DOI:
10.1039/D3SC02436A
(Edge Article)
Chem. Sci., 2023,
14, 7944-7955
Ammonolytic transfer dehydrogenation of amines and amides: a versatile method to valorize nitrogen compounds to nitriles†
Received
12th May 2023
, Accepted 3rd July 2023
First published on 4th July 2023
Abstract
The dehydrogenation of amines has been identified as an efficient method for nitrile synthesis. At present, this approach is restricted to (oxidative) dehydrogenations of primary amines, most often with specialized homogeneous catalysts. In this work, amines were transfer dehydrogenated to nitriles using simple and cheap alkenes (e.g. ethylene or propene) as hydrogen scavengers. The scope was expanded to secondary amines, tertiary amines and even aldehydes. Additional nitrogen is built in from NH3. The versatility of the process was proven by coupling it to the ammonolysis of secondary amides. This enabled us to recycle long-chain polyamides (LCPA) into monomeric compounds, i.e. α,ω-amidonitriles and dinitriles. Reactions were performed with a recyclable heterogeneous Pt catalyst, at 200 °C and with limited addition of NH3 and ethylene. High yields of up to 94% were obtained for the corresponding nitriles.
Introduction
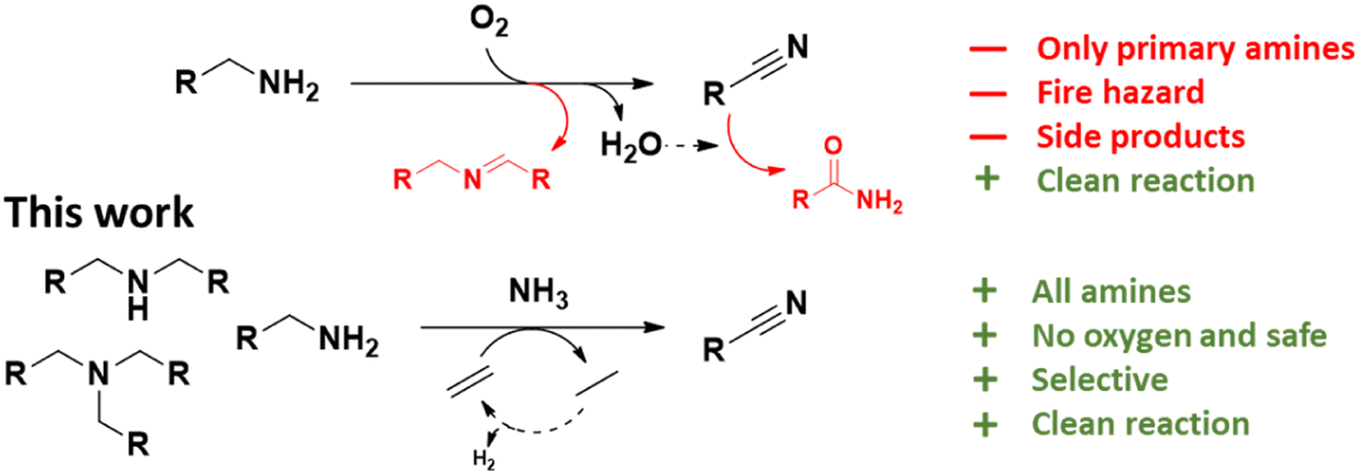
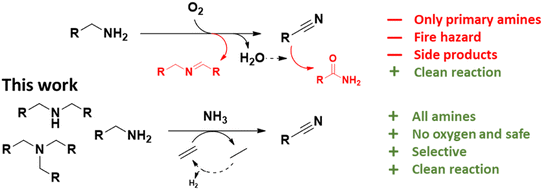
Nitriles are an important class of nitrogen containing molecules that are widely used in the chemical industry. The molecules' cyano groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) can be readily converted to or reacted with other functional groups. This makes them very versatile and desirable starting molecules for the synthesis of polymers, pharmaceuticals and pesticides.1–4 For this reason, nitriles have become a critical area of study in organic chemistry, and contemporary research is focused on developing new synthesis methods.5,6 In recent years, the dehydrogenation of amines was identified as and proven to be a clean and efficient route for nitrile synthesis.7–16 A minority of these reports concern the acceptorless dehydrogenation of amines. Although no additional chemicals are required, these methods often lead to low nitrile yields and/or require a continuous removal of generated H2.7,8 For this reason, most researchers studied the oxidative synthesis of nitriles, often using oxygen as the hydrogen acceptor.10–16 At first sight this appears as a very clean reaction, generating only water as a side product, with reported yields of over 80%. Nevertheless, this method has drawbacks. Firstly, suppressing side reactions remains challenging, with common side products such as aldimines, secondary and tertiary amines. In addition, the formed water can hydrate the nitriles, leading to primary or even secondary amides.10–13,17–21 Secondly, while oxygen is a green reagent, it is associated with serious fire hazards, especially if the reaction would occur on a large industrial scale.22 Thirdly, so far only primary amines can be successfully converted to nitriles. To react secondary or tertiary amines, additional nitrogen must be built in, ideally from ammonia (NH3). The combination of O2 and NH3 is challenging to work with and easily leads to the generation of NOx compounds in the presence of a noble metal catalyst at elevated temperatures.23–28 To our knowledge, only in the recent work of Olivares et al., an effective autoxidation of primary amines to nitriles was achieved with a specialized homogeneous Ru catalyst in the presence of NH3 and with high product yields (i.e. >90%).10 Nevertheless, technology that is able to convert secondary and tertiary amines to nitriles, would be a valuable addition to current synthesis methods. The most common amine synthesis methods (i.e. amination of alcohols, reductive amination of carbonyl compounds and nitrile hydrogenation) typically yield a mixture of primary, secondary and tertiary amines. Unfortunately, in most cases only one or two of the latter are truly desired.29–38 A method to convert the by-products back to nitriles can help to upgrade and reintegrate them into the chemical industry. For this reason, we explored a fairly new approach, viz. the dehydrogenation of amines to nitriles with alkenes as hydrogen acceptors (Scheme 1). Over the years, such (transfer) dehydrogenations have been proven to be a powerful technique for activating seemingly unreactive molecules and/or synthesizing a variety of pharmaceutically relevant compounds.59–62 However the dehydrogenation of amines to nitriles has quite often been overlooked. This approach was previously explored solely in the work of Bernskoetter et al.9 Although successful, they only converted a small scope of primary amines to the corresponding nitriles and used a specialized homogeneous Ir catalyst with a liquid sacrificial alkene (i.e. tert-butylethylene). In this work, we developed an ammonolytic transfer dehydrogenation process with simple alkenes, i.e. ethylene and propene. These gases are cheap, readily available, fire safe and compatible with NH3. Additionally, no water is generated during the reaction, which benefits the product yield. At present, ethylene is primarily sourced by thermal cracking of fossil feedstock.39 However, an increasing number of renewable production methods are being developed, e.g. dehydration of bio-ethanol or even electrochemical CO2-to-ethylene conversion.39–43 Even well-designed ethane crackers, e.g. with use of the H2 produced for heating, have a significantly reduced footprint.43 Aside from using readily available reagents, we report recyclable heterogeneous Pt catalysts as highly efficient catalysts for the (ammonolytic) transfer dehydrogenation of primary, secondary and tertiary amines. High yields of the corresponding nitriles were achieved (i.e. up to 94%) under fairly mild reaction conditions, i.e. limited addition of NH3 and ethylene. The versatility of our process was illustrated by coupling it to the ammonolysis of secondary amides. This enabled us to depolymerize long-chain polyamides (LCPA; PA11 and PA12) into monomers that can easily be reintegrated into the chemical industry. To our knowledge, this is the first report of such an ammonolytic transfer dehydrogenation process.
N) can be readily converted to or reacted with other functional groups. This makes them very versatile and desirable starting molecules for the synthesis of polymers, pharmaceuticals and pesticides.1–4 For this reason, nitriles have become a critical area of study in organic chemistry, and contemporary research is focused on developing new synthesis methods.5,6 In recent years, the dehydrogenation of amines was identified as and proven to be a clean and efficient route for nitrile synthesis.7–16 A minority of these reports concern the acceptorless dehydrogenation of amines. Although no additional chemicals are required, these methods often lead to low nitrile yields and/or require a continuous removal of generated H2.7,8 For this reason, most researchers studied the oxidative synthesis of nitriles, often using oxygen as the hydrogen acceptor.10–16 At first sight this appears as a very clean reaction, generating only water as a side product, with reported yields of over 80%. Nevertheless, this method has drawbacks. Firstly, suppressing side reactions remains challenging, with common side products such as aldimines, secondary and tertiary amines. In addition, the formed water can hydrate the nitriles, leading to primary or even secondary amides.10–13,17–21 Secondly, while oxygen is a green reagent, it is associated with serious fire hazards, especially if the reaction would occur on a large industrial scale.22 Thirdly, so far only primary amines can be successfully converted to nitriles. To react secondary or tertiary amines, additional nitrogen must be built in, ideally from ammonia (NH3). The combination of O2 and NH3 is challenging to work with and easily leads to the generation of NOx compounds in the presence of a noble metal catalyst at elevated temperatures.23–28 To our knowledge, only in the recent work of Olivares et al., an effective autoxidation of primary amines to nitriles was achieved with a specialized homogeneous Ru catalyst in the presence of NH3 and with high product yields (i.e. >90%).10 Nevertheless, technology that is able to convert secondary and tertiary amines to nitriles, would be a valuable addition to current synthesis methods. The most common amine synthesis methods (i.e. amination of alcohols, reductive amination of carbonyl compounds and nitrile hydrogenation) typically yield a mixture of primary, secondary and tertiary amines. Unfortunately, in most cases only one or two of the latter are truly desired.29–38 A method to convert the by-products back to nitriles can help to upgrade and reintegrate them into the chemical industry. For this reason, we explored a fairly new approach, viz. the dehydrogenation of amines to nitriles with alkenes as hydrogen acceptors (Scheme 1). Over the years, such (transfer) dehydrogenations have been proven to be a powerful technique for activating seemingly unreactive molecules and/or synthesizing a variety of pharmaceutically relevant compounds.59–62 However the dehydrogenation of amines to nitriles has quite often been overlooked. This approach was previously explored solely in the work of Bernskoetter et al.9 Although successful, they only converted a small scope of primary amines to the corresponding nitriles and used a specialized homogeneous Ir catalyst with a liquid sacrificial alkene (i.e. tert-butylethylene). In this work, we developed an ammonolytic transfer dehydrogenation process with simple alkenes, i.e. ethylene and propene. These gases are cheap, readily available, fire safe and compatible with NH3. Additionally, no water is generated during the reaction, which benefits the product yield. At present, ethylene is primarily sourced by thermal cracking of fossil feedstock.39 However, an increasing number of renewable production methods are being developed, e.g. dehydration of bio-ethanol or even electrochemical CO2-to-ethylene conversion.39–43 Even well-designed ethane crackers, e.g. with use of the H2 produced for heating, have a significantly reduced footprint.43 Aside from using readily available reagents, we report recyclable heterogeneous Pt catalysts as highly efficient catalysts for the (ammonolytic) transfer dehydrogenation of primary, secondary and tertiary amines. High yields of the corresponding nitriles were achieved (i.e. up to 94%) under fairly mild reaction conditions, i.e. limited addition of NH3 and ethylene. The versatility of our process was illustrated by coupling it to the ammonolysis of secondary amides. This enabled us to depolymerize long-chain polyamides (LCPA; PA11 and PA12) into monomers that can easily be reintegrated into the chemical industry. To our knowledge, this is the first report of such an ammonolytic transfer dehydrogenation process.
