
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTethered together: DASA design towards aqueous compatibility†
Julie A.
Peterson
,
Natalia M.
Neris
and
Javier
Read de Alaniz
 *
*
Department of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, 93106 CA, USA. E-mail: javier@chem.ucsb.edu
First published on 27th October 2023
Abstract
Donor–acceptor Stenhouse adducts (DASAs) are an exciting class of photoswitches due to their facile tunability, visible light absorbance, and negative photochromism. While they have shown use in a variety of applications, to date all reported DASA derivatives have low equilibrium and/or poor photoswitching in polar protic solvents, which is vital for moving towards applications in biological systems. We demonstrate a strategy to introduce a substitution on the DASA triene that results in derivatives that are stable and have high dark equilibrium of the open form in polar protic solvents. Decreasing the charge separation of these new derivatives also allows for reversible switching in polar and protic solvents including THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixtures.
:water mixtures.
Introduction
Donor–acceptor Stenhouse adducts (DASAs) are a class of photoswitches that can be activated with visible to near-IR light.1–3 DASA derivatives consist of an amine donor and carbon acid acceptor connected through a triene with a hydroxyl group at the C2 position (Fig. 1a). The linear open form (A) absorbs visible light with a high extinction coefficient and a relatively narrow absorbance profile, while the cyclic closed isomer (C) does not absorb visible light, giving them the advantage of being negatively photochromic molecules. The lack of absorbance of their closed form allows for the use of broadband visible light sources. The combination of visible to NIR activation and large property changes upon irradiation (e.g., shape, polarity, absorbance) make them exciting candidates for photochromic applications such as surface wettability,4 photothermal agents,5 photoactuation,6 and drug delivery.7 Of particular interest is their use in biological studies due to their long wavelength activation wavelengths, which would avoid the cytotoxic UV activation that the most commonly used photoswitches (e.g., azobenzene, diarylethene) require.8,9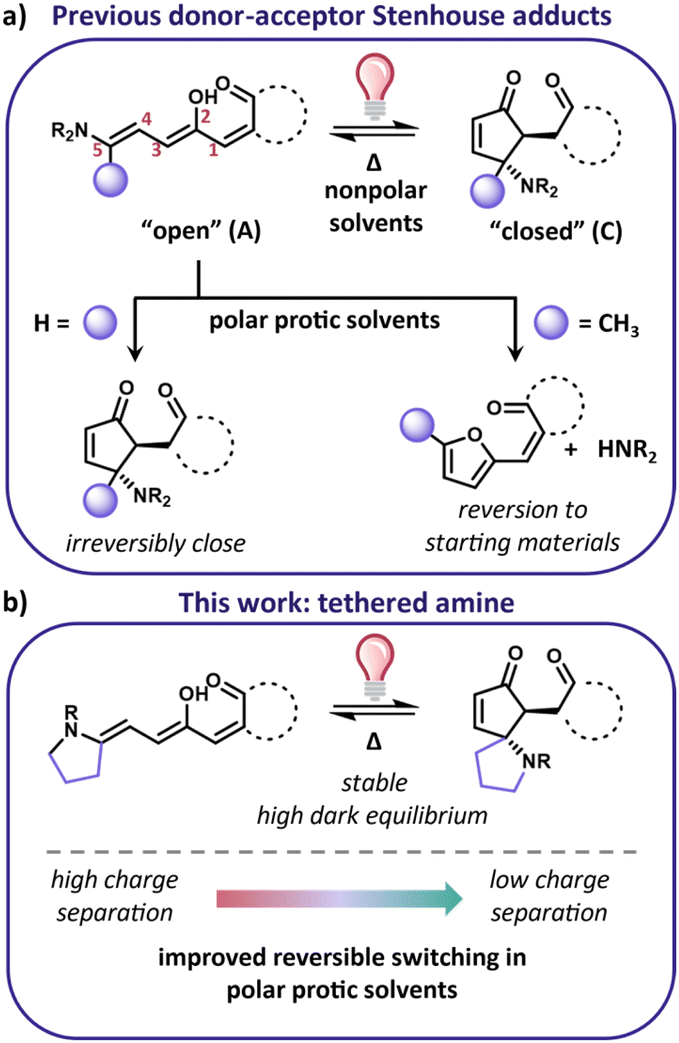 | ||
| Fig. 1 (a) Previously studied DASA derivatives either prefer the cyclic closed form (enol form is shown, but the closed form can be enol, keto, or zwitterionic) or revert to the DASA building blocks in polar protic solvents; (b) this work shows tethered amine derivatives with high equilibrium and reversible switching in polar protic solvents. | ||
To move to increasingly complex systems, such as biological systems, photoswitches must operate in polar protic solvents. Unfortunately, this is a feature not available for current DASA based photoswitches. The dark equilibrium and the switching kinetics of DASA are both sensitive to the environment and become compromised in increasingly polar environments.3,10–13 Due to the environmental sensitivity, thus far no DASA small molecule has been developed with both a high dark equilibrium and reversible switching in aqueous solutions. Developing strategies to address the aqueous compatibility of DASA derivatives is vital for designing derivatives that can be used for biological applications.
