
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemically assisted morphing of shape shifting polymers†
Rui
Tang
a,
Wenli
Gao
 a,
Yulin
Jia
a,
Kai
Wang
a,
Barun Kumar
Datta
a,
Wei
Zheng
b,
Huan
Zhang
a,
Yuanze
Xu
a,
Yangju
Lin
*c and
Wengui
Weng
*a
a,
Yulin
Jia
a,
Kai
Wang
a,
Barun Kumar
Datta
a,
Wei
Zheng
b,
Huan
Zhang
a,
Yuanze
Xu
a,
Yangju
Lin
*c and
Wengui
Weng
*a
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, 422 South Siming Road, Xiamen, Fujian 361005, P. R. China. E-mail: wgweng@xmu.edu.cn
bCollege of Materials Science, Xiamen University, 422 South Siming Road, Xiamen, Fujian 361005, P. R. China
cDepartment of Chemical Engineering, Stanford University, 443 Via Ortega, Stanford, California 94305, USA. E-mail: yl2020@stanford.edu
First published on 10th August 2023
Abstract
Morphing in creatures has inspired various synthetic polymer materials that are capable of shape shifting. The morphing of polymers generally relies on stimuli-active (typically heat and light active) units that fix the shape after a mechanical load-based shape programming. Herein, we report a strategy that uses a mechanochemically active 2,2′-bis(2-phenylindan-1,3-dione) (BPID) mechanophore as a switching unit for mechanochemical morphing. The mechanical load on the polymer triggers the dissociation of the BPID moiety into stable 2-phenylindan-1,3-dione (PID) radicals, whose subsequent spontaneous dimerization regenerates BPID and fixes the temporary shapes that can be effectively recovered to the permanent shapes by heating. A greater extent of BPID activation, through a higher BPID content or mechanical load, leads to higher mechanochemical shape fixity. By contrast, a relatively mechanochemically less active hexaarylbiimidazole (HABI) mechanophore shows a lower fixing efficiency when subjected to the same programing conditions. Another control system without a mechanophore shows a low fixing efficiency comparable to the HABI system. Additionally, the introduction of the BPID moiety also manifests remarkable mechanochromic behavior during the shape programing process, offering a visualizable indicator for the pre-evaluation of morphing efficiency. Unlike conventional mechanical mechanisms that simultaneously induce morphing, such as strain-induced plastic deformation or crystallization, our mechanochemical method allows for shape programming after the mechanical treatment. Our concept has potential for the design of mechanochemically programmable and mechanoresponsive shape shifting polymers.
Introduction
The capabilities of morphing (or shape shifting) have enabled creatures to perform various challenging tasks. Cephalopods, for instance, can adapt their body to navigate through narrow spaces or to seamlessly blend with their surroundings.1 The Venus flytrap utilizes a closable leaf structure to ensnare prey.2 Pufferfish can inflate their body to more than twice their normal size when threatened.3 Similarly, the shameplant can rapidly fold its leaves inward to avoid potential harm.4 These smart morphing behaviors have inspired a variety of synthetic shape shifting polymer materials, many of which have been applied in soft actuators,5,6 soft robotics,7–9 adaptive materials,10–12 and biomedical devices.13,14Morphing of synthetic polymer materials is performed in either a reversible or irreversible manner. In an irreversible morphing process, polymer materials are often subjected to plastic deformation and so lose the elastic energy to return to their original shape.15,16 By contrast, polymers with reversible morphing behavior (e.g., shape memory polymers) carry the memory of initial/permanent shape after being morphed into other desired temporary shapes, and an on-demand external stimulus is often required to trigger the shape recovery.17 In a more sophisticated design that utilizes two or more distinct molecular mechanisms for temporary morphing (e.g., crystallinity and dynamic covalent/noncovalent bonds),12,18–20 polymers exhibit multi-stage shape recovering behavior.21,22
The morphing of polymer materials often requires prior stimulus activation of the switching units followed by subsequent mechanical loading to create deformation or temporary shapes. Typical documented switching units include reversible crystallization,23 glass transition,24 liquid crystal anisotropic/isotropic transition,25 supramolecular association,12 dynamic covalent bonds,26 and so forth. Considering that most potential applications of shape shifting polymers are as structural materials, direct application of mechanical force is an interesting trigger mechanism. However, examples have only been achieved through mechanically created plastic deformation15 and strain-induced crystallization,27 and the morphing occurs simultaneously upon applying the mechanical load. It would be desirable to have a mechanical mechanism that enables subsequent customized shape programming.
