
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acid-driven self-assembly of diiridium macrocyclic catalysts imparts substrate selectivity and glutathione tolerance†
Hieu D.
Nguyen
 ,
Rahul D.
Jana
,
Dylan T.
Campbell
,
Thi V.
Tran
and
Loi H.
Do
*
,
Rahul D.
Jana
,
Dylan T.
Campbell
,
Thi V.
Tran
and
Loi H.
Do
*
Department of Chemistry, University of Houston, 4800 Calhoun Road, Houston, Texas, USA. E-mail: loido@uh.edu
First published on 4th September 2023
Abstract
Molecular inorganic catalysts (MICs) tend to have solvent-exposed metal centers that lack substrate specificity and are easily inhibited by biological nucleophiles. Unfortunately, these limitations exclude many MICs from being considered for in vivo applications. To overcome this challenge, a strategy to spatially confine MICs using Lewis acid-driven self-assembly is presented. It was shown that in the presence of external cations (e.g., Li+, Na+, K+, or Cs+) or phosphate buffered saline, diiridium macrocycles spontaneously formed supramolecular iridium-cation species, which were characterized by X-ray crystallography and dynamic light scattering. These nanoassemblies selectively reduced sterically unhindered C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups via transfer hydrogenation and tolerated up to 1 mM of glutathione. In contrast, when non-coordinating tetraalkylammonium cations were used, the diiridium catalysts were unable to form higher-ordered structures and discriminate between different aldehyde substrates. This work suggests that in situ coordination self-assembly could be a versatile approach to enable or enhance the integration of MICs with biological hosts.
O groups via transfer hydrogenation and tolerated up to 1 mM of glutathione. In contrast, when non-coordinating tetraalkylammonium cations were used, the diiridium catalysts were unable to form higher-ordered structures and discriminate between different aldehyde substrates. This work suggests that in situ coordination self-assembly could be a versatile approach to enable or enhance the integration of MICs with biological hosts.
Introduction
Bioorthogonal chemistry has allowed researchers to study and manipulate living systems with high precision and efficiency.1–4 It has extensive biomedical applications, from the labelling of disease targets to the in vivo generation of therapeutic drugs. Metal-free click reactions such as strain-promoted azide–alkyne cycloaddition5 and inverse electron-demand Diels–Alder6 are among the most widely used methods. There is growing interest in expanding the bioorthogonal chemistry library by leveraging metal catalysis.7–11 For example, molecular inorganic catalysts (MICs) have been used to mediate intracellular C–C bond cross-coupling,12 olefin metathesis,13,14 protecting group cleavage,15–18 transfer hydrogenation,19,20 and others.21–23 Unfortunately, since MICs lack the ability to restrict access to their active sites, they are prone to inhibition by endogenous thiols21,24,25 and have poor substrate selectivity. These deficiencies severely limit their practical applications in living environments.To enhance their biocompatibility, MICs can be spatially confined26–30 within macromolecular hosts31–33 or designer ligands34–36 (Chart 1A). For example, Ward and coworkers reported that organoiridium complexes incorporated into human carbonic anhydrase II were capable of catalyzing transfer hydrogenation reactions in the periplasm of E. coli bacteria.37 These artificial metalloenzymes achieved turnover numbers of >90. In another example, Zimmerman and coworkers constructed metal–organic nanoparticles by crosslinking single polymer chains containing copper ions.38,39 These complexes have low toxicity and were used to synthesize anti-microbial agents inside E. coli. In recent work, Martinez, Colomban and coworkers developed caged catalysts comprising Cu(I) centers supported by hemicryptophane ligands. These caged MICs catalyzed azide–alkyne cycloaddition in the presence of excess glutathione (GSH).40 A caveat, however, is that these reactions were not performed under biologically relevant conditions (e.g., they were conducted in MeOH/CH2Cl2 solutions). Other researchers have reported the use of inorganic nanoparticle catalysts for in-cell catalysis, but these materials are not based on MICs.41–44 Although the confinement strategies above have been demonstrated to be effective in some cases, they also have limitations. For example, macromolecular catalysts may not be able to cross the blood–brain barrier due to their large size45 and some MICs may not be easily converted into caged species.