The difficulty of designing DASA derivatives compatible with water stems from the interplay between the charge separation of the DASA isomers and the barriers for the multistep isomerization mechanism. The isomerization mechanism from the open to closed form has been experimentally and computationally elucidated by the Beves,14,15 Feringa,11,16,17 Jacquemin,18 and Martinez19,20 groups. It relies on a photochemical Z/E isomerization followed by a subsequent thermal Z/E isomerization and cyclization towards a metastable cyclic “closed form” isomer. The barriers for the bond rotations are linked to the single vs. double bond character of the triene which is in turn governed by the charge separation of the DASA open form.21 The charge separation is highly interconnected to both the strength of the donor and acceptor as well as the polarity of the solvent. Increasing charge separation changes the barriers such that the pathway to the closed form is no longer preferred. For this reason, DASA derivatives that have weaker donors and acceptors, such as “2nd generation” DASAs, have shown the most promise for switching in polar environments. However, the dark equilibrium of these derivatives favors the closed form. For example, 1 is approximately 30% open in solvents such as dichloromethane and toluene and less than 1% open in methanol (Fig. 2b).2,3,11 Castanga et al. recently demonstrated the switching of one such derivative in aqueous solution. While reversible switching was observed in water, the solution required pH 5 and 10 wt/vol% cyclodextrin additive to stabilize the open form.22 Even under those conditions, the equilibrium was low (a 500 μM solution showed an absorbance around 0.3, whereas a 10 μM solution that is predominantly in the open form will show an absorbance around 1). Finding structural modifications that can improve the equilibrium will allow for a more general strategy to investigate switching in a wider range of conditions without requiring additives or pH ranges that may be incompatible with systems of interest. To perform well in water, derivatives must be designed such that they are stable, have a high equilibrium of the open form, and have a low charge separation indicated by a less negative solvatochromic slope.
 | ||
| Fig. 2 (a) Strategy for the synthesis of tethered amine derivatives; (b) normalized UV-Vis following the evolution of 1 after injection into methanol; (c) normalized UV-Vis data of 2 after injection into methanol, showing the reversion to furan adduct; (d) normalized UV-Vis data of 3 after injection into methanol showing a stable absorbance of the open form. (e) Irradiation of a 10 μM solution of 3 in toluene tracking the absorbance of A (560 nm) and B/B′ (640 nm) over time. The UV-Vis is tracked for 5 points before irradiation, then irradiated for 15 minutes with a 595 nm LED (9 mW), after which the light is turned off and the recovery is monitored in the dark. (f) UV-Vis data of a 10 μM solution of 3 in toluene before irradiation (solid line) and during irradiation after 1 minute (dotted line), both normalized to 1 to show the long-lived presence of the B/B′. Data was normalized by dividing by the absorbance of the λmax at time zero. | ||
Previously our group discovered that 2, with substitution on the 5-position of the triene (alpha to the donor), has an improved equilibrium compared to 1 (>96% open for 2 in dichloromethane and toluene), with the same donor and acceptor pair.23 While this derivative has a high dark equilibrium, it thermally reverts to the amine and furan adduct starting materials in polar solvents (Fig. 1a and 2c). In this work, we demonstrate that tethering the amine to the triene backbone is a successful strategy to design derivatives that have a high equilibrium of the open form and are stable in polar protic solvents. Furthermore, decreasing the charge separation character of these derivatives results in a new DASA that is also able to reversibly switch in protic solvents including 60:40 THF:H2O. In contrast to the poor aqueous compatibility of previous derivatives, this work presents a straightforward strategy for the design of DASA photoswitches that can be used for applications in aqueous environments.
