS NPs-2. (B) Time-dependent release level of Ca2+ by using a calcium colorimetric assay kit in the presence of various pH environments (5.0, 6.0, and 7.4). (C) Time-dependent release of Pt ion from Pt(IV)SS@CaCO3@Biotin by using ICP-MS at pH 5.0 without GSH, and pH 5.0 with GSH (100 μM). (D) Energy dispersive spectroscopy mapping of Pt(IV)SS@CaCO3. (E) The broad unknown relative chromatogram of Pt(IV)SS NPs-2 determined by gel permeation chromatography in tetrahydrofuran. (F) TEM image of Pt(IV)SS@CaCO3@Biotin upon incubation in various pH environments (7.4 and 5.0) and GSH (100 μM). Illustration: enlarged TEM image of Pt(IV)SS NPs-2 from Fig. 1F (pH = 5.0). (G) Schematic illustration of the dialysis device and reaction mechanism for Pt(IV)SS@CaCO3@Biotin. | ||
As biotin is primarily internalized by the sodium multivitamin transporter, which is overexpressed in various cancers, the functionalization with biotin could improve cancer cell selectivity.33 Thus, the nanomaterial was coated on the surface with DSPE-PEG2000-Biotin. The sizes of (CaCO3@Biotin: 101 ± 4.8 nm, 108 ± 3.2 nm, Pt(IV)-2SH@CaCO3@Biotin: 108 ± 3.2 nm, Pt(IV)SS@CaCO3@Biotin: 115 ± 5.7 nm) were measured by dynamic laser scattering (Fig. S13†). The TEM images of the encapsulated nanoparticles showed a polymeric layer on the surface of the particles (Fig. S14†). Using inductively coupled plasma mass spectrometry (ICP-MS), the Pt content of the particles was determined to be 19.8% for Pt(IV)SS@CaCO3@Biotin and 21.4% for Pt(IV)-2SH@CaCO3@Biotin (Table S1†), This loading efficiency is associated with previous literature.36 As a crucial property for any biological application, the stability of the encapsulated nanoparticles was studied upon incubation in water, an aqueous 0.9% sodium chloride solution, cell medium, or serum for 24 h. No aggregation or change in the size of the particles was observed, indicating their high physiological stability (Fig. S15†).
pH response and GSH depletion
Research shows that as endosomes/lysosomes are characterized by an acidic environment (pH 4.5–5.0), the specific nature of this cell organelle could be utilized for a selective drug release.39 Therefore, the stability and potential release of the therapeutic metal complexes from Pt(IV)SS@CaCO3@Biotin were studied at pH 5.0, 6.0, and 7.4. While the particles remained intact upon incubation at pH 6.0 and 7.4 using a specific Ca2+ detection kit, a quick degradation of the supramolecular structure at pH 5.0 was observed and 85.5 ± 1.8% (12 h) Ca2+ content was released (Fig. 1B). A TEM analysis of Pt(IV)SS@CaCO3@Biotin after incubation at pH 5.0 revealed that the particles disintegrated and released small uniform nanoparticles (Fig. 1F). Interestingly, using ICP-MS analysis, no Pt ion was observed in the solution, indicating that the nanoparticles contain the encapsulated Pt nanoprodrug, Pt(IV)SS NPs-2 (Fig. 1C). While no Pt ion was detected in an acidic solution of Pt(IV)SS@CaCO3@Biotin, a high amount of Pt ion was observed upon the addition of GSH (Fig. 1C and F). Otherwise, a large amount of Pt ion was observed after the Pt(IV)SS NPs-2 dissociation upon the addition of GSH (pH 7.4) (Fig. S16†). Overall, these findings indicated that Pt(IV)SS NPs-2 was released after the tumor-selective degradation of Pt(IV)SS@CaCO3@Biotin, and the disulfide bonds are then further completely degraded into thiols in the presence of GSH in micromolar concentrations (0.1–600 μM).40 Ulteriorly, the reduction of the Pt(IV) center to Pt(II) is caused by reducing agents such as glutathione triggers.22,41 These properties could allow for a multimodal treatment through intracellular Ca2+ overload and Pt2+ sensitivity through dual GSH depletion (Fig. 1G). These results suggest that GSH could be efficiently depleted through a multimodal mechanism.Cellular uptake and localization
For an understanding of the biological effects of Pt nanoprodrugs, the cellular uptake of Pt(IV)-2SH@CaCO3@Biotin, Pt(IV)SS@CaCO3@Biotin, Pt(IV)SS NPs-2 and free cisplatin was studied in non-small cell cisplatin-resistant lung cancer (A549R) cells by ICP-MS. Both Pt(IV)-2SH@CaCO3@Biotin and Pt(IV)SS@CaCO3@Biotin demonstrated a time-dependent high cellular uptake in comparison to cisplatin (Fig. S17A†). Similarly, for analysis of intracellular Ca2+ levels, both nanoparticles had almost the same time-dependent cellular uptake as CaCO3@Biotin (Fig. S17B†). While the overall amount of Pt inside the cancer cells increased with a prolongation of the incubation time, the Pt content in the lysosomes decreased with longer incubation times, indicating that the Pt complexes are released in the acidic environment of the lysosomes. Following the release, the Pt complexes accumulated selectively in the nucleus as the final subcellular target, where the most significant fraction of the compound was detected (Fig. S18†). For a deeper understanding, the amount of Pt–DNA adducts was determined by ICP-MS. After A549R cells were treated with different samples, both Pt(IV)-2SH@CaCO3@Biotin and Pt(IV)SS@CaCO3@Biotin showed a drastically higher amount of Pt–DNA adducts than cisplatin (Fig. S19†) due to the enhanced cellular uptake and depletion of GSH which could prevent the binding of the Pt complex to DNA. Overall, these findings indicate that Pt(IV)SS@CaCO3@Biotin is efficiently internalized in cancer cells where the Pt complex is released in the acidic environment of the lysosomes and ultimately accumulates in the nucleus upon generation of Pt–DNA adducts.Cytotoxicity