In contrast to conventional stimuli, mechanical force is clean, directional and can be harnessed at the molecular level to facilitate otherwise prohibitive or inaccessible chemical reactions with high selectivity and fewer side reactions.28–30 In the last decade, mechanical force has been widely used as a productive tool in the study of polymer mechanochemistry and mechanoresponsive materials,31–34 in which the mechanically responsive moieties are termed mechanophores (MPs). Among the many MPs developed so far,35 reversible scissile ones (covalent or non-covalent) are able to dissociate upon mechanical loading and further recombine under appropriate conditions after relaxation.36–41 We therefore envisaged that the breaking–reformation capability of reversible scissile MPs can be exploited as a switching mechanism for the morphing of polymers. As illustrated in Fig. 1, the polymer network is first subjected to mechanical force to dissociate embedded scissile MPs, and the subsequent morphing can be fixed by the spontaneous recombination of the mechanochemically activated MPs at new sites, under either strained or strain-free states. Upon application of another stimulus, the reorganization of activated MPs and entropic gain recover the initial shape.
 | ||
| Fig. 1 Illustrated mechanochemical activation of a reversible scissile mechanophore for morphing followed by stimulus triggered shape recovery. | ||
Herein, we report the first example of mechanochemically assisted morphing of shape shifting polymers with a dual crosslinking structure, in which a thermally and mechanochemically sensitive 2,2′-bis(2-phenylindan-1,3-dione) MP (BPID, Scheme 1a) together with a conventional covalent bond are used as dynamic and permanent cross-links, respectively.
 | ||
| Scheme 1 (a) Activation of reversible BPID into chromic radical species through heat or mechanical force; (b) the HABI mechanophore undergoes reversible cleavage upon light or force treatment; (c) synthetic route of polyurethane networks used in this study. The total crosslinking density is kept constant and the ratio of different crosslinkers is varied. | ||
Results and discussion
We first synthesized 2,2′-bis(2-phenylindan-1,3-dione)tetraol (BPID-TO) (Scheme S1 and Fig. S1–S9†), the hydroxyl functionalized BIPD, to be used as the dynamic cross-linker. Our previous studies on the mechanochromic behavior of BIPD,42–44 along with the reversible thermochromism of a BPID-TO solution in DMF (Fig. S10†) as well as the mechanochromism of the BPID-TO powder (Fig. S11†) validated BPID as an effective dynamic covalent moiety that is both thermally and mechanochemically active. Further analysis from electron spin resonance (ESR) results (Fig. S12 and S13†) indicated free radical species of 2-phenylindan-1,3-dione (PID), which can recombine rapidly upon removal of external stimuli. The use of BPID as a dynamic motif for thermally activated shape memory polymers has been reported,42 while we addressed its application for an alternative mechanism that is triggered mechanically.Two polyurethane (PU) shape shifting polymers consisting of dynamic BPID and conventional triethanolamine (TEOA) crosslinkers were prepared (PUB1 and PUB2, Scheme 1c), and the BPID/TEOA molar ratio was varied while the overall crosslinking density was kept constant (Table S1†). For comparison, we also synthesized two control polyurethanes: one without BPID MP (PUc); the other one with a dynamic hexaarylbiimidazole (HABI, with its activated form TPIR, Schemes 1b and S2, and Fig. S14–S20†), which has been experimentally and theoretically scrutinized as a relatively less active mechanophore,35,41 in place of BPID (PUH). The preserved dynamic feature of BIPD in PUB1 and PUB2 was evidenced by the reversible thermochromism and ESR study at different temperatures (Fig. S21 and S22†). On the other hand, the two controls (PUc and PUH) showed no thermochromism behavior (Fig. S21 and S23†), and the sluggish thermal activity in PUH can be attributed to the high energy barrier of HABI (>30 kcal mol−1).45,46 Alternatively, HABI has been well documented as a photochromic compound (Fig. S29†),47 which makes it possible for PUH to act as a photoresponsive but mechanochemically and thermally inert control.
