
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-induced palladium–carbon bond weakening in catalytically relevant T-shaped complexes†
Peter M.
Waddell
a,
Lei
Tian
 a,
Anthony R.
Scavuzzo
a,
Lalu
Venigalla
b,
Gregory D.
Scholes
a and
Brad P.
Carrow
*b
a,
Anthony R.
Scavuzzo
a,
Lalu
Venigalla
b,
Gregory D.
Scholes
a and
Brad P.
Carrow
*b
aDepartment of Chemistry, Princeton University, Princeton, NJ 08544, USA
bDepartment of Chemistry, University of Houston, Houston, TX 77204, USA. E-mail: bcarrow@uh.edu
First published on 28th November 2023
Abstract
Triggering one-electron redox processes during palladium catalysis holds the potential to unlock new reaction mechanisms and synthetic methods not previously accessible in the typical two-electron reaction manifolds that dominate palladium catalysis. We report that T-shaped organopalladium(II) complexes coordinated by a bulky monophosphine, a class of organometallic intermediate featured in a range of contemporary catalytic reactions, undergo blue light-promoted bond weakening leading to mild and efficient homolytic cleavage of strong Pd(II)–C(sp3) bonds under ambient conditions. The origin of light-triggered radical formation in these systems, which lack an obvious ligand-based chromophore (i.e., π-systems), was investigated using a combination of DFT calculations, photoactinometry, and transient absorption spectroscopy. The available data suggest T-shaped organopalladium(II) complexes manifest unusual blue light-accessible Pd-to-C(sp3) transition. The quantum efficiency and excited state lifetime of this process were unexpectedly superior compared to a prototypical (α-diimine)Pd(II) complex featuring a low-lying, ligand-centered LUMO (π*). These results suggest coordinatively-unsaturated organopalladium(II) compounds, catalysts in myriad catalytic processes, have untapped potential for one-electron reactivity under visible light excitation.
Introduction
The photophysical properties of transition metal complexes have for many years been the subject of intense interest and have been exploited for a myriad of applications, from synthetic and preparative chemistry to artificial photosynthesis.1–9 A variety of electronic transitions are possible when a transition metal complex absorbs light. For metal complexes with 1 to 9 d electrons, d–d transitions can occur.10,11 Another important class of electronic transition is metal-to-ligand charge transfer (MLCT), which involves transitions from a d-orbital to low-lying antibonding orbitals on the ligands (e.g., π*) and generally feature intense absorption bands. The resulting charge separation can engender reactivity not accessible in the ground state. For example, [Ru(2,2′-bipyridine)3]2+ is both a better oxidant and better reductant in the excited state, and this characteristic is an important feature of the photocatalysts used in photoredox processes.4,6,12–14Depending on the nature of the orbitals involved in the electronic transition (i.e. bonding, antibonding or nonbonding), excitation may strengthen or weaken bonds in the complex and lead to chemical reactivity. A classic example of this phenomenon is photoinduced CO dissociation from metal carbonyl complexes (e.g., M(CO)6, M = Cr, Mo, W).15–20 In this case, absorption of a photon promotes a transition from an orbital that is π-bonding to an orbital that is σ-antibonding with respect to the M–L bond. Excited state reactivity in metal complexes has emerged as a broadly useful concept in synthetic chemistry.2,21 As a corollary, the discovery of previously unknown excited state reactivity, particularly with complexes with established utility in catalysis, holds the potential to enable novel and interesting transformations. In this regard, palladium is one of the most used transition metals in catalysis yet the large majority of processes developed using Pd catalysts operate under ground state conditions. While several photoredox systems have been developed which use Ru or Ir photosensitizers to absorb visible light and subsequently perform electron or energy transfer with Pd or Ni catalysts, transformations that take advantage of direct photoexcitation of Pd catalysis are less explored.22–38
The interaction of visible light with Pd(0) complexes has recently been advanced by the groups of Gevorgyan, Shang and Fu, Ryu and others as a method for one-electron reduction of organic halides that departs from the typical strong preference of Pd(0) to react by two-electron oxidative addition mechanisms (Scheme 1).39 This manifold has been used in a wide range of reactions including Heck and Negishi-type couplings, the coupling of alkyl halides with C(sp2)–H and C(sp3)–H sites, desaturations, tandem reactions involving the cyclization of radical intermediates with carbonylation, C–C coupling, and others.40–56 However, these examples only alter catalysis at the oxidative addition step.
 | ||
| Scheme 1 Photochemical excitation of Pd(0) versus Pd(II). | ||
Light-promoted reactivity from organo–Pd(II) species, on the other hand, represents a less explored aspect that could expand the utility of one-electron reactivity in Pd catalysis,57–59 especially considering these intermediates can be accessed not just by oxidative addition but other elementary reactions as well (e.g., transmetalation, migratory insertion, C–H activation, etc.). Prior studies have established the potential for organopalladium complexes to undergo photochemical reactions (Scheme 2). Complexes featuring tri- or tetradentate ancillary ligands, such as PNP or PCP pincer ligands in independent studies by Ozerov and Goldberg or N4 ligands by Mirica undergo Pd–C cleavage under UV irradiation.60–63 Photochemical Pd(II)–C scission in (bisphosphine)Pd complexes has also been observed, which occurred using visible light in cases of weaker metal–acyl bonds in work by Arndtsen while stronger Pd(II)–alkyl bonds required UV light.64–66
 | ||
| Scheme 2 Representative examples of Pd(II)–C photocleavage from organo–Pd(II) complexes. | ||
Recently, Harth and co-workers reported that Pd catalysts featuring a redox active α-diimine ligand were able to liberate free radicals during olefin polymerization upon irradiation with blue LEDs. This phenomenon was exploited to prepare block copolymers by light-induced switching from isomerizing coordination-insertion polymerization of 1-hexene in the dark to radical polymerization of an acrylate under visible light irradiation.67–69 Accessing mild, visible light-inducted photochemistry with organo–Pd complexes coordinated by privileged ligand classes would be desirable to more broadly exploit the utility of light-induced one-electron chemistry in Pd catalysis.
Photochemical reactions of coordinatively unsaturated organopalladium complexes lacks precedent even though sterically hindered alkylphosphines that generate such species are some of the most frequently used ancillary ligands in modern Pd chemistry. The high donicity and the steric pressure of these ligands facilitates catalyst stability by resisting deactivation via deligation and accelerates reductive elimination, respectively.70–76 Tri-tert-butylphosphine in particular has been applied extensively across many contemporary synthetic organic methods, including C–C bond-forming reactions such as Migita–Kosugi–Stille, Suzuki–Miyaura, Sonogashira, Murahashi and Mizoroki–Heck reactions, as well as Buchwald–Hartwig amination and related C–O cross-coupling reactions.77–85 A related bulky monophosphine Ad3P (Ad = 1-adamantyl) has also emerged recently as an effective ligand for challenging Pd- and Ni-catalysed reactions.86–92 Furthermore, complexes such as (tBu3P)Pd(CH3)Cl (1a) have been demonstrated as catalysts for alkene insertion (co)polymerizations in a living fashion.93–95 In this study we report on a serendipitous discovery that T-shaped organo–Pd complexes ligated by tBu3P or Ad3P undergo efficient visible light-induced bond weakening, which induces homolysis of otherwise strong Pd(II)–C(sp3) bonds under ambient conditions. Experiments and computations in this study shed light on the nature and fate of the relevant excited state involved in these processes.