The cell viability of LLC, A549 and A549R cells upon treatment with CaCO3@Biotin, cisplatin, Pt(IV)SS NPs-2, Pt(IV)-2SH@CaCO3@Biotin, or Pt(IV)SS@CaCO3@Biotin was studied using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based assay. The cytotoxicity profiles are shown in Table S2.† As expected, cisplatin was found to be therapeutically active in A549 cells but highly deactivated in A549R cells (IC50, A549 = 16.3 ± 1.2 μM, IC50, A549R = 86.1 ± 3.4 μM). The nanoformulations Pt(IV)-2SH@CaCO3@Biotin (IC50, A549 = 1.71 ± 0.54 μM, IC50, A549R = 4.87 ± 0.98 μM, based on Pt) and Pt(IV)SS@CaCO3@Biotin (IC50, A549 = 0.77 ± 0.18 μM, IC50, A549R = 2.82 ± 0.87 μM, based on Pt) demonstrated enhanced cytotoxicity towards the cancer cells. Nonetheless, the cytotoxicity of Pt(IV)SS NPs-2 (IC50, A549 = 6.4 ± 0.81 μM, IC50, A549R = 14.9 ± 1.4 μM, based on Pt) was slightly lower due to the lower cellular uptake than both nanoformulations. Complementarily, the ability of the compounds to trigger cell death in A549R cells was evaluated with the dual annexin V-FITC/PI stain. Pt(IV)SS@CaCO3@Biotin showed the highest cytotoxic effect with approximately 82.4% of cancer cells in an early or late apoptotic stage after incubation for 24 h (Fig. S20†), indicative of an apoptotic cell death mechanism. Overall, these results suggest that Pt(IV)SS@CaCO3@Biotin has a high cytotoxic effect on (cisplatin-resistant) cancer cells and could therefore overcome cisplatin resistance.Multimodal mechanism of action
As the nanoparticles have demonstrated to quickly degrade in an acidic environment and release Ca2+ ions from the nanoparticle scaffold, the Ca2+ content in A549R cells upon treatment with Pt(IV)-2SH@CaCO3@Biotin, or Pt(IV)SS@CaCO3@Biotin was studied using the specific Ca2+-probe Fluo-4 AM. While no fluorescence signal corresponding to the sensor was observed upon incubation with cisplatin, a strong emission in the confocal laser scanning microscopy (CLSM) image was noticed upon incubation with both nanoformulations (Fig. S21A†), verifying the release of Ca2+ ions. Quantitatively, flow cytometric analysis showed higher levels of Ca2+ ions for Pt(IV)SS@CaCO3@Biotin and similar results to CLSM (Fig. S21B†). Mitochondrial Ca2+ overload induces the opening of the mitochondrial permeability transition pore (mPTP), disrupting the electron transport chain, generating excessive ROS, and eventually leading to apoptosis.30 As a result of the opening of the mPTP, the characteristic green/red (aggregates/monomer) ratio of JC-1 showed the loss of mitochondrial membrane potential after treatment with Pt(IV)-2SH@CaCO3@Biotin or Pt(IV)SS@CaCO3@Biotin by confocal imaging (Fig. S22A†). Similar results were also confirmed for the quantitative analysis of flow cytometry (Fig. S22B†). As an additional hallmark of the misregulation of the mitochondria, the change of intracellular adenosine triphosphate (ATP) levels was monitored. While the treatment with cisplatin only slightly influenced the levels of ATP, the concentration drastically decreased during the treatment with Pt(IV)-2SH@CaCO3@Biotin or Pt(IV)SS@CaCO3@Biotin compared with CaCO3@Biotin (Fig. S23†). Moreover, A549R cells were treated and afterward incubated with the reactive oxygen species specific dye 2′,7′-dichlorodihydrofluorescein diacetate. While no fluorescence signal corresponding to the probe was observed upon incubation with cisplatin and the other three groups, a strong DCF emission in the CLSM images was noticed upon incubation with Pt(IV)SS@CaCO3@Biotin. Secondly, in the case of Pt(IV)-2SH@CaCO3@Biotin (Fig. S24A†), the quantitative analysis by flow cytometry showed the same results (Fig. S24B†). The ability to deplete GSH in A549R cells was investigated using a GSH/GSSG extraction kit. The incubation of the cancer cells with nanoformulations strongly decreased the intracellular levels of GSH, especially Pt(IV)SS@CaCO3@Biotin with 3.68-fold contrast to cisplatin attributed to dual GSH depletion (Fig. S25†).18 Combining these findings suggests that the nanoparticles could interact in the cancer cells through a multimodal mechanism of action by overloading the mitochondria with Ca2+ ions, amplifying reactive oxygen species, and dual depletion of GSH.Cell death mechanism
