
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-controlled alternating block polychalcogenophenes: synthesis, structural characterization, molecular properties, and transistors for bromine detection†
Kuo-Hsiu
Huang
a,
Huai-Hsuan
Liu
a,
Kuang-Yi
Cheng
a,
Chia-Lin
Tsai
a and
Yen-Ju
Cheng
 *ab
*ab
aDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, 1001 University Road, Hsinchu, 30010, Taiwan, China. E-mail: yjcheng@nycu.edu.tw
bCenter for Emergent Functional Matter Science, National Chiao Tung University, 1001 University Road, Hsinchu, 30010, Taiwan, China
First published on 14th July 2023
Abstract
Sequence-controlled polychalcogenophenes have attracted much interest in terms of synthesis, structure and function in polymer science. For the first time, we developed a new class of alternating block conjugated copolymers denoted as poly(alt-AB)x-b-(alt-AC)y where both blocks are constituted by an alternating copolymer. 3-Hexylthiophene (S), 3-hexylselenophene (Se) and 3-hexyltellurophene (Te) are used as A, B and C units to assemble three sequence-controlled polychalcogenophenes P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) which are prepared by adding two different Grignard monomers in sequence to carry out Ni(dppp)Cl2-catalyzed Kumada polymerization. The molecular weight, dispersity, and length of each block (x = y) and main-chain sequence can be synthetically controlled via the catalyst transfer polycondensation mechanism. The polymer structures, i.e. alternating block main chain with high side-chain regioregularity, are unambiguously confirmed by 1H-NMR and 13C-NMR. The optical and electrochemical properties of the polymers can be systematically fine-tuned by the composition and ratio of the chalcogenophenes. From GIWAXS measurements, all the polymers exhibited predominantly edge-on orientations, indicating that the packing behaviors of the alternating block polychalcogenophenes with high regioregularity are inherited from the highly crystalline P3HT. P(SSe)b(STe) exhibited a hole OFET mobility of 1.4 × 10−2 cm2 V−1 s−1, which represents one of the highest values among the tellurophene-containing polychalcogenophenes. The tellurophene units in the polymers can undergo Br2 addition to form the oxidized TeBr2 species which results in dramatically red-shifted absorption due to the alternating arrangement to induce strong charge transfer character. The OFET devices using the tellurophene-containing polychalcogenophenes can be applied for Br2 detection, showing high sensitivity, selectivity and reversibility.
Introduction
Biopolymers with perfectly ordered sequences exhibit unique structural properties and fascinating functions unmatched by synthetic polymers. The recent advances in organometallic chemistry in organic synthesis have paved the way to access various types of sequence-controlled polymers with novel structural features.1–3 The synthesis of sequence-controlled conjugated polymers (SCCPs) is one of the emerging and challenging topics in polymer science.4–9 SCCPs with structural regularity are prone to self-assembly into ordered nanostructures, which is essential for realizing high-performance organic optoelectronics, such as organic photovoltaics (OPVs)10,11 and field-effect transistors (OFETs).12,13 Regioregular poly(3-alkylthiophene) (P3AT) is one of the most representative semiconducting polymers widely applied in various organic optoelectronics due to its high crystallinity, ordered packing structure in thin film and good chemical stability.14,15 However, the limited absorption window of 300 to 550 nm with fixed energy levels is the main deficiency of P3ATs. Despite the fact that aliphatic side-chain modification has been implemented to tailor the solubility and morphological properties of P3AT, it has little effect on the optical and electrochemical properties. Consequently, donor–acceptor (D–A) conjugated copolymers prepared by metal-catalyzed polymerization of an electron-deficient monomer with an electron-rich monomer have been considerably developed to tailor HOMO/LUMO energy levels with a tunable optical bandgap.16,17 Nevertheless, incorporation of two discrepant units with different sizes, geometric shapes, polarities and side-chain substituents dramatically alters the intermolecular interactions, resulting in unpredictable solid-state molecular packing and morphology.Selenophene and tellurophene in the chalcogenophene family using heavier selenium and tellurium elements in group 16 have similar structural geometries to thiophene but different electronic properties.18,19 Compared to thiophene, selenophene and tellurophene have lower aromaticity and higher polarizability resulting in a reduced HOMO/LUMO bandgap and enhanced intermolecular interactions.20–27 Tellurophene has even more distinct properties and pronounced impacts due to the metalloid character of tellurium.28,29
Poly(3-hexylselenophene) P3HSe and poly(3-hexyltellurophene) P3HTe have been synthesized by using traditional catalyst transfer polymerization (CTP).30–32 The strong intermolecular interactions of P3HSe, especially P3HTe, were found to unfavorably lead to reduced solubility, thereby limiting the molecular weight growth during polymerization and deteriorating the thin-film formation directly related to the charge transport, unless the high-carbon aliphatic side chains are introduced.33
It is envisaged that integration of selenophene or tellurophene analogs with thiophene in a polymer backbone provides a practical and effective approach to fine-tune optical and electronic properties without substantially altering the ordered packing behavior and sacrificing solution processability. Consequently, the assembling 3-hexylthiophene, 3-hexylselenophene and 3-hexyltellurophene monomers with controlled sequence, ratio, combination and regioregularity will lead to a new class of structurally and functionally appealing polychalcogenophenes.
Block copolymers with controlled morphology are of great significance in nanomaterials and nanotechnology.34–37 Block polychalcogenophenes such as poly(3-hexylthiophene-block-3-hexylselenophene) P(S)b(Se)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38–43 and poly(3-hexylthiophene-block-3-hexyltellurophene) P(S)b(Te)44 have been synthesized by controlling the addition sequence of the two monomers during the CTP process (Fig. 1).
38–43 and poly(3-hexylthiophene-block-3-hexyltellurophene) P(S)b(Te)44 have been synthesized by controlling the addition sequence of the two monomers during the CTP process (Fig. 1).
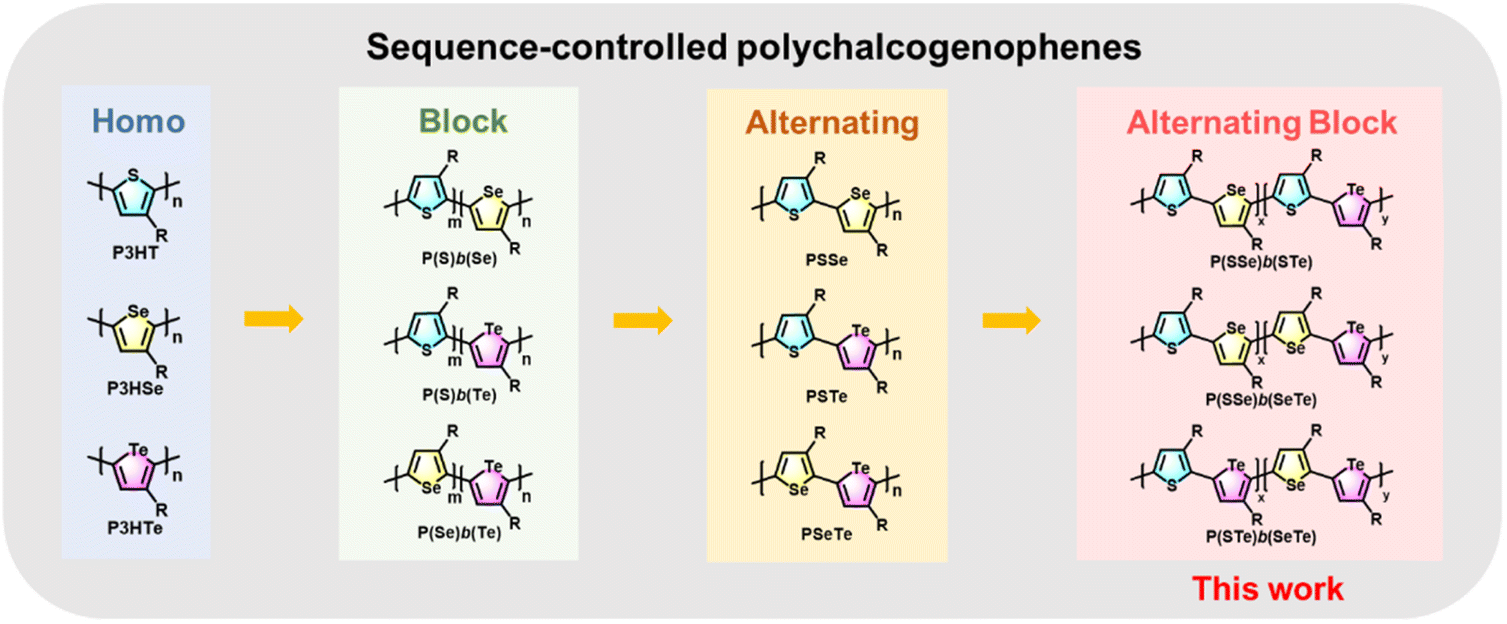 | ||
| Fig. 1 Homopolychalcogenophenes P3HT, P3HSe, and P3HTe; block polychalcogenophenes P(S)b(Se), P(S)b(Te) and P(Se)b(Te); alternating polychalcogenophenes PSSe, PSTe, and PSeTe; and alternating block polychalcogenophenes P(SSe)b(STe), P(SSe)b(SeTe), and P(STe)b(SeTe) in this work. | ||
On the other hand, synthesis of AB-type alternating polychalcogenophenes with high side-chain regioregularity is more challenging.45–47 Recently, we developed a new series of well-designed AB-type monomers for the preparation of poly(3-hexylthiophene-alt-3-hexylselenophene) PSSe, poly(3-hexylthiophene-alt-3-hexyltellurophene) PSTe and poly(3-hexylselenophene-alt-3-hexyltellurophene) PSeTe (Fig. 1).48 Development of a block conjugated polymer where one block is comprised of an alternating copolymer is also of great interest in polymer science. For example, poly[3-hexylthiophene-block-(alt-donor–acceptor)n], where P3HT is the first block followed by covalent extension with the second block of an alternating D–A conjugated copolymer, was designed and synthesized.49,50 The purpose of such a polymer arrangement is to induce microphase separation between the donor block (P3HT) and the acceptor block (D–A) which can be used as a single-component active layer for organic solar cells.51,52 To the best of our knowledge, block conjugated polymers in which both blocks are constituted by alternating copolymers denoted as poly(alt-AB)x-b-(alt-AC)y have seldom been reported and explored.53 We envision that 3-hexylthiophene, 3-hexylselenophene and 3-hexyltellurophene could be used as A, B and C units to create such unprecedented sequence-controlled polychalcogenophenes with high side-chain regioregularity. To this end, we report the first synthesis of a new class of poly(alt-AB)x-b-(alt-AC)y type polychalcogenophenes where each block consists of an alternating copolymer, including poly(3-hexylthiophene-alt-3-hexylselenophene)-block-(3-hexylthiophene-alt-3-hexyltellurophene) denoted as P(SSe)b(STe), poly(3-hexylthiophene-alt-3-hexylselenophene)-block-(3-hexylselenophene-alt-3-hexyltellurophene) denoted as P(SSe)b(SeTe) and poly(3-hexylthiophene-alt-3-hexyltellurophene)-block-(3-hexylselenophene-alt-3-hexyltellurophene) denoted as P(STe)b(SeTe) (Fig. 1). Their structural features, intrinsic molecular and morphological properties, Br2 oxidation and OFETs for Br2 sensors have been thoroughly investigated.