Results and discussion
Visible light photochemistry of T-shaped complexes
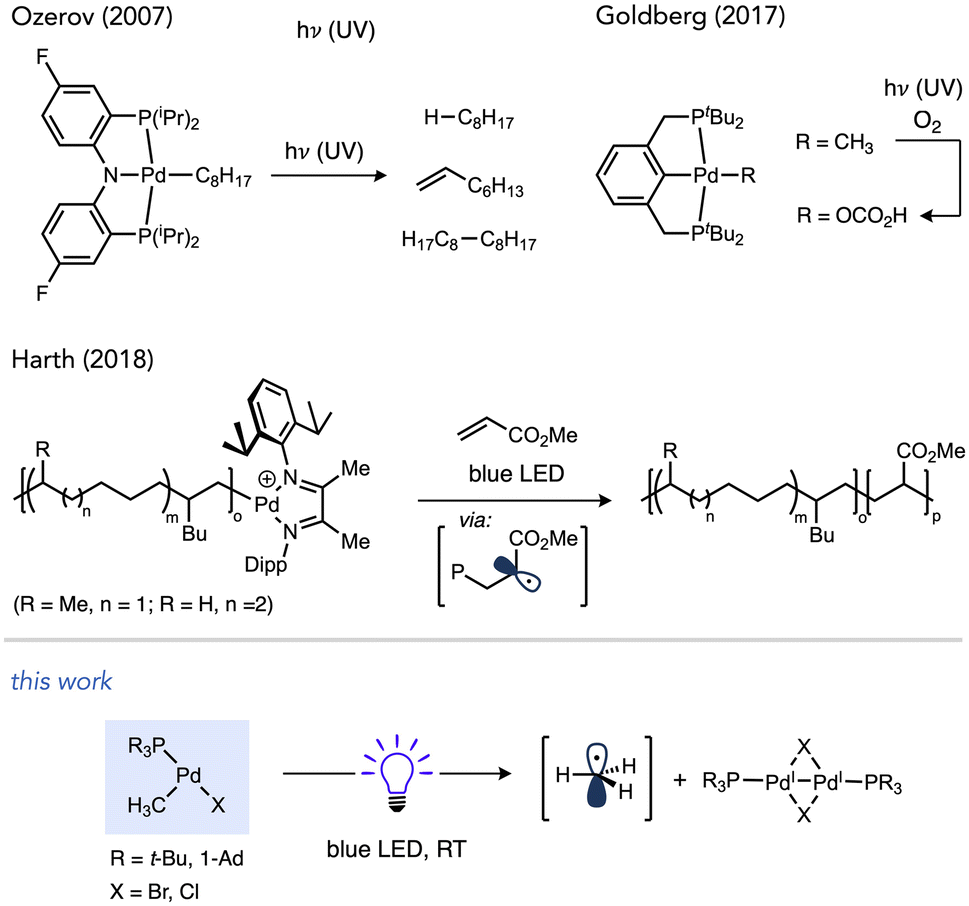
The known T-shaped complex (tBu3P)Pd(CH3)Cl (1a)96 is a bench-stable solid under ambient light. It is also indefinitely stable in CDCl3 in the dark. However, we found that irradiation of a CDCl3 solution of 1a with blue LED resulted in the formation of CH4 (16%) and CH3D (44%) over 12 h at RT, as determined by 1H NMR versus cyclohexane as an internal standard (Scheme 3). This observation is indicative of the formation of methyl radicals and their subsequent trapping by hydrogen atom abstraction, likely from the phosphine ligand and/or solvent. The addition of 1,4-cyclohexadiene as an H-atom donor led to an increase in the yield of CH4 (33%) and is also consistent with methyl radical formation.97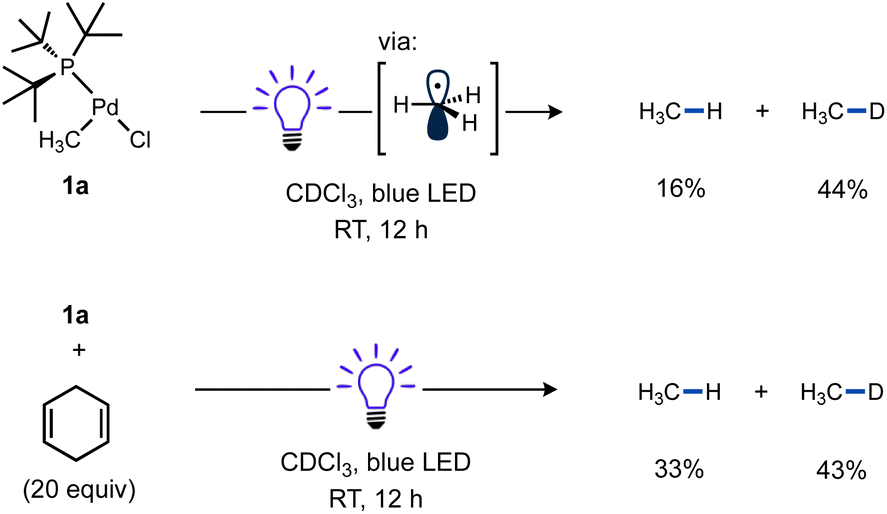 | ||
| Scheme 3 Reactivity of T-shaped complexes under blue LED irradiation. Cyclohexane was included as a 1H NMR standard. In the reaction with 1,4-cyclohexadiene (20 equiv.), Bu4P+BF4− was also included as a 31P NMR standard. A fan was used to cool the reaction mixtures. Yields determined by NMR. See ESI† (p. S28) for full details. | ||
A reaction mixture of 1a and excess TEMPO, a common trap for radicals, was used to monitor evolution of methyl radical over time. Reaction progress of this mixture upon irradiation with blue LEDs was monitored using 1H and 31P NMR spectroscopy. Consumption of the starting material occurred cleanly along with formation of TEMPO–CH3 as the sole observable organic product in all cases (Fig. 1). No reactivity was observed in the absence of light when irradiation was intermittently ceased, demonstrating that the system is well-behaved and inconsistent with a sustained radical chain process.
 | ||
| Fig. 1 Light on–off study of 1a in the presence of excess TEMPO; white indicates blue LED irradiation while grey indicates dark. Reaction performed using fiber optic insert in NMR probe, see ESI page S47† for full details. | ||
Additional experiments were performed to test the possibility of thermal reaction caused by heat from the LEDs, despite the use of cooling fans in these reactions. A mixture of 1a and TEMPO remained unchanged in the dark at RT after 5 h as observed by 1H and 31P NMR. Even heating at 60 °C for 1.5 h gave no significant spectroscopic changes (Scheme 4, top). In contrast, another sample of 1a and TEMPO kept at RT while irradiating for 1.5 h yielded a comparable amount of TEMPO–CH3 as the previous reactions (Scheme 4, bottom). These results support the requirement for visible light to form methyl radicals from 1a and the absence of a significant thermal background reaction.
 | ||
| Scheme 4 Reactivity of T-shaped complexes under blue LED irradiation with TEMPO. 1,3,5-(CF3)3C6H3 and Bu4P+BF4− were included as 1H and 31P NMR standards. A fan was used to cool the reaction mixture. Yields determined by NMR. The photochemical reaction was run to 64% yield, as determined by 31P NMR. | ||
Considering the strong Pd(II)–CH3 bond98–100 was effectively cleaved, it should be expected other (weaker) Pd(II)–C(sp3) bonds would also be susceptible to homolysis under blue light irradiation. To test this idea, a new T-shaped complex (4) bearing a neosilyl group was prepared. Complex 4 did indeed react with TEMPO upon blue LED irradiation and generated the corresponding radical trapping product TEMPO–CH2SiMe3 in nearly quantitative yield as determined by 1H NMR (Scheme 5). Formation of an unidentified Pd product 5a (vide infra) in high yield was also observed by 31P NMR.