Results and discussion
We designed derivatives inspired by our intramolecular aza-Piancatelli rearrangement,24 with the amine donor tethered to the triene via a three-carbon linkage, resulting in a 5-membered ring (Fig. 2a). We expected 3, with a Meldrum's acid acceptor and aniline donor to have improved equilibrium compared to 1 and improved stability compared to 2. Indeed, when a 10 μM solution of 3 is prepared in methanol it neither reverts to the starting materials (as does 2) nor equilibrates to the closed form (as does 1) as can be observed by UV-Vis (Fig. 2b–d). Additionally, 1H NMR spectra taken after equilibration for at least 24 hours in toluene, DMSO, and methanol show only the open form (Fig. S1–S4†). This is a remarkable improvement over other DASA derivatives which exhibit less than 1% dark equilibrium in polar protic solvents.3,10 Encouraged by this result, we initially tested the photoswitching of a 10 μM solution of 3 in toluene to compare with previous DASA derivatives. This derivative shows slow forward switching (∼10 minutes to reach 62% conversion to the closed form) upon irradiation with a 595 nm LED (9 mW). Upon irradiation, a red-shifted shoulder appears, which was previously shown to be indicative of the metastable open isomers B and B′ (Fig. 2e and f).25 The ratio of the main peak and the B/B′ shoulder is consistent throughout the irradiation period (Fig. S5†), indicating that a photostationary state between A and B/B′ is quickly reached. This photostationary state is then slowly drained to the closed form over prolonged irradiation time (Fig. 2e). This behavior is similar to recently published DASA derivatives with sterically hindered donors and acceptors, which allowed for multi-wavelength addressability.25 Next we tested the photoswitchability in a range of non-polar and polar solvents including THF, acetonitrile and DMSO. While 3 shows little or no visible switching in more polar solvents (Fig. S6†), similar structures may lead towards designing new multi-wavelength addressable derivatives. The lack of switching in polar solvents is partially due to high charge separation, indicated by the highly negative solvatochromic slope (−56 nm, Fig. S7†) compared to other aryl amine derivatives (−7 nm for 1).12 Such a highly negative solvatochromic slope is reminiscent of previously reported 1st generation derivatives with alkyl amine donors which also do not switch in polar environments. In the case of 3, the aryl amine is likely a better donor than traditional aryl amines due to the tethered alkyl chain locking the lone pair of electrons on the nitrogen into place as well as the aryl group being rotated out of plane. Large charge separation contributes to lower A–B barriers and higher B–B′ barriers due to increased single bond character around