For a deeper understanding of the therapeutic effect of the nanoparticles, the cell death mechanism in A549R cells was studied by monitoring the cell viability upon co-incubation with necrosis (necrostatin-1), autophagy (3-methyladenine), apoptosis (Z-VAD-FMK), iron chelation/antioxidation (deferoxamine), and ferroptosis/peroxyradical (ferrostatin-1) inhibitors.33,42 As the incubation with necrosis and autophagy inhibitors had a negligible effect on cell survival, surprisingly, the co-incubation with apoptosis, iron chelation/antioxidation, and ferroptosis/peroxyradical inhibitors strongly enhanced the cell viability (Fig. 2A), indicating the involvement of apoptosis and ferroptosis in cell death. Previous studies have indicated that the depletion of GSH may inactivate GPX4 and inhibit the lipid repair system, resulting in ferroptosis.12 Based on this, the expression of GPX4 upon treatment of A549R cells was investigated. As shown in Fig. 2C, the incubation with Pt(IV)-2SH@CaCO3@Biotin strongly reduced the GPX4 expression, especially in Pt(IV)SS@CaCO3@Biotin. The specific lipid peroxide probe C11 BODIPY, as a hallmark of ferroptosis, detected the generation of intracellular lipid peroxides (LPOs) via CLSM images. While the cells treated with cisplatin did not show any LPO, the formation of LPO was observed upon treatment with Pt(IV)-2SH@CaCO3@Biotin, especially Pt(IV)SS@CaCO3@Biotin, which was much stronger than Pt(IV)SS NPs-2 (Fig. 2E). Complementarily, the cell death pathway was further studied by flow cytometry using C11 BODIPY in combination with the ferroptosis inhibitors DFO and Fer-1. This generation can be effectively suppressed by Fer-1 or DFO treatment (Fig. S26†). Furthermore, the amount of malondialdehyde (MDA), an important end-metabolite of LPO, was quantified as a measure of LPO. The result is consistent with CLSM images, in which Pt(IV)SS@CaCO3@Biotin was found to have a remarkable MDA level with 7.2-fold contrast to control (Fig. 2B). Combining these results suggests that the nanoparticles cause cell death by ferroptosis.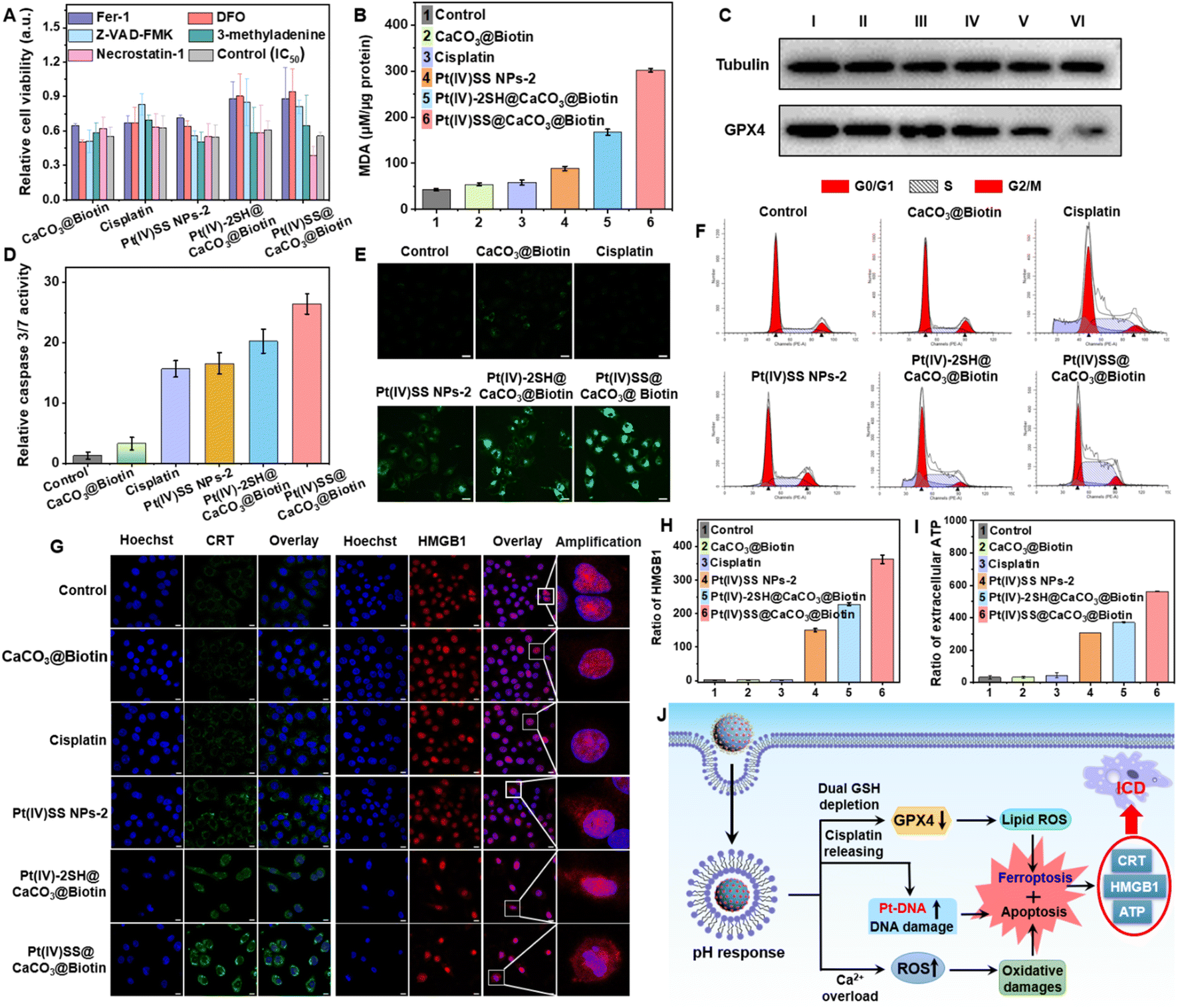 | ||
| Fig. 2 Mechanism action and ICD hallmarks of Pt(IV)SS@CaCO3@Biotin in A549R cells. (A) Cell viability upon pre-incubation of necrosis (necrostatin-1, 60 μM), autophagy (3-methyladenine, 100 μM), apoptosis (Z-VAD-FMK, 20 μM), iron chelation/antioxidation (deferoxamine (DFO), 100 μM), and ferroptosis/peroxyradical (ferrostatin-1 (Fer-1), 10 μM) inhibitors with various samples for 24 h. (B) The intracellular level of malondialdehyde of A549R cells was incubated with various samples for 24 h. (C) Expression of GPX4 upon treatment with the IC50 dose of the therapeutic species after 24 h. I: control, II: CaCO3@Biotin, III: cisplatin, IV: Pt(IV)SS NPs-2, V: Pt(IV)-2SH@CaCO3@Biotin, VI: Pt(IV)SS@CaCO3@Biotin. (D) Caspase 3/7 activity on treatment with IC50 dose of various samples for 24 h. (E) Confocal laser scanning microscopy images of A549R cells incubated with various samples and the specific lipid peroxide probe C11 BODIPY. C11 BODIPY: λex/em = 488/520 ± 10 nm. Scale bar: 20 μm. (F) Cell cycle analysis of A549R cells incubated with the IC50 dose of various samples for 24 h. (G) The release of CRT and HMGB1 was imaged through immunostaining of A549R cells. Scale bar: 10 μm. (H) Release of HMGB1 and (I) analysis of ATP levels in the cell culture supernatant. A549R cells were incubated with various samples for 24 h. (J) Schematic illustration of combining apoptosis and ferroptosis which triggered the release of DAMPs, causing the ICD effect. | ||
Next, we performed the caspase-3/7 assay to determine the potential route by which apoptosis occurs in A549R cells (Fig. 2D). As expected, the treatment with Pt(IV)SS NPs-2, Pt(IV)-2SH@CaCO3@Biotin, and Pt(IV)SS@CaCO3@Biotin demonstrated to activate the caspase 3/7 pathways, which were similar to cisplatin. Previous studies have indicated that resistant cancer cells contain a subpopulation of dormant persistent cells that reversibly enter the G0 cell cycle arrest to avoid the therapeutic effect of the drug.43 As shown in Fig. 2F, while the treatment with Pt(IV)-2SH@CaCO3@Biotin showed the same cell cycle (A549R) distribution as cisplatin, the treatment with Pt(IV)SS@CaCO3@Biotin demonstrated a decrease in the population in the G0/G1 phase and an increase in the population in the S phase. These findings may be attributed to the ability of Pt(IV)SS@CaCO3@Biotin to cause more efficient ferroptosis than Pt(IV)-2SH@CaCO3@Biotin.44 Furthermore, the bio-TEM images of A549R cells treated with both nanoparticles showed obvious nuclear pyknosis, while mitochondrial fragmentation and cristae alterations were observed, which are the characteristics of cell apoptosis and ferroptosis, respectively (Fig. S27†), while the cisplatin group was similar to the control. Overall, these results suggest that the nanoparticles trigger cell death combined with apoptosis and ferroptosis.