Results and discussion
Synthesis of block copolymer
The block copolymers were synthesized via Grignard metathesis followed by Kumada catalyst transfer polycondensation (KCTP). Three kinds of unsymmetrical diiodobichalcogenophene monomers SSeI2, STeI2, and SeTeI2 were designed and synthesized according to our previous work.48 Due to the favorable electronic and steric effects, the Grignard metathesis of SSeI2, STeI2, and SeTeI2 with one equivalent of iPrMgCl·LiCl occurs regioselectively at the lighter chalcogenophenes to form MgSSeI, MgSTeI, and MgSeTeI, respectively (Scheme 1).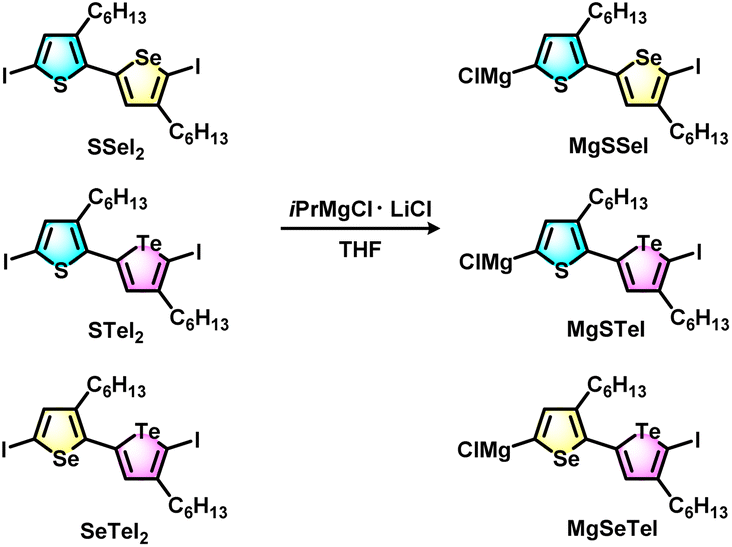 | ||
| Scheme 1 Regioselective Grignard metathesis of SSeI2, STeI2, and SeTeI2 monomers with iPrMgCl·LiCl to form unsymmetrical active monomer species MgSSeI, MgSTeI, and MgSeTeI. | ||
For the synthesis of P(SSe)b(STe), the SSeI2 was reacted with iPrMgCl·LiCl to form MgSSeI which undergoes Ni(dppp)Cl2-catalyzed Kumada polymerization at room temperature. After the MgSSeI monomer was completely consumed to form the first PSSe block for 30 min, a second freshly prepared MgSTeI monomer was introduced to continue elongating the second block for 2 h, leading to the formation of P(SSe)b(STe). The molar ratio of Ni(dppp)Cl2/MgSSeI/MgSTeI is set as 1:50:50. High-temperature gel permeation chromatography with trichlorobenzene as eluent was used to monitor the polymerization reaction. As soon as the polymerization was quenched by 6 M HCl/MeOH solution after 30 min, the molecular weight of the first block PSSe was determined to be Mn = 11.4 kDa with a dispersity (Đ) of 1.20 (Fig. 2). The resultant block polymer P(SSe)b(STe) exhibited an increased Mn = 24.2 kDa with Đ = 1.30, confirming the continuous growth of the second PSTe block on the first PSSe block.
 | ||
| Fig. 2 (a) Synthesis of alternating block copolymers P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) by two-step sequential Ni(dppp)Cl2-catalyzed Kumada polymerization; GPC curves with molecular weight and dispersity for (b) PSSe/P(SSe)b(STe), (c) PSSe/P(SSe)b(SeTe), and (d) PSTe/P(STe)b(SeTe). | ||
In a similar manner, P(SSe)b(SeTe) and P(STe)b(SeTe) were successfully prepared by Grignard metathesis/Kumada Ni(dppp)Cl2-catalyzed sequence-controlled polymerization of MgSSeI (first block, 30 min)/MgSeTeI (second block, 2 h) and MgSTeI (first block, 30 min)/MgSeTeI (second block, 2 h), respectively (Fig. 2). The molecular weights of the first block and block polymers are shown in Fig. 2. Because the molar ratio of Ni(dppp)Cl2/first monomer/second monomer is kept as 1:50:50, the conjugated length of the two blocks in the three polymers is approximately the same due to the CTP character. As a result, the ratio of thiophene/selenophene/tellurophene composition is roughly S50%/Se25%/Te25% for P(SSe)b(STe), S25%/Se50%/Te25% for P(SSe)b(SeTe), and S25%/Se25%/Te50% for P(STe)b(SeTe).
1H- and 13C-NMR spectroscopy was used to investigate the chemical structures of the three block alternating poly(bichalcogenophene)s (Fig. 3 and S1†). The 1H-NMR spectra of the P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) polymers all showed four well-defined singlet peaks in the aromatic region, confirming four kinds of protons in four different 3-hexylchalcogenophene environments (two from one block and two from the other), which is consistent with the block structures with high side-chain regioregularity. The chemical shifts of protons on thiophene (Ha)/selenophene (Hb) of the SSe block and thiophene (Hc)/tellurophene (Hd) of the second STe block are determined to be 6.93/7.19 and 6.83/7.65 ppm, respectively, for P(SSe)b(STe). In a similar manner, the chemical shifts of protons on thiophene (Ha)/selenophene (Hb) of the SSe block and selenophene (He)/tellurophene (Hf) of the second SeTe block are determined to be 6.92/7.18 and 7.00/7.57 ppm for P(SSe)b(SeTe). Furthermore, the chemical shifts of protons on thiophene (Hc)/tellurophene (Hd) of the STe block and selenophene (He)/tellurophene (Hf) of the second SeTe block are 6.82/7.64 and 7.00/7.57 ppm for P(STe)b(SeTe). It should be noted that the chemical shifts of the protons of the SSe, STe and SeTe blocks in the three different polymers are essentially identical, clearly demonstrating the formation of regioregular alternating block architectures. On the other hand, the integration ratio of Ha + Hb:Hc + Hd for P(SSe)b(STe), Ha + Hb:He + Hf for P(SSe)b(SeTe) and Hc + Hd:He + Hf for P(STe)b(SeTe) is approximately 1.00:1.01, 1.00:1.04 and 1.00:1.00, respectively, indicating that the two blocks in the three polymers have essentially the same conjugated length. In other words, the molar ratio of the thiophene:selenophene:tellurophene units in P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) is 2:1:1, 1:2:1 and 1:1:2, respectively. This result confirms the character of catalyst transfer polymerization so that the length of the two blocks can be synthetically controlled by the feed ratio of the two monomers (1 to 1). On the other hand, the 13C-NMR spectra of the P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) show sixteen aromatic peaks derived from four different 3-hexylchalcogenophene environments, which is also in good agreement with the block structures of the polymers (see the ESI†). The peaks appearing at 2.6–2.8 ppm come from the protons on the first carbon atoms of the hexyl side-chains.
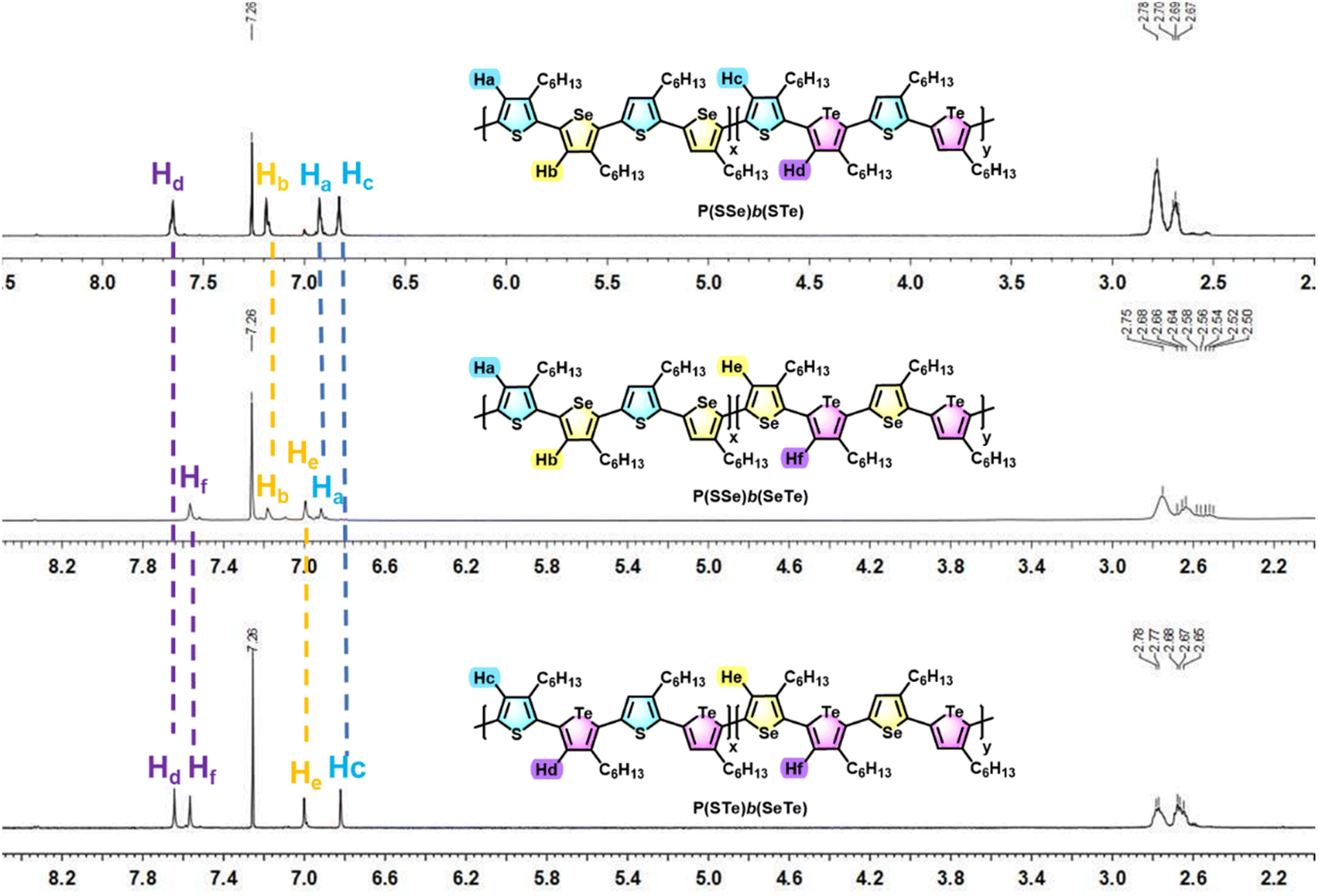 | ||
| Fig. 3 Comparison of the aromatic protons of P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) in 1H NMR spectra. | ||
Reversing the synthetic sequence of the block copolymer was also attempted. Using MgSeTeI as the first monomer and MgSSeI as the second monomer resulted in the formation of P(SeTe)b(SSe) with Mn = 25.4 kDa and Đ = 1.37 which have the same characteristics as P(SSe)b(SeTe). This experiment revealed that changing the monomer addition sequence does not affect the polymerization. The Ni species can undergo reductive elimination to form the carbon–carbon bond at the block–block junction and subsequently migrate (ring walk) to the chain end to propagate the second block.