C2–C3 and increased double bond character between C3–C4.21 To confirm this, we calculated the switching pathway from A–C (the first closed form isomer) using M06-2X/6-31+g(d,p) in chloroform with an SMD solvent model (Fig. 3).9,11 Indeed, for 3, the barrier from B′–C is calculated to be higher than the barrier from B–A for 3 in chloroform which supports that any B generated photochemically is more likely to drain back to A rather than move forward to C in solvents with similar or higher polarity than chloroform. | ||
| Fig. 3 (a) Resonance isomers of DASA derivatives; (b) simplified mechanism of isomerization from the open form A to the first closed form isomer C; (c) calculated ΔG values in kcal mol−1 of the isomers A–C on the isomerization pathway from the open form to the closed form of 3 and 4 calculated using M06-2X/6-31+G(d,p) in chloroform using an SMD solvent model. 4 shows a higher energy transition state from A–B and lower energy barriers from B–B′ and B′–C. | ||
In order to design derivatives with improved properties in polar solvents, it is important to lower the B′–C barrier while keeping the B–A barrier sufficiently high to allow for thermal conversion to the colorless closed form after the initial photo driven Z/E isomerization. Pyrazalone acceptors have previously been shown to have increased rates of forward switching compared to their Meldrum's acid counterparts.3 Furthermore, decreasing the charge separation of DASA derivatives has been shown to allow for photoswitching in an increased range of environments.10,12,26,27 Guided by these principles, we chose to target a pyrazalone bearing a methyl substituent in place of the traditionally used CF3 to render the acceptor less electron withdrawing. To our delight, the calculated barriers from B′–C and B–B′ are lower while the barrier from B–A is higher for 4 compared to 3 (Fig. 3), indicating the potential for improved switching in polar solvents.
Encouraged by these computational results and stability of the tethered amine scaffold, we synthesized 4 with methyl pyrazalone carbon acid and a tethered aniline donor. As predicted, a 10 μM solutions of this derivative switches well in toluene (Fig. S11†). Given this promising result, we investigated switching in increasingly polar solvents (Fig. 4b). In tetrahydrofuran and dichloromethane 4 reached a photostationary state of 92% and 16% conversion to the closed form, respectively, upon irradiation for 95 s with a 617 nm LED. However, in acetonitrile negligible switching was still observed. The behavior in protic solvents mimics that of aprotic solvents, with the extent of switching depending largely on the polarity of the solvent. Irradiation of 4 for 95 s resulted in 55% and 12% conversion to the closed form in tert-butanol (t-BuOH) and isopropanol (IPA), respectively; however, showed less than 5% conversion in ethanol (EtOH) and methanol (MeOH). We envisioned that we could further improve the switching in these more polar solvents by decreasing the charge separation of DASA through the introduction of electron withdrawing groups on the aniline donor. Unfortunately, synthetic access to these derivatives proved challenging. For example, we did not observe any formation of the 4-cyano or 4-nitro aniline analogues after 3 days due to their poor nucleophilicity. To our delight, we were able to synthesize 5 with a 4-iodoaniline donor. Introducing the iodine on the aniline sufficiently lowers the zwitterionic nature of the open form as evident by the solvatochromic slope of −20 nm compared to −32 nm without the iodine, Table S1 and Fig. S7.† With this new derivative in hand, we tested the photoswitching properties in a range of solvents (Fig. 4e and f). DASA 5 has dramatically improved switching in polar solvents with 42% conversion to the closed form in ACN after 95 s irradiation as well as 15% conversion to the closed form in EtOH. 5 showed only 4% conversion to the closed form in MeOH after 95 s of irradiation; however, even this is a major improvement over some other derivatives that irreversibly close in methanol.11
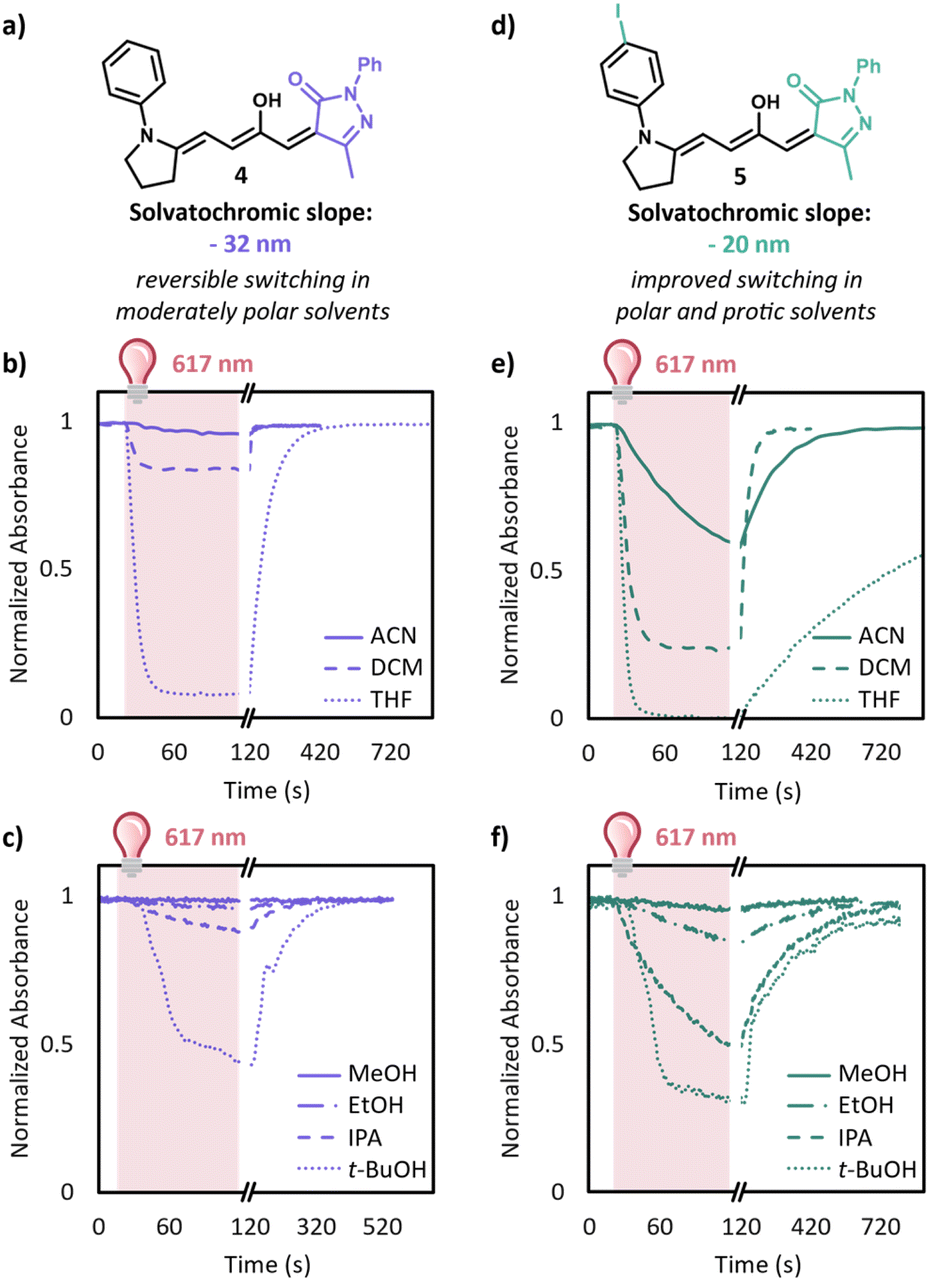 | ||
| Fig. 4 (a) Design of 4 with a methyl-substituted pyrazalone acceptor; (b) switching of 10 μM solutions of 4 in tetrahydrofuran (THF), dichloromethane (DCM), and acetonitrile (ACN); (c) switching of 10 μM solutions of 4 in tert-butanol (t-BuOH), isopropanol (IPA), ethanol (EtOH), and methanol (MeOH); (d) design of 5 with an iodine substituent that decreases the solvatochromic slope and allows for switching in increasingly polar solvents; (e) switching of 10 μM solutions of 5 in THF, DCM, and ACN (f) switching of 10 μM solutions of 5 in t-BuOH, IPA, EtOH, and MeOH. Switching studies were conducted monitoring the absorbance at the λmax at 1 s intervals for 25 points before irradiation, 95 points during irradiation, and at 10 s (1 s for ACN and DCM) after irradiation until recovery was complete. Irradiation was done with a 10 mW 617 nm LED. Data was normalized by dividing by the absorbance of the λmax at time zero. | ||
We next turned to 1H NMR and UV-Vis spectroscopy to investigate their dark equilibrium. Equilibrated 1H NMR solutions of these derivatives in CD2Cl2 show only the open form. Due to poor solubility at 1H NMR concentrations we estimated their equilibria in methanol using UV-Vis spectroscopy (Table S2 and Fig. S10†). While these derivatives have a lower equilibrium than 3, they are still a remarkable improvement over previous DASA derivatives (1 is less than 1% open in methanol) with 4 being 70% open in methanol and 5 being 24% open in methanol.