 | ||
| Scheme 5 Reactivity of T-shaped Pd complex with a weaker Pd–C bond. 1,3,5-(CF3)3C6H3 and Bu4P+BF4− were included as 1H and 31P NMR standards. A fan was used to cool the reaction mixture. Yield determined by NMR. | ||
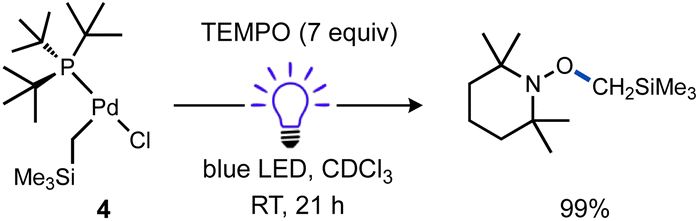
The fate of the Pd product generated after Pd(II)–C bond cleavage was also investigated, which presumably involves a one-electron reduction of Pd(II) upon alkyl radical ejection. Conveniently, [(tBu3P)PdI(μ-X)]2 (X = Br, I) species are stable diamagnetic complexes, although the chloride-bridged congener is unknown.101,102 For this reason, the T-shaped complex with a bromide ligand (1b) was also prepared and investigated. Formation of these stable dimers from the putative mononuclear Pd(I) generated under photochemical conditions should be spectroscopically observable, but consumption of Pd(I) species prior to dimerization might also occur, such as by disproportionation. Examination of the 31P NMR spectrum of the reaction of 1a (δP 70.0 ppm) with TEMPO under blue light irradiation (Scheme 6a) indicated clean formation of a new phosphorus-containing species 5a (δP 88.1 ppm) in 96% yield. Analogous experiments using either 1b (δP 68.4 ppm) or the Ad3P-coordinated congener 2 (δP 53.2 ppm) also generated new 31P NMR resonances at δP 90.4 (5b) and 76.9 ppm (S2) in high yields (96% and 95%), respectively. The yield of TEMPO–CH3 was also high in all cases (70–89%). Importantly, the 31P NMR resonance of product 5b does not match the reported shift for [(tBu3P)PdI(μ-Br)]26 (δP 86.3 ppm)103 indicating a unique Pd product formed under these conditions.
 | ||
| Scheme 6 Identity of Pd product of irradiation of T-shaped complexes in the absence and presence of excess TEMPO. In (a), cyclohexane and Bu4P+BF4− were included as NMR standards, the reactions were cooled with a fan. Yields were determined by NMR for (a) and (c). In (b), yield was determined gravimetrically. Yield of 6 based on total Pd. | ||
We subsequently found that the combination of Pd(I) complex 6 and TEMPO led to an immediate colour change upon mixing and formed 5b in 97% yield (Scheme 6b). The solid-state structure of 5b subsequently revealed this complex to be a Pd complex coordinated by TEMPO in an η2-N,O fashion, which has been observed in a few other cases and comparable to known η2-nitrosoarene adducts.69,104–106 The solid-state structures of 5a (as illustrated in Fig. 2) and an analogous Ad3P-coordinated complex S2 (see Fig. S206†) were obtained from photochemical reactions of 1a and 2 respectively. Each of these complexes is indefinitely stable under air at room temperature. Based on the similarity of Pd–N and Pd–O bond lengths (±0.04 Å) and N–Pd–O bond angles (±0.2°) in the η2-TEMPO complexes 5a, 5b, and S2, and the reference compounds with three membered rings of Pd(II), N and O reported by Figueroa and Harth, we likewise assign these as Pd(II) complexes.69,106 This is consistent with our hypothesis that a Pd(I) species forms as the immediate transition metal product following light-induced Pd(II)–C homolytic cleavage, which then undergoes rapid one-electron oxidation by the excess TEMPO present under these reaction conditions.
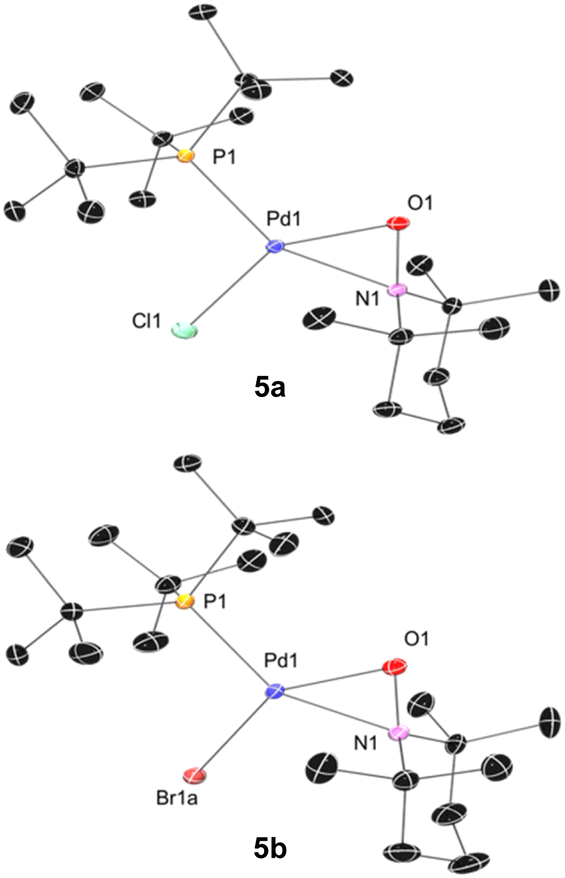 | ||
| Fig. 2 Solid state structure of 5a (left) and 5b (right) (50% thermal ellipsoids; H atoms omitted). Selected bond lengths (Å) and angles (deg) for 5a: Pd1–N1 = 2.0882(6); Pd1–O1 = 2.0415(6); N1–O1 = 1.3611(8); Pd1–P1 = 2.3082(4); Pd1–Cl1 = 2.3461(5); P1–Pd1–O1 = 112.09(2); O1–Pd1–N1 = 38.47(2); N1–Pd1–Cl1 = 106.56(2); Cl1–Pd1–P1 = 102.83(1). For 5b: Pd1–N1 = 2.109(1); Pd1–O1 = 2.036(1); N1–O1 = 1.3642(7); Pd1–P1 = 2.3099(6); Pd1–Br1A = 2.482(2); P1–Pd1–O1 = 110.77(3); O1–Pd1–N1 = 38.38(3); N1–Pd1–Br1a = 106.28(5); Br1a–Pd1–P1 = 104.34(4) (disorder in Br and P methyl carbons omitted). | ||
A different product distribution was formed when 1b was irradiated in the absence of TEMPO. Under these conditions formation of the Pd(I) complex 6 can be detected by 31P NMR (Scheme 6c). Complex 6 gradually disappears with concomitant formation of (tBu3P)2Pd(0) over time, possibly due to a disproportionation side reaction. The latter complex eventually becomes the dominant species observable by 31P NMR.
We were curious to test if alkyl radicals generated from Pd–C photocleavage could be trapped not only by radical scavengers but by another organometallic complex leading eventually to formation of a C–C cross-coupling product from the putative higher-valent metal species generated upon radical attack. We selected a candidate complex (Ad3P)Pd(p-C6H4F)Br (7) featuring a stronger Pd–C(sp2) bond that should be less susceptible to photolysis. Upon irradiation of 1b in the presence of 7, formation of 4-fluorotoluene (54%) was observed (Scheme 7). Interestingly, the yield of the Pd(I) dimer 6 was significantly higher (86%) under these conditions, likely due to complex 7 being a much weaker oxidant than TEMPO. Because no 4-fluorotoluene formation was observed in the absence of light, we propose methyl radical generated from photoexcited 1b is captured by 7 to generate a transient Pd(III) intermediate that undergoes facile reductive elimination of 4-fluorotoluene. This light-enabled Pd-to-Pd transmetalation highlights the potential for visible light to stimulate intermetallic reactivity not observed in ground state Pd chemistry. This experiment also validates that the reduced Pd species can be generated efficiently under certain photolytic conditions, which bodes well for the potential for integrating this light-driven chemistry into novel catalytic reactions considering the established intermediacy of Pd(I) species in numerous Pd-catalysed reactions.107–112
 | ||
| Scheme 7 Observation of light-induced, Pd-to-Pd radical-based transmetalation. Additionally, 1,4-dioxane and Bu4P+BF4− were included as NMR standards. Yields determined by NMR. Yield of 6 based on total Pd. | ||
Methyl radical generation from these monophosphine-ligated T-shaped complexes might be expected to occur with lower photochemical efficiency due to the lack of typical low-lying ligand-based acceptor orbitals (e.g., π*) commonly associated with MLCT in transition metal photochemistry.13,113–117 Nevertheless, quantum yield measurements for reactions of 1a and related T-shaped complexes revealed light-induced Pd(II)–C homolysis from these species can in fact be more efficient as compared to prototypical photoactive compounds, such as (α-diimine)Pd(II) complexes.67
Quantum yields for Pd(II)–C photocleavage were determined using photo-NMR actinometry (see ESI† for full experimental details) according to the method reported by Ji and co-workers.118 Reactions were performed using either of two representative complexes, 1a or [ArN = C(Me)C(Me) = NAr]Pd(CH3)Cl 3 (Ar = 2,6-iPr2C6H3). The similarity of the reaction rates conducted at two different concentrations ([Pd]o = 60 mM or 120 mM for 1a, [Pd]o = 30 mM or 60 mM for 3) in the presence of TEMPO (7 equiv.) were consistent with zeroth-order dependence on [Pd] and consequently occurs within a photon-limited kinetic regime inside this concentration range. The quantum yield (Φobs = kpc,0/I0 where kpc,0 is the observed initial rate of Pd–C photocleavage, and I0 is the photon flux determined by actinometry, see ESI† for full details) for 1a under these conditions was calculated to be 0.67 (Table 1, entry 1). Analogous experiments conducted in C6D6 or MeCN-d3 indicated a weak dependence of quantum yield on solvent identity (see ESI,† page S86). The related Ad3P–ligated complex 2 gave Φ = 0.36 for formation of TEMPO–CH3 in CDCl3 (Table 1, entry 3). Importantly, the quantum yields for both T-shaped complexes are higher compared to the reference complex 3 (Φ = 0.11) under otherwise identical conditions (Table 1, entry 5). Note that repetition of these measurements in the absence of TEMPO, which absorbs light in the visible region119 and might interfere with quantum yield measurements, gave only a modest increase for T-shaped 1a (Φ = 0.67–0.73) or α-diimine complex 3 (Φ = 0.11–0.16) as shown in Table 1, entries 1 vs. 2 or 4 vs. 5. We conducted several additional experiments and computations to understand the origin of the counterintuitive trend of greater quantum efficiency for the T-shaped complexes compared to the diimine–ligated complex.