Optical properties
To investigate the chalcogen effects on the optical absorption behavior of the block polychalcogenophenes, the UV-visible absorption spectra were recorded both in solution state (Fig. 4a) and thin-film state (Fig. 4b). The absorption data are summarized in Table 1. Upon increasing the amount of heavier chalcogenophenes (selenophene and tellurophene) in the polymers, the absorption is gradually red-shifted with accompanying broadening of the full width at half maximum (FWHM) of the band. In solution, the λmax of the P(SSe)b(STe) block copolymer was observed at 488 nm which is in between those of the PSSe (471 nm) and PSTe (504 nm). Similarly, the absorption maximum of P(SSe)b(SeTe) at 490 nm is also in between those of PSSe (471 nm) and PSeTe (541 nm). With a heavier chalcogen involved, P(STe)b(SeTe) (S25%/Se25%/Te50%) exhibited a strong absorption peak in the 400–600 nm range, extending the λmax to 514 nm (in between PSTe and PSeTe) as shown in Fig. 4a. All three polymers exhibited red-shifted absorption profiles both in λmax and λonset values in thin-film states: 584 and 743 nm for P(SSe)b(STe) (S50%/Se25%/Te25%); 594 and 830 nm for P(SSe)b(SeTe) (S25%/Se50%/Te25%); 600 and 830 nm for P(STe)b(SeTe) (S25%/Se25%/Te50%). It is interesting to find that block alternating copolymers P(SSe)b(STe) (S50%/Se25%/Te25%), P(SSe)b(SeTe) (S25%/Se50%/Te25%), and P(STe)b(SeTe) (S25%/Se25%/Te50%) turn out to have more red-shifted λmax values than the single block of alternating copolymers PSTe (S50%/Te50%, 576 nm) and PSeTe (Se50%/Te50%, 589 nm) which actually contain a higher percentage of heavier chalcogenophenes. This result implies that block alternating copolymers might have more ordered π–π stacking than the single block alternating copolymers. In particular, P(SSe)b(SeTe) showed the most red-shifted λmax from solution to solid state (more than 100 nm shift), implying that P(SSe)b(SeTe) might have the highest order of polymeric packing among the three polymers. We envisage that the SSe block in one P(SSe)b(SeTe) polymer might prefer to interact with the SeTe block in another polymer to form a more balanced π-stacking distance.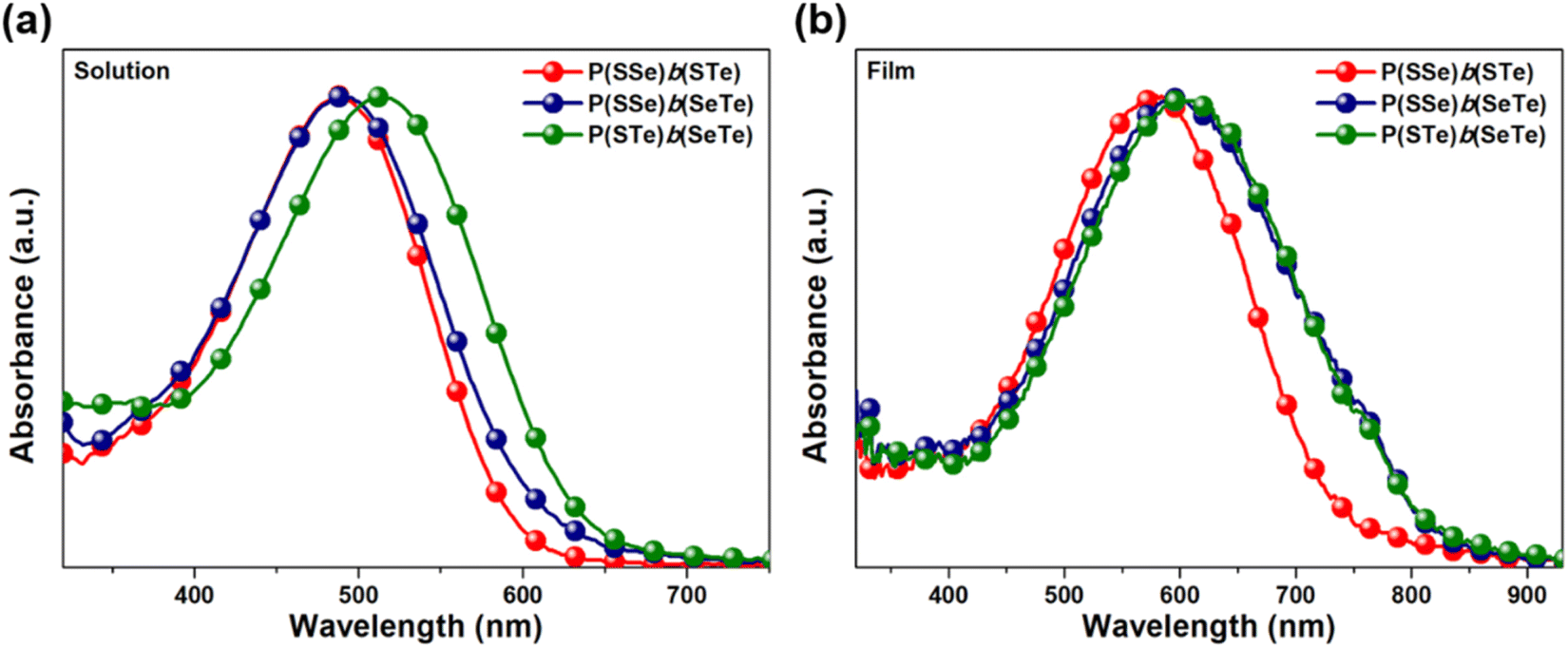 | ||
| Fig. 4 Absorption spectra of P(SSe)b(STe), P(SSe)b(SeTe), and P(STe)b(SeTe) in (a) o-DCB solution and (b) the thin-film state. | ||
Electrochemical properties
Cyclic voltammetry (CV) was evaluated to examine the electrochemical properties of the polymers and determine their HOMO and LUMO energy levels and electrochemical bandgaps (Fig. 5 and Table 1). Increasing the amount of heavier chalcogenophenes in the polymers resulted in the gradually elevated HOMO levels and reduced LUMO levels, thereby narrowing the band gaps, which is consistent with the optical properties.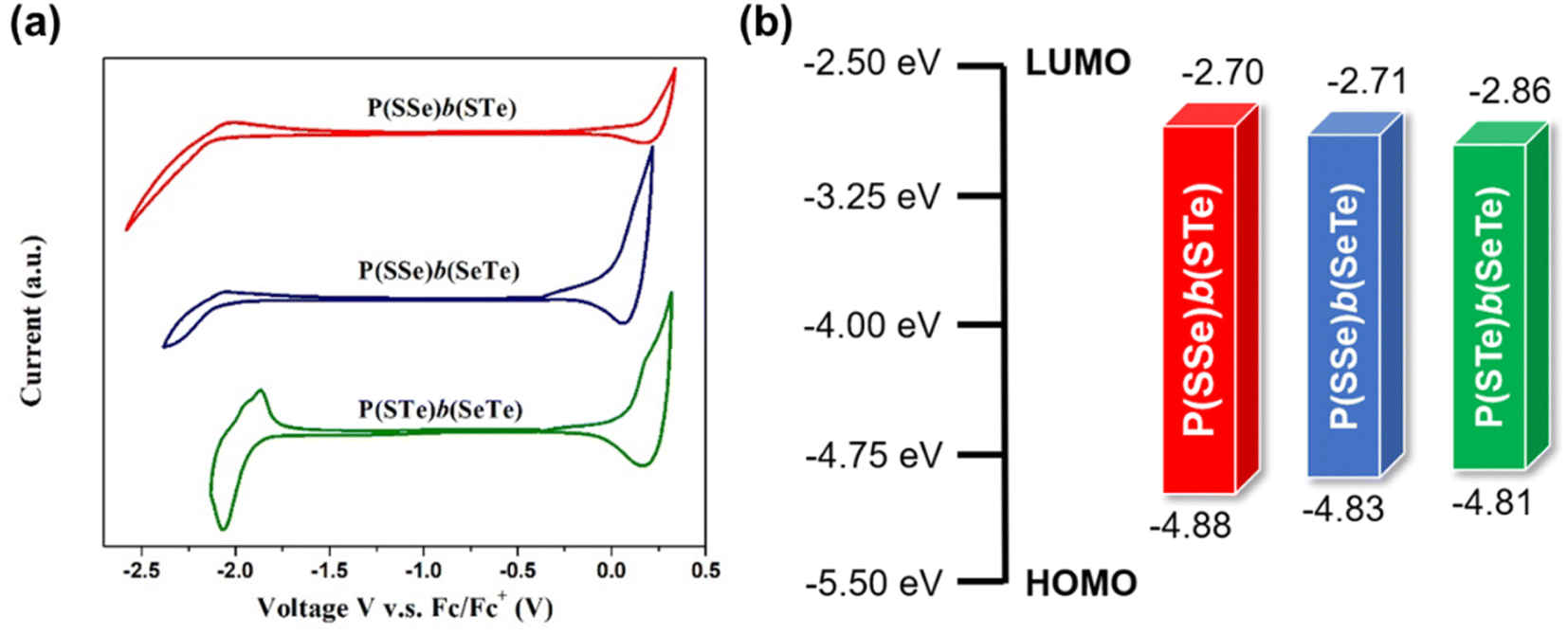 | ||
| Fig. 5 (a) Cyclic voltammetry plots of P(SSe)b(STe), P(SSe)b(SeTe), and P(STe)b(SeTe) in thin films and (b) the HOMO/LUMO energy levels. | ||
To gain insight into the molecular orientation and packing of polymers, grazing incidence wide-angle X-ray scattering (GIWAXS) was performed to investigate the morphology of thin films prepared on silicon wafers. The 2D GIWAXS images and the corresponding 1-dimensional out-of-plane (qz) and in-plane (qxy) diffraction patterns of the polymer thin films annealed at 100 °C for 10 minutes and as-cast are illustrated in Fig. 6 and Table 2, respectively. The polymers exhibited obvious (010) in-plane peaks corresponding to periodic π–π stacking between the two facing conjugated backbones, indicating that the polymers are arranged in predominantly edge-on orientations with the backbone plane nearly perpendicular to the substrate, which is beneficial for horizontal charge transfer.54,55 The π–π stacking distances (dπ) of P(SSe)b(STe) and P(SSe)b(SeTe) showed the same packing distance of 3.84 Å similar to that of lighter block PSSe while the dπ of 3.92 Å for the P(STe)b(SeTe) is between those of the two blocks PSTe and PSeTe (3.87 Å for PSeTe, and 3.94 Å for PSTe). Besides, the P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) polymer films exhibited out-of-plane (h00) signals assigned to a lamellar spacing (dl) corresponding to side-chain interdigitation. Interestingly, the lamellar spacing dl decreased upon increasing the tellurophene content (16.02 Å for P(STe)b(SeTe), 16.08 Å for P(SSe)b(STe), and 16.44 Å for P(SSe)b(SeTe)). This observation can be rationalized by the fact that the increased π–π stacking distance allows more space for the interdigitation of hexyl groups, thus leading to the shortened lamellar distance.56,57 After thermal annealing at 100 °C for 10 min, the dπ distance of P(SSe)b(STe) and P(SSe)b(SeTe) slightly decreases with enhanced signal intensities, indicating the thermal-induced formation of more compact structures.