 |
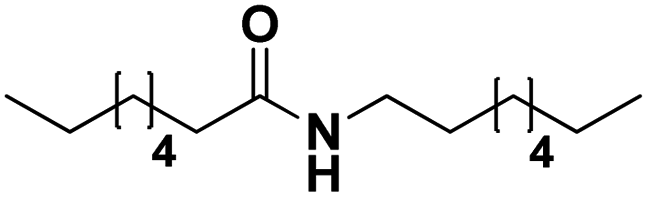
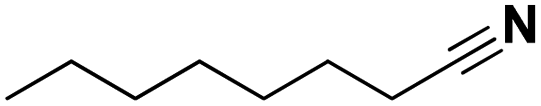
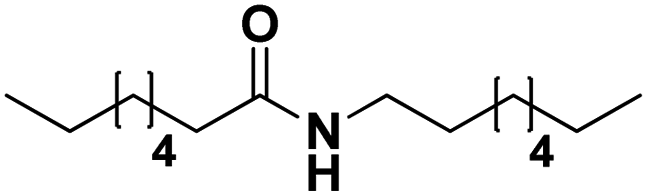
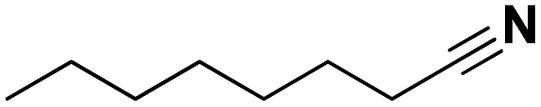
| | Scheme 1 Dehydrogenation of amines to nitriles. Top: available technology. Bottom: approach in this work. | |
Results and discussion
Catalyst evaluation
The catalytic dehydrogenation of alkanes with supported noble metals, especially platinum, is well described in literature.44 We therefore hypothesized that they might also be able to selectively dehydrogenate amines to nitriles. Calculations revealed that using an alkene as hydrogen scavenger, this reaction would be highly exergonic, although this would not be the case if an aromatic compound is used as a hydrogen acceptor (e.g. benzene, see ESI†). Therefore, as a starting point of this research, the catalytic activity of supported platinum (Pt), palladium (Pd), ruthenium (Ru) and rhodium (Rh) was evaluated for the transfer dehydrogenation of amines with ethylene as hydrogen scavenger (Fig. 1). In these experiments, the primary amine octylamine was selected as the model compound; initial promising results were obtained for secondary and tertiary amine as well (i.e. dioctylamine and trioctylamine). Of the four noble metals, Ru and Rh showed the least promise. Reactions with Ru/C and Rh/C only resulted in approximately 12% octanenitrile yield, with di- and trioctylamine as the main side products. Supported Pt appeared to be the most performing catalyst, with already 70% octanenitrile yield for Pt/C (Escat 2431) in this initial screening. A remarkable side product, detectable in low quantities in nearly all samples, is octanamide. Palladium had a mediocre catalytic performance, with the best result for Pd/C, with 41% nitrile yield. Nevertheless, these experiments were performed with 2.5 mol% metal. For industrial applications it is usually not important which metal is the most active, but which catalyst is the cheapest. Pd is only half as heavy as Pt, and about 1.5 times as expensive. For this reason, catalytic experiments were also performed with the same weight content of metal (Fig. S2†). In these conditions, Pd and Pt have similar catalytic activities; however, Pt/C is more selective. A reaction with Pd/C leads to a higher content of 2-hexyldecanenitrile, which is a condensation dead-end product. The ability of Pt to more rapidly dehydrogenate intermediate products was already clear in Fig. 1; nearly no intermediate products were observed for Pt/C whereas for Pd/C a total yield of 9% N-octylideneoctylamine was obtained. We therefore selected Pt/C as the best transfer dehydrogenation catalyst.
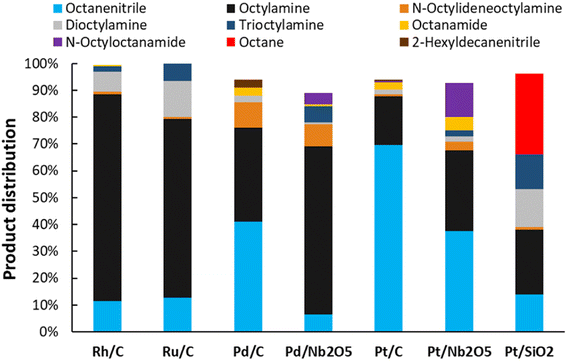 |
| | Fig. 1 Catalyst evaluation for the transfer dehydrogenation of n-octylamine. Reaction conditions: octylamine (1 mmol), 200 °C, 6 bar NH3, 2 bar ethylene, 2.5 mol% PGM (commercial PGM/C catalysts contain 5 wt% PGM, all other catalysts contain 4 wt% PGM), dodecane (20 μL), CPME (10 mL), 16 h. Before reaction, the catalysts were pre-activated under 10 bar H2, 1 bar NH3, at 160 °C for 15 minutes. | |
Simultaneously with the catalyst evaluation, we also explored the most effective catalyst pretreatment method. In ambient air, the outer surface of noble metal nanoparticles is partially oxidized.45 To remove this passive layer, the catalyst must be treated with H2 (Fig. 2). Without any pre-treatment, low octanenitrile yields are obtained. After a short pre-activation of 15 minutes starting from a temperature of 150 °C at 10 bar H2, the catalyst reaches its maximal catalytic activity. For the catalysts with a carbon support, we also noticed a clear mass loss of more than 10%, even if the catalyst wasn't properly activated. This mass loss is more prominent if the dehydrogenation was performed with a high catalyst loading over a short reaction time, than with a low catalyst loading over a long reaction time (Fig. S2†). It is therefore evident that this is the result of a selective adsorption or, more likely, a chemical reaction with the amine substrate. Activated carbon typically consists of 4% to 20% oxygen, trapped in various functional groups (e.g. ketones and (aromatic) alcohols).46–49 At temperatures above 100 °C, Maillard reactions start to occur spontaneously, trapping part of the amine substrate in the process.50 We found two effective methods to greatly improve the global mass balance. The first method involves the replacement of old with fresh reaction medium after the pre-activation (Fig. S2†). This way, the majority of the amine chemisorption sites have been saturated. Alternatively, the addition of 3 bar NH3 during pre-activation has a similar effect (Fig. 2). Due to the simplicity of this method, the latter was selected.