|
|
|||
|---|---|---|---|
| Entry | Complex | X | Φ obs |
| a 1,3,5-Tris(trifluoromethyl)benzene was included as NMR standard. Reactions monitored by NMR. In the reactions with TEMPO, the rate of TEMPO–CH3 evolution was used to calculate Φobs, while in the reactions without TEMPO, the rate of consumption of Pd–CH3 was used to calculate Φobs. | |||
| 1 | 1a | 7 | 0.67 |
| 2 | 1a | 0 | 0.73 |
| 3 | 2 | 7 | 0.36 |
| 4 | 3 | 7 | 0.11 |
| 5 | 3 | 0 | 0.16 |
It could be the case that differences in the excited state energy profiles of 1a and 3 might give rise to a discrepancy in activation energy for metal–carbon bond homolysis in these two representative species. To probe this possibility, the temperature dependence of photocleavage quantum yield was used to calculate an activation energy barrier for each photochemical reaction.
 | (1) |
An Arrhenius-type plot (Fig. 3) was constructed according to eqn (1) for reactions of 1a or 3 with TEMPO monitored by photo-NMR spectroscopy (440 nm LED) at six different temperatures.117 The calculated activation energies for these reactions were Ea = 4 kcal mol−1 and 3 kcal mol−1 for 1a and 3, respectively. The similarity of these small energy barriers for Pd(II)–C cleavage at the excited state therefore does not account for the observed differences in quantum yield between the two types of organo–Pd complex. Additional computational and spectroscopic investigations were informative in parsing other possible origins of efficiency differences.
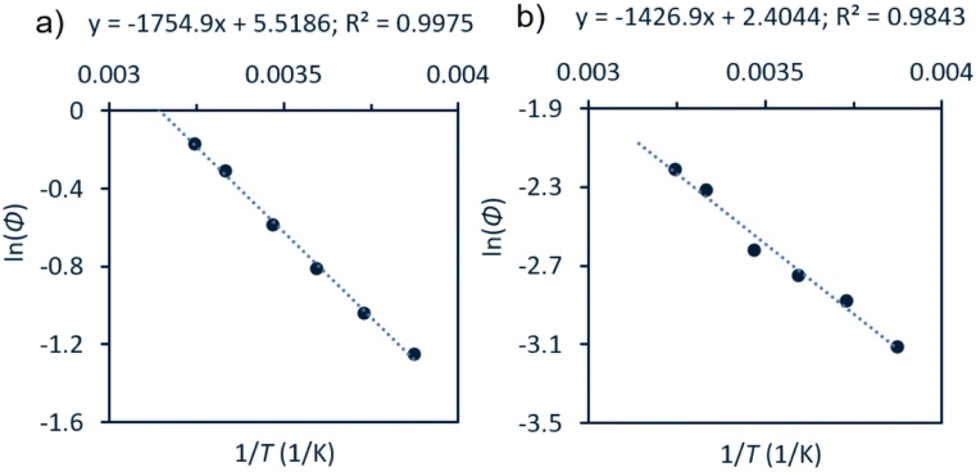 | ||
| Fig. 3 Arrhenius-type analyses for photochemical reaction of 1a or 3 with TEMPO. Conditions: a solution of Pd complex (60 mM for 1a, (a); 30 mM for 3, (b)) and TEMPO (7 equiv.) in CDCl3 were irradiated at room temperature using photo-NMR (440 nm LED). The initial rate was taken from each reaction and used to calculate Φ. See ESI,† page S111 for full details. | ||
Excited state analysis by DFT calculations
The electronic transitions that could facilitate Pd(II)–C homolytic cleavage were investigated by density functional theory (DFT) calculations using complex 1a as a representative case. Calculations were performed using various functionals (vide infra), the 6-311G++(d,p)/LANL2DZ(Pd) GD3 basis set and solvent corrected with the SMD(CHCl3) solvent model. Agreement between simulated and experimental (Fig. S172†) UV-vis spectra was used as a criterion for computational method optimization.120 The general features of the spectrum are matched qualitatively in the simulated spectrum using CAM-B3LYP, including the absorbance in the 400–500 nm region,121 that overlaps the spectral range of blue LED output. A similar absorption spectrum was observed for 2 (Fig. S187†). The HOMO and LUMO of 1a are illustrated in Fig. 4, which suggests the HOMO orbital is predominantly dz2(Pd) in character and the LUMO features dx2−y2(Pd) and σ*(Pd(II)–CH3) character. | ||
| Fig. 4 Simplified energy level diagram of HOMO (two views) and LUMO of 1a. | ||
Linear time-dependent density-functional theory (TD-DFT) calculations were also performed. From these calculations the HOMO–LUMO gap was calculated to be ca. 2.72 eV, equivalent to the energy of a 456 nm photon. We postulate the relatively low energy of a dx2−y2/σ* orbital in these complexes might be facilitated by the T-shaped geometry due to the absence of a ligand trans to the methyl ligand (and associated destabilizing trans influence). Excitation leading to population of an orbital with anti-bonding character would be expected to weaken the metal–ligand bond.
Population analysis using TD-DFT calculations indicates that excitation of 1a at 456 nm is accompanied by a decrease of the Pd(II)–C bond order by 0.43. The difference density isosurface for this electronic transition (Fig. 5a) illustrates that the decrease in bond order is correlated to a decrease in electron density at Pd with a concomitant increase in electron density at the CH3 group by 0.32 e and 0.25 e, respectively (see ESI,† Table S22 for tabular data). We interpret these data as indicative of an uncommon Pd-to-CH3 MLCT-like process. We also note that extinction coefficient measurements at λmax for this absorption (370 nm) indicate ε = 2.2 × 103 M−1 cm−1, which is within a generally accepted range for transitions involving significant charge separation character (for 3, ε = 4.3 × 103 M−1 cm−1, see ESI page S184†).122–127 This contrasts what has been proposed for the electronic transition underpinning the photochemistry of (α-diimine)Pd complexes, such as 3,67,68via traditional d-to-π* MLCT that populates an ancillary ligand-centred orbital. The potential to stimulate charge transfer to unactivated (i.e. lacking groups that stabilize radicals) alkyl groups of an organometallic complex highlights that π-based ancillary ligands are not essential for achieving mild, efficient photochemistry in Pd complexes. It may also be inferred that there may exist a reservoir of untapped photochemical reactivity in organo–Pd(II) complexes previously presumed to be photochemically inert due to a lack of ancillary ligand-based chromophores.