Uniaxial tensile tests of all four elastomers showed typical sigmoid shaped stress–strain curves with strain hardening occurring at around 300–350% strain (Fig. 2a). A color change from pale yellow to green in PUB1 and PUB2 occurred beyond a critical strain, suggesting the mechanochemical activation of BPID into PID radicals (Fig. 2b and S24†). Further stretching stimulated intensification of green color, indicating a greater extent of BPID activation. We then applied an RGB imaging analysis method48,49 to quantitatively evaluate the activation of BPID in the strained samples. As shown in Fig. 2c, the G/R ratio at each 10%-strain interval was recorded for all samples during the tensile tests, and the relative change, i.e., (G/R) − (G/R)0, was calculated. A growth of (G/R) − (G/R)0 over strain was observed in PUB1 and PUB2, while there were no observable changes in PUc and PUH. Further, the onset strains of BIPD activation were estimated to be ∼350% for PUB1 and ∼250% for PUB2. Despite the different activation strains, the corresponding activation stress appeared to be similar (2.4 MPa vs. 2.6 MPa) and correlated to the strain-hardening regions. We reasoned that the activation of BPID requires enthalpic stretching of polymer chains and so exhibits a critical activation onset, whereas, in some supramolecular mechanophore systems,50,51 the activation can occur during the entropic stretching of polymer chains and is more correlated with strain change. At 500% strain, the activation of BPID in PUB2 is ∼6-fold that in PUB1.
 | ||
| Fig. 2 (a) Stress–strain curves of PU polymer films in this study. (b) Image recording of polymer films at various strains. The black dots are marks for strain calculation. (c) RGB imaging analysis of polymer films at different strains, and the data were retrieved at a strain interval of 10%. The increase in G/R value, i.e., (G/R) − (G/R)0, was applied to remove the initial background and to evaluate the color change. | ||
We verified the activation of BPID into PID radical species in the PUB1 films through in situ ESR experiments. In alignment with the RGB analysis, a rise in the intensity of ESR signals indicated more BPID activation over strain (Fig. S24†). Upon the release of strain/stress, a rapid discoloration happened within 3 min, as revealed by the rapid decay of both G/R and B/R ratios (Fig. S25†). Moreover, when the film was maintained at 500% strain at room temperature, color fading also took place in hours (Fig. S26†), suggesting the relaxation of chains bearing PID moieties and recombination of PID free radicals. Therefore, the mechanochemically generated free radicals can successfully reorganize and recombine under both strain-free and strained states.