 |
| | Fig. 2 Evaluation of the optimal pre-activation procedure. The catalyst was pretreated under the specified conditions, after which a transfer dehydrogenation reaction took place. Reaction conditions: octylamine (1 mmol), 200 °C, 6 bar NH3, 1.5 bar ethylene, 2.5 mol% Pt (commercial Pt/C, 5 wt% Pt), dodecane (20 μL), CPME (10 mL), 16 h. | |
Optimization of the reaction conditions
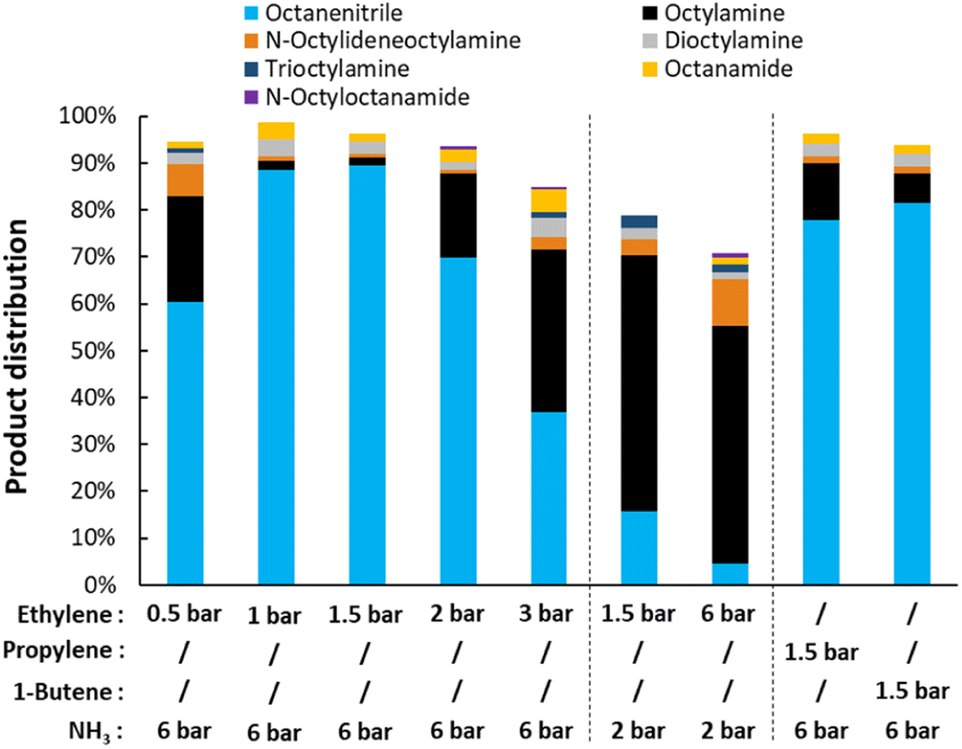
Starting with the variation of the gas mixture, the reaction conditions were optimized (Fig. 3). Although no net NH3 is consumed during the dehydrogenation of octylamine, its presence seems to be crucial for the reaction to succeed. It appears that ammonia stabilizes the catalyst, possibly by occupying part of the ethylene adsorption sites. Starting from an ethylene pressure of 1.5 bar (with 6 bar NH3), the final octanenitrile yield decreases notably with increasing ethylene pressure. This effect is particularly prominent when the NH3 content is low. After a dehydrogenation reaction at 6 bar ethylene and 2 bar NH3 (at 200 °C), a low yield of only 5% octanenitrile is obtained. The catalyst was completely deactivated and could not be reused afterwards. Most likely, the Pt nanoparticles were completely coked up. As is known, on a Pt surface at elevated temperature, ethylene is gradually converted to ethylidyne species (i.e. Pt3C–CH3) via a cascade of reaction steps.51 These species are fairly stable and tend to coke up by forming new C–C bonds, effectively deactivating the catalyst.52 Under the optimal conditions of 6 bar NH3 and 1 to 1.5 bar ethylene, a yield of 90% octanenitrile is obtained with a nearly perfect mass balance. Here, ethylene reacts more slowly, which gives it more time to capture hydrides originating from the dehydrogenation of an amine. An FTIR analysis of the final gas mixture clearly shows the formation of ethane (Fig. S3†). The available data also indicate that some dimerization occurred, leading to the formation of butene.52 While this dimerization results in the loss of one unsaturation, the reaction seems to be effective with various alkenes (e.g. propylene and butene).
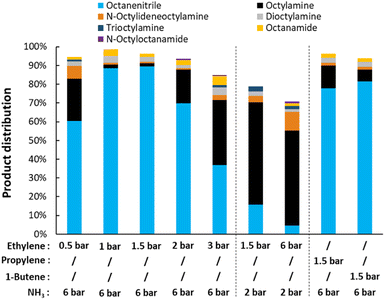 |
| | Fig. 3 Variation of the gas composition for the transfer dehydrogenation of n-octylamine. Reaction conditions: octylamine (1 mmol), 200 °C, 2.5 mol% Pt (commercial Pt/C, 5 wt% Pt), dodecane (20 μL), CPME (10 mL), 16 h. Before reaction, the catalysts was pre-activated under 10 bar H2, 3 bar NH3, at 160 °C for 15 minutes. | |
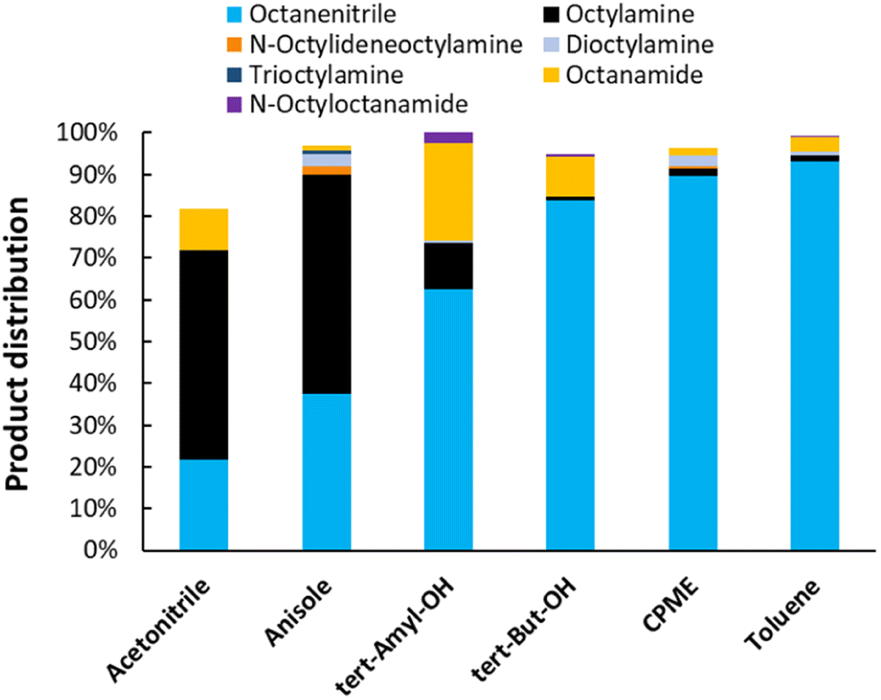
Next, the reaction solvent was varied (Fig. 4). In general, the reaction could be performed in a variety of solvents (i.e. ethers, hydrocarbons and alcohols). Anisole and acetonitrile are the least suitable reaction media. They dissolve a lot of NH3 at an equilibrium partial pressure of 6 bar, to the point where this prevents other reactants from adsorbing on the catalytic surface.31 A reaction in acetonitrile also led to the formation of multiple side products. In tertiary alcohol solvents such as tert-amylalcohol and tert-butylalcohol, high conversions are reached. However, the solvent is partially dehydrated, which ultimately leads to the formation of hydrated products (i.e. octanamide, corresponding yields of 23% and 9%). Reactions in cyclopentyl methyl ether (CPME) and toluene resulted in the highest octanenitrile yield of 90% and 93% respectively. As a result, subsequent reactions were performed in one of those solvents.
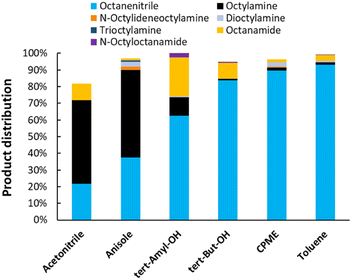 |
| | Fig. 4 Variation of the reaction solvent for the transfer dehydrogenation of n-octylamine. Reaction conditions: octylamine (1 mmol), 200 °C, 6 bar NH3, 1.5 bar ethylene, 2.5 mol% Pt (commercial Pt/C, 5 wt% Pt), dodecane (20 μL), CPME (10 mL), 16 h. Pre-activation as in Fig. 3. tert-Amyl-OH: tert-amylalcohol. tert-But-OH: tert-butylalcohol. | |
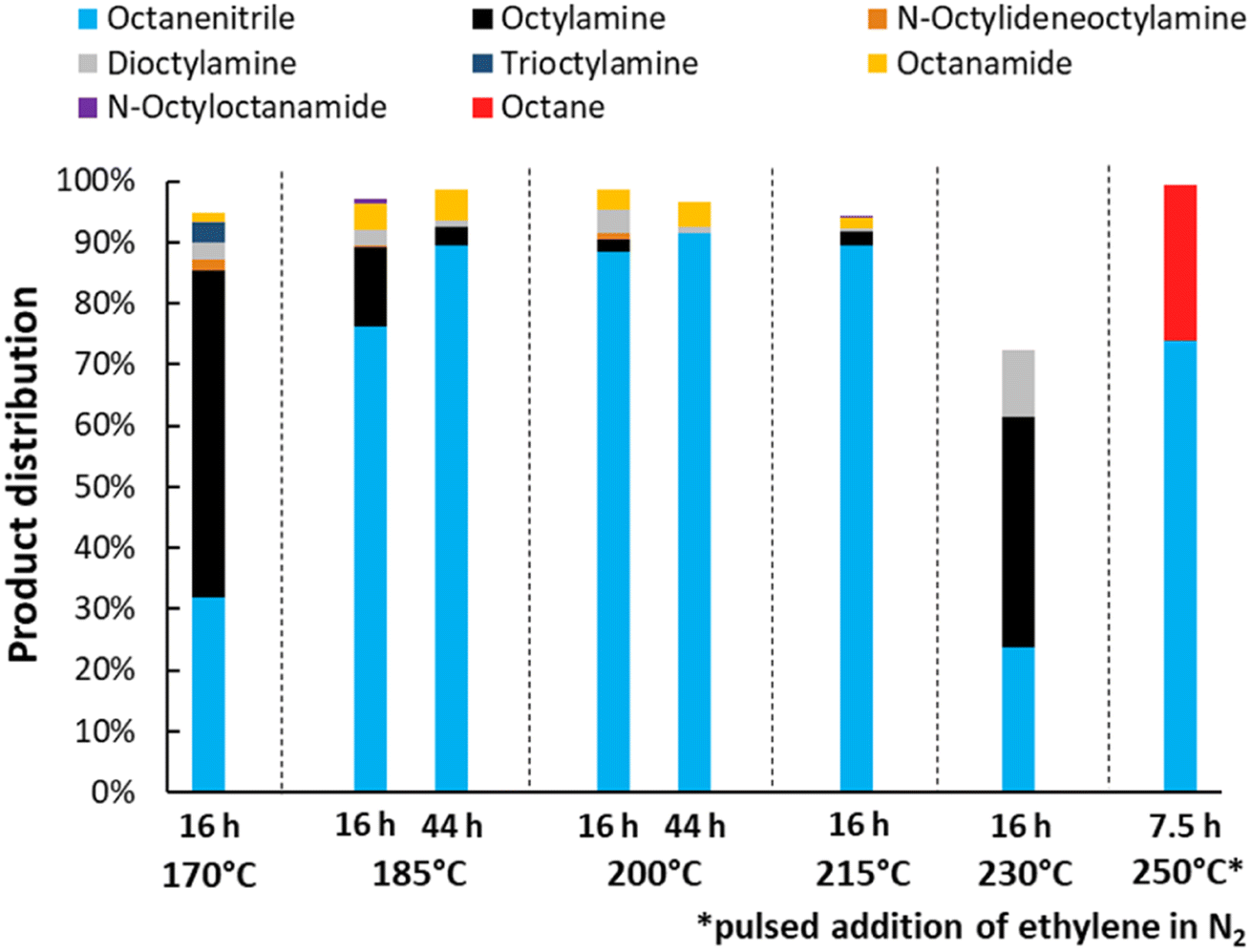
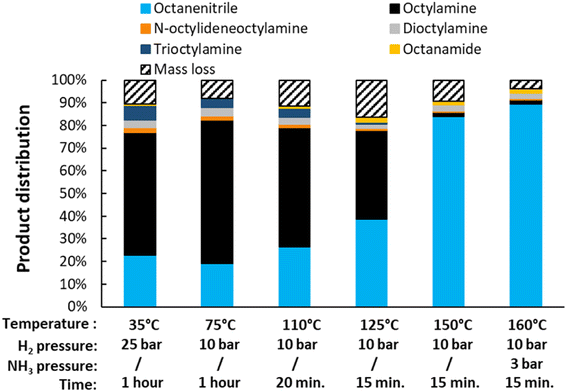
Lastly, the influence of the reaction temperature was studied (Fig. 5). Ideally, the dehydrogenation is performed at 200 °C. At lower temperatures, a high selectivity for the desired product can still be obtained, but the reaction takes a long time to complete (e.g. 44 h at 185 °C, with 89% nitrile yield). With increasing temperature, the catalyst also deactivates more easily, resulting in a low yield at 230 °C under the applied conditions. This can be improved by gradually adding ethylene over the course of the reaction. Using this method at a temperature of 250 °C, the reaction time could be shortened drastically and a final yield of 74% octanenitrile was obtained along with 26% octane. We did not further optimize this gradual addition, but simply performed the experiment as a proof of concept. Furthermore, it is worth noting that previous experiments were conducted with a substrate concentration of 0.1 M. The reaction, however, can be performed in an at least 5 times more concentrated solution (Fig. S4†). Nonetheless, more concentrated reaction mixtures require frequent replenishments of the reaction atmosphere in order not to run out of alkene reagent. For simplicity, we therefore chose to maintain the substrate concentration at 0.1 M in subsequent experiments.