 | ||
| Fig. 5 Difference density isosurfaces for transitions occurring at 459 nm and 458 nm for (a) 1a and (b) at 456 nm for S3. Units for scale: electrons per au3 (au is Hartree atomic units). Negative/red represents electron loss, positive/blue represents electron gain. | ||
For comparative purposes the truncated complex [PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)C(Me)NPh]Pd(CH3)Cl (S3) was also examined. The optimal functional, based again on best agreement of experimental and calculated absorption spectra (see ESI,† Fig. S171), was M06 in this case. Difference density isosurface calculations for S3 confirmed an expected d-to-π* MLCT transition at 459 nm along with another transition at 458 nm which appears to have Cl-to-π* character (Fig. 5b). Population analysis of this (α-diimine)Pd(II) system showed an average loss of 0.29 e at Pd and gain of 0.5 e on the diimine ligand (see ESI,† Table S20 for tabular data). A significant change in Pd(II)–C bond order was not observed for this complex, which is not yet understood. One rationalization could be the need for thermalization of the Franck–Condon state, intersystem crossing and/or a change in geometry needed to reach another excited state (e.g., S1 or T1) prior to weakening of the Pd(II)–C bond. If true this would shorten the window of time where Pd–C cleavage is possible, and would therefore contribute to the difference in quantum yield between 3 and 1a.
C(Me)C(Me)NPh]Pd(CH3)Cl (S3) was also examined. The optimal functional, based again on best agreement of experimental and calculated absorption spectra (see ESI,† Fig. S171), was M06 in this case. Difference density isosurface calculations for S3 confirmed an expected d-to-π* MLCT transition at 459 nm along with another transition at 458 nm which appears to have Cl-to-π* character (Fig. 5b). Population analysis of this (α-diimine)Pd(II) system showed an average loss of 0.29 e at Pd and gain of 0.5 e on the diimine ligand (see ESI,† Table S20 for tabular data). A significant change in Pd(II)–C bond order was not observed for this complex, which is not yet understood. One rationalization could be the need for thermalization of the Franck–Condon state, intersystem crossing and/or a change in geometry needed to reach another excited state (e.g., S1 or T1) prior to weakening of the Pd(II)–C bond. If true this would shorten the window of time where Pd–C cleavage is possible, and would therefore contribute to the difference in quantum yield between 3 and 1a.
Examination of excited state optimized geometries of representative T-shaped (1a) and α-diimine (S3) complexes revealed considerable differences in the type and degree of distortion between the two classes. The optimized geometry for the triplet excited state of 1a only subtly distorts from the ground state T-shaped geometry (α = 172°, β = 97° and γ = 91°)128 toward Y-shaped (α = 150°, β = 112° and γ = 98°). On the other hand, S3 shows a significant geometry change from square planar in the ground state (τ4 = 0.10) toward tetrahedral (τ4 = 0.58).129 The triplet state geometries and overlaid higher energy SOMO orbitals are illustrated in Fig. 6. The change in bond order between the S0 and T1 states was calculated to decrease by 0.49 and 0.11 for 1a and S3, respectively (see ESI,† Table S19 for tabular data). While the bond order change is modest for the (α-diimine)Pd complex, it may be consistent with the notion that bond weakening in these complexes may occur after thermalization from the Franck–Condon state. Unfortunately, extensive efforts to optimize the geometries of the lowest singlet excited state for 1a and S3 were unsuccessful. Considering the experimentally small activation barrier of Pd(II)–C homolysis upon photoexcitation, a conical intersection between the potential energy surfaces between S1 and S0 may exist, which is a documented challenge for TD-DFT calculations.130,131 In any case, the optimized triplet states were subsequently used to estimate the free energy of bond weakening.
| 1a or 1b → tBu3PdIX + H3C˙ | (2) |
 | (3) |
 | (4) |
 | ||
| Fig. 6 Optimized triplet excited state geometries (T1) for 1a (left) and S3 (right) showing the higher-energy SOMO. | ||
Based on homolytic Pd(II)–C cleavage from the ground state T-shaped complexes summarized in eqn (2), the bond dissociation free energy and enthalpy were calculated to be BDFE (BDE) = 34.3 (47.0) and 33.5 (46.5) kcal mol−1 for T-shaped complexes 1a and 1b, respectively (DLPNO-CCSD(T)/def2-TZVPP//B3LYP-D3/6-311G++(d,p)/LANL2DZ(Pd); see ESI† for full details). These data are consistent with experimental observations that homolysis does not occur from the ground state under thermal conditions alone. Isodesmic reactions (eqn (3) and (4)) were also used to estimate the free energy change in the Pd(II)–CH3 bond upon excitation from the ground to triplet state (see ESI,† page S139 for full details). These calculations used the experimentally determined BDFE for [Pd(II)–CH3]+ (59 kcal mol−1) based on ion beam reactive scattering.132 Experimental BDFEs of more closely related Pd(II)–CH3 complexes were not found, but relative energy changes benchmarked against this gas phase species are still informative. By this method the Pd(II)–C BDFE in T-shaped 1a is estimated to weaken by 20 kcal mol−1 in the triplet excited state. Similarly, the Pd(II)–C bond in α-diimine complex S3 is estimated to weaken by 28 kcal mol−1 in the triplet excited state. These data further corroborate the feasibility of facile homolysis of otherwise strong Pd(II)–C(sp3) bonds upon visible light excitation at ambient temperature.
Transient absorption spectroscopic measurements
The relevant excited state for T-shaped and α-diimine Pd complexes can potentially decay through radiative (luminescence) or other nonradiative decay (NRD) pathways (i.e., solvent-mediated) that do not lead to radical formation, in addition to the pathway resulting in Pd(II)–C homolytic cleavage. One of the former two processes might explain the discrepancies in quantum yield we observe for methyl radical formation from either 1a or 3 if its relative rate is fast enough to dominate the fate of the complex's excited state. We investigated the luminescence properties of these complexes to test this hypothesis. Luminescence measurements were complicated by sample decomposition due to the Pd(II)–C homolysis process, as determined by changes in the absorption spectra even after a single scan. As such, data were collected using low beam power and the sample solution was replenished between each scan to minimize the influence of sample decomposition.133 Features consistent with luminescence emission were not detected from either 1a or the α-diimine complex 3. These data suggest radiative decay is a negligible relaxation pathway for both types of complexes.Transient absorption (TA) spectroscopy was used to study the excited state lifetimes of 1a and 3. These measurements were used to assess the relative rates of NRD between the T-shaped and α-diimine Pd complexes; since both complexes appear to have similar barriers to Pd–C cleavage and do not undergo significant luminescence decay, we hypothesize that differences in their excited state lifetimes are likely due to different rates of unproductive NRD. In addition, TA measurements can also provide information about the magnitude of charge transfer between ground and excited states.134,135 To ensure that the amount of photodegradation was minimized to the extent possible, the intensity of the laser was optimized and pre- and post-scan absorption measurements were performed. A ground state bleach from 375–410 nm is apparent for 1a (Fig. 7a) coinciding with the absorptive region in the absorption spectrum. A broad excited state absorption is also evident from 410 to beyond 610 nm, which decays within ca. 50 ps (Fig. 7b). This latter feature was assigned as the excited state decay of 1a. A long-lived feature peaked at 450 nm follows, which persists out to the nanosecond timescale. This is consistent with the difference absorption obtained from pre- and post-scan steady-state measurements (Fig. S199†). This feature is consequently assigned to a photodegradation process.
 | ||
| Fig. 7 Transient absorption data for 1a (ΔOD = change in optical density). (a) TA difference spectrum for 1a in deoxygenated CDCl3, pumping at 400 nm (100 μW) at various delay times. Note the scattering feature around 400 nm. (b) Transient behaviour at 450 nm from 0 to 50 ps. | ||
A ground state bleach around 360–480 nm is apparent in the TA spectrum of 3 (Fig. 8a), which represents absorption into the MLCT band. The MLCT absorption is convoluted with a broad excited state absorption across the spectral range. The excited state absorption decays to baseline within 25 ps (Fig. 8b) suggesting a relatively low degree of photodegradation and is consistent with the modest quantum yield for Pd(II)–C homolysis in 3vs.1a. This fast absorption decay for 3 also indicates a large proportion of the sample returns to the ground state.