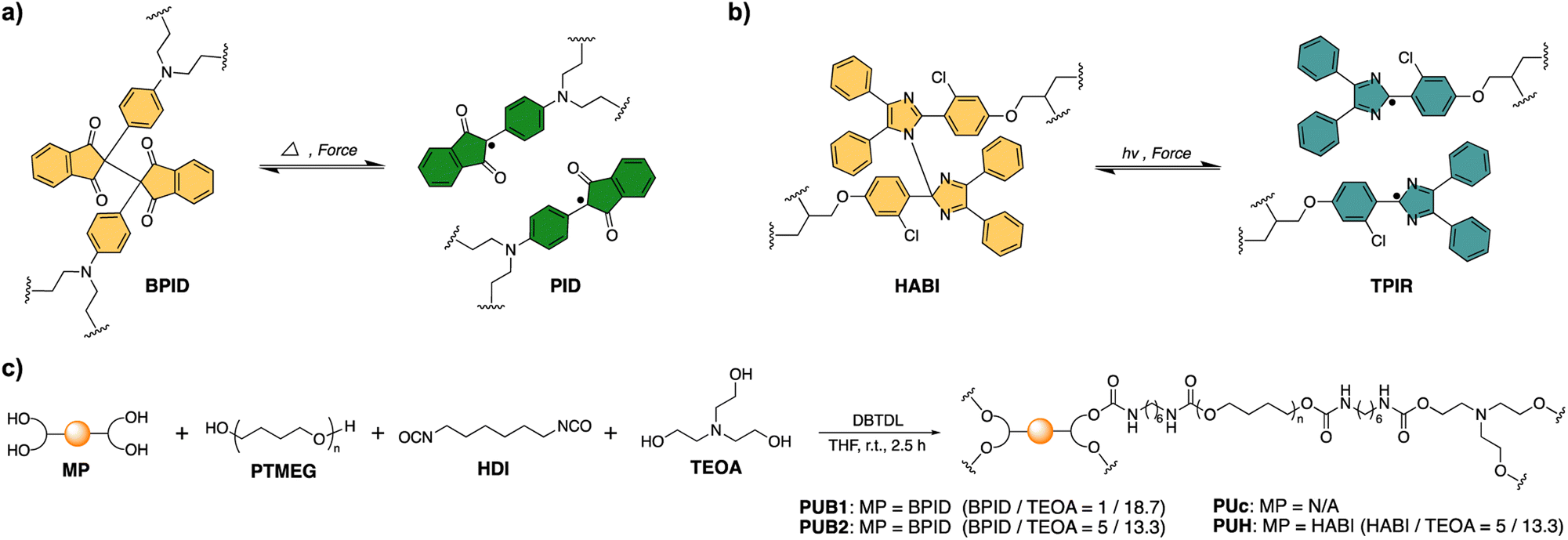
The remarkable dynamic behaviors of BPID moieties allow for mechanochemical morphing and thermal recovery of the shape shifting polymers. As illustrated in Fig. 3a, polymer films were manually stretched to 500% to activate the BPID moieties followed by strain release. Subsequently, the films were wrapped around a glass rod, after which the fixed sample was set at 25 °C for various times to assess the morphing efficiency. A semi-quantitative but visualizable parameter, i.e., the curvature of the resulting films, was applied to evaluate the efficiency. As shown in Fig. 3b, the curvature of both PUB1 and PUB2 films reached a maximum after 6 h of fixing time, suggesting the completion of morphing. As expected, PUB2 showed a higher curvature than PUB1 owing to the greater amount of activated BPID that acts as a fixing unit in the reshaped film. Notably, the polymer before and after strain release exhibited nearly the same WXRD pattern (Fig. S27†), indicating that no residual crystallinity is induced by the strain process.
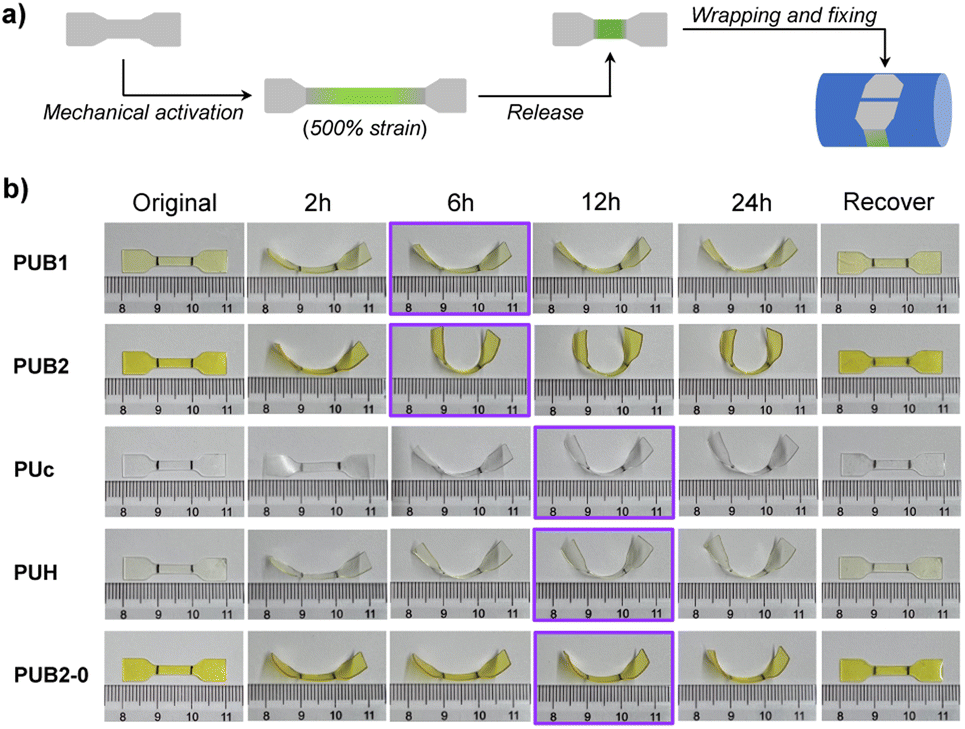 | ||
| Fig. 3 (a) Schematic illustration of the morphing of polymer film through mechanochemical activation of dynamic covalent bonds. (b) Images showing the morphing efficiency of mechanically treated polymer films after various fixing times, and PUB2-0 is a reference sample without mechanical treatment. The purple boxes highlight the time when fixing reaches a steady state. The shape recovery was performed by heating at 80 °C for 8 min. The unit of the displayed ruler is cm. | ||
The paramount role of mechanochemically activated BPID was further validated by inspecting the morphing efficiency of three control samples: (1) a PUc film which does not contain the BIPD mechanophore, (2) a PUH film containing a relatively mechanically inert HABI mechanophore, and (3) a PUB2 film without pre-straining treatment (denoted as PUB2-0). Interestingly, PUc and PUH exhibited a similar morphing efficiency to PUB1, but lower than PUB2, and the time to reach maximum fixity is longer (12 h vs. 6 h). The presence of a certain morphing efficiency in PUc could be attributed to the reorganization of hydrogen bonding in the films (as will be discussed later), and the longer time in morphing might be a consequence of the more dynamic feature in hydrogen bonding, which fixes polymer chains less efficiently. As such, the morphing depends more on the slow network relaxation. A similar result was also observed in PUH film, in which there is no observable mechanochemically activated TPIR (Fig. 2c and S28†). It should be mentioned that PUB1 presented a shorter morphing time despite the similar curvature to PUc and PUH, suggesting the fast kinetics of PID combination