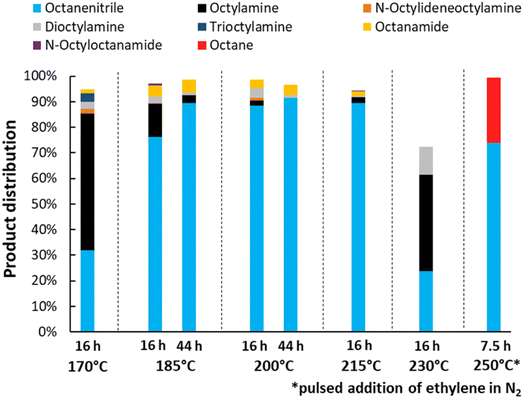 |
| | Fig. 5 Variation of the reaction temperature and time for the transfer dehydrogenation of n-octylamine. Reaction conditions: octylamine (1 mmol), 6 bar NH3, 1 bar ethylene, 2.5 mol% Pt (commercial Pt/C, 5 wt% Pt), dodecane (20 μL), CPME (10 mL). Pre-activation as in Fig. 3. The reaction at 250 °C was performed in toluene since CPME starts to drastically degrade above 230 °C. Over the course of this particular reaction, every half hour the pressure inside the reactor was increased with approximately 1.5 bar, by adding a pulse of 15% ethylene in N2. | |
Unravelling the reaction network
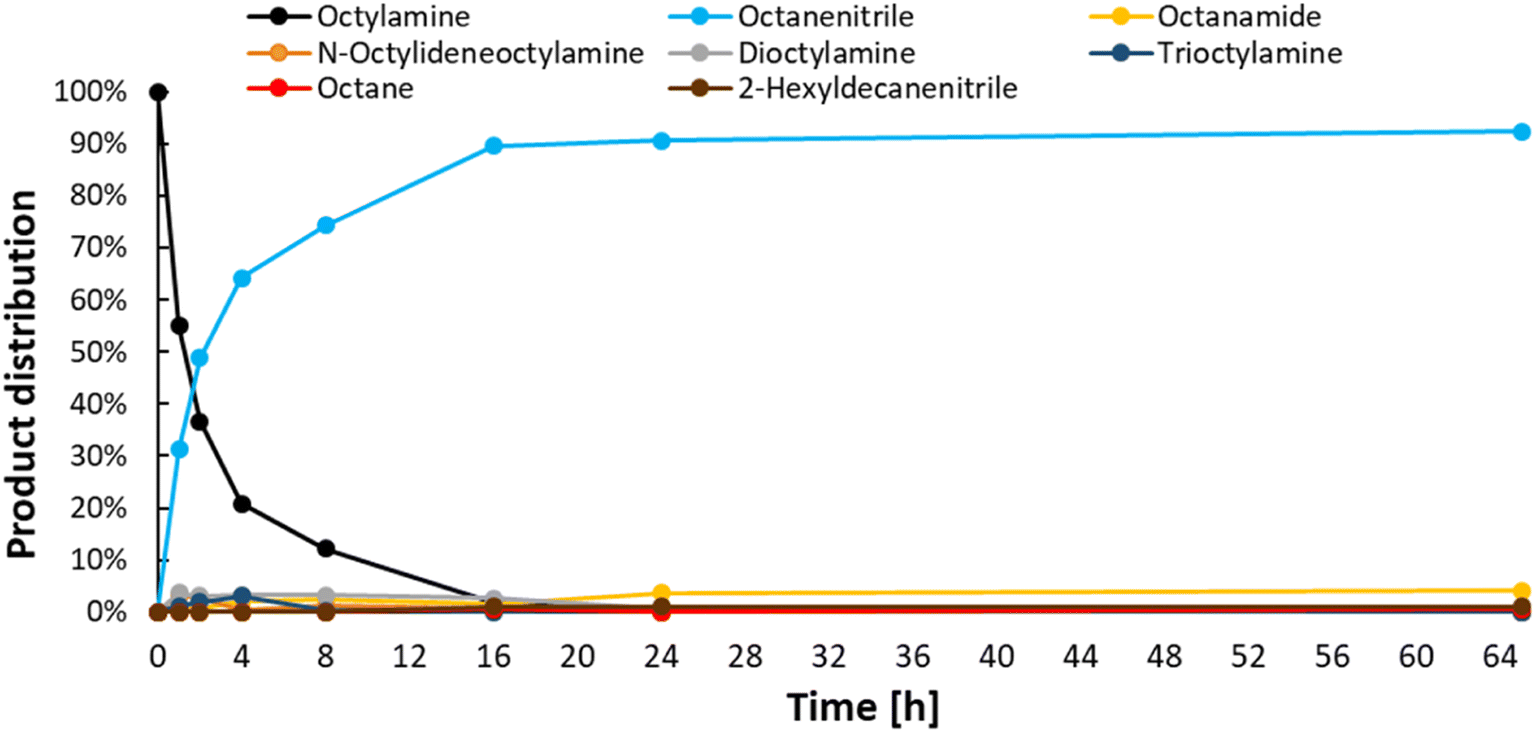
After obtaining the optimal reaction conditions (i.e. 200 °C, 1.5 bar ethylene, 6 bar NH3 in CPME or toluene), a time profile was made (Fig. 6). This helped us unravel the reaction network (Scheme 2). In the early stages of the reaction, octylamine is directly converted to octanenitrile, while minor amounts of several by-products are also formed. However, after their initial formation, the concentrations of secondary amine, tertiary amine and intermediate aldimine decrease rapidly over time. Small concentrations of the permanent dead-end products 2-hexyldecanenitrile and n-octane are also generated in the initial phase of the reaction. Once the reaction reaches a complete conversion, the nitrile product remains very stable over time. Traces of water that were present in the reaction vessel led to the hydration of small quantities of product. Ultimately, a product mixture was obtained that comprised mainly octanenitrile (92% yield) and octanamide (4% yield; 65 hours reaction time). This allowed us to construct a reaction scheme (Scheme 2). When starting from a primary amine, such as octylamine (1), 2 alkene molecules (e.g. ethylene) are needed in order to form octanenitrile (3). The intermediate imine (2) can, however, condensate with leftover amines leading to N-octylideneoctylamine (7), dioctylamine (6) or even trioctylamine (8). These amine condensation reactions are reversible. As was hinted to earlier, this enables us to also convert these secondary and tertiary molecules to nitriles. Unfortunately, some condensation reactions are non-reversible. The α-carbon of a nitrile is fairly acidic and could behave as a nucleophile after deprotonation. A condensation between octanenitrile (3) and octan-1-imine (2) ultimately leads to the dead-end compound 2-hexyldecanenitrile (9). The formation of octane is another irreversible reaction observed. Based on our findings, we believe that most of the octane is generated during the (intramolecular) degradation of N-octylidene-octylamine (7) into one molecule of octanenitrile (3) and one molecule of octane. Such a degradation is unlikely to occur with a tertiary enamine. As was clear from the time profile (Fig. 6), once all octanenitrile (3) is formed and no leftover substrate remains, the product is very stable. However, the presence of small quantities of water leads to the formation of octanamide (4). If the catalyst has significant (Lewis) acidic properties as well, octanamide (4) can even be transaminated with octylamine (1) to a secondary amide (i.e. N-octyloctanamide (5)). Like the condensation of amines, this reaction is also reversible.