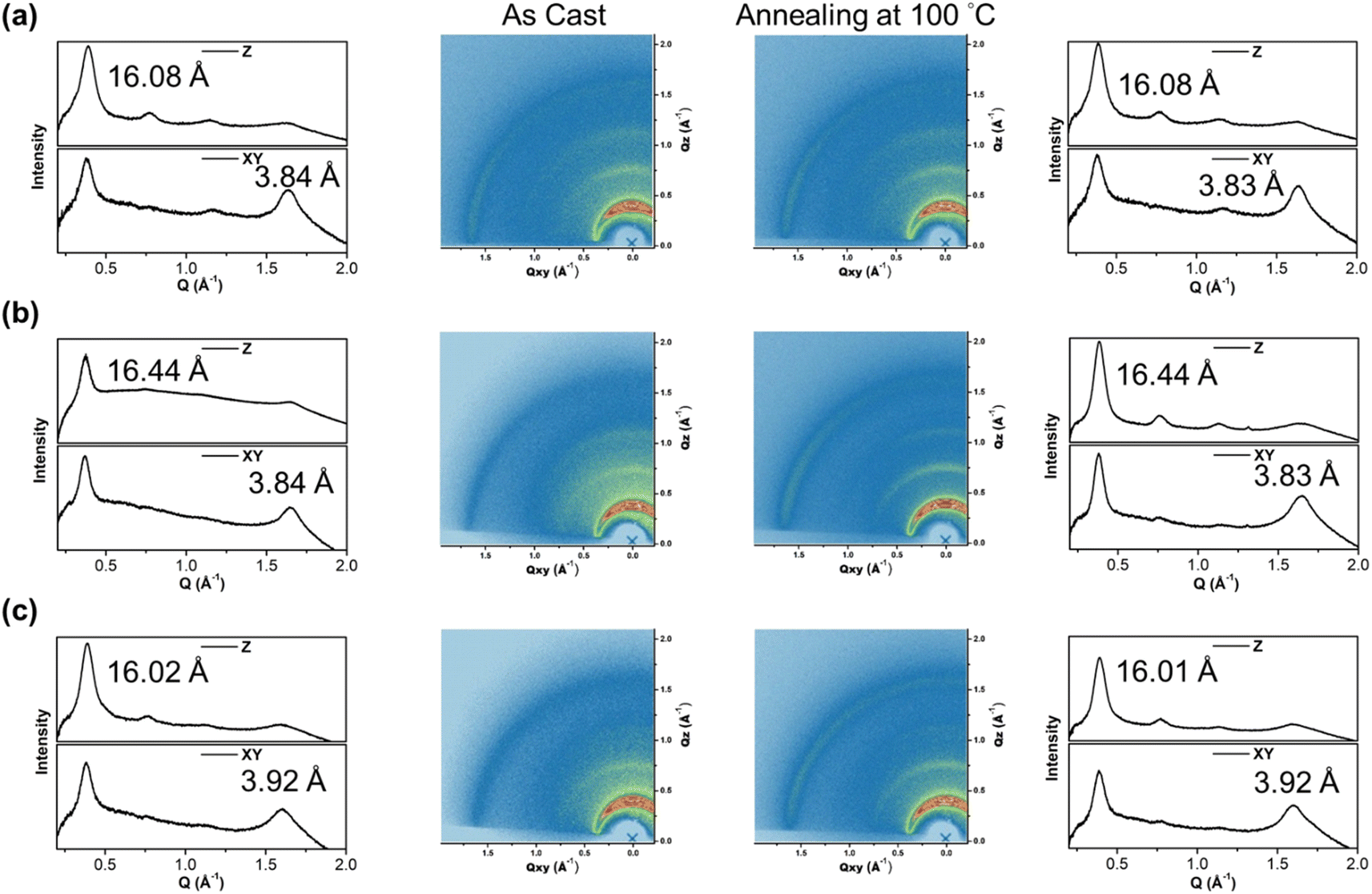 | ||
| Fig. 6 (a) P(SSe)b(STe), (b) P(SSe)b(SeTe), and (c) P(STe)b(SeTe) 2D-GIWAXS images of the polymer thin films measured before and after thermal annealing at 100 °C and their corresponding 1D in-plane (bottom) and out-of-plane (up) linecuts. | ||
| As-cast | After thermal annealing at 100 °C | |||
|---|---|---|---|---|
| Lamellar spacing (dl) (Å) | π-Stacking spacing (dπ) (Å) | Lamellar spacing (dl) (Å) | π-Stacking spacing (dπ) (Å) | |
| P(SSe)b(STe) | 16.08 | 3.84 | 16.08 | 3.83 |
| P(SSe)b(SeTe) | 16.44 | 3.84 | 16.44 | 3.83 |
| P(STe)b(SeTe) | 16.02 | 3.92 | 16.01 | 3.92 |
| PSSe | 16.51 | 3.84 | 16.51 | 3.83 |
| PSTe | 15.95 | 3.94 | 15.92 | 3.92 |
| PSeTe | 16.33 | 3.87 | 16.28 | 3.87 |
Morphological characterization
Atomic force microscopy (AFM) was conducted to investigate the surface morphology of the block copolymers in the thin film state (Fig. 7). Because of the relatively low solubility of the polymers, o-dichlorobenzene was chosen as the processing solvent. The root-mean-square roughness (Rq) is associated with the content of chalcogenophene units. P(SSe)b(STe) (S50%/Se25%/Te25%) with 50% thiophene content exhibited a relatively smooth and uniform surface with a small root-mean-square roughness (Rq) of 1.88 nm. The Rq of P(SSe)b(SeTe) (S25%/Se50%/Te25%) and P(STe)b(SeTe) (S25%/Se25%/Te50%) with increasing heavier chalcogenophene content is increased to 2.97 and 3.39 nm, respectively. As the ratio of heavier chalcogenophenes increases in a polymer, the strength of intermolecular interaction also increases. These changes lead to the formation of larger aggregates with higher Rq.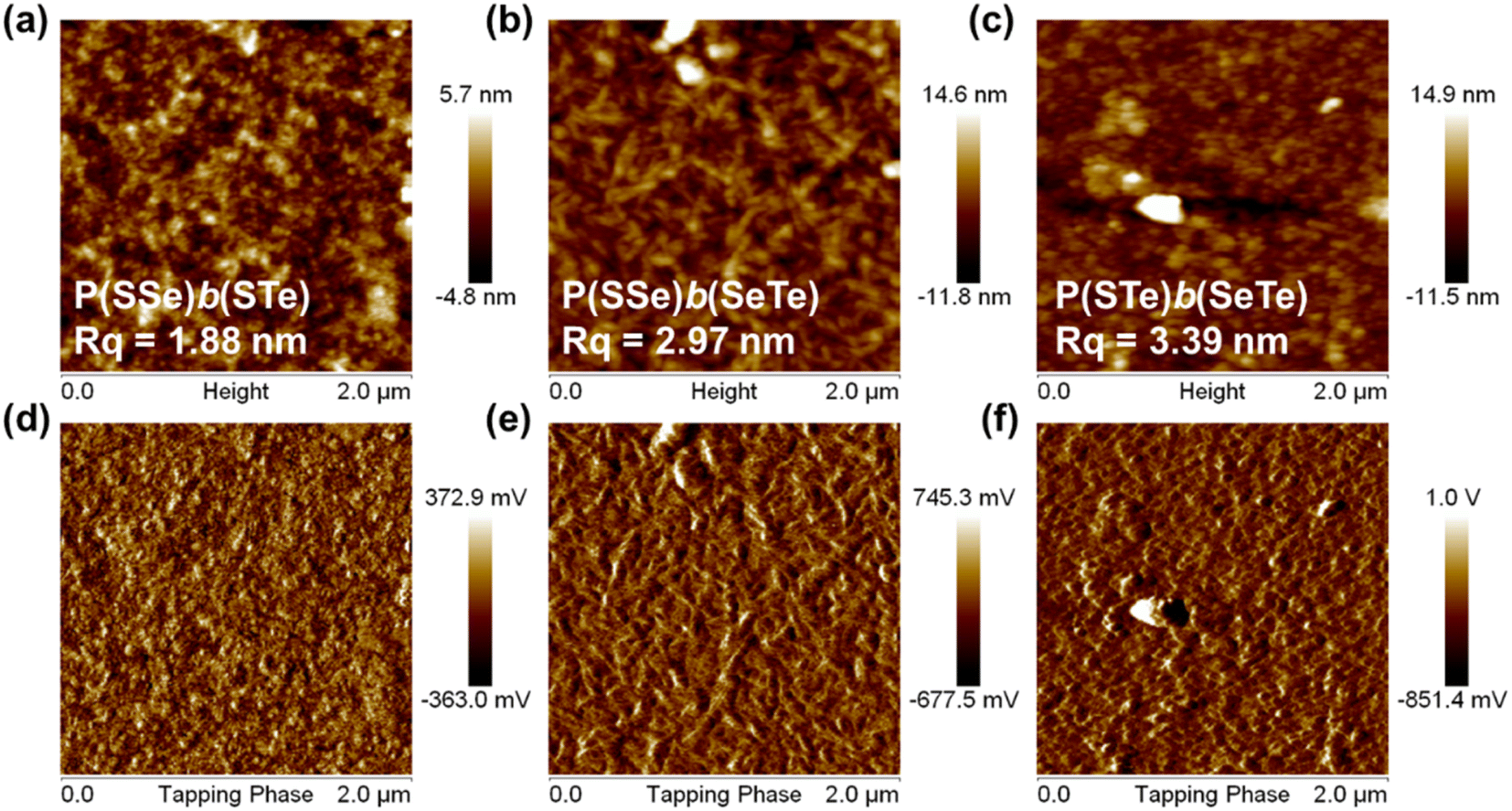 | ||
| Fig. 7 Height (a–c) and phase (d–f) images of atomic force microscopy: P(SSe)b(STe) (a and d), P(SSe)b(SeTe) (b and e), and P(STe)b(SeTe) (c and f). | ||
Transistor characterization
To investigate the charge mobility of three p-type polymers, organic field-effect transistors (OFETs) were fabricated by spin-coating P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe) solutions on the incorporating ODTS-treated SiO2/Si dielectric substrates in a bottom-gate/top-contact structure. The polymeric thin films were thermally annealed at 100 °C for 10 min. The output characteristics and transfer characteristics are shown in Fig. 8. Due to the edge-on π–π stacking orientation for efficient charge transport in OFETs, P(SSe)b(STe) exhibited a hole mobility of 1.4 × 10−2 cm2 V−1 s−1, which represents one of the highest values among the tellurophene-containing polychalcogenophenes. It is also interesting to find that the mobility of block P(SSe)b(STe) is higher than that of the single block polymer (7.2 × 10−3 cm2 V−1 s−1 for PSSe and 2.7 × 10−3 cm2 V−1 s−1 for PSTe) due to further reduced packing distances.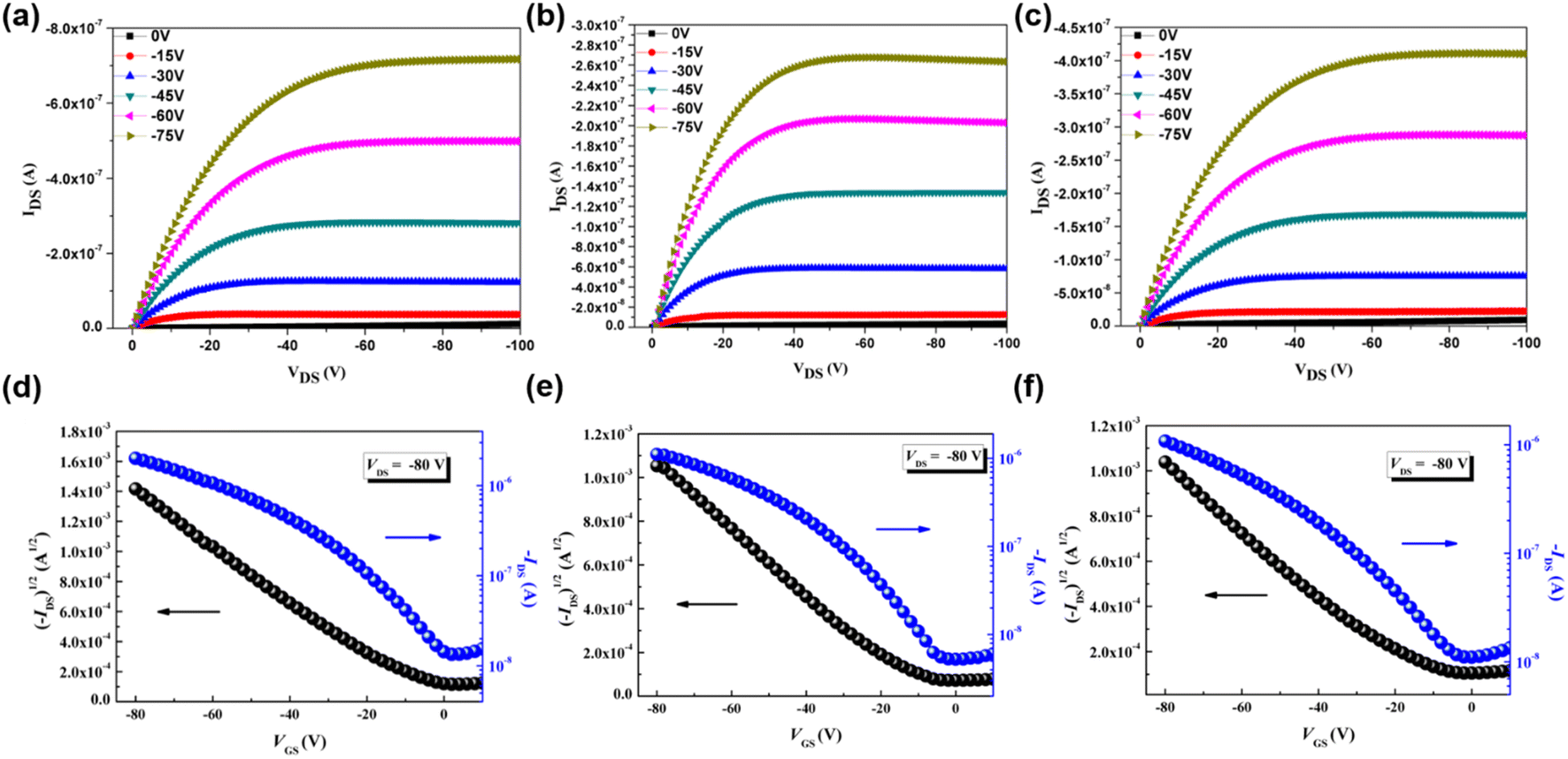 | ||
| Fig. 8 Output and transfer characteristics of OFETs based on P(SSe)b(STe) (a and d), P(SSe)b(SeTe) (b and e), and P(STe)b(SeTe) (c and f). | ||