Immunogenic cell death (ICD)
The ability to trigger ICD was investigated upon determination of the release of specific damage-associated molecular patterns (DAMPs).22 The release of the high mobility group box 1 (HMGB1) is able to induce the myeloid differentiation primary response signaling cascade, enhancing antigen processing and presentation to T-cells.31 Recent studies have indicated that the secretion of HMGB1 supports cell death by ICD as well as ferroptosis.32 In contrast to ferroptosis, ICD-inducing therapeutic agents could eradicate distant, metastatic tumors.45 While the treatment with cisplatin did not influence the localization of HMBG1, the translocation of HMBG1 from the cell nucleus into the cytoplasm was observed by CLSM upon treatment with Pt(IV)SS@CaCO3@Biotin (Fig. 2G). For a quantitative insight, the release of HMGB1 was determined using an HMGB1-specific commercially available enzyme-linked immunosorbent (ELISA) assay kit. The release of HMGB1 from the nucleus was 363-fold enhanced upon treatment of A549R cells with Pt(IV)SS@CaCO3@Biotin in comparison to the untreated cells (Fig. 2H). Besides, another ICD marker, calreticulin (CRT) from the endoplasmic reticulum was translocated to the cell surface upon treatment with Pt(IV)SS@CaCO3@Biotin by CLSM images (Fig. 2H). Complementarily, the migration of CRT was quantified using flow cytometry (Fig. S28†). Additionally, the treatment of the cancerous cells with Pt(IV)SS@CaCO3@Biotin was demonstrated to increase the secretion of ATP approximately 18 times in comparison to untreated cells (Fig. 2I). Combined, these findings indicate that Pt(IV)SS@CaCO3@Biotin is able to therapeutically intervene by a combination of apoptosis, ferroptosis, and ICD (Fig. 2J).Evaluation in a cisplatin-resistant tumor mouse model
Based on the biotin group on the surface of nanoparticles, we first investigated the tumor-targeting properties. A549R tumor-bearing mice were injected in the tail vein with Pt(IV)-2SH@CaCO3@Biotin (5 mg kg−1) or Pt(IV)SS@CaCO3@Biotin (5 mg kg−1) and the biodistribution determined by measurement of the Pt content by ICP-MS. Strikingly, the comparison between the nanoparticles showed a higher tumor accumulation of Pt(IV)SS@CaCO3@Biotin than of Pt(IV)-2SH@CaCO3@Biotin (Fig. 3B and S29†), which was attributed to preventing drug leakage from Pt(IV)SS@CaCO3@Biotin. While cisplatin (1.4 h) was found with a low blood-circulation half-times, the nanoparticles Pt(IV)-2SH@CaCO3@Biotin (11.7 h) and Pt(IV)SS@CaCO3@Biotin (24.1 h) showed an order of magnitude longer blood-circulation half-times (Fig. S30†). Combined, this indicates that the in situ oxidative polymerization of the metal complex could improve the biodistribution and blood circulation profile upon evaluation in an animal model.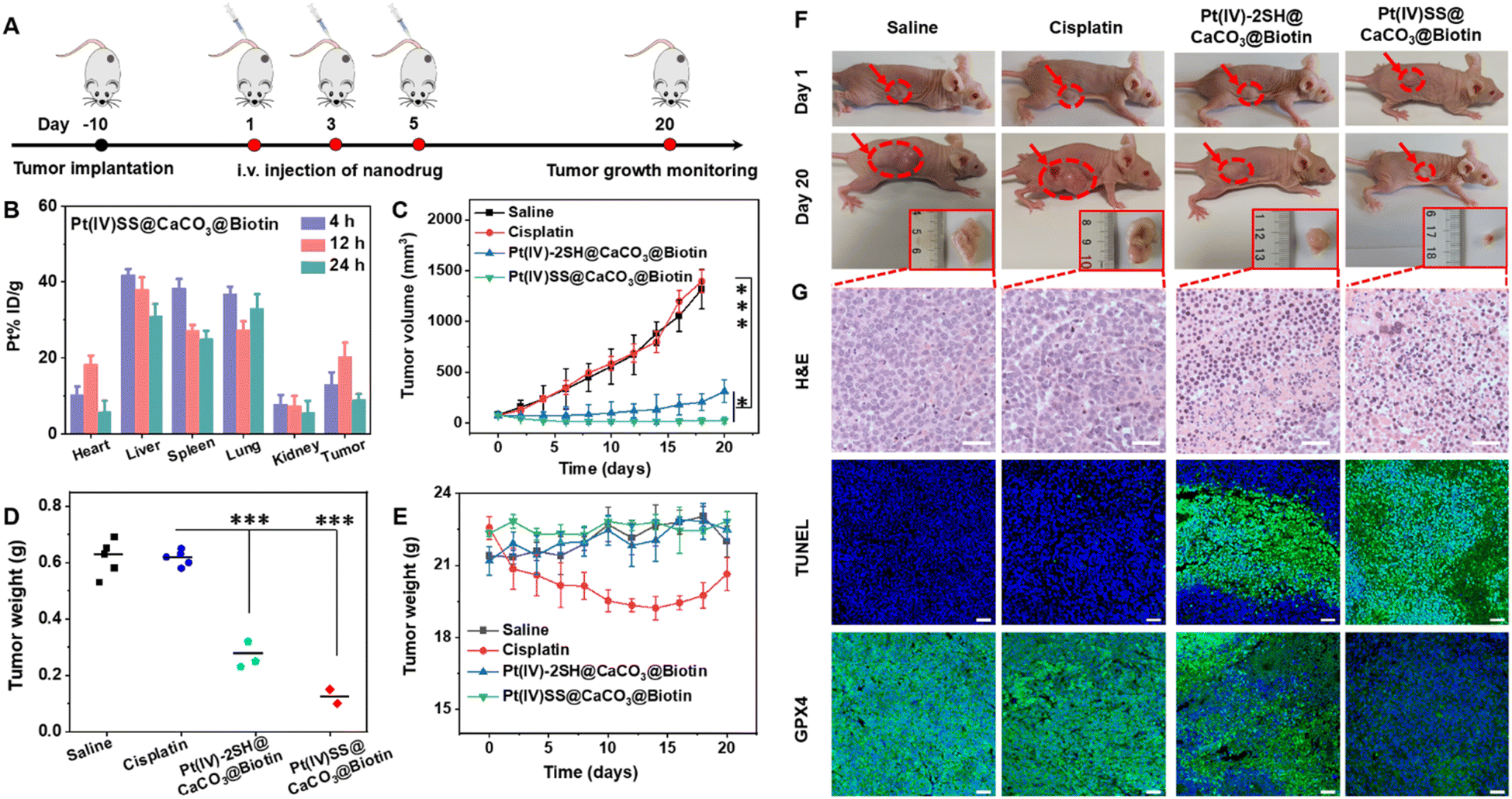 | ||
| Fig. 3 Evaluation of the therapeutic efficiency upon intravenous injection of cisplatin, Pt(IV)-2SH@CaCO3@Biotin, and Pt(IV)SS@CaCO3@Biotin in the tail vein of an A549R tumor-bearing mouse model. (A) Schematic illustration of the timeline for tumor model establishment and cancer therapy. (B) Time-dependent biodistribution of Pt(IV)SS@CaCO3@Biotin by determination of the Pt content inside the major organs and the tumor by ICP-MS (n = 3). (C) The tumor volume over the treatment period, (D) the weight of the tumor after treatment for 20 days, (E) the weight of the animal model over the treatment period, (F) photographs of the animal models at the beginning and the end of the treatment, (G) histological analysis of the tumor tissue by (top) hematoxylin and eosin (H&E) stain, (middle) TdT-mediated dUTP nick end labeling (TUNEL) stain, or (bottom) GPX4 stain upon injection in the tail vein on the days 1, 3 and 5 with saline, cisplatin (5 mg kg−1, based on 1.67 mM Pt), Pt(IV)-2SH@CaCO3@Biotin (5 mg kg−1, based on 0.548 mM Pt), or Pt(IV)SS@CaCO3@Biotin (5 mg kg−1, based on 0.507 mM Pt). Statistical analysis was carried out using Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001, mean ± SD; n = 5. | ||