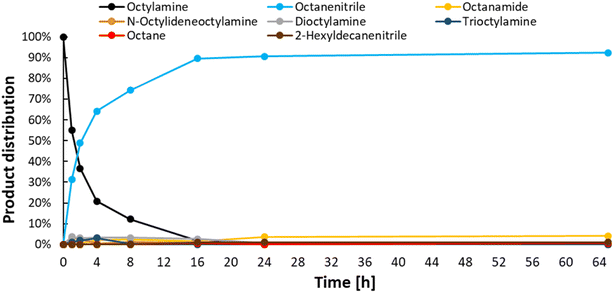 |
| | Fig. 6 Time profile for the transfer dehydrogenation of n-octylamine. Reaction conditions: octylamine (1 mmol), 200 °C, 6 bar NH3, 1.5 bar ethylene, 2.5 mol% Pt (commercial Pt/C, 5 wt% Pt), dodecane (20 μL), CPME (10 mL). Pre-activation as in Fig. 3. | |
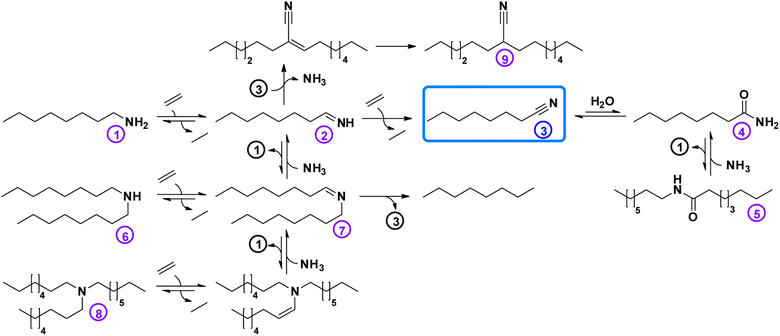 |
| | Scheme 2 Reaction network for the (ammonolytic) transfer dehydrogenation of amines and amides to nitriles. | |
Ammonolytic transfer dehydrogenation of secondary amides
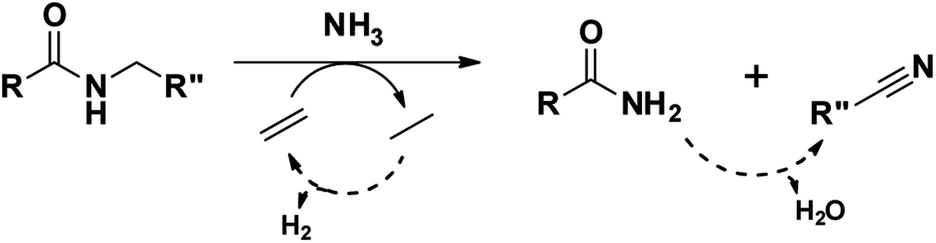
To illustrate the versatility of our process, we coupled the dehydrogenation of amines with a preceding ammonolysis of amides. In previous work, we demonstrated that niobia (i.e. ortho-Nb2O5) is an excellent catalyst for the ammonolysis of secondary amides.30 This ammonolysis is an equilibrium reaction and to drive it to completion, it needs to be linked to a subsequent irreversible reaction. Whereas in our aforementioned report, we aimed to produce monomeric compounds by hydrogenating the generated primary amide to a primary amine, we now choose a different approach by transforming the generated amine into a nitrile. In the case where a secondary amide is split into two molecules with the same carbon chain length, this could also lead to a convergent upgrading of secondary amides to nitriles, if the primary amides in the product mixture are (catalytically) dehydrated in the same reaction (Scheme 3).53,54
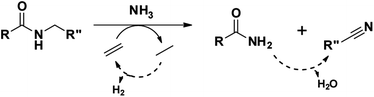 |
| | Scheme 3 The ammonolytic transfer dehydrogenation of secondary amides to nitriles (and primary amides). | |
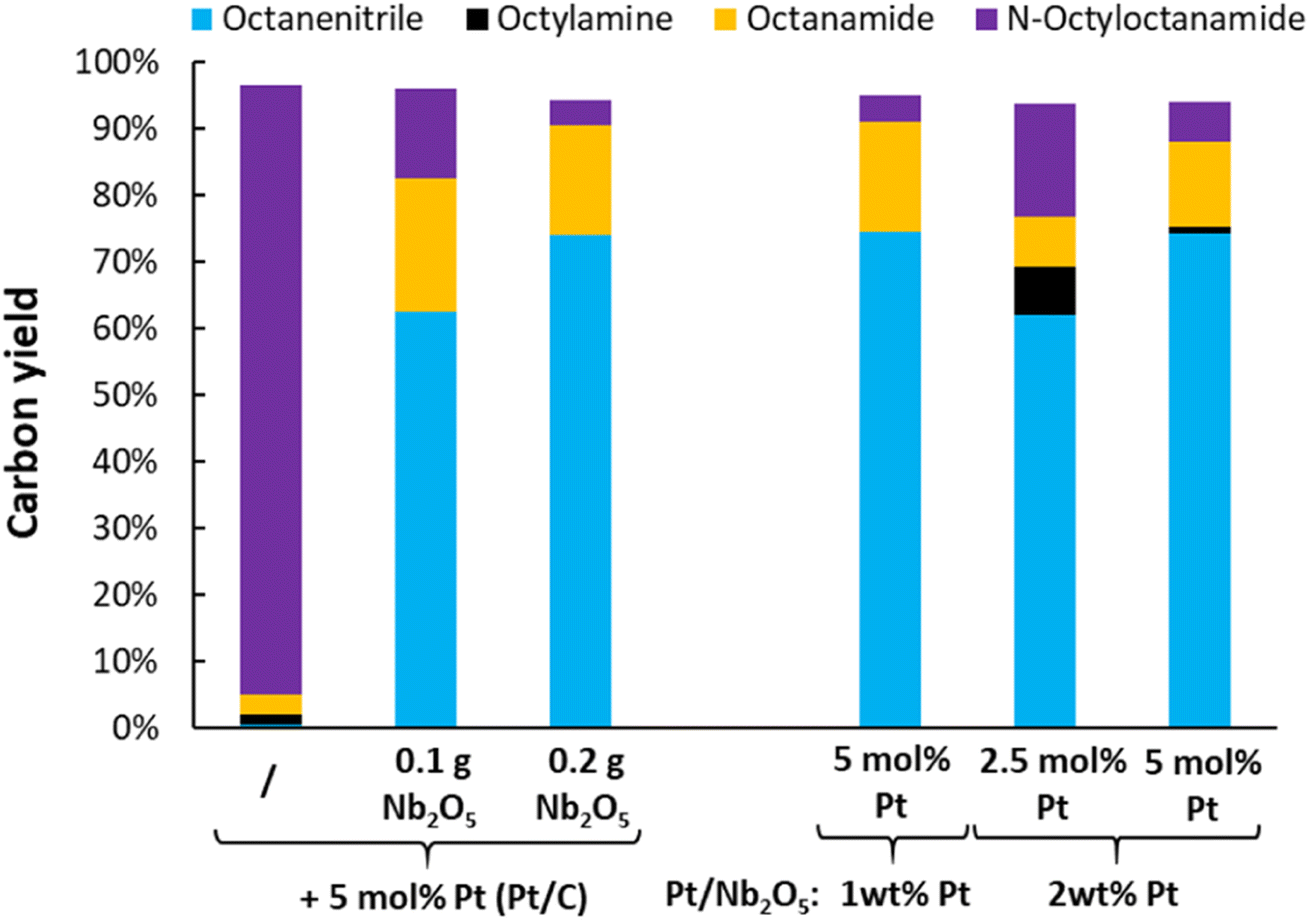
Upon switching from octylamine to N-octyloctanamide as the model reactant, the ammonolytic dehydrogenation reactions were initially performed by simply adding 0.2 g of niobia as an ammonolysis catalyst (Fig. 7). After 16 h (5 mol% Pt), the reaction reaches nearly full conversion (i.e. 96%). The resulting carbon yields were 74% octanenitrile and 17% octanamide, with 4% of unconverted N-octyloctanamide. Clearly, a significant fraction of the in situ generated octanamide has been dehydrated. Instead of combining the two catalysts (i.e. Pt/C and Nb2O5), Pt was also loaded directly on a ortho-Nb2O5 support. These catalysts were less active than the dual catalyst system. However, a reaction with Pt/Nb2O5 containing 1 wt% Pt (reaction with 5 mol% Pt) led to virtually the same result (i.e. 75% octanenitrile, 16% octanamide and 4% N-octyloctanamide). Just like the dehydrogenation of octylamine, this ammonolytic dehydrogenation of secondary amides was carried out successfully at 250 °C with the gradual addition of ethylene (Fig. S6†). A final carbon yield of 87% octanenitrile was obtained, with only small quantities of octanamide (i.e. 2%) and octane (i.e. 5%).
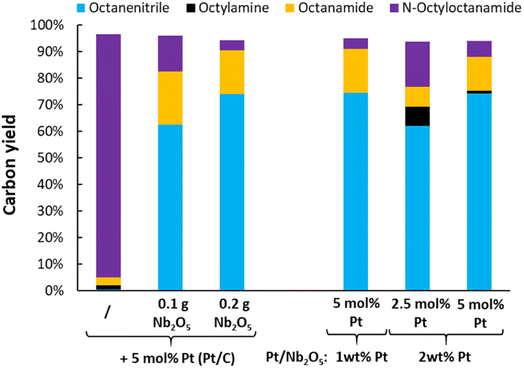 |
| | Fig. 7 Evaluation of the catalyst and catalyst composition for the ammonolytic transfer dehydrogenation of N-octyloctanamide. Reaction conditions: N-octyloctanamide (0.5 mmol), 200 °C, 6 bar NH3, 2 bar ethylene, dodecane (20 μL), toluene (10 mL), 16 h. Pre-activation as in Fig. 3. | |
Characterization and recyclability of the catalyst
ATR-FTIR was used to examine the surface of the Pt/C catalyst (Fig. S8†). The spectra of the fresh and used sample are essentially the same, with no clear peaks that can be distinguished (as is typical for activated carbon samples).55–57 Nevertheless, the absence of an absorption band between 3000 and 3500 cm−1 illustrates that the catalysts contain very little water. The catalysts increasingly absorb IR light with decreasing wavenumber, indicating the presence of (unsaturated) C–C, C–O and/or C–N bonds.