While introducing heavier chalcogenophenes, P(SSe)b(SeTe) and P(STe)b(SeTe) exhibited slightly reduced mobilities of 5.0 × 10−3 cm2 V−1 s−1 and 4.6 × 10−3 cm2 V−1 s−1. Similarly, the values are also higher than that of single block PSeTe (5.2 × 10−4 cm2 V−1 s−1).
In particular, P(STe)b(SeTe) containing 50% Te has larger π–π stacking spacing (3.92 Å) due to the larger radius of the Te atom, which might affect the intermolecular charge hopping transportation. Moreover, P(SSe)b(SeTe) and P(STe)b(SeTe) exhibit relatively low solubility due to the presence of strong Te–Te, Se–Se, or Te–Se interactions. This characteristic poses a challenge in forming high-quality thin films suitable for OFETs. We envisage that the OFET mobility of tellurophene-containing polychalcogenophenes can be improved by replacing the hexyl group with longer branched aliphatic side chains.
Bromine addition for PSTe and P(SSe)b(PSTe)
The metalloid character of tellurium enables tellurophene to undergo chemical coordination to form hyper-valence species.58–61 In particular, previous studies conducted by Seferos et al. and others have demonstrated that small molecules and conjugated polymers incorporating tellurophene units can undergo bromine addition at the tellurium (Te) center.62–67 However, bromine addition for tellurophene-incorporated polychalcogenophenes in an alternating fashion and the resulting properties have never been explored. STePh2, a monomer containing a single (3-hexylthiophene-3-hexyltellurophene) unit, was designed and synthesized as a reference compound for carefully studying the bromine coordination (Fig. 9 and S2†). 1H-NMR of STePh2 is used to monitored the progress of bromine addition. The two triplet peaks at around 2.5–3.0 ppm are from the characteristic protons of the first carbon (CH2) in the two different hexyl groups (Fig. 9). Upon titration of 0.5 eq. of bromine in the solution, two new triplet peaks emerge and the integration ratio between the original and new triplet peaks is about 1:1, implying that 50% of STePh2 undergoes bromine addition at the tellurophene moiety. Upon titration of 1 eq. of bromine in the solution, the original peaks are completely replaced by the new triplet peaks. Similar behaviors can be observed in the aromatic region, ruling out the possibility of bromo-substitution at the β-position of thiophene or tellurophene. The absorption spectrum of STePh2 was also measured upon addition of bromine (Fig. 10). The absorption band with λmax at 376 nm is gradually decreased with concomitant appearance of a new band at 462 nm. The absorption profile is saturated when 1.4 eq. of bromine is introduced. The observation of an isosbestic point at 425 nm clearly indicates that only two STePh2 and STePh2Br2 species are directly interconverted without formation of other intermediates. Mass spectrometry showing the molecular weight of 742 m/z again confirms the formation of STePh2Br2 (Fig. S3†).