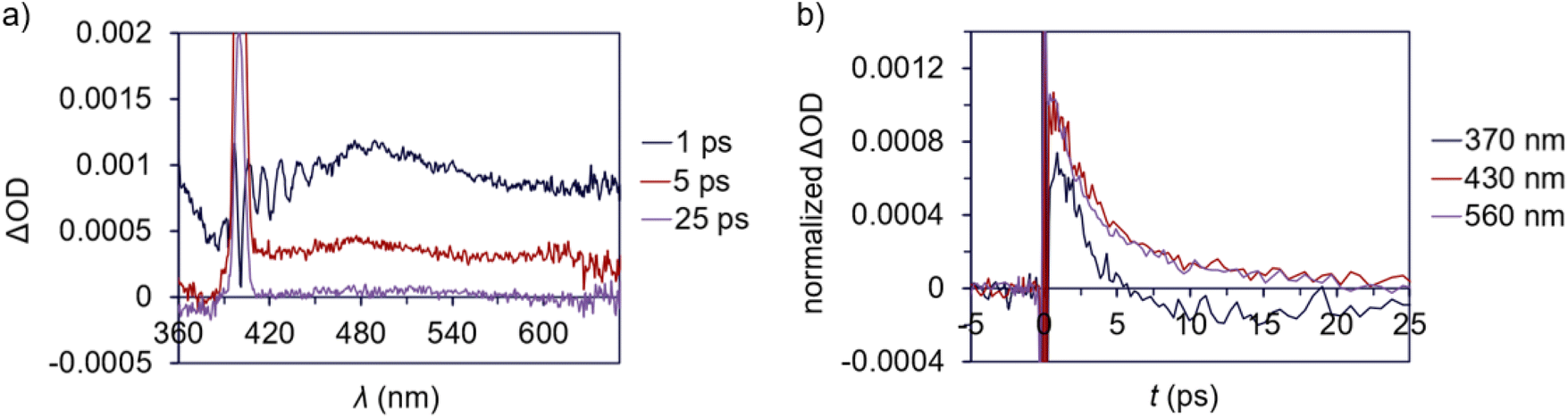 | ||
| Fig. 8 Transient absorption data for 3 (ΔOD = change in optical density). (a) TA difference spectrum for 3 in deoxygenated CDCl3, pumping at 400 nm (100 μW) at various delay times. Note the scattering feature around 400 nm. (b) Transient behaviour at 370, 430 and 560 nm from 0 to 25 ps. | ||
The excited state lifetime of T-shaped complex 1a is about twofold longer than for α-diimine complex 3 based on the time constants of 6.4 and 3.1 ps, respectively, determined from TA analyses in CDCl3. Measurements taken in less polar solvent (benzene) yielded time constants of 14 ps for 1a and 3.9 ps for 3 (Fig. S197 and S195† respectively). There are no significant differences in the absorption spectra for either complex in CDCl3 compared to C6H6 (Fig. S198†). The higher sensitivity of the excited state lifetime of 1a to solvent polarity could reflect significant charge separation between the ground and excited state,134–136 which would be consistent with the substantial charge transfer suggested by difference density isosurface calculations for this complex (vide supra). Considering the absence of significant radiative decay for either complex and similarity of energy barriers toward Pd(II)–C homolysis, we postulate the longer excited state lifetime of 1a may result from lower rates of unproductive NRD compared to 3 (i.e., NRD that does not result in Pd(II)–C homolysis). Longer excited state persistence would then account for higher quantum yield in the T-shaped complexes by extending the window of time in which thermal homolysis of the Pd(II)–C bond can occur.
A qualitative diagram shown in Fig. 9 summarizes our current understanding of the evolution of the excited states for representative T-shaped and α-diimine organo–Pd(II) complexes. Spin state and associated intersystem crossing details are omitted from this diagram; the dynamics of the TA transients are well-described by single time constants and are thus consistent with Pd(II)–C cleavage from one excited state, but unambiguous assignment of the spin state based on the currently available data was precluded. Visible light excitation of 1a promotes an electron from a nonbonding orbital to one with σ*(Pd(II)–C) character. Rupture of the Pd(II)–C bond constitutes ca. 73% of the decay from the [Pd d → C(sp3) σ*] excited state based on quantum yield (vide supra). For this T-shaped complex the productive NRD effectively outcompetes other unproductive NRD pathways (ca. 27%) that recover the ground state.
 | ||
| Fig. 9 Proposed excited state potential energy curves for 1a and 3. | ||
Excitation of the α-diimine complex 3 into the MLCT band (∼350–500 nm) produces an excited state by population of the π* orbitals on the diimine. At present it is not definitively established whether this state leads to Pd–C cleavage, or whether this occurs after conversion to a lower-lying excited state, as has been proposed for (bpy)Ni aryl complexes.122,137 Our computational investigations suggest the Pd–C bond is not weakened in the Franck–Condon state of 3. Rather, some other processes such as thermalization, intersystem crossing and/or geometric changes occur prior to bond cleavage. We did not find this to be the case for 1a, which could be another contributing factor rationalizing the quantum yield differences between the two types of complexes. Homolysis of the Pd(II)–C bond represents ca. 16% of the excited state decay based on quantum yield data with the remainder assigned to other unproductive NRD (ca. 84%). The origin of the discrepancy in unproductive NRD between the two types of organo–Pd(II) complexes will require further investigation to understand. It is plausible 3 could possess stronger vibronic coupling to the solvent molecules relative to 1a, which would facilitate the dissipation of the energy of its excited state by unproductive NRD. It would also be interesting to understand what aspects of the ligand choice (alkylphosphine vs. diimine) and/or Pd(II) geometry (T-shaped vs. square planar) influence the extent of unproductive NRD, which could guide the design of improved quantum efficiency in photochemical reactions of catalytically relevant organo–Pd(II) complexes.
Conclusions
We have found that visible light can promote mild yet efficient homolytic cleavage of the strong Pd(II)–C bond in T-shaped organo–Pd(II) compounds. Stoichiometric reactions and quantum yield measurements are consistent with free radical formation by a blue light-promoted, non-chain process. Kinetic data from photo-NMR analyses indicate the photophysical process occurs with a small thermal barrier (3–4 kcal mol−1). Electronic structure calculations of a model T-shaped (tBu3P)Pd(CH3)Cl complex point to a mechanism involving charge transfer from Pd to the CH3 ligand. The strength of the metal–carbon bond in the resulting [Pd d → C(sp3) σ*] excited state is predicted to significantly weakened such that homolytic cleavage of an otherwise a strong organometallic bond occurs easily under ambient conditions. The result of this photochemical pathway is the formation of an alkyl radical along with a reduced Pd(I) intermediate.A comparative analysis using a prototypical photoactive square planar (α-diimine)Pd(II) complex also yielded the surprising observation of significantly lower quantum yield for Pd(II)–C homolytic cleavage even though it possesses conjugated dative ligands typically associated with easily accessible MLCT states (i.e., [Pd d → diimine π*]) previously shown to facilitate free radical formation. The higher quantum efficiency in the T-shaped complexes that otherwise lack the classic ligand-based chromophores typically found in photoactive late transition metal complexes was a surprising finding. These data thus demonstrate a latent one-electron reaction manifold is readily accessible in T-shaped organo–Pd(II) complexes – some of the most active organometallic species in modern Pd catalysis – using visible light as an external stimulus. The findings of this study may help to expand the synthetic toolbox of this central catalytic metal by shunting Pd away from its strong preference toward two-electron mechanisms.