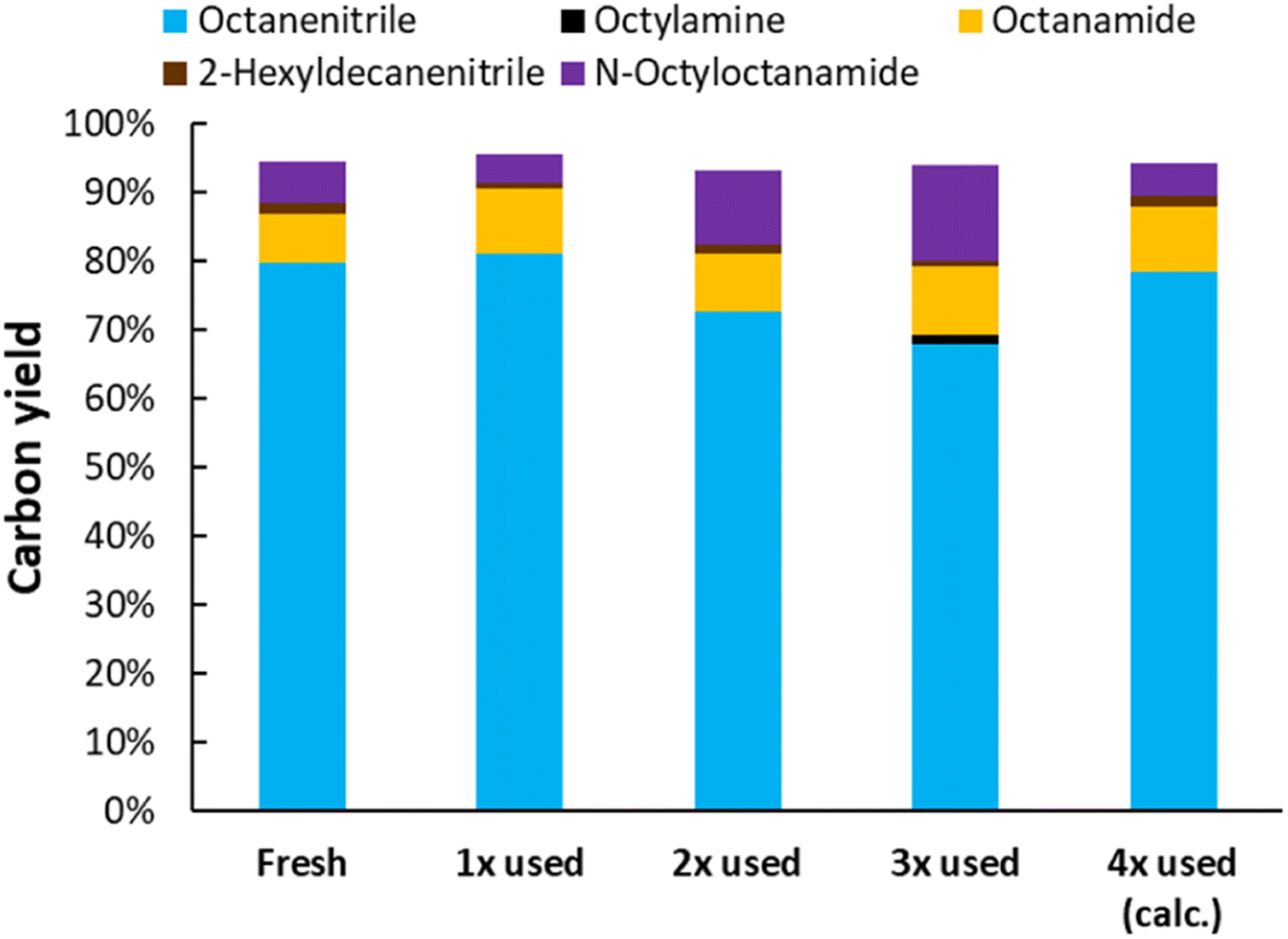
The dispersion of Pt was investigated for both Pt/C and Pt/Nb2O5 (Table 1). The commercial Pt/C has the highest Pt dispersion (29%); the dispersion of Pt is lower for Pt/Nb2O5 (viz. 15%) than for Pt/C. This is most likely the reason why for Pt/Nb2O5 additional Nb2O5 was required (in comparison with Pt/C + Nb2O5), to compensate for the slower dehydrogenation of the in situ generated amine (see 1 wt% Pt, Pt/Nb2O5, Fig. 7). A recycling test was performed with Pt/Nb2O5 for the ammonolytic dehydrogenation of N-octyloctanamide (Fig. 8). Although the catalytic activity slightly drops after each run, the catalyst can be fully reactivated by recalcining the material. Thus, the catalyst demonstrated outstanding recyclability.
Table 1 CO chemisorption and N2 physisorption results for Pt/C and Pt/Nb2O5
| Catalyst |
Pt/C(Escat 2431) |
Pt/Nb2O5 |
| Metal content |
5 wt% |
1 wt% |
| Average crystal size |
3.9 nm |
7.4 nm |
| Dispersion |
29% |
15% |
| BET surface area |
669 m2 g−1 |
223 m2 g−1 |
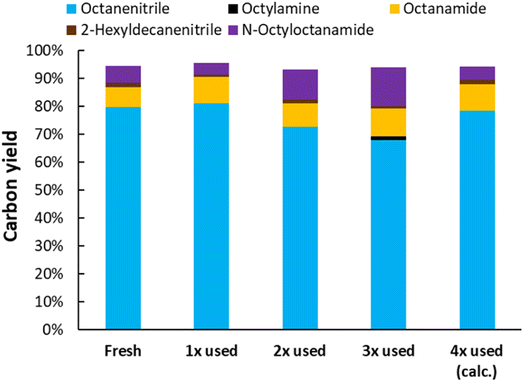 |
| | Fig. 8 Recycling test of Pt/Nb2O5 (1 wt% Pt) for the ammonolytic transfer dehydrogenation of N-octyloctanamide. Reaction conditions: N-octyloctanamide (0.5 mmol), 200 °C, 6 bar NH3, 1.5 bar ethylene, dodecane (20 μL), toluene (10 mL), 16 h. Pre-activation as in Fig. 3. After reusing the material 3 times, the material was recalcined at 400 °C. | |
Substrate scope
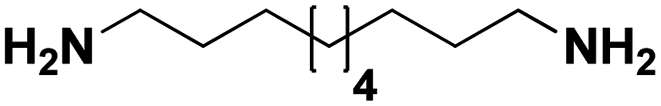
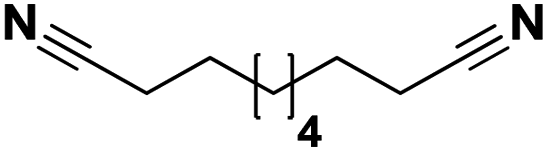
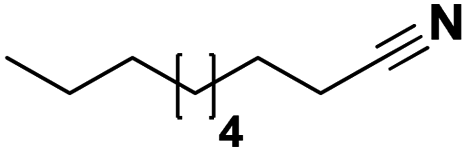
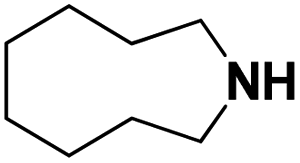
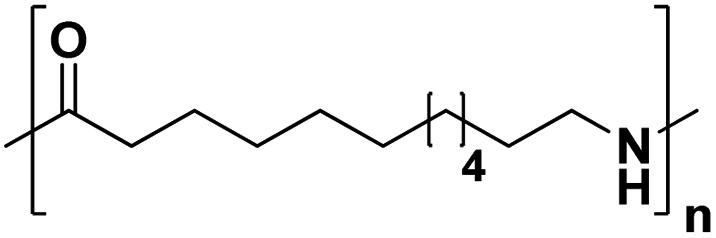
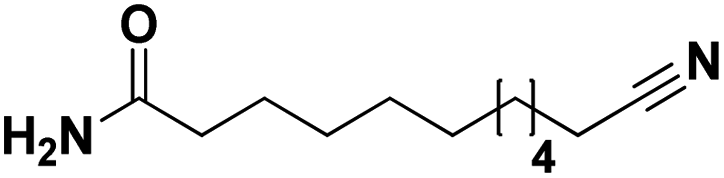
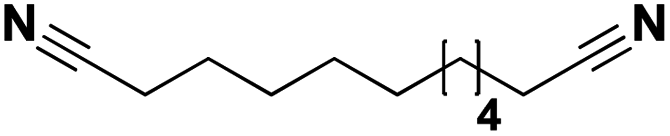
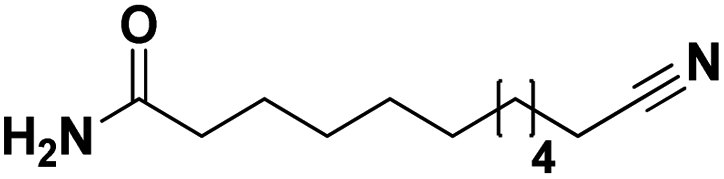
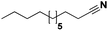
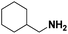
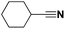
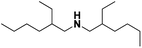
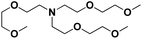
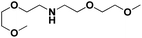
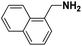
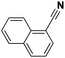
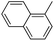
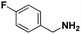
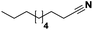
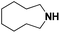
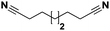
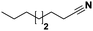
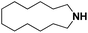
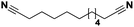
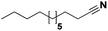
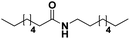
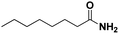
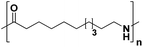
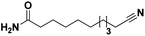
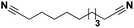
Lastly, a substrate scope investigation was performed to illustrate the range of applicability (Tables 2 and S2†). Nearly full conversions and high yields were obtained for all linear primary amines (up to 93% yield, entries 1–3). For hexahydrobenzylamine, the conversion was slightly lower (i.e. 88%), but with a similar selectivity (i.e. 93%, entry 4). Common side products are nitriles that underwent alkylation at the α-carbon (e.g. 2-hexyldecanenitrile) and primary amides (due to traces of water). The scope was successfully extended to most aliphatic secondary and tertiary amines, with yields of up to 94% (entries 5–9). The catalytic system, however, is not suitable for methyl-substituted amines (entry 10). A reaction with N,N-dimethylhexylamine resulted in a low conversion (i.e. 33%), with formation of only 6% hexanenitrile and some demethylated side products (e.g. N-methylhexylamine and hexylamine). Since this reaction worked flawlessly with mono-, di and trihexylamine (or even triethylamine, see ESI†), it is most probable that the methylamines, as logical splitting products, were dehydrogenated to hydrogen cyanide, which adsorbs strongly on a Pt surface, effectively poisoning the catalyst.58 While the quantities of cyanide were too low for us to actually detect, precaution should be taken when attempting a similar experiment. For a sterically hindered molecule, like tris(2-ethylhexyl)amine, a considerably slower reaction is observed (entry 11). Similarly, a reaction with an amine containing alkoxy groups, resulted in a low nitrile yield (entry 12). For instance, although tris[2-(2-methoxyethoxy)ethyl]amine easily undergoes ammonolysis to the corresponding primary and secondary amine, the dehydrogenation to 2-(2-methoxyethoxy)acetonitrile proceeded at a much slower rate (only 16% yield). An ammonolytic dehydrogenation was also performed with a linear alcohol and aldehyde (entry 13 and 14). While the catalyst struggles to dehydrogenate octanol, resulting in a low conversion of 8% (with 5% octanenitrile yield), octanal readily reacts with NH3 and is dehydrogenated to the nitrile. During this reaction, one equivalent of water is formed, leading to an increased amount of hydrated product (i.e. octanamide). Next, ammonolytic transfer dehydrogenations were performed with several benzylic amines (entries 15–21). High yields of the corresponding nitriles were obtained for all tested primary amines, secondary amines and aldehydes (entry 15–19, 21), ranging from 71% to 91% yield. The dehydrogenation reaction was tolerant for halides, like fluorine, and amino groups directly substituted on the aromatic ring (entries 17 and 19). Polycyclic aromatic molecules, however, tend to be defunctionalized more easily (e.g. 1-naphthylmethylamine, entry 16). A reaction with the tertiary amine tribenzylamine was less successful (entry 20). Unlike many other amines, this molecule cannot be easily cleaved since it cannot form an enamine, leading to only 30% conversion and 15% benzonitrile yield. Reactions with α,ω-diamines and cyclic secondary amines resulted in similar product distributions (entries 22–24). Approximately 65% to 88% of the substrate is converted to the corresponding α,ω-dinitrile. However, some defunctionalization is observed as well. We suspect that the cyclic aldimine intermediate may be somewhat constrained, leading more often to the splitting of the cyclic molecule into an aliphatic tail and a nitrile group (see defunctionalization, Scheme 2). Nevertheless, our process could valorize several side products during synthesis of polymer building blocks into useful monomeric compounds.30 This reaction, however, was not successful with azepane (not in the table). Finally, some amide substrates were ammonolytically dehydrogenated (entries 25–30). Our method works excellently for secondary amides, but not for tertiary amides (entry 27). A reaction with N,N-dioctyloctanamide resulted in a conversion of only 11%. It is clear that the Nb2O5 catalyst lacks activity for splitting tertiary amides with NH3. Long-chain polyamides (LCPAs), i.e. nylon 11 and nylon 12, were successfully depolymerized into monomeric compounds (entries 28–30). The reaction is ideally performed with the dual catalyst system Pt/C & Nb2O5, leading to full depolymerization. The Pt/Nb2O5 lacked catalytic activity and only resulted in a monomer yield of 57%. Although part of the α,ω-amidonitriles were dehydrated to α,ω-dinitriles, virtually no defunctionalization to monofunctionalized molecules was observed (in contrast to entry 22–24). Thus, our process enables the complete recycling of these high value LCPAs.