Encouraged by the high tumor accumulation inside the animal mode, the therapeutic efficiency of an A549R tumor-bearing mouse model was studied and is shown in Fig. 3A. Once the tumors of the mice reached approximately 75 mm3, the animals were divided into five groups and treated upon injection in the tail vein (5 mg kg−1) on days 1, 3, and 5 with saline, cisplatin, Pt(IV)-2SH@CaCO3@Biotin, or Pt(IV)SS@CaCO3@Biotin. As expected, the tumors of the animals treated with cisplatin grew exponentially in a similar manner to the mice injected with saline, while the treatment with Pt(IV)SS@CaCO3@Biotin showed a remarkable tumor growth inhibition effect in contrast to Pt(IV)-2SH@CaCO3@Biotin (Fig. 3C and S31†). The therapeutic efficacy was further highlighted by a comparison of the weight of the tumors (Fig. 3F) or visual inspection of the tumors (Fig. 3F and S32†). For a deeper understanding of the therapeutic effect, the tumorous tissue was histologically examined using hematoxylin and eosin (H&E) and TdT-mediated dUTP nick end labeling (TUNEL) stain. The tumors of the mice treated with the nanoparticles showed apoptotic characteristics such as nuclei shrinkage and fragmentation (top/middle panel of Fig. 3G). Based on the ability of Pt(IV)SS@CaCO3@Biotin to efficiently trigger cell death by ferroptosis, the tumor tissue slices were stained for the presence of GPX4. While the animals treated with cisplatin showed normal levels of GPX4, the treatment with both nanoformulations demonstrated a strong reduction of GPX4 (bottom panel of Fig. 3G). These findings confirm the ability of Pt(IV)SS@CaCO3@Biotin to cause a therapeutic effect combined with apoptosis and ferroptosis in the animal model.
Besides the therapeutic efficiency, the biosafety of the nanoparticles was thoroughly assessed. The treatment with cisplatin showed a drastic reduction in the body weight of the animal models, indicating severe side effects. In contrast, the treatment with Pt(IV)-2SH@CaCO3@Biotin, or Pt(IV)SS@CaCO3@Biotin did not change the body weight of the mouse model (Fig. 3E). Moreover, a histological analysis of the main organs (heart, liver, spleen, lung, kidney, brain, and intestines) using H&E revealed serious damage to the liver and the kidneys of the animals treated with cisplatin. Promisingly, the treatment with both nanoformulations showed no organ damage (Fig. S33†). For a deeper insight into biosafety, the blood of the animals was screened for various biochemical markers. The mice treated with cisplatin were found to have a decreased amount of hematocrit, hemoglobin, and red blood cells as well as an increased number of white blood cells. In addition, the animals showed abnormal levels of kidney function biomarkers (BUN, CREA, CR) and liver function biomarkers (ALT, AST, ALP), indicating severe bone marrow hematopoietic toxicity, hepatotoxicity, and nephrotoxicity. In contrast, the animals treated with both nanoformulations demonstrated normal levels of these biomarkers (Fig. S34 and Table S3†), suggestive of high biocompatibility and biosafety. Overall, these results suggest that Pt(IV)SS@CaCO3@Biotin has a strong antitumor efficiency towards cisplatin-resistant tumors and high biosafety, therefore overcoming the limitations of the clinically applied drug cisplatin.