that assists morphing, but the abundance of PID is too low to improve the fixity. Lastly, the contribution of equilibrium between BPID dissociation and combination to shape fixing at 25 °C was also ruled out based on the similar fixity of PUB2-0 to PUc. At a high temperature of 90 °C, PUB2-0 showed excellent shape fixity and shape recovery (Fig. S29†). Collectively, the mechanochemical activation of BPID led to the improvement of morphing efficiency, both in time and fixity.
The shape memory effect of morphed polymer films was verified by applying heat. All samples recovered their original shape without any observable hysteresis (Fig. 3b). Alternatively, shape recovery achieved by applying mechanical force was scrutinized. The morphed PUB1 and PUB2 samples were stretched to 500% strain to, potentially, activate the recombined BPID unit in the morphing procedure. However, the films uncurved marginally (Fig. S30 and S31†). Two potential scenarios account for the observation: (1) the recombined BPID in the morphing process is not activated in the stretching; (2) the stretching does reactivate BPID, but the PID recombines or combines with nearby PID species due to the absence of fixing force.
Hydrogen bonding has been previously explored as a dynamic motif for shape memory polymers.12,52 Similarly, the certain shape fixity exhibited in PUc and PUH could be attributed to the carbamate hydrogen bonding (Fig. 3b). To differentiate the contribution of hydrogen bonding to the fixity in PUB2, we studied the temperature-dependent shape recovery of PUc and PUB2 at both 60 °C and 70 °C. Because the activation energy of BPID is much higher than that of the carbamate hydrogen bond (∼20 kcal mol−1 (ref. 53) vs. 5.4 kcal mol−1 (ref. 54)), the rate of BPID dissociation is more dependent on temperature and, therefore, the recovery rate of PUB2 would be more different at these two temperatures than that of PUc. Consistent with our expectation, PUc recovered quickly within a similar time of ∼16 s at both temperatures (Fig. S32 and S33†). By contrast, the recovery time was >180 s at 60 °C and ∼40 s at 70 °C for the PUB2 sample (Fig. S32 and S33†). This temperature-dependency highly suggested the significant role of activated PID in the shape programming of PUB2 samples. By comparing the shape fixity of PUB2 and PUc in Fig. 3b, we can qualitatively estimate the relative contribution of BPID and hydrogen bonding, through the bending angle of the strip end (∼90° vs. ∼45° against the horizontal direction).
Taken together, mechanochemically assisted morphing has been demonstrated in shape shifting polymers containing BPID mechanophores as the dynamic crosslink, and the subsequent shape recovery can be achieved by heat treatment.