With these improvements in mind, we moved to test the compatibility of 5 in THF:water mixtures. Remarkably, even with 40% water in THF a 10 μM solution of 5 exhibits an absorbance of 0.2 after equilibration (Fig. 5a). This is at least an order of magnitude improvement in equilibrium above unsubstituted DASA derivatives, such as 1, which has no visible absorbance band after equilibration of 10 μM solutions in 75:25 THF:water (Fig. S10†). Even more remarkably, 5 switches well in 60:40 THF:water, with irradiation resulting 5 in a 20% loss of absorbance after 400 s of irradiation with 617 nm light and 95% recovery with a half-life of 240 s (Fig. 5b). Furthermore, 5 demonstrates reversible switching in 60:40 THF:water over at least 10 cycles with 500 s irradiation and 3000 s recovery. Studies on improving the synthetic accessibility of similar derivatives with low charge separation and increased solubility are underway.
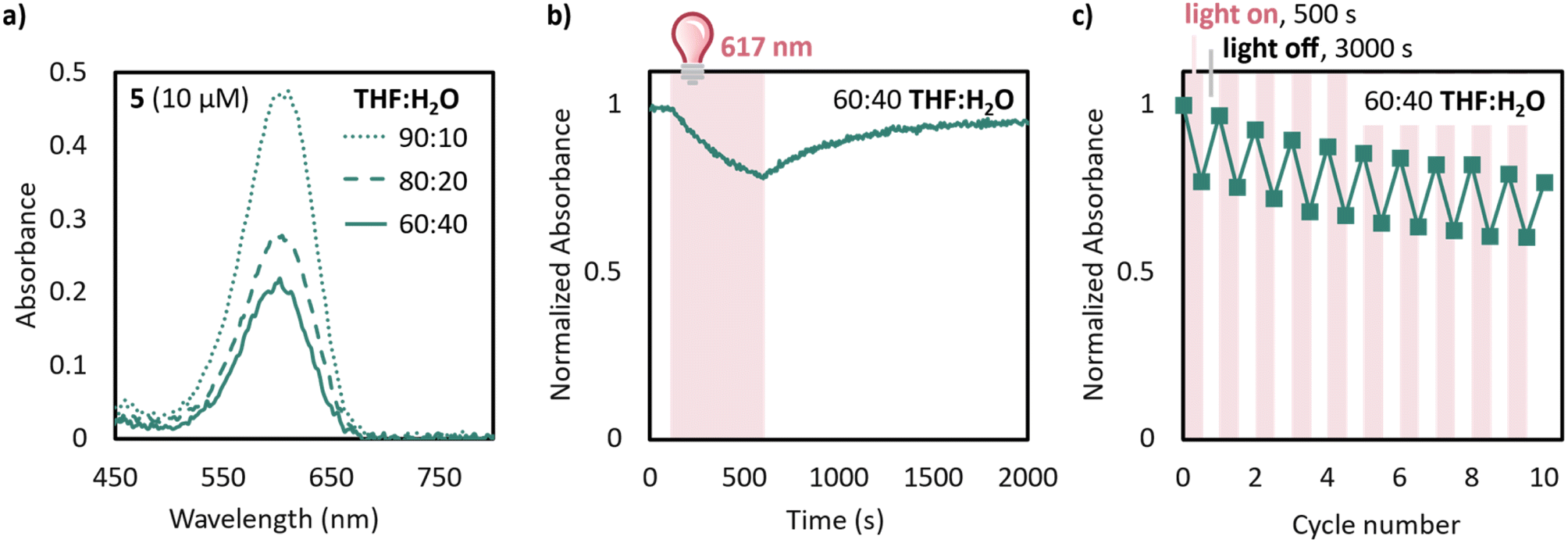 | ||
| Fig. 5 (a) UV-Vis of 10 μM solutions of 5 in THF:water mixtures irradiated until a stationary state was reached and allowed to equilibrate in the dark for 2 h; (b) time-dependent UV-Vis of 5 in 60:40 THF:water following the normalized peak at 575 nm, corresponding to; (c) cycling of a solution of 5 in 60:40 THF:water solvent mixture with irradiation for 500 s followed by thermal recovery for 3000 s following the normalized absorbance at 575 nm. Data was normalized by dividing by the absorbance of the λmax at time zero. | ||
Conclusions
In conclusion, we have shown the design of new DASA derivatives which address problems with low equilibrium and stability in polar protic solvents. Tethering the donor to the DASA via an alkyl linkage is an effective means to improve the stability while retaining substitution on the 5-position of the DASA triene to increase the dark equilibrium of the open form. This strategy is an exciting step towards the design of derivatives with high equilibrium and reversible switching in polar protic environments. Going forward, designing derivatives with lower ground state charge separation and increasing water solubility using this architecture will lead towards derivatives that reversibly switch in physiological conditions to be used in biological studies.Data availability