Table 2 Substrate scope investigation for the (ammonolytic) transfer dehydrogenation to nitrilesa
|
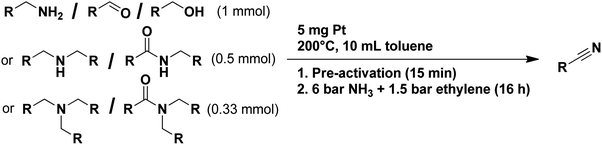
|
|
|
Catalyst |
Substrate |
X
[%] |
Nitrile product |
Y
[%] |
Main side product |
Y
[%] |
|
Before each run, the catalysts were pre-activated under 10 bar H2, 3 bar NH3, at 160 °C for 15 minutes. In case of an aromatic molecule, the substrate was added after the pre-activation step to prevent hydrogenation of the aromatic ring structure.
Conversion.
Yield or carbon yield in case of an amide substrate.
Reaction performed in CPME instead of toluene.
With 0.2 g Nb2O5, 5 mg Pt in the form of Pt/C (5 wt% Pt).
With 1 mmol instead of the above specified 0.5 mmol.
Reaction time of 72 h.
This reaction possibly generates (ammonium) cyanide.
|
| 1 |
Pt/C |

|
96 |
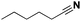
|
91 |
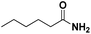
|
3 |
| 2 |
Pt/C |

|
96 |

|
93 |
|
1 |
| 3 |
Pt/C |
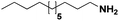
|
93 |
|
88 |
— |
— |
| 4 |
Pt/C |
|
88 |
|
82 |
— |
— |
| 5 |
Pt/C |
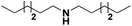
|
98 |
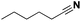
|
81 |

|
8 |
| 6 |
Pt/C |
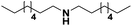
|
>99 |

|
90 |

|
5 |
| 7 |
Pt/C |
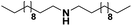
|
>99 |
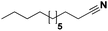
|
94 |
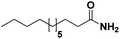
|
1 |
| 8 |
Pt/C |
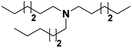
|
>99 |
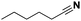
|
93 |
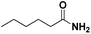
|
2 |
| 9 |
Pt/C |
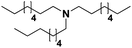
|
>99 |

|
92 |
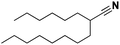
|
2 |
| 10 |
Pt/Ch |
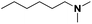
|
33 |
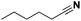
|
6 |
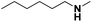
|
11 |
| 11 |
Pt/C |
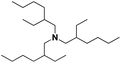
|
31 |
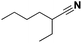
|
18 |
|
10 |
| 12 |
Pt/C |
|
80 |
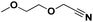
|
16 |
|
19 |
| 13 |
Pt/C |
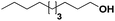
|
8 |

|
5 |
— |
— |
| 14 |
Pt/C |

|
>99 |

|
77 |

|
10 |
| 15 |
Pt/C |
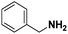
|
>99 |
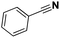
|
89 |
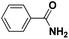
|
4 |
| 16 |
Pt/C |
|
>99 |
|
76 |
|
13 |
| 17 |
Pt/C |
|
>99 |
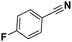
|
90 |
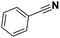
|
4 |
| 18 |
Pt/C |
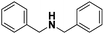
|
>99 |
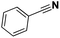
|
91 |
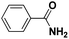
|
2 |
| 19 |
Pt/Cf |
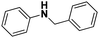
|
66 |
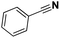
|
63 |
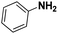
|
64 |
| 20 |
Pt/C |
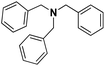
|
30 |
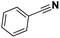
|
15 |
|
4 |
| 21 |
Pt/C |
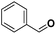
|
>99 |
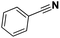
|
71 |
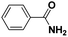
|
9 |
| 22 |
Pt/Cd |
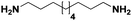
|
>99 |
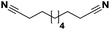
|
65 |
|
14 |
| 23 |
Pt/C |
|
98 |
|
65 |
|
15 |
| 24 |
Pt/Cd |
|
>99 |
|
88 |
|
3 |
| 25 |
Pt/C + Nb2O5e |
|
96 |

|
74 |
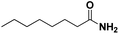
|
17 |
| 26 |
Pt/Nb2O5 |
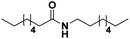
|
96 |

|
75 |
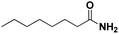
|
16 |
| 27 |
Pt/C + Nb2O5e |
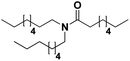
|
11 |

|
7 |
|
3 |
| 28 |
Pt/C + Nb2O5d,e,g |
|
>99 |
|
54 |
|
39 |
| 29 |
Pt/Nb2O5d,g |
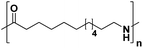
|
n.d. |
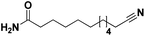
|
22 |
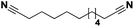
|
30 |
| 30 |
Pt/C + Nb2O5d,e,g |
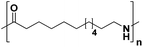
|
>99 |
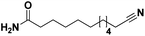
|
52 |
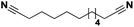
|
43 |
Conclusion
In conclusion, the ammonolytic transfer dehydrogenation is an excellent method to convert primary, secondary and tertiary amines into nitriles, with simple and cheap alkenes, e.g. ethylene, as hydrogen scavengers. The process requires a recyclable heterogeneous Pt catalyst, is robust and can utilize a wide range of alkenes. This would allow a possible industrial process to always make use of the cheapest available option. In principle, the generated alkane side product could be recycled to the olefin in an (electrified) steam cracker, making it a very green process with energy as the only input. The reaction conditions were optimized to 200 °C with 6 bar NH3 and 1.5 bar ethylene. We also demonstrated that the reaction could be performed in concentrated mixtures and with the continuous addition of ethylene. The versatility of our process was proven in two ways. Firstly, we coupled the dehydrogenation of amines to the ammonolysis of amides. This required the addition of a Nb2O5 catalyst. Secondly, an extensive substrate scope investigation was performed. Good yields for the corresponding nitriles, with values up to 94%, were obtained for a wide variety of substrates. We were even able to completely depolymerize the high-value long-chain polyamides nylon 11 and nylon 12 into monomeric building blocks that can easily be reintegrated into the chemical industry. To our knowledge, this is the first report of an ammonolytic transfer dehydrogenation process that is generally applicable to amides and/or amides.