BPID and HABI showed distinct stimuli-responsive properties in our film matrices, where BPID is mechanically and thermally active while HABI is only photo-responsive (Fig. S34–S36†). We therefore sought to use mechanical force and light as orthogonal stimuli for multiple-state morphing. A laminated film sample consisting of PUB2 and PUH layers was applied to demonstrate this concept (Fig. 4a). As shown in Fig. 4b, when stretched up to a strain of 500%, only the bottom PUB2 layer showed mechanochromism. The sample was then firstly kept at 50% strain (at which there is no observable activation of BPID, Fig. 2c and 4b), and the PUH layer was irradiated with 405 nm light for 2 min to activate HABI into TPIR. Following setting at 50% strain for an additional 2 min allows for HABI reformation, yielding morphing state S1 that bends toward the PUB2 side. The second step includes the aforementioned mechanochemical morphing procedure with the PUH side wrapped around the rod. Consequently, a new state S2 that bends toward the PUH side was obtained. Upon thermal heating at 80 °C for 8 min, the selective thermal activation of BPID enables recovery to the S1* state. Further activation of HABI by 405 nm light at 80 °C (high temperature was applied simultaneously to ensure the fast chain relaxation) recovered the sample to its original state in 30 min.
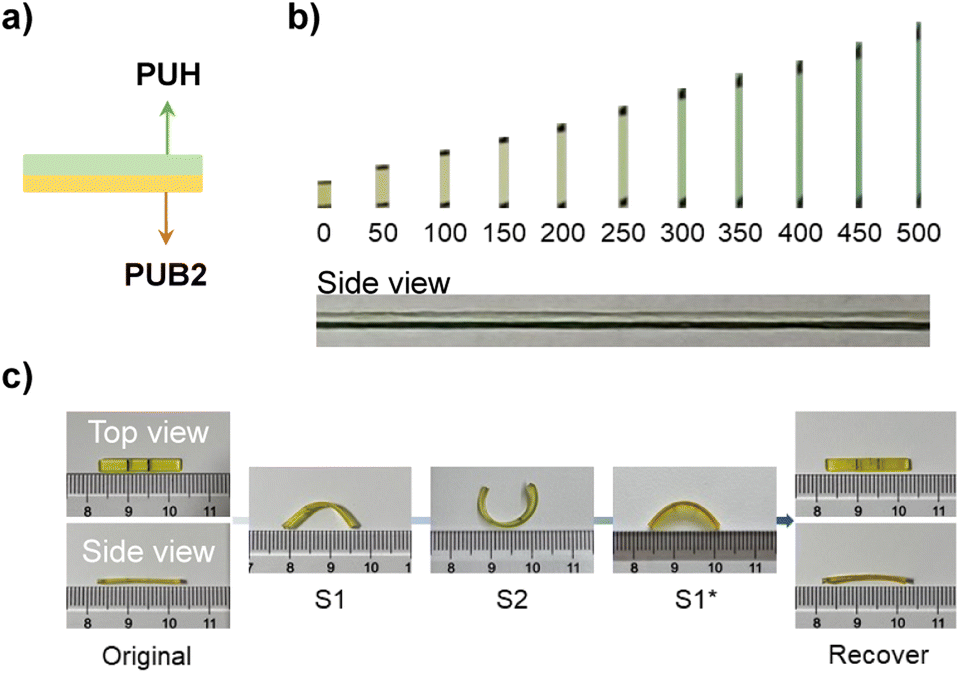 | ||
| Fig. 4 (a) Illustrated side view of a laminated film containing PUB2 and PUH polymers. (b) Imaging record of mechanochemical activation in the laminated film at various strains. The side view image shows activation in the PUB2 layer (bottom) at 500% strain. (c) Demonstration of orthogonal stimuli triggered multiple-state shape memory behavior using the laminated polymer film shown in (a). S1 was obtained by fixing PUH using photo-activated morphing, and S2 was obtained by fixing PUB2 through mechanochemical morphing. The shape recovery state S1* was achieved by heating at 80 °C for 8 min, and the final recovery state was obtained through photo-activation coupled with heating (80 °C). | ||
Conclusions
In summary, we have demonstrated the concept of mechanochemically assisted morphing of shape shifting polymers by taking advantage of mechanochemically stimulated dissociation of BPID moieties into PID radicals and their rapid, spontaneous reassociation ability at room temperature. Complementary to the commonly known mechanisms of mechanically induced plastic deformation or crystallization, the mechanochemically triggered morphing allows for shape programming post the mechanical treatment. The morphing efficiency was found to be dependent42 on the amount of activated BPID, where a higher content of activated BPID led to a better shape fixity. In addition, the mechanochemically activated