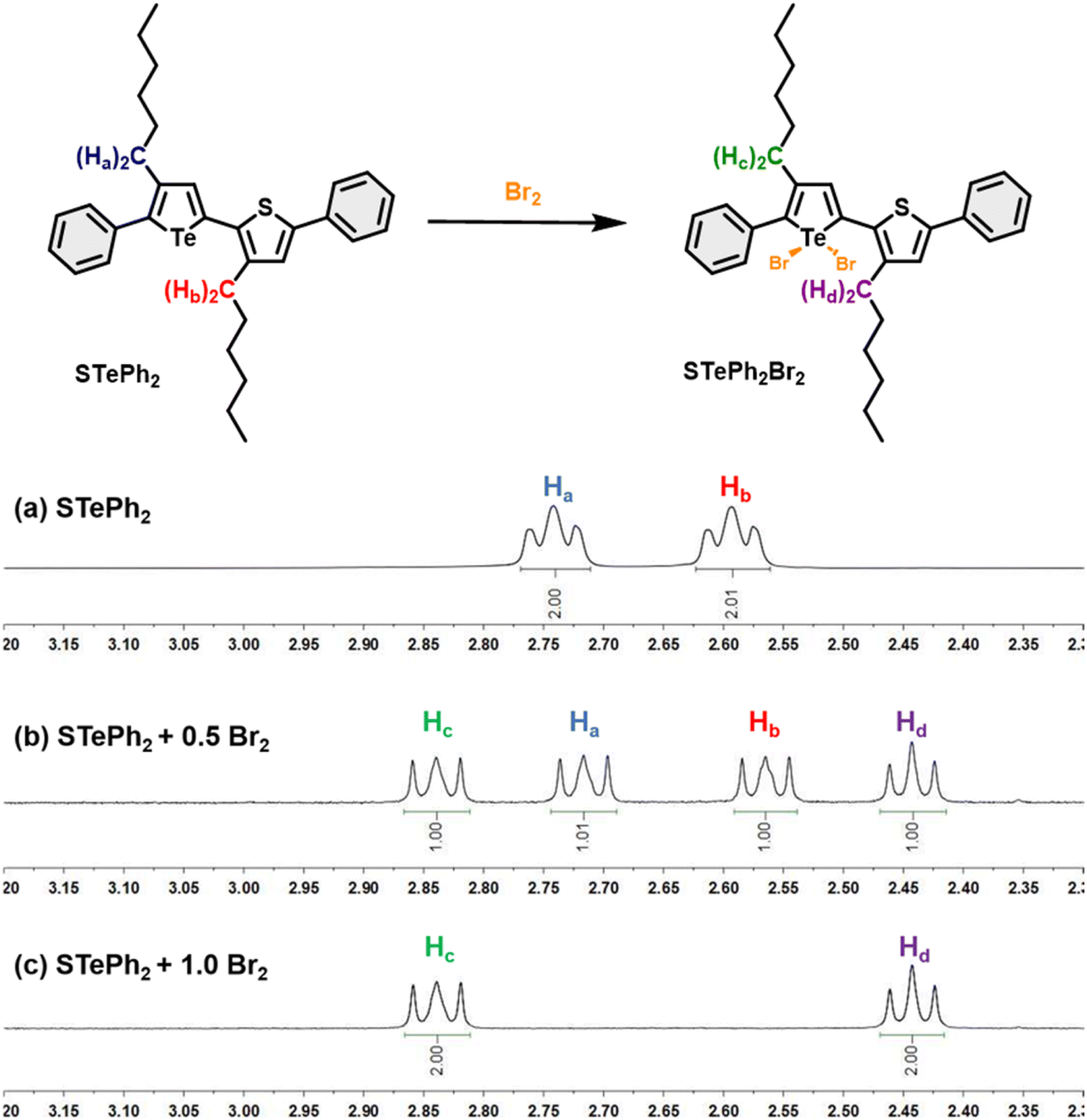 | ||
| Fig. 9 Bromine addition for the STePh2 model compound. 1H NMR spectra of (a) STePh2 and (b) STePh2 with 0.5 eq. of Br2 and (c) STePh2 with 1 eq. of Br2 in the aliphatic region (2.3–3.2 ppm). Ha/Hb are protons on the alpha carbons of the hexyl groups in STePh2 and Hc/Hd are the corresponding protons in STePh2Br2 after bromination. | ||
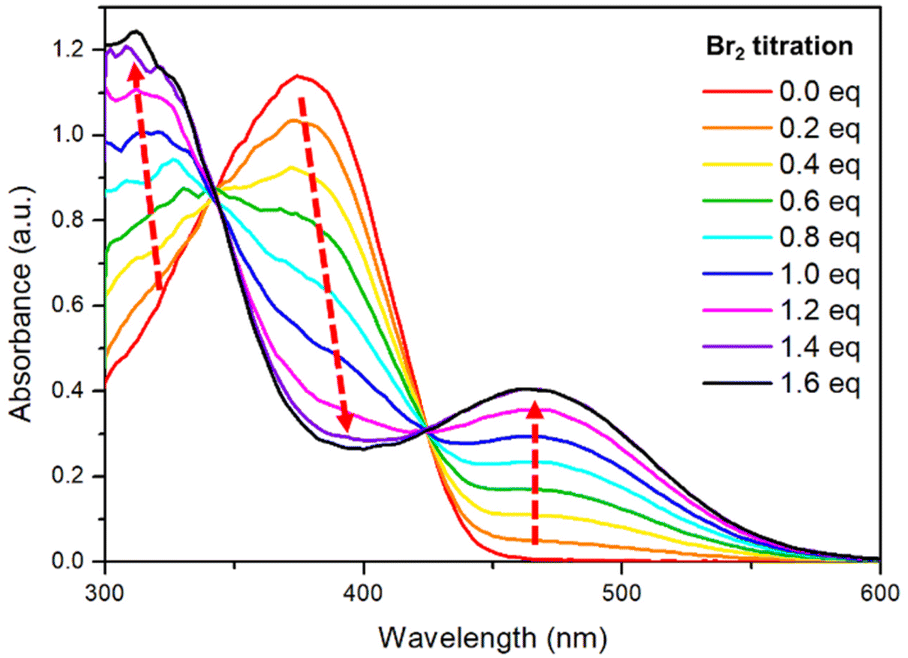 | ||
| Fig. 10 Absorption spectra of STePh2 in CHCl3 upon bromine titration. | ||
After the structure of STePh2Br2 has been confirmed, we further monitor the absorption change of the alternating PSTe copolymer with 50% of tellurophene with respect to bromine addition. To be more precise, for titration, the concentration of PSTe (M) in o-DCB is calculated based on the molecular weight of the STe repeating unit (428.15 m/z). When bromine (from low to high: 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, and 2.0 equivalents) is introduced, the λmax band at 504 nm is gradually reduced and hypsochromically shifted while a new band at longer wavelengths steadily intensifies along with the bathochromically shifted λmax (Fig. 11a). The absorption spectrum is saturated at 1.8 eq. of bromine with a final λmax located at 768 nm. The overall λmax red-shift of 264 nm, the largest value ever observed in the literature, is rather significant, indicating that strong and efficient photo-induced intramolecular charge transfer takes place between the [thiophene/electron-deficient TeBr2 (IV)] units in an alternating manner. The same bromine-addition absorption experiments were carried out for P(SSe)b(PSTe) whose concentration is based on the molecular weight of SSeSTe (908 m/z) on the condition that the SSe and STe blocks have the same length. P(SSe)b(PSTe) exhibited similar optical behaviors with conversion of the original band at λmax of 488 nm to a new band at λmax of 769 nm when reaching saturation at 1.8 eq. of bromine addition (Fig. 11b). Although only the PSTe block can react with bromine, the overall λmax red-shift of P(SSe)b(PSTe) is 281 nm which is even larger than that of PSTe. Furthermore, unlike PSTe, the original band of P(SSe)b(PSTe) does not completely vanish upon addition of bromine, which should be ascribed to the absorption of the non-Te PSSe block. These results demonstrate that the block and alternating arrangement of P(SSe)b(PSTe) with the coordination nature of Te enable us to precisely modulate the optical absorption from the UV-Vis to near-infrared region. It should be noted that after bromine addition, the solubility of the P(SSe)b(STeBr2) decreases, which implies stronger intermolecular interactions ascribed to electrostatic interactions or dipole–dipole interactions induced by the more electron-deficient TeBr2 (IV) species.