Antitumor immunity in the tumor mouse model
Based on the promising antitumor response inside an A549R tumor-bearing mouse model, the immunotherapeutic properties of the nanoparticles were studied in an in vivo vaccination study. Healthy C57BL/6J female mice were randomly divided into four groups, with 5 mice in each group. The animal models were injected with (1) saline, (2) LLC cells that were treated with cisplatin (5.96 mg kg−1), (3) LLC cells that were treated with Pt(IV)-2SH@CaCO3@Biotin (1.95 mg per kg Pt4+, 0.14 mM Ca2+), and (4) LLC cells that were treated with (1.95 mg per kg Pt4+, 0.14 mM Ca2+) for 24 h, washed with PBS to remove the non-penetrating nanoparticles, and re-suspended in free fetal bovine serum containing DMEM medium. The aforementioned dying cancer cells were subcutaneously injected into immunocompetent homogenic mice on the right side, as shown in Fig. 4A. On day 8, living cancer cells were subcutaneously inoculated into the left side and the mice were monitored for palpable neoplastic lesions every two days for 27 days. While the tumor of the mice that were injected with cisplatin-treated cancer cells was found to grow exponentially in a similar manner to the control group, the tumor of the mice models that were injected with nanoparticle treated cancer cells was nearly fully eradicated. After 27 days, the tumor growth inhibition rate of Pt(IV)SS@CaCO3@Biotin was approximately 22 times higher than for the control group (Fig. 4B, photographs of the tumors: Fig. 4C). Importantly, no changes in body weight (Fig. S35†) or histological damage for healthy tissue, including heart, liver, spleen, lung, and kidneys (Fig. S36†) were observed after the treatment period. On day 27, the mice models were sacrificed and the tumors were collected. H&E staining of the tumorous tissue showed nuclear shrinkage and necrosis upon treatment with Pt(IV)SS@CaCO3@Biotin, indicative of the immunotherapeutic effect of the nanoparticles (Fig. 4F). These results demonstrate that the injection with Pt(IV)SS@CaCO3@Biotin-treated-cells was able to protect the animal model from new tumor growth and, therefore could find application for the treatment of distant or metastatic tumors.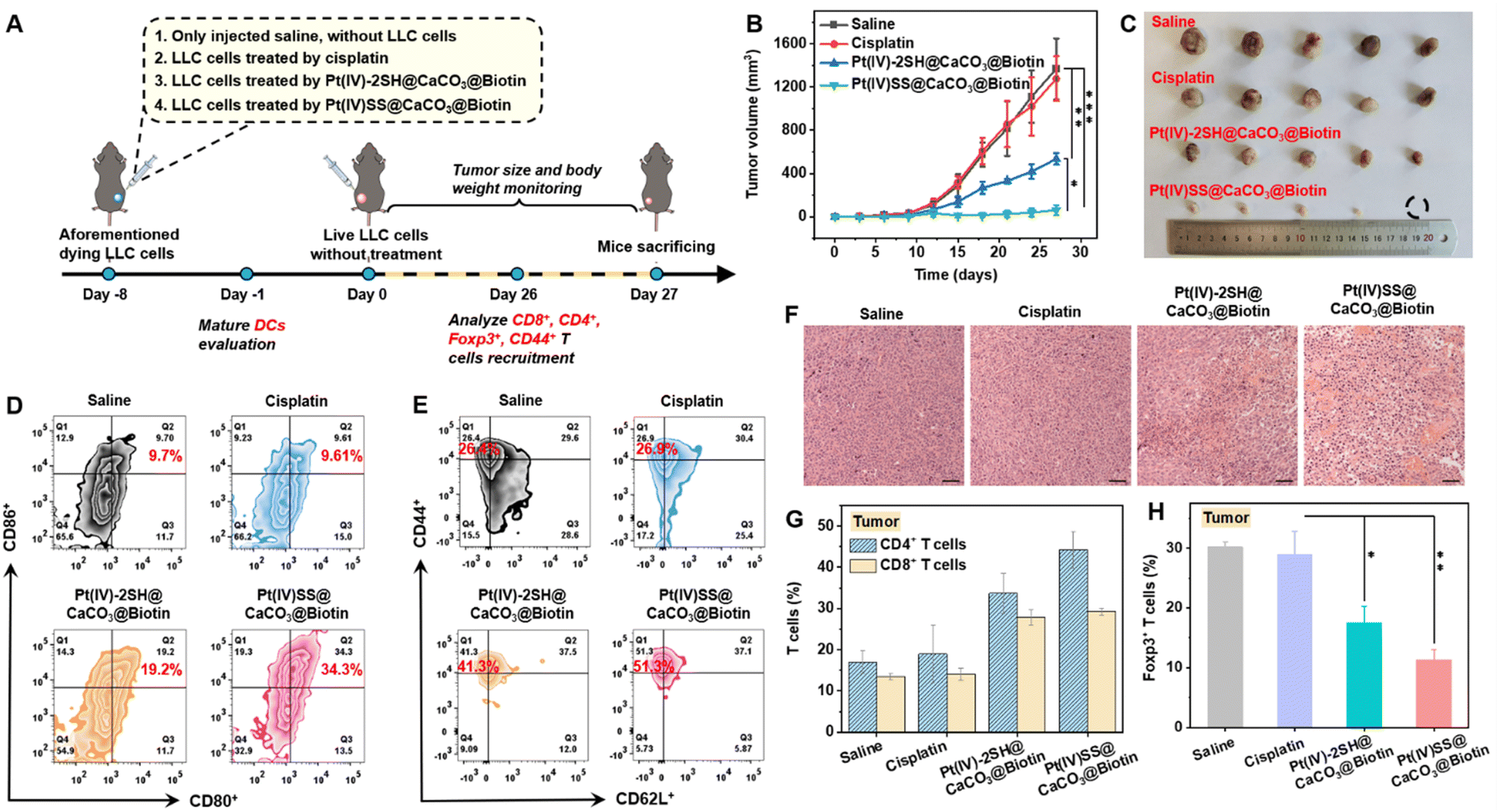 | ||
| Fig. 4 Evaluation of the antitumor immunity in a mouse model. (A) Schematic diagram of the ICD vaccine experiment. The LLC cells were incubated with cisplatin (5.96 mg kg−1), Pt(IV)-2SH@CaCO3@Biotin (1.95 mg per kg Pt4+, 0.14 mM Ca2+) and Pt(IV)SS@CaCO3@Biotin (1.95 mg per kg Pt4+, 0.14 mM Ca2+), respectively. (B) The plots of tumor volumes for the different groups versus the time post-live LLC cancer cell inoculation (n = 5). (C) Photographs of the tumors removed from the mice 27 days after treatment. (D) Flow cytometric plots of mature DCs in the tumors after treatment for 26 days, gated by CD11c+ cells. (E) Flow cytometric analysis of memory T cells (CD44+, CD62L+, gating on CD4+ T cells) in the tumor after treatment for 26 days. (F) H&E staining of tumor sections 27 days after treatment. Scale bar: 50 μm. Representative flow cytometric quantification of intratumoral infiltration of (G) CD4+ and CD8+ T cells (gating on CD3+ T cells) and (H) Foxp3+ T cells (gating on CD3+CD4+ T cells) after treatment for 26 days (n = 4). Statistical analysis was carried out using Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001, mean ± SD; n = 5. | ||
Dendritic cells (DCs) are antigen-presenting cells that play a crucial role in the T cell-mediated immune response.26 To investigate the immunogenic effect of the nanomaterials, the levels of CD80+CD86+ (gating on CD11c+ cells) DCs of the tumor-draining lymph nodes (TDLNs) of tumor-bearing mouse models upon treatment were studied by flow cytometry. While the treatment with cisplatin did not significantly influence the levels of matured DCs (9.6%), the amount of matured DCs was strongly enhanced upon treatment with Pt(IV)-2SH@CaCO3@Biotin (19.2%) or Pt(IV)SS@CaCO3@Biotin (34.3%) (Fig. 4D and S37†). These findings confirm the observed immunogenic therapeutic response of the Pt(IV)SS@CaCO3@Biotin nanoparticles.