PID radical species exhibited remarkable chromism, providing a useful and visualizable indicator for the preassessment of morphing efficiency. Furthermore, utilization of a photoresponsive but mechanochemically and thermally inert HABI to fabricate a composite layered structure enabled orthogonal morphing. We envisage that our concept is applicable to other mechanophore, both covalent and non-covalent, cross-linked shape shifting polymers and may open up new opportunities for polymer morphing and design of shape shifting polymers.Author contributions
R. Tang and W. Weng conceived the study. R. Tang, W. Gao, and Y. Jia designed and conducted the experiment. K. Wang, B. K. Datta and W. Zheng helped collect the data. R. Tang, H. Zhang, Y. Xu, Y. Lin and W. Weng analyzed the data. Y. Lin and W. Weng supervised the work, drafted and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 21774106).Notes and references
- J. J. Allen, G. R. Bell, A. M. Kuzirian, S. S. Velankar and R. T. Hanlon, J. Morphol., 2014, 275, 371–390 CrossRef PubMed.
- G. M. Durak, R. Thierer, R. Sachse, M. Bischoff, T. Speck and S. Poppinga, Adv. Sci., 2022, 9, e2201362 CrossRef.
- S. Zhao, J. K. Song and X. J. Wang, Dongwuxue Yanjiu, 2010, 31, 539–549 Search PubMed.
- J. Braam, New Phytol., 2005, 165, 373–389 CrossRef PubMed.
- V. Lutz-Bueno, S. Bolisetty, P. Azzari, S. Handschin and R. Mezzenga, Adv. Mater., 2020, 32, e2004941 CrossRef PubMed.
- C. Yuan, D. J. Roach, C. K. Dunn, Q. Mu, X. Kuang, C. M. Yakacki, T. J. Wang, K. Yu and H. J. Qi, Soft Matter, 2017, 13, 5558–5568 RSC.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, ACS Appl. Mater. Interfaces, 2015, 7, 22431–22435 CrossRef CAS PubMed.
- Y. Li, Y. Teixeira, G. Parlato, J. Grace, F. Wang, B. D. Huey and X. Wang, Soft Matter, 2022, 18, 6857–6867 RSC.
- D. S. Shah, J. P. Powers, L. G. Tilton, S. Kriegman, J. Bongard and R. Kramer-Bottiglio, Nat. Mach. Intell., 2021, 3, 51–59 CrossRef.
- C. Xu, G. T. Stiubianu and A. A. Gorodetsky, Science, 2018, 359, 1495–1500 CrossRef CAS PubMed.
- J. H. Pikul, S. Li, H. Bai, R. T. Hanlon, I. Cohen and R. F. Shepherd, Science, 2017, 358, 210–214 CrossRef CAS PubMed.
- W. Peng, G. Zhang, Q. Zhao and T. Xie, Adv. Mater., 2021, 33, e2102473 CrossRef PubMed.
- A. Kirillova and L. Ionov, J. Mater. Chem. B, 2019, 7, 1597–1624 RSC.
- W. Zhao, L. Liu, F. Zhang, J. Leng and Y. Liu, Mater. Sci. Eng., C, 2019, 97, 864–883 CrossRef CAS PubMed.
- F. Zhang, D. Li, C. Wang, Z. Liu, M. Yang, Z. Cui, J. Yi, M. Wang, Y. Jiang, Z. Lv, S. Wang, H. Gao and X. Chen, Nat. Commun., 2022, 13, 7294 CrossRef CAS PubMed.
- A. Rath, P. M. Geethu, S. Mathesan, D. K. Satapathy and P. Ghosh, Soft Matter, 2018, 14, 1672–1680 RSC.
- J. Zhou and S. S. Sheiko, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1365–1380 CrossRef CAS.