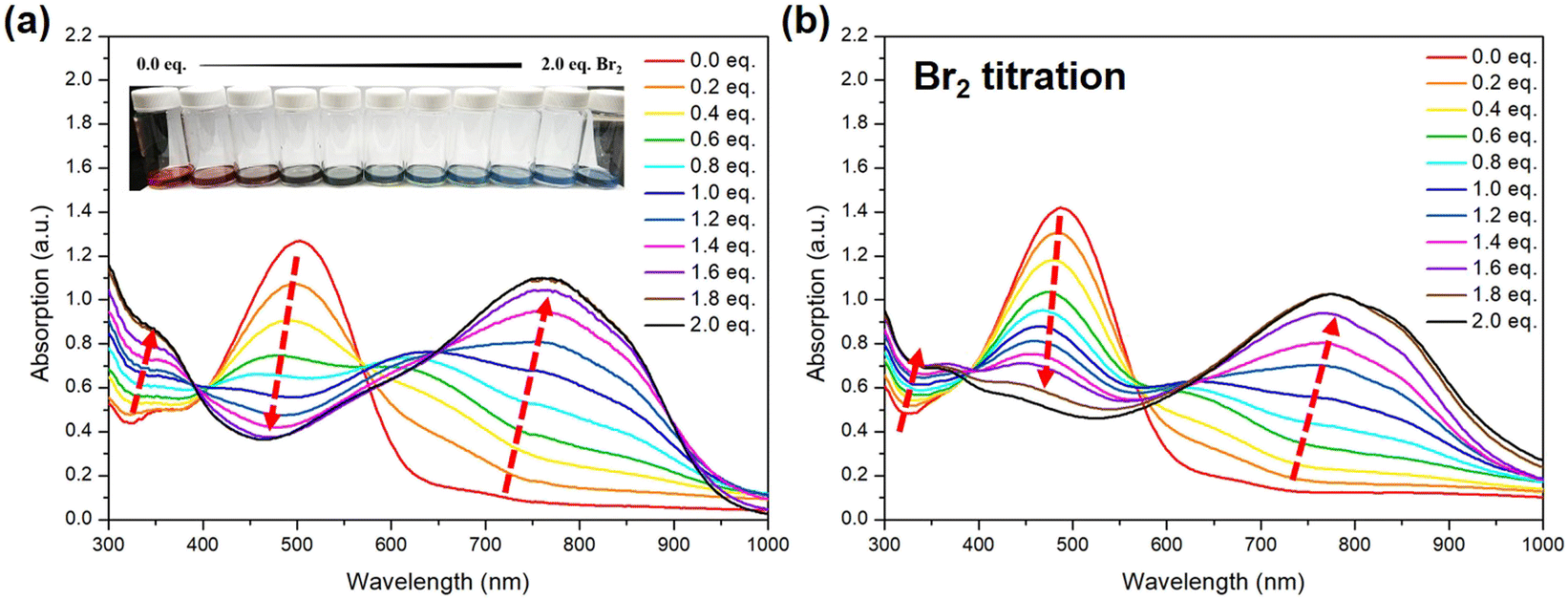 | ||
| Fig. 11 Absorption spectra of (a) PSTe and (b) P(SSe)b(PSTe) in o-DCB upon bromine titration. | ||
OFET devices as bromine sensors
After understanding the Br2 coordination ability of the tellurophene-containing polychalcogenophenes, it is envisaged that OFET devices using these organic semiconducting materials can be utilized for detecting bromine gas. We first fabricated the bottom-gate-top-contact OFET devices made of PSTe thin films that exhibited an obvious p-type transistor behavior in the transfer curves to deliver a hole mobility of 2.7 × 10−3 cm2 V−1 s−1. After the devices were exposed to Br2 vapor with different concentration of 4, 12, 20, 28 and 40 ppm, the source-to-drain current (ISD) is steadily increased when the VGS and VDS are kept at −80 V (Fig. S4†). When the surface of the PSTe thin film is partially oxidized by Br2, the oxidized PSTeBr2 species with electron-deficient character can similarly function as a p-dopant to increase the hole carrier concentration.68–71 As a result, the magnitude of ΔISD/pristine ISD (VDS and VGS of −80 V) is linearly proportional to the Br2 concentration (Fig. 12a). The limit of detection can be extrapolated to be as low as 2.8 ppm. The devices were also exposed to the vapor of common organic solvents including acetone, chloroform, THF, methanol, ethanol, and dichloromethane. None of the devices showed the enhancement of ISD, demonstrating that the devices are only selectively responsive to Br2 vapor (Fig. S5†). Interestingly, thermal annealing of the brominated PSTe thin film at 150 °C for 10 min led to the elimination of the bromine. TGA measurement of the brominated PSTe showed a first weight loss of 27% at a temperature of 150 °C, which is consistent with the molecular weight of Br2 (Fig. S6†). The second weight loss takes place at ca. 380 °C which is similar to the Td of pristine PSTe. Furthermore, the absorption spectrum of the PSTeBr2 thin film completely shifts back to its original profile of PSTe after thermal annealing, confirming the thermally induced reduction of the TeBr2 (IV) species (Fig. S7†). The reversibility between Br2 addition and elimination endows the Br2 sensor devices with reusability. After thermal annealing at 150 °C for 10 min, the oxidized polymer in the OFET device is reduced back to the neutral pristine state so that the increased ISD returns back to the original level. As shown in Fig. 12b, the bromination/heating cycle can be applied to the OFET devices repetitively. In a similar manner, the OFET devices using P(SSe)b(STe), P(SSe)b(SeTe), and P(STe)b(SeTe) block copolymers can also be used for Br2 detection (Fig. S8†).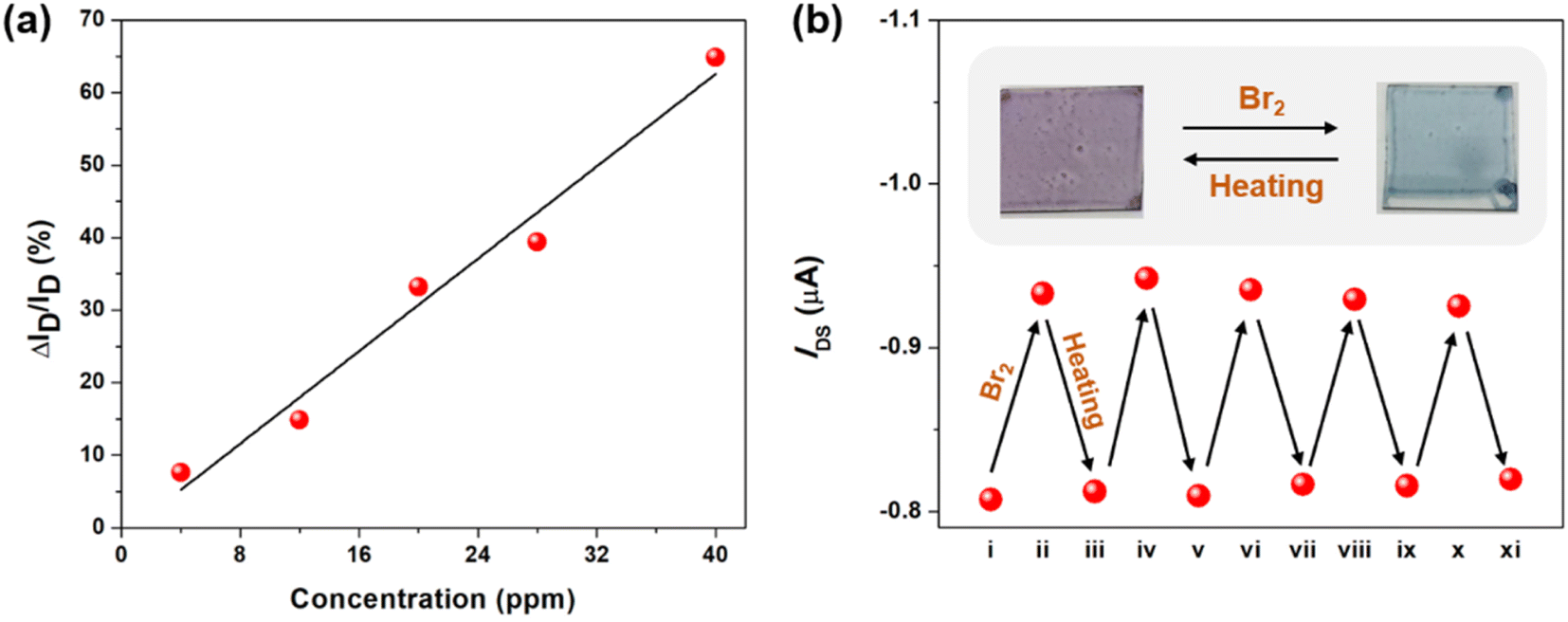 | ||
| Fig. 12 (a) ΔISD/ISD of the PSTe-based OFET device as a function of Br2 vapor concentration. (b) IDS variation of the PSTe-OFET device with VGS and VDS at −80 V during five cycles of Br2 exposure followed by thermal annealing at 150 °C for 10 min. | ||
Conclusions
For the first time, we developed a new class of sequence-controlled conjugated polymers denoted as poly(alt-AB)x-b-(alt-AC)y in which 3-hexylthiophene, 3-hexylselenophene and 3-hexyltellurophene units are used as A, B and C units to assemble three alternating block polychalcogenophenes P(SSe)b(STe), P(SSe)b(SeTe) and P(STe)b(SeTe). The block copolymers were prepared by sequential addition of two asymmetrical bichalcogenophene Grignard monomers under the Ni(dppp)Cl2-catalyzed Kumada polymerization. The molecular weight, dispersity, length of two blocks (x = y), side chain regioregularity and main-chain sequence can be synthetically controlled via the catalyst transfer polycondensation mechanism. The polymer structures are unambiguously confirmed by 1H-NMR and 13C-NMR spectra. The optical and electrochemical parameters of the polychalcogenophenes can be precisely fine-tuned by the composition and ratio of the different chalcogenophenes. GIWAXS analysis showed that all the polymers exhibited predominantly edge-on orientations. P(SSe)b(STe) exhibited a hole OFET mobility of 1.4 × 10−2 cm2 V−1 s−1, which represents one of the highest values among the tellurophene-containing polychalcogenophenes. The tellurophene units in PSTe and P(SSe)b(PSTe) can undergo Br2 addition to form the oxidized TeBr2 species which results in dramatically red-shifted absorption (>280 nm) as a result of the alternating arrangement to induce strong charge transfer character. The OFET devices using these tellurophene-containing polychalcogenophenes as semiconductors can be used for Br2 detection. The oxidized TeBr2 species on the surface of the OFET device can serve as a p-type dopant to increase the source–drain hole current, showing high sensitivity and selectivity to Br2. More importantly, the oxidized TeBr2 species in the Br2 sensors can be thermally reduced back to the neutral state at 150 °C, demonstrating excellent reusability.Data availability
The details for general measurement and characterization, fabrication of devices, measurements, thermal properties, synthetic procedures, and 1H and 13C NMR spectra are included in the ESI.†Author contributions
The project was conceived and conceptualized by Y.-J. Cheng. K.-H. Huang and H.-H. Liu synthesized the polymers and measured all the optical and electrochemical properties and GIWAXS data. K.-Y. Cheng and C.-L. Tsai fabricated the OFETs and analyzed the transistor characterization and bromine sensor experiments. The manuscript was written by K.-H. Huang and Y.-J. Cheng.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science and Technology Council, Taiwan (grant no. 110-2628-M-A49-001-MY3 and 111-2221-E-A49-002) and the Center for Emergent Functional Matter Science of National Yang Ming Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. We thank the National Synchrotron Radiation Research Center (NSRRC) in Taiwan, and Prof. U-Ser Jeng and Dr Chun-Jen Su at the BL23A1 station.References
- T.-Y. Luh and Y.-J. Cheng, J. Chin. Chem. Soc., 2022, 69, 1223–1232 CrossRef CAS.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- J. Rodriguez, H. H. Dhanjee, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 11706–11712 CrossRef CAS PubMed.
- C. Xu, C. He, N. Li, S. Yang, Y. Du, K. Matyjaszewski and X. Pan, Nat. Commun., 2021, 12, 5853 CrossRef CAS PubMed.
- C. Xu, J. Dong, C. He, J. Yun and X. Pan, Giant, 2023, 14, 100154 CrossRef CAS.
- S. Cheng, R. Zhao and D. S. Seferos, Acc. Chem. Res., 2021, 54, 4203–4214 CrossRef CAS.
- M. A. Baker, C.-H. Tsai and K. J. T. Noonan, Chem.–Eur. J., 2018, 24, 13078–13088 CrossRef CAS.