Data available in the ESI: general methods, synthetic procedures, and material characterization.†Author contributions
The project was conceptualized by J. P. and J. R. d. A. Synthesis and data collection was done and analyzed by J. P. and N. N. with supervision from J. R. d. A. The manuscript was written by J. P. and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Elings Prize Fellowship from the California NanoSystem Institute at UCSB. This work was also partially supported by the Office of Naval Research through a MURI on Photomechanical Material Systems (Grant No. ONR N00014-18-1-2624). Views and conclusions are those of the authors and should not be interpreted as representing official policies, either expressed or implied, of the US Government. Use was made of computational facilities purchased with funds from the National Science Foundation (CNS-1725797) and administered by the Center for Scientific Computing (CSC). The CSC is supported by the California NanoSystems Institute and the Materials Research Science and Engineering Center (MRSEC; NSF DMR 2308708) at University of California, Santa Barbara (UCSB). We would also like to thank Dr Livius Muff for helpful discussion.References
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- J. R. Hemmer, Z. A. Page, K. D. Clark, F. Stricker, N. D. Dolinski, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2018, 140, 10425–10429 CrossRef CAS PubMed.
- H. Zhao, D. Wang, Y. Fan, M. Ren, S. Dong and Y. Zheng, Langmuir, 2018, 34, 15537–15543 CrossRef CAS PubMed.
- S. Seshadri, L. F. Gockowski, J. Lee, M. Sroda, M. E. Helgeson, J. Read de Alaniz and M. T. Valentine, Nat. Commun., 2020, 11, 2599–2607 CrossRef CAS PubMed.
- J. Lee, M. M. Sroda, Y. Kwon, S. El-Arid, S. Seshadri, L. F. Gockowski, E. W. Hawkes, M. T. Valentine and J. Read de Alaniz, ACS Appl. Mater. Interfaces, 2020, 12, 54075–54082 CrossRef CAS PubMed.
- J. E. Yap, L. Zhang, J. T. Lovegrove, J. E. Beves and M. H. Stenzel, Macromol. Rapid Commun., 2020, 41, 2000236 CrossRef CAS PubMed.