- W. Zou, B. Jin, Y. Wu, H. Song, Y. Luo, F. Huang, J. Qian, Q. Zhao and T. Xie, Sci. Adv., 2020, 6, eaaz2362 CrossRef CAS PubMed.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- Q. Zhao, W. Zou, Y. Luo and T. Xie*, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- G. Zhang, Q. Zhao, L. Yang, W. Zou, X. Xi and T. Xie, ACS Macro Lett., 2016, 5, 805–808 CrossRef CAS PubMed.
- N. Zheng, J. Hou, Y. Xu, Z. Fang, W. Zou, Q. Zhao and T. Xie, ACS Macro Lett., 2017, 6, 326–330 CrossRef CAS PubMed.
- L. K. Jang, M. K. Abdelrahman and T. H. Ware, ACS Appl. Mater. Interfaces, 2021, 14, 22762–22770 CrossRef PubMed.
- D. M. Feldkamp and I. A. Rousseau, Macromol. Mater. Eng., 2010, 295, 726–734 CrossRef CAS.
- H. M. van der Kooij, S. A. Semerdzhiev, J. Buijs, D. J. Broer, D. Liu and J. Sprakel, Nat. Commun., 2019, 10, 3501 CrossRef PubMed.
- G. Zhang, Q. Zhao, L. Yang, W. Zou, X. Xi and T. Xie, ACS Macro Lett., 2016, 5, 805–808 CrossRef CAS PubMed.
- Y. Qiu, D.-R. Munna, F. Wang, J. Xi, Z. Wang and D. Wu, Macromolecules, 2021, 54, 5694–5704 CrossRef CAS.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- C. L. Brown, B. H. Bowser, J. Meisner, T. B. Kouznetsova, S. Seritan, T. J. Martinez and S. L. Craig, J. Am. Chem. Soc., 2021, 143, 3846–3855 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, M. T. Ong, H. M. Klukovich, A. L. Rheingold, T. J. Martinez and S. L. Craig, Nat. Chem., 2015, 7, 323–327 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef PubMed.
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. J. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2021, 6, 84–98 CrossRef CAS.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- I. M. Klein, C. C. Husic, D. P. Kovacs, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS PubMed.
- H. M. Klukovich, Z. S. Kean, S. T. Iacono and S. L. Craig, J. Am. Chem. Soc., 2011, 133, 17882–17888 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- H. Sakai, T. Sumi, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1359–1363 CrossRef CAS PubMed.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem.–Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed.
- F. Verstraeten, R. Gostl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- L. Yang, P. Ouyang, Y. Chen, S. Xiang, Y. Ruan, W. Weng, X. He and H. Xia, Giant, 2021, 8, 100069 CrossRef CAS.
- W. Gao, R. Tang, M. Bai, H. Yu, Y. Ruan, J. Zheng, Y. Chen and W. Weng, Polym. Chem., 2022, 13, 2173–2177 RSC.
- Y. Chen, L. Yang, W. Zheng, P. Ouyang, H. Zhang, Y. Ruan, W. Weng, X. He and H. Xia, ACS Macro Lett., 2020, 9, 344–349 CrossRef CAS PubMed.
- D. M. White and J. Sonnenberg, J. Am. Chem. Soc., 1966, 88, 3825–3829 CrossRef CAS.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1965, 38, 685–686 CrossRef CAS.
- M. Bagheri, M. Mirzaee, S. Hosseini and P. Gholamzadeh, Dyes Pigm., 2022, 203, 110322 CrossRef CAS.
- Y. Lin, M. H. Barbee, C. C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. M. Wang, G. W. Welshofer, X. H. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS PubMed.
- H. Traeger, Y. Sagara, J. A. Berrocal, S. Schrettl and C. Weder, Polym. Chem., 2022, 13, 2860–2869 RSC.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS PubMed.
- J. Li, J. A. Viveros, M. H. Wrue and M. Anthamatten, Adv. Mater., 2007, 19, 2851–2855 CrossRef CAS.
- I. V. Khudyakov, A. I. Yasmenko and V. A. Kuzmin, Int. J. Chem. Kinet., 1979, 11, 621–633 CrossRef CAS.
- Q. Li, H. Zhou, D. A. Wicks, C. E. Hoyle, D. H. Magers and H. R. McAlexander, Macromolecules, 2009, 42, 1824–1833 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02404k |
| This journal is © The Royal Society of Chemistry 2023 |