Data availability
Crystallographic data for 1b, 2, 4, 5a, 5b and S2 have been deposited at the CCDC under 2264687–2264692 and can be obtained from https://www.ccdc.cam.ac.uk/structures. Energies and Cartesian coordinates for computational datasets supporting this article have been uploaded as part of the ESI.†Author contributions
P. M. W. and B. P. C. conceived the idea for this work. P. M. W. and L. V. carried out experiments. B. P. C. provided direction for the scientific strategy. L. T. and G. D. S. designed and carried out transient absorption spectroscopic and computational analyses. A. R. S. performed coupled cluster calculations. P. M. W. and B. P. C. wrote the manuscript and ESI† with input from all authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
BPC acknowledges support from the National Institutes of Health (R35GM128902) and the National Science Foundation (CAREER CHE-1654664). GDS acknowledges support from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant No. DE-SC0019370. Philip D. Jeffrey is thanked for help with X-ray crystallography. Simulations were performed using computational resources of Princeton Research Computing, a consortium including the Princeton Institute for Computational Science and Engineering (PICSciE) and the Office of Information Technology's High Performance Computing Center and Visualization Laboratory at Princeton University and the Research Computing Data Core at the University of Houston.Notes and references
- I. McConnell, G. H. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. A. Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878 CrossRef CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS.
- Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed.
- L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119, 4628 CrossRef CAS PubMed.
- T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- G. A. Crosby, W. G. Perkins and D. M. Klassen, J. Chem. Phys., 2004, 43, 1498 CrossRef.
- P. A. Anderson, F. Richard Keene, T. J. Meyer, J. A. Moss, G. F. Strouse and J. A. Treadway, J. Chem. Soc., Dalton Trans., 2002, 3820 RSC.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117 CrossRef CAS.
- A. Vlcek, Coord. Chem. Rev., 1998, 177, 1 CrossRef CAS.
- J. K. McCusker, Acc. Chem. Res., 2003, 36, 876 CrossRef CAS PubMed.
- M. Wrighton, Chem. Rev., 1974, 74, 401 CrossRef CAS.
- G. L. Geoffroy, J. Chem. Educ., 1983, 60, 861 CrossRef CAS.
- N. A. Beach and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 5713 CrossRef CAS.
- M. A. Graham, M. Poliakoff and J. J. Turner, J. Chem. Soc. A, 1971, 2939 RSC.
- I. W. Stolz, R. K. Sheline and G. R. Dobson, J. Am. Chem. Soc., 1962, 84, 3589 CrossRef CAS.
- W. Strohmeier and K. Gerlach, Chem. Ber., 1961, 94, 398 CrossRef CAS.
- O. S. Wenger, Chem. –Eur. J., 2021, 27, 2270 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. F. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481 CrossRef CAS PubMed.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Adv. Synth. Catal., 2012, 354, 3517 CrossRef CAS PubMed.
- J. Zoller, D. C. Fabry, M. A. Ronge and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13264 CrossRef CAS PubMed.
- H. H. Zhang, J. J. Zhao and S. Y. Yu, ACS Catal., 2020, 10, 4710 CrossRef CAS.
- J. Zheng, A. Nikbakht and B. Breit, ACS Catal., 2021, 11, 3343 CrossRef CAS.
- J. Zheng, N. N. Tang, H. Xie and B. Breit, Angew. Chem., Int. Ed., 2022, 61, e202200105 CrossRef CAS PubMed.
- C. H. Song, H. H. Zhang and S. Y. Yu, ACS Catal., 2022, 12, 1428 CrossRef CAS.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830 CrossRef CAS PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380 CrossRef CAS PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef CAS PubMed.
- L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555 CrossRef CAS PubMed.
- M. Kudisch, C. H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479 CrossRef CAS PubMed.
- C. Zheng, W. M. Cheng, H. L. Li, R. S. Na and R. Shang, Org. Lett., 2018, 20, 2559 CrossRef CAS PubMed.
- K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467 CrossRef CAS PubMed.
- N. Toriumi, K. Shimomaki, J. Caner, K. Murata, R. Martin and N. Iwasawa, Bull. Chem. Soc. Jpn., 2021, 94, 1846 CrossRef CAS.
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586 CrossRef CAS PubMed.
- M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340 CrossRef CAS PubMed.
- M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712 CrossRef CAS PubMed.
- G. Z. Wang, R. Shang, W. M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS PubMed.
- D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212 CrossRef CAS PubMed.
- G. Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888 CrossRef CAS PubMed.
- M. Koy, F. Sandfort, A. Tlahuext-Aca, L. Quach, C. G. Daniliuc and F. Glorius, Chem. –Eur. J., 2018, 24, 4552 CrossRef CAS PubMed.
- R. Kancherla, K. Muralirajan, B. Maity, C. Zhu, P. E. Krach, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3412 CrossRef CAS PubMed.
- I. Abdiaj, L. Huck, J. M. Mateo, A. de la Hoz, M. V. Gomez, A. Diaz-Ortiz and J. Alcazar, Angew. Chem., Int. Ed., 2018, 57, 13231 CrossRef CAS PubMed.
- W. J. Zhou, G. M. Cao, G. Shen, X. Y. Zhu, Y. Y. Gui, J. H. Ye, L. Sun, L. L. Liao, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683 CrossRef CAS PubMed.
- Z. W. Jiao, L. H. Lim, H. Hirao and J. S. Zhou, Angew. Chem., Int. Ed., 2018, 57, 6294 CrossRef CAS PubMed.
- G. Z. Wang, R. Shang and Y. Fu, Synthesis, 2018, 50, 2908 CrossRef CAS.
- M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227 RSC.
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563 CrossRef CAS PubMed.
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586 CrossRef CAS PubMed.
- K. S. Mühlfenzl, M. Sardana, T. Skrydstrup and C. S. Elmore, ChemistrySelect, 2022, 7, e202203582 CrossRef.
- G. Zhao, W. Yao, J. N. Mauro and M. Y. Ngai, J. Am. Chem. Soc., 2021, 143, 1728 CrossRef CAS PubMed.
- K. Pak Shing Cheung, J. Fang, K. Mukherjee, A. Mihranyan and V. Gevorgyan, Science, 2022, 378, 1207 CrossRef CAS PubMed.
- A. D. Marchese, A. G. Durant, C. M. Reid, C. Jans, R. Arora and M. Lautens, J. Am. Chem. Soc., 2022, 144, 20554 CrossRef CAS PubMed.
- M. L. Czyz, G. K. Weragoda, T. H. Horngren, T. U. Connell, D. Gomez, R. A. J. O'Hair and A. Polyzos, Chem. Sci., 2020, 11, 2455 RSC.
- N. Toriumi, T. Inoue and N. Iwasawa, J. Am. Chem. Soc., 2022, 144, 19592 CrossRef CAS PubMed.
- C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318 CrossRef CAS PubMed.
- K. A. Smoll, W. Kaminsky and K. I. Goldberg, Organometallics, 2017, 36, 1213 CrossRef CAS.
- C. T. Burns, H. Shen and R. F. Jordan, J. Organomet. Chem., 2003, 683, 240 CrossRef CAS.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303 CrossRef CAS PubMed.
- G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318 CrossRef CAS PubMed.
- P. W. N. M. Vanleeuwen, C. F. Roobeek and R. Huis, J. Organomet. Chem., 1977, 142, 233 CrossRef CAS.
- Y. Liu, C. Zhou, M. Jiang and B. A. Arndtsen, J. Am. Chem. Soc., 2022, 144, 9413 CrossRef CAS PubMed.
- A. Keyes, H. E. B. Alhan, U. Ha, Y. S. Liu, S. K. Smith, T. S. Teets, D. B. Beezer and E. Harth, Macromolecules, 2018, 51, 7224 CrossRef CAS.
- A. Keyes, H. Dau, H. E. B. Alhan, U. Ha, E. Ordonez, G. R. Jones, Y. S. Liu, E. Tsogtgerel, B. Loftin, Z. L. Wen, J. I. Wu, D. B. Beezer and E. Harth, Polym. Chem., 2019, 10, 3040 RSC.
- H. Dau, A. Keyes, H. E. B. Alhan, E. Ordonez, E. Tsogtgerel, A. P. Gies, E. Auyeung, Z. Zhou, A. Maity, A. Das, D. C. Powers, D. B. Beezer and E. Harth, J. Am. Chem. Soc., 2020, 142, 21469 CrossRef CAS PubMed.
- U. Christmann and R. Vilar, Angew Chem. Int. Ed. Engl., 2005, 44, 366 CrossRef CAS PubMed.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141 CrossRef CAS PubMed.