- J.-W. Lee, C. Sun, S.-W. Lee, G.-U. Kim, S. Li, C. Wang, T.-S. Kim, Y.-H. Kim and B. J. Kim, Energy Environ. Sci., 2022, 15, 4672–4685 RSC.
- S. Seo, J.-W. Lee, D. J. Kim, D. Lee, T. N. Phan, J. Park, Z. Tan, S. Cho, T.-S. Kim and B. J. Kim, Adv. Mater., 2023, 35, e2300230 CrossRef PubMed.
- S. Jung, Y. Cho, Y. Ji, J. Oh, G. Park, W. Kim, S. Jeong, S. M. Lee, S. Chen, Y. Zhang and C. Yang, Nano Energy, 2023, 106, 108059 CrossRef CAS.
- D. Gao, J. Hollinger, A. A. Jahnke and D. S. Seferos, J. Mater. Chem. A, 2014, 2, 6058–6063 RSC.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2020, 6, 7–26 CrossRef.
- S. Chen, L. Li, D. Zhai, Y. Yin, X. Shang, B. Ni and J. Peng, ACS Appl. Mater. Interfaces, 2020, 12, 58094–58104 CrossRef CAS.
- M. Brinkmann and J. C. Wittmann, Adv. Mater., 2006, 18, 860–863 CrossRef CAS.
- D.-H. Kim, J.-T. Han, Y.-D. Park, Y. Jang, J.-H. Cho, M. Hwang and K. Cho, Adv. Mater., 2006, 18, 719–723 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- S. Ye, V. Lotocki, H. Xu and D. S. Seferos, Chem. Soc. Rev., 2022, 51, 6442–6474 RSC.
- S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
- J. Hollinger, D. Gao and D. S. Seferos, Isr. J. Chem., 2014, 54, 440–453 CrossRef CAS.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS.
- C.-E. Tsai, R.-H. Yu, F.-J. Lin, Y.-Y. Lai, J.-Y. Hsu, S.-W. Cheng, C.-S. Hsu and Y.-J. Cheng, Chem. Mater., 2016, 28, 5121–5130 CrossRef CAS.
- F.-Y. Cao, C.-C. Tseng, F.-Y. Lin, Y. Chen, H. Yan and Y.-J. Cheng, Chem. Mater., 2017, 29, 10045–10052 CrossRef CAS.
- F.-Y. Cao, F.-Y. Lin, C.-C. Tseng, K.-E. Hung, J.-Y. Hsu, Y.-C. Su and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 11674–11683 CrossRef CAS PubMed.
- S.-Y. Chen, Y.-C. Pao, S.-K. Sahoo, W.-C. Huang, Y.-Y. Lai and Y.-J. Cheng, Chem. Commun., 2018, 54, 1517–1520 RSC.
- H.-H. Chang, C.-E. Tsai, Y.-Y. Lai, W.-W. Liang, S.-L. Hsu, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2013, 46, 7715–7726 CrossRef CAS.
- S.-Y. Chen, S.-K. Sahoo, C.-L. Huang, T.-H. Chan and Y.-J. Cheng, Org. Lett., 2020, 22, 2318–2322 CrossRef CAS.
- E. Rivard, Chem. Lett., 2015, 44, 730–736 CrossRef CAS.
- E. I. Carrera and D. S. Seferos, Macromolecules, 2014, 48, 297–308 CrossRef.
- A. M. Ballantyne, L. Chen, J. Nelson, D. D. C. Bradley, Y. Astuti, A. Maurano, C. G. Shuttle, J. R. Durrant, M. Heeney, W. Duffy and I. McCulloch, Adv. Mater., 2007, 19, 4544–4547 CrossRef CAS.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. Buchaca Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC.
- S. Ye, M. Steube, E. I. Carrera and D. S. Seferos, Macromolecules, 2016, 49, 1704–1711 CrossRef CAS.
- X. Li, P. J. Wolanin, L. R. MacFarlane, R. L. Harniman, J. Qian, O. E. C. Gould, T. G. Dane, J. Rudin, M. J. Cryan, T. Schmaltz, H. Frauenrath, M. A. Winnik, C. F. J. Faul and I. Manners, Nat. Commun., 2017, 8, 15909 CrossRef CAS PubMed.
- J. Peng and Y. Han, Giant, 2020, 4, 100039 CrossRef.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 CrossRef PubMed.
- X. Lin, S. Ye, C. Kong, K. Webb, C. Yi, S. Zhang, Q. Zhang, J. T. Fourkas and Z. Nie, J. Am. Chem. Soc., 2020, 142, 17282–17286 CrossRef CAS PubMed.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- J. Hollinger, P. M. DiCarmine, D. Karl and D. S. Seferos, Macromolecules, 2012, 45, 3772–3778 CrossRef CAS.
- J. A. Amonoo, A. Li, G. E. Purdum, M. E. Sykes, B. Huang, E. F. Palermo, A. J. McNeil, M. Shtein, Y.-L. Loo and P. F. Green, J. Mater. Chem. A, 2015, 3, 20174–20184 RSC.
- E. L. Kynaston, Y. Fang, J. G. Manion, N. K. Obhi, J. Y. Howe, D. F. Perepichka and D. S. Seferos, Angew. Chem., Int. Ed., 2017, 56, 6152–6156 CrossRef CAS PubMed.
- E. L. Kynaston, K. J. Winchell, P. Y. Yee, J. G. Manion, A. D. Hendsbee, Y. Li, S. Huettner, S. H. Tolbert and D. S. Seferos, ACS Appl. Mater. Interfaces, 2019, 11, 7174–7183 CrossRef CAS PubMed.
- M. Zhu, S. Pan, Y. Wang, P. Tang, F. Qiu, Z. Lin and J. Peng, Angew. Chem., Int. Ed., 2018, 57, 8644–8648 CrossRef CAS PubMed.
- G. E. J. Hicks, C. N. Jarrett-Wilkins, J. R. Panchuk, J. G. Manion and D. S. Seferos, Chem. Sci., 2020, 11, 6383–6392 RSC.
- Y.-Y. Lai, T.-C. Tung, W.-W. Liang and Y.-J. Cheng, Macromolecules, 2015, 48, 2978–2988 CrossRef CAS.
- C.-H. Tsai, A. Fortney, Y. Qiu, R. R. Gil, D. Yaron, T. Kowalewski and K. J. T. Noonan, J. Am. Chem. Soc., 2016, 138, 6798–6804 CrossRef CAS PubMed.
- Y. Qiu, A. Fortney, C.-H. Tsai, M. A. Baker, R. R. Gil, T. Kowalewski and K. J. T. Noonan, ACS Macro Lett., 2016, 5, 332–336 CrossRef CAS PubMed.
- H.-H. Liu, W.-W. Liang, Y.-Y. Lai, Y.-C. Su, H.-R. Yang, K.-Y. Cheng, S.-C. Huang and Y.-J. Cheng, Chem. Sci., 2020, 11, 3836–3844 RSC.
- S. Y. Ku, M. A. Brady, N. D. Treat, J. E. Cochran, M. J. Robb, E. J. Kramer, M. L. Chabinyc and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 16040–16046 CrossRef CAS PubMed.
- M. Sommer, H. Komber, S. Huettner, R. Mulherin, P. Kohn, N. C. Greenham and W. T. S. Huck, Macromolecules, 2012, 45, 4142–4151 CrossRef CAS.
- K. Nakabayashi and H. Mori, Macromolecules, 2012, 45, 9618–9625 CrossRef CAS.
- C. Guo, Y. H. Lin, M. D. Witman, K. A. Smith, C. Wang, A. Hexemer, J. Strzalka, E. D. Gomez and R. Verduzco, Nano Lett., 2013, 13, 2957–2963 CrossRef CAS PubMed.
- H. Kim, J. Lee, T. Kim, M. Cho and T.-L. Choi, Angew. Chem., Int. Ed., 2022, 61, e202205828 CAS.
- J. Rivnay, S. C. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- S. Y. Son, T. Park and W. You, Chem. Mater., 2021, 33, 4541–4550 CrossRef CAS.
- L. Li, J. Hollinger, A. A. Jahnke, S. Petrov and D. S. Seferos, Chem. Sci., 2011, 2, 2306–2310 RSC.
- Y. Wang, H. Cui, M. Zhu, F. Qiu, J. Peng and Z. Lin, Macromolecules, 2017, 50, 9674–9682 CrossRef CAS.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 3542–3548 CrossRef CAS.
- M. Kaur, D.-S. Yang, K. Choi, M.-J. Cho and D.-H. Choi, Dyes Pigm., 2014, 100, 118–126 CrossRef CAS.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed.
- K. Wen, L. Wu, X. Wu, Y. Lu, T. Duan, H. Ma, A. Peng, Q. Shi and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 12756–12761 CrossRef CAS PubMed.
- A. A. Jahnke, G. W. Howe and D. S. Seferos, Angew. Chem., Int. Ed., 2010, 49, 10140–10144 CrossRef CAS PubMed.
- M. Kaur, D.-S. Yang, J. Shin, T.-W. Lee, K. Choi, M.-J. Cho and D.-H. Choi, Chem. Commun., 2013, 49, 5495–5497 RSC.
- M. Kaur, D.-H. Lee, D.-S. Yang, H.-A. Um, M.-J. Cho, J.-S. Kang and D.-H. Choi, Chem. Commun., 2014, 50, 14394–14396 RSC.
- M. Kaur, D.-H. Lee, D.-S. Yang, H.-A. Um, M.-J. Cho, J.-S. Kang and D.-H. Choi, Dyes Pigm., 2015, 123, 317–322 CrossRef CAS.
- E. I. Carrera, A. E. Lanterna, A. J. Lough, J. C. Scaiano and D. S. Seferos, J. Am. Chem. Soc., 2016, 138, 2678–2689 CrossRef CAS PubMed.
- L. Lv, X. Wang, X. Wang, L. Yang, T. Dong, Z. Yang and H. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 34620–34629 CrossRef CAS PubMed.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T. Q. Nguyen, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- E. H. Suh, J. G. Oh, J. Jung, S. H. Noh, T. S. Lee and J. Jang, Adv. Energy Mater., 2020, 10, 2002521 CrossRef CAS.
- J. Li, J. Liu and C. Gao, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 125–128 CrossRef CAS.
- J. H. Schön, C. Kloc, E. Bucher and B. Batlogg, Nature, 2000, 403, 408–410 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02289g |
| This journal is © The Royal Society of Chemistry 2023 |