For a deeper insight into the immunogenic mechanism, the levels of cytotoxic CD8+ T cells and CD4+ helper T cells inside tumor-bearing mouse models upon treatment were investigated by flow cytometry. The results showed a 2.1- and 2.8-fold enhancement of CD8+ T cells and a 3.1- and 1.7-fold increase of CD4+ T cells in the tumor and spleen upon treatment with Pt(IV)SS@CaCO3@Biotin (Fig. 4G and S38†), respectively. Previous studies have indicated that the depletion of CD3+CD4+Foxp3+ T regulatory cells is able to reduce the immunosuppressive properties and therefore enhance the immunotherapeutic effect.22 Capitalizing on this, the level of CD3+CD4+Foxp3+ T regulatory cells in the tumor and spleen was determined by flow cytometry. While the treatment with cisplatin was found not to influence the amount of CD3+CD4+Foxp3+ T cells (30%) inside the animal model, the treatment with Pt(IV)-2SH@CaCO3@Biotin (20.7%) and Pt(IV)SS@CaCO3@Biotin (10.2%) demonstrated strong reduction of the percentage of CD3+CD4+Foxp3+ T cells in the tumor (Fig. 4H and S39†), indicating the necessity for the reprogramming of the tumor microenvironment. To investigate whether the treatment can trigger a long-term antitumor immune response, the levels of memory T cells (CD3+CD4+CD44+CD62L+) in the tumor and spleen of the animal models were investigated by flow cytometry. The treatment with Pt(IV)SS@CaCO3@Biotin showed a strong enhancement of the amount of memory T cells (Fig. 4E and S40†), suggestive of long-term immune protection of the mouse model. Overall, these findings confirm that the treatment with Pt(IV)SS@CaCO3@Biotin is able to provide long-term immune protection against the development of new/distant tumors.
Conclusions
In summary, herein, the physical adsorption and oxidative disulfide polymerization of Pt(IV) prodrugs Pt(IV)-2SH inside of CaCO3 nanoparticles are presented. To enhance aqueous solubility and improve tumor targeting, the nanoparticles were post-translationally functionalized with DSPE-PEG2000-Biotin Pt(IV)SS@CaCO3@Biotin. While the nanoparticle scaffold remained stable in an aqueous solution, the nanomaterial quickly degraded into calcium ions in the presence of acid and into cisplatin in the presence of glutathione. Studies of the biological properties revealed that Pt(IV)SS@CaCO3@Biotin interacted in cisplatin-resistant non-small lung cancer cells by a multimodal mechanism of action involving the disruption of the Ca2+ homeostasis and Ca2+ overloading of the mitochondria, dual depletion of intracellular levels of glutathione, binding to nuclear DNA, and amplification of the generation of reactive oxygen species and lipid peroxides. These cellular misregulations resulted in the triggering of cell death by a combination of apoptosis, ferroptosis and ICD in A549R cells. Based on this multimodal treatment, the nanoparticles demonstrated a strong tumor growth inhibition effect towards a non-small cisplatin-resistant lung cancer tumor inside the animal model. Additionally, within a tumor-bearing animal model, Pt(IV)SS@CaCO3@Biotin was able to trigger the maturation of dendritic cells in the tumor-draining lymph nodes and enhance tumor T-cell infiltration. Using a tumor re-challenging experiment, the (long-term) immune response towards a specific kind of cancer was demonstrated. The ability of the nanoparticles to therapeutically intervene by a multimodal mechanism of action could pave the way to overcome tumor drug resistance. The generated immune response inside the animal model could find application for the treatment of distant or metastatic tumors. Capitalizing on this, the presented design strategy could circumvent the current major challenges in clinics.Statistical analysis
The significance of several experimental results was analyzed by using the analysis of T-test. Probabilities p < 0.05 (*) and p < 0.01 (**), and ***P < 0.001 were marked in figures and 0.05 was chosen as the significance level.Data availability
ESI† is available and includes experimental materials and methods for the synthesis, preparation, and characterization of the nanoparticles, in vitro antitumor efficiency, immune activation, and in vivo animal experiments and vaccine trials.Author contributions
H. Chao oversaw and designed all experiments; F. Wei and L. Ke synthesized the materials and performed characterization of materials and properties in vitro; F. Wei, S. Gao, J. Wang and Y. Chen performed the animal experiments; F. Wei, J. Karges, L. Ji and H. Chao analyzed the data and wrote the manuscript. All authors discussed the results and commented on and proofread the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22120102002 and 22277013), and the Science and Technology Innovation Program of Hunan Province of China (No. 2021RC5028).Notes and references
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef CAS PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- Z. Wang, Z. Xu and G. Zhu, Angew. Chem., Int. Ed., 2016, 55, 15564–15568 CrossRef CAS PubMed.
- X. Zeng, Y. Wang, J. Han, W. Sun, H.-J. Butt, X.-J. Liang and S. Wu, Adv. Mater., 2020, 32, 2004766 CrossRef CAS.
- Y. Min, C.-Q. Mao, S. Chen, G. Ma, J. Wang and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS.
- L. P. Martin, T. C. Hamilton and R. J. Schilder, Clin. Cancer Res., 2008, 14, 1291–1295 CrossRef CAS PubMed.
- W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, J. Am. Chem. Soc., 2005, 127, 1382–1383 CrossRef CAS.
- K.-C. Tong, P.-K. Wan, C.-N. Lok and C.-M. Che, Chem. Sci., 2021, 12, 15229–15238 RSC.
- Y. Dai, T. Li, Z. Zhang, Y. Tan, S. Pan, L. Zhang and H. Xu, J. Am. Chem. Soc., 2021, 143, 10709–10717 CrossRef CAS.
- B. R. Stockwell, Nature, 2019, 575, 597–598 CrossRef CAS PubMed.
- P. A. Kiberstis, Science, 2020, 368, 42–44 CrossRef.
- F. Wu, Y. Du, J. Yang, B. Shao, Z. Mi, Y. Yao, Y. Cui, F. He, Y. Zhang and P. Yang, ACS Nano, 2022, 16, 3647–3663 CrossRef CAS.
- E. M. Bolitho, C. Sanchez-Cano, H. Shi, P. D. Quinn, M. Harkiolaki, C. Imberti and P. J. Sadler, J. Am. Chem. Soc., 2021, 143, 20224–20240 CrossRef CAS PubMed.
- G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef CAS PubMed.