- A. F. Littke, C. Y. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- P. Surawatanawong and M. B. Hall, Organometallics, 2008, 27, 6222 CrossRef CAS.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694 RSC.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936 CrossRef CAS PubMed.
- M. Yamashita and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 5344 CrossRef CAS PubMed.
- H. K. Shaughnessy, Curr. Org. Chem., 2020, 24, 231 CrossRef.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS PubMed.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694 RSC.
- S. R. Dubbaka, Synlett, 2005, 709 CrossRef CAS.
- K. H. Shaughnessy, Isr. J. Chem., 2020, 60, 180 CrossRef CAS.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118 CrossRef CAS PubMed.
- B. Seifinoferest, A. Tanbakouchian, B. Larijani and M. Mahdavi, Asian J. Org. Chem., 2021, 10, 1319 CrossRef CAS.
- L. Y. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392 CrossRef CAS PubMed.
- L. Chen, H. Francis and B. P. Carrow, ACS Catal., 2018, 8, 2989 CrossRef CAS.
- S. H. Lau, P. Yu, L. Chen, C. B. Madsen-Duggan, M. J. Williams and B. P. Carrow, J. Am. Chem. Soc., 2020, 142, 20030 CrossRef CAS PubMed.
- S. Zhao, T. Gensch, B. Murray, Z. L. Niemeyer, M. S. Sigman and M. R. Biscoe, Science, 2018, 362, 670 CrossRef CAS PubMed.
- A. L. Kocen, M. Brookhart and O. Daugulis, Nat. Commun., 2019, 10, 438 CrossRef PubMed.
- K. Kubota, R. Takahashi, M. Uesugi and H. Ito, ACS Sustain. Chem. Eng., 2020, 8, 16577 CrossRef CAS.
- R. Takahashi, T. Seo, K. Kubota and H. Ito, ACS Catal., 2021, 11, 14803 CrossRef CAS.
- M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, Organometallics, 2006, 25, 4588 CrossRef CAS.
- D.-G. Kim, A. Bell and R. A. Register, ACS Macro Lett., 2015, 4, 327 CrossRef CAS PubMed.
- M. V. Bermeshev and P. P. Chapala, Prog. Polym. Sci., 2018, 84, 1 CrossRef CAS.
- M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, Organometallics, 2006, 25, 4588 CrossRef CAS.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. Phys. Chem., 1995, 99, 12723 CrossRef CAS.
- M. E. Evans, T. Li, A. J. Vetter, R. D. Rieth and W. D. Jones, J. Org. Chem., 2009, 74, 6907 CrossRef CAS PubMed.
- D. D. Wick and W. D. Jones, Organometallics, 1999, 18, 495 CrossRef CAS.
- R. Vilar, D. M. P. Mingos and C. J. Cardin, J. Chem. Soc., Dalton Trans., 1996, 4313 RSC.
- A. D. Burrows, D. M. P. Mingos, S. Menzer, R. Vilar and D. J. Williams, J. Chem. Soc., Dalton Trans., 1995, 2107 RSC.
- V. Durà-Vilà, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, J. Organomet. Chem., 2000, 600, 198 CrossRef.
- M. Okunaka, G. Matsubayashi and T. Tanaka, Bull. Chem. Soc. Jpn., 1977, 50, 907 CrossRef CAS.
- N. C. Tomson, L. A. Labios, T. Weyhermuller, J. S. Figueroa and K. Wieghardt, Inorg. Chem., 2011, 50, 5763 CrossRef CAS PubMed.
- B. R. Barnett, L. A. Labios, C. E. Moore, J. England, A. L. Rheingold, K. Wieghardt and J. S. Figueroa, Inorg. Chem., 2015, 54, 7110 CrossRef CAS PubMed.
- K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434 RSC.
- I. Kalvet, K. J. Bonney and F. Schoenebeck, J. Org. Chem., 2014, 79, 12041 CrossRef CAS PubMed.
- F. Proutiere, M. Aufiero and F. Schoenebeck, J. Am. Chem. Soc., 2012, 134, 606 CrossRef CAS PubMed.
- M. Aufiero, F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2012, 51, 7226 CrossRef CAS PubMed.
- C. C. C. J. Seechurn, T. Sperger, T. G. Scrase, F. Schoenebeck and T. J. Colacot, J. Am. Chem. Soc., 2017, 139, 5194 CrossRef PubMed.
- C. Fricke, T. Sperger, M. Mendel and F. Schoenebeck, Angew. Chem., Int. Ed., 2021, 60, 3355 CrossRef CAS PubMed.
- A. Gabrielsson, A. M. Blanco-Rodriguez, P. Matousek, M. Towrie and A. Vlcek, Organometallics, 2006, 25, 2148 CrossRef CAS.
- A. Vlcek, Coord. Chem. Rev., 2000, 200, 933 CrossRef.
- I. G. Virrels, M. W. George, J. J. Turner, J. Peters and A. Vlcek, Organometallics, 1996, 15, 4089 CrossRef CAS.
- C. J. Kleverlaan, D. J. Stufkens, I. P. Clark, M. W. George, J. J. Turner, D. M. Martino, H. van Willigen and A. Vlcek, J. Am. Chem. Soc., 1998, 120, 10871 CrossRef CAS.
- J. Vichova, F. Hartl and A. Vlcek, J. Am. Chem. Soc., 1992, 114, 10903 CrossRef CAS.
- Y. N. Ji, D. A. DiRocco, C. M. Hong, M. K. Wismer and M. Reibarkh, Org. Lett., 2018, 20, 2156 CrossRef CAS PubMed.
- D. A. Wilcox, J. Snaider, S. Mukherjee, L. Yuan, L. B. Huang, B. M. Savoie and B. W. Boudouris, Soft Matter, 2019, 15, 1413 RSC.
- For example of computational benchmarking based on absorption spectra, see: B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035 CrossRef CAS PubMed.
- For the simulated UV-vis spectrum of 3, see the ESI†.
- S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800 CrossRef CAS PubMed.
- P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083 RSC.
- F. S. Mancilha, L. Barloy, F. S. Rodembusch, J. Dupont and M. Pfeffer, Dalton Trans., 2011, 40, 10535 RSC.
- S.-W. Lai, T.-C. Cheung, M. C. W. Chan, K.-K. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 2000, 39, 255 CrossRef CAS PubMed.
- C. A. Craig and R. J. Watts, Inorg. Chem., 1989, 28, 309 CrossRef CAS.
- R. von der Stück, M. Krause, D. Brünink, S. Buss, N. L. Doltsinis, C. A. Strassert and A. Klein, Z. Anorg. Allg. Chem., 2022, 648, e202100278 CrossRef.
- T. L. Davis, J. L. Watts, K. J. Brown, J. S. Hewage, A. R. Treleven, S. V. Lindeman and J. R. Gardinier, Dalton Trans., 2015, 44, 15408 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- S. Gozem, F. Melaccio, A. Valentini, M. Filatov, M. Huix-Rotllant, N. Ferre, L. M. Frutos, C. Angeli, A. I. Krylov, A. A. Granovsky, R. Lindh and M. Olivucci, J. Chem. Theory Comput., 2014, 10, 3074 CrossRef CAS PubMed.
- B. G. Levine, C. Ko, J. Quenneville and T. J. Martinez, Mol. Phys., 2006, 104, 1039 CrossRef CAS.
- M. L. Mandich, L. F. Halle and J. L. Beauchamp, J. Am. Chem. Soc., 1984, 106, 4403 CrossRef CAS.
- It was still the case that no signals consistent with luminescent emission were observed even when higher beam power and greater concentrations were used.
- K. S. Kjaer, et al. , Phys. Chem. Chem. Phys., 2018, 20, 4238 RSC.
- N. Kitamura and E. Sakuda, J. Phys. Chem. A, 2005, 109, 7429 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583 CrossRef CAS.
- D. A. Cagan, D. Bím, B. Silva, N. P. Kazmierczak, B. J. McNicholas and R. G. Hadt, J. Am. Chem. Soc., 2022, 144, 6516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2264687–2264692. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02588h |
| This journal is © The Royal Society of Chemistry 2023 |