- M. Londoño-Berrío, S. Pérez-Buitrago, I. C. Ortiz-Trujillo, L. M. Hoyos-Palacio, L. Y. Orozco, L. López, D. G. Zárate-Triviño, J. A. Capobianco and P. Mena-Giraldo, Polymers, 2022, 14, 3119 CrossRef PubMed.
- J. Pang, Z. Gao, L. Zhang, H. Wang and X. Hu, Front. Chem., 2018, 6, 217 CrossRef PubMed.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. C. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Chem. Commun., 2016, 52, 13576–13579 RSC.
- M. M. Lerch, M. Di Donato, A. D. Laurent, M. Medved’, A. Iagatti, L. Bussotti, A. Lapini, W. J. Buma, P. Foggi, W. Szymański and B. L. Feringa, Angew. Chem. Int. Ed., 2018, 57, 8063–8068 CrossRef CAS PubMed.
- M. M. Sroda, F. Stricker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Chem.–Eur. J., 2021, 27, 4183–4190 CrossRef CAS PubMed.
- D. Wang, L. Zhao, H. Zhao, J. Wu, M. Wagner, W. Sun, X. Liu, M. sheng Miao and Y. Zheng, Commun. Chem., 2019, 2, 118 CrossRef.
- J. N. Bull, E. Carrascosa, N. Mallo, M. S. Scholz, G. da Silva, J. E. Beves and E. J. Bieske, J. Phys. Chem. Lett., 2018, 9, 665–671 CrossRef CAS PubMed.
- N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242–8252 RSC.
- H. Zulfikri, M. A. J. J. Koenis, M. M. Lerch, M. Di Donato, W. Szymański, C. Filippi, B. L. Feringa and W. J. Buma, J. Am. Chem. Soc., 2019, 141, 7376–7384 CrossRef CAS PubMed.
- M. Di Donato, M. M. Lerch, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, S. P. Ihrig, M. Medved, D. Jacquemin, W. Szymański, W. J. Buma, P. Foggi and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 15596–15599 CrossRef CAS PubMed.
- A. D. Laurent, M. Medveď and D. Jacquemin, ChemPhysChem, 2016, 17, 1846–1851 CrossRef CAS PubMed.
- D. M. Sanchez, U. Raucci and T. J. Martínez, J. Am. Chem. Soc., 2021, 143, 20015–20021 CrossRef CAS PubMed.
- U. Raucci, D. M. Sanchez, T. J. Martínez and M. Parrinello, J. Am. Chem. Soc., 2022, 144, 19265–19271 CrossRef CAS PubMed.
- F. Stricker, J. Peterson, S. K. Sandlass, A. de Tagyos, M. Sroda, S. Seshadri, M. Gordon and J. Read de Alaniz, Chem, 2023, 9(7), 1994–2005 CAS.
- R. Castagna, G. Maleeva, D. Pirovano, C. Matera and P. Gorostiza, J. Am. Chem. Soc., 2022, 144, 15595–15602 CrossRef CAS PubMed.
- J. A. Peterson, F. Stricker and J. Read de Alaniz, Chem. Commun., 2022, 58, 2303–2306 RSC.
- L. I. Palmer and J. Read de Alaniz, Angew. Chem., Int. Ed., 2011, 50, 7167–7170 CrossRef CAS PubMed.
- F. Stricker, D. M. Sanchez, U. Raucci, N. D. Dolinski, M. S. Zayas, J. Meisner, C. J. Hawker, T. J. Martínez and J. Read de Alaniz, Nat. Chem., 2022, 14, 942–948 CrossRef CAS PubMed.
- R. McDonough, N. Rudgley, O. Majewski, M. V. Perkins, R. A. Evans and D. A. Lewis, ChemPhotoChem, 2022, 6, e202200076 CrossRef CAS.
- M. Clerc, F. Stricker, S. Ulrich, M. Sroda, N. Bruns, L. F. Boesel and J. Read de Alaniz, Angew. Chem., Int. Ed., 2021, 60, 10219–10227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02835f |
| This journal is © The Royal Society of Chemistry 2023 |