Data availability
All additional data regarding the work was included in the ESI.†
Author contributions
R. C. and D. E. D. V. were responsible for the concept and interpretation of the experiments. Additionally, R. C. was responsible for catalyst synthesis, characterization and design of the experiments. M. H. assisted in the execution and interpretation of the results. All authors contributed to writing the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
R. C. thanks VLAIO for research funding. M. H. and D. E. D. V. thank the FWO for research project funding. We would also like to thank Paul Van der Aerschot, Sam Van Minnebruggen, Aline Lauwers & Hendrik Van Dessel, Frederick Martens and Tim de la Croix for technical support, CO chemisorption experiments, assistance during FTIR experiments, assistance during N2 physisorption experiments and discussions about Pt-based dehydrogenation catalysts respectively.
References
-
P. Pollak, G. Romeder, F. Hagedorn and H.-P. Gelbke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, vol. 11, pp. 7672–7684 Search PubMed.
-
D. R. Axel Kleeman, J. Engel and B. Kutscher, Pharmaceutical substances syntheses, patents and applications of the most relevant AIPs, NY Thieme, Stuttgart New York, 2009 Search PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
-
A. Al-mamun and J. Y. Chen, Industrial Applications Biopolymers Environmental Impact, CRC Press, 2020 Search PubMed.
- J. Dallenes, J. Wuyts, N. Van Velthoven, A. Krajnc, G. Mali, O. A. Usoltsev, A. L. Bugaev and D. De Vos, Nat. Catal., 2023, 8, 16274–16282 Search PubMed.
- G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- S. Kamiguchi, A. Nakamura, A. Suzuki, M. Kodomari, M. Nomura, Y. Iwasawa and T. Chihara, J. Catal., 2005, 230, 204–213 CrossRef CAS.
- I. Dutta, S. Yadav, A. Sarbajna, S. De, M. Hölscher, W. Leitner and J. K. Bera, J. Am. Chem. Soc., 2018, 140, 8662–8666 CrossRef CAS PubMed.
- W. H. Bernskoetter and M. Brookhart, Organometallics, 2008, 27, 2036–2045 CrossRef CAS.
- M. Olivares, P. Knörr and M. Albrecht, Dalton Trans., 2020, 49, 1981–1991 RSC.
- L. Claes, J. Verduyckt, I. Stassen, B. Lagrain and D. E. De Vos, Chem. Commun., 2015, 51, 6528–6531 RSC.
- P. Capdevielle, A. Lavigne, D. Sparfel, J. Baranne-Lafont, K. C. Nguyen and M. Maumy, Tetrahedron Lett., 1990, 31, 3305–3308 CrossRef CAS.
- R. D. Patil and M. K. Gupta, Adv. Synth. Catal., 2020, 362, 3987–4009 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Synlett, 2010, 2365–2382 CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480–1483 CrossRef CAS PubMed.
- J. S. Belew, C. Garza and J. W. Mathieson, J. Chem. Soc. D, 1970, 634–635 RSC.
- J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 9249–9251 CrossRef CAS PubMed.
- R. Ray, A. S. Hazari, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2018, 24, 1067–1071 CrossRef CAS PubMed.
- S. K. Klitgaard, K. Egeblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419–442 RSC.
- S. Biswas, B. Dutta, K. Mullick, C. H. Kuo, A. S. Poyraz and S. L. Suib, ACS Catal., 2015, 5, 4394–4403 CrossRef CAS.
- L. Aschwanden, T. Mallat, M. Maciejewski, F. Krumeich and A. Baiker, ChemCatChem, 2010, 2, 666–673 CrossRef CAS.
-
J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 1–103 Search PubMed.
- Y. Goto, K. Shimizu, K. Kon, T. Toyao, T. Murayama and W. Ueda, J. Catal., 2016, 344, 346–353 CrossRef CAS.
- M. Lin, B. An, N. Niimi, Y. Jikihara, T. Nakayama, T. Honma, T. Takei, T. Shishido, T. Ishida, M. Haruta and T. Murayama, ACS Catal., 2019, 9, 1753–1756 CrossRef CAS.
- W. W. Scott and W. D. Leech, Ind. Eng. Chem., 1927, 19, 170–173 CrossRef CAS.
- J. H. Shin, G. J. Kim and S. C. Hong, Appl. Surf. Sci., 2020, 506, 144906 CrossRef CAS.
- L. Chmielarz and M. Jabłońska, RSC Adv., 2015, 5, 43408–43431 RSC.
- L. S. Kibis, D. A. Svintsitskiy, A. I. Stadnichenko, E. M. Slavinskaya, A. V. Romanenko, E. A. Fedorova, O. A. Stonkus, V. A. Svetlichnyi, E. D. Fakhrutdinova, M. Vorokhta, B. Šmíd, D. E. Doronkin, V. Marchuk, J.-D. Grunwaldt and A. I. Boronin, Catal. Sci. Technol., 2021, 11, 250–263 RSC.
-
P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2015, vol. 11, pp. 1–55 Search PubMed.
- R. Coeck, A. De Bruyne, T. Borremans, W. Stuyck and D. E. De Vos, ACS Sustainable Chem. Eng., 2022, 10, 3048–3056 CrossRef CAS.
- R. Coeck, J. Meeprasert, G. Li, T. Altantzis, S. Bals, E. A. Pidko and D. E. De Vos, ACS Catal., 2021, 11, 7672–7684 CrossRef CAS.
- R. Coeck and D. E. De Vos, Green Chem., 2020, 22, 5105–5114 RSC.
- R. Coeck, S. Berden and D. E. De Vos, Green Chem., 2019, 21, 5326–5335 RSC.
- D. Ruiz, A. Aho, T. Saloranta, K. Eränen, J. Wärnå, R. Leino and D. Y. Murzin, Chem. Eng. J., 2017, 307, 739–749 CrossRef CAS.
- F. Niu, S. Xie, M. Bahri, O. Ersen, Z. Yan, B. T. Kusema, M. Pera-Titus, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2019, 9, 5986–5997 CrossRef CAS.
- H. Qi, J. Yang, F. Liu, L. Zhang, J. Yang, X. Liu, L. Li, Y. Su, Y. Liu, R. Hao, A. Wang and T. Zhang, Nat. Commun., 2021, 12, 3295 CrossRef CAS PubMed.
- J. Wang, Q. Tang, S. Jin, Y. Wang, Z. Yuan, Q. Chi and Z. Zhang, New J. Chem., 2019, 44, 549–555 RSC.
- Y. Lv, J. Li, S. Feng, P. Liu, F. Hao, W. Xiong and H. Luo, Chem. Eng. J., 2018, 346, 203–216 CrossRef CAS.
-
H. Zimmermann and R. Walzl, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- J. Gu, H. Kim and H. Lim, Energy Convers. Manage., 2022, 270, 116256 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
-
Ineos, Project One [online], 2023, https://project-one.ineos.com/en, accessed April 2023 Search PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- R. Banerjee, D. A. Chen, S. Karakalos, M.-L. C. Piedboeuf, N. Job and J. R. Regalbuto, ACS Appl. Nano Mater., 2018, 1, 5876–5884 CrossRef CAS.
- W.-J. Lee, S. Bera, C. M. Kim, E.-K. Koh, W.-P. Hong, S.-J. Oh, E. Cho and S.-H. Kwon, NPG Asia Mater., 2020, 12, 40 CrossRef CAS.
- A. Plavniece, G. Dobele, A. Volperts, A. Zhurinsh and I. Kruusenberg, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 503, 012011 CAS.
- P. Kamedulski, P. A. Gauden, J. P. Lukaszewicz and A. Ilnicka, Materials, 2019, 12, 3354 CrossRef CAS PubMed.
-
F. S. Baker, C. E. Miller, A. J. Repik and E. D. Tolles, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, vol. 1914 Search PubMed.
- H. Yu, R. Zhang, F. Yang, Y. Xie, Y. Guo, W. Yao and W. Zhou, Trends Food Sci. Technol., 2021, 112, 795–807 CrossRef CAS.
- Z.-J. Zhao, L. V. Moskaleva, H. A. Aleksandrov, D. Basaran and N. Rösch, J. Phys. Chem. C, 2010, 114, 12190–12201 CrossRef CAS.
-
M. Pruski, X. Wu, M. W. Smale, B. C. Gerstein and T. S. King, in Studies in Surface Science and Catalysis, 1991, vol. 68, pp. 699–706 Search PubMed.
- A. Mekki-Berrada, S. Bennici, J. P. Gillet, J. L. Couturier, J. L. Dubois and A. Auroux, J. Catal., 2013, 306, 30–37 CrossRef CAS.
-
K. Visek, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2003, vol. 306, pp. 30–37 Search PubMed.
- I. Isik-Gulsac, Braz. J. Chem. Eng., 2016, 33, 1021–1030 CrossRef CAS.
- J. Shu, S. Cheng, H. Xia, L. Zhang, J. Peng, C. Li and S. Zhang, RSC Adv., 2017, 7, 14395–14405 RSC.
- A. Chafidz, W. Astuti, V. Augustia, D. T. Novira and N. Rofiah, IOP Conf. Ser.: Earth Environ. Sci., 2018, 167, 012013 CrossRef.
- B. Ren, D.-Y. Wu, B.-W. Mao and Z.-Q. Tian, J. Phys. Chem. B, 2003, 107, 2752–2758 CrossRef CAS.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- X. Zhang, A. Fried, S. Knapp and A. S. Goldman, Chem. Commun., 2003, 3, 2060 RSC.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional experimental data, characterization data, GC-MS and/or NMR spectra of all compounds (PDF). See DOI: https://doi.org/10.1039/d3sc02436a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Margot
Houbrechts
,
Margot
Houbrechts