- Z. Zhou, K. Maxeiner, P. Moscariello, S. Xiang, Y. Wu, Y. Ren, C. J. Whitfield, L. Xu, A. Kaltbeitzel, S. Han, D. Mücke, H. Qi, M. Wagner, U. Kaiser, K. Landfester, I. Lieberwirth, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2022, 144, 12219–12228 CrossRef CAS PubMed.
- X.-Q. Zhou, M. Mytiliniou, J. Hilgendorf, Y. Zeng, P. Papadopoulou, Y. Shao, M. P. Dominguez, L. Zhang, M. B. S. Hesselberth, E. Bos, M. A. Siegler, F. Buda, A. M. Brouwer, A. Kros, R. I. Koning, D. Heinrich and S. Bonnet, Adv. Mater., 2021, 33, 2008613 CrossRef CAS.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- D. Wei, Y. Yu, X. Zhang, Y. Wang, H. Chen, Y. Zhao, F. Wang, G. Rong, W. Wang, X. Kang, J. Cai, Z. Wang, J.-Y. Yin, M. Hanif, Y. Sun, G. Zha, L. Li, G. Nie and H. Xiao, ACS Nano, 2020, 14, 16984–16996 CrossRef CAS PubMed.
- K. Ganesh, Z. K. Stadler, A. Cercek, R. B. Mendelsohn, J. Shia, N. H. Segal and L. A. Diaz, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 361–375 CrossRef PubMed.
- T. Yang, S. Zhang, H. Yuan, Y. Wang, L. Cai, H. Chen, X. Wang, D. Song, X. Wang, Z. Guo and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202213337 CrossRef CAS.
- X. Su, B. Liu, W.-J. Wang, K. Peng, B.-B. Liang, Y. Zheng, Q. Cao and Z.-W. Mao, Angew. Chem., Int. Ed., 2023, 62, e202216917 CAS.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- D. Wei, Y. Huang, B. Wang, L. Ma, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2022, 61, e202201486 CAS.
- J. Li, H. Wang, Y. Wang, X. Gong, X. Xu, X. Sha, A. Zhang, Z. Zhang and Y. Li, Adv. Mater., 2020, 32, 2002380 CrossRef CAS PubMed.
- X. Wang, M. Li, K. Ren, C. Xia, J. Li, Q. Yu, Y. Qiu, Z. Lu, Y. Long, Z. Zhang and Q. He, Adv. Mater., 2020, 32, 2002160 CrossRef CAS.
- D. Y. Q. Wong, W. W. F. Ong and W. H. Ang, Angew. Chem., Int. Ed., 2015, 54, 6483–6487 CrossRef CAS PubMed.
- K.-B. Huang, F.-Y. Wang, H.-W. Feng, H. Luo, Y. Long, T. Zou, A. S. C. Chan, R. Liu, H. Zou, Z.-F. Chen, Y.-C. Liu, Y.-N. Liu and H. Liang, Chem. Commun., 2019, 55, 13066–13069 RSC.
- M. Sabbatini, I. Zanellato, M. Ravera, E. Gabano, E. Perin, B. Rangone and D. Osella, J. Med. Chem., 2019, 62, 3395–3406 CrossRef CAS PubMed.
- V. Novohradsky, L. Markova, H. Kostrhunova, J. Kasparkova, J. Hoeschele and V. Brabec, J. Inorg. Biochem., 2022, 226, 111628 CrossRef CAS PubMed.
- L. Wang, R. Guan, L. Xie, X. Liao, K. Xiong, T. W. Rees, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2021, 60, 4657–4665 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- W. Wang, M. Green, J. E. Choi, M. Gijón, P. D. Kennedy, J. K. Johnson, P. Liao, X. Lang, I. Kryczek, A. Sell, H. Xia, J. Zhou, G. Li, J. Li, W. Li, S. Wei, L. Vatan, H. Zhang, W. Szeliga, W. Gu, R. Liu, T. S. Lawrence, C. Lamb, Y. Tanno, M. Cieslik, E. Stone, G. Georgiou, T. A. Chan, A. Chinnaiyan and W. Zou, Nature, 2019, 569, 270–274 CrossRef CAS PubMed.
- S. Kuang, F. Wei, J. Karges, L. Ke, K. Xiong, X. Liao, G. Gasser, L. Ji and H. Chao, J. Am. Chem. Soc., 2022, 144, 4091–4101 CrossRef CAS.
- P. Zheng, B. Ding, G. Zhu, C. Li and J. Lin, Angew. Chem., Int. Ed., 2022, 61, e202204904 CAS.
- Y. Xiao, Z. Li, A. Bianco and B. Ma, Adv. Funct. Mater., 2023, 33, 2209291 CrossRef CAS.
- Z. Dong, L. Feng, Y. Hao, Q. Li, M. Chen, Z. Yang, H. Zhao and Z. Liu, Chem, 2020, 6, 1391–1407 CAS.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- B. A. Trofimov, L. M. Sinegovskaya and N. K. Gusarova, J. Sulfur Chem., 2009, 30, 518–554 CrossRef CAS.
- S. H. Kim, Z. Madak-Erdogan, S. C. Bae, K. E. Carlson, C. G. Mayne, S. Granick, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Am. Chem. Soc., 2015, 137, 10326–10335 CrossRef CAS.
- C. G. Lee, C. Lee, J. Lee, J. S. Nam, B.-S. Kim and T.-H. Kwon, Angew. Chem., Int. Ed., 2022, 61, e202210623 CAS.
- Z. Xu, Z. Wang, Z. Deng and G. Zhu, Coord. Chem. Rev., 2021, 442, 213991 CrossRef CAS.
- H. Yuan, Z. Han, Y. Chen, F. Qi, H. Fang, Z. Guo, S. Zhang and W. He, Angew. Chem., Int. Ed., 2021, 60, 8174–8181 CrossRef CAS PubMed.
- A. Roesch, M. Fukunaga-Kalabis, E. C. Schmidt, S. E. Zabierowski, P. A. Brafford, A. Vultur, D. Basu, P. Gimotty, T. Vogt and M. Herlyn, Cell, 2010, 141, 583–594 CrossRef CAS.
- X. Meng, D. Li, L. Chen, H. He, Q. Wang, C. Hong, J. He, X. Gao, Y. Yang, B. Jiang, G. Nie, X. Yan, L. Gao and K. Fan, ACS Nano, 2021, 15, 5735–5751 CrossRef CAS PubMed.
- J. P. Friedmann Angeli, D. V. Krysko and M. Conrad, Nat. Rev. Cancer, 2019, 19, 405–414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02264a |
| This journal is © The Royal Society of Chemistry 2023 |