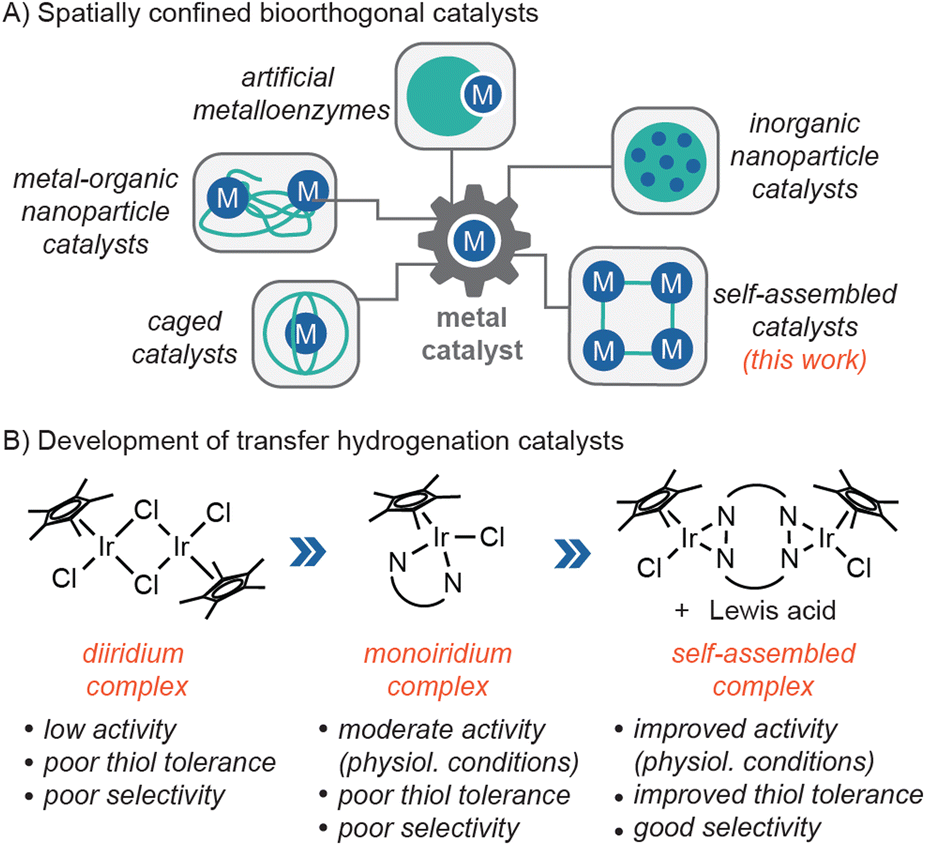 | ||
| Chart 1 Examples of spatially confined bioorthogonal catalysts (A) and chemical optimization of transfer hydrogenation catalysts (B). | ||
In our bioorthogonal catalyst discovery program, we use an iterative approach to engineer MICs with improved biocompatibility. In previous work, we found that [Cp*IrCl2]2 (Cp* = pentamethylcyclopentadienyl anion) was a poor transfer hydrogenation catalyst under physiologically relevant conditions (Chart 1B).46 This complex is too kinetically labile to form single component species necessary for efficient catalysis. When the Cp*Ir unit was ligated with N-phenylpicolinamidate to give [Cp*Ir(N-phenylpicolinamidate)Cl] (Ir1), high catalytic activity was observed in aqueous solutions.46,47 Surprisingly, other bidentate donors such as 2,2′-bipyridine or 2-phenylpyridine did not produce viable Ir catalysts. Despite its high intrinsic activity, Ir1 is prone to catalyst inhibition by thiols due to its unprotected half-sandwich structure. Although Ir1 was used successfully to perform transfer hydrogenation in living cells,19 its catalytic efficiency is likely low due to their potential to be deactivated in the biological milieu.
In the present work, to protect Ir1, we created a macrocyclic platform that would accommodate two Cp*Ir units to give Ir2 (Scheme 1). In addition to being easier to synthesize than a caged catalyst, Ir2 also contains backbone amides that are capable of binding external Lewis acids, reminiscent of amino acid side chains inside biological ion channels.48,49 We discovered that combining Ir2 with alkali ions led to the formation of higher-ordered structures, which exhibited improved tolerance towards glutathione and reacted preferentially with small aldehyde substrates under physiologically relevant conditions. Although supramolecular metal–organic complexes have been used in biological applications,50–52 bioorthogonal catalysts formed in situ via metal–ligand coordination are unprecedented to the best of our knowledge. Our work suggests that coordination self-assembly may complement other MIC confinement strategies26 and provide a convenient way to endow catalysts with emergent properties.53,54 In particular, the ability to discriminate between sterically hindered vs. unhindered carbonyl groups could be useful in biological aldehyde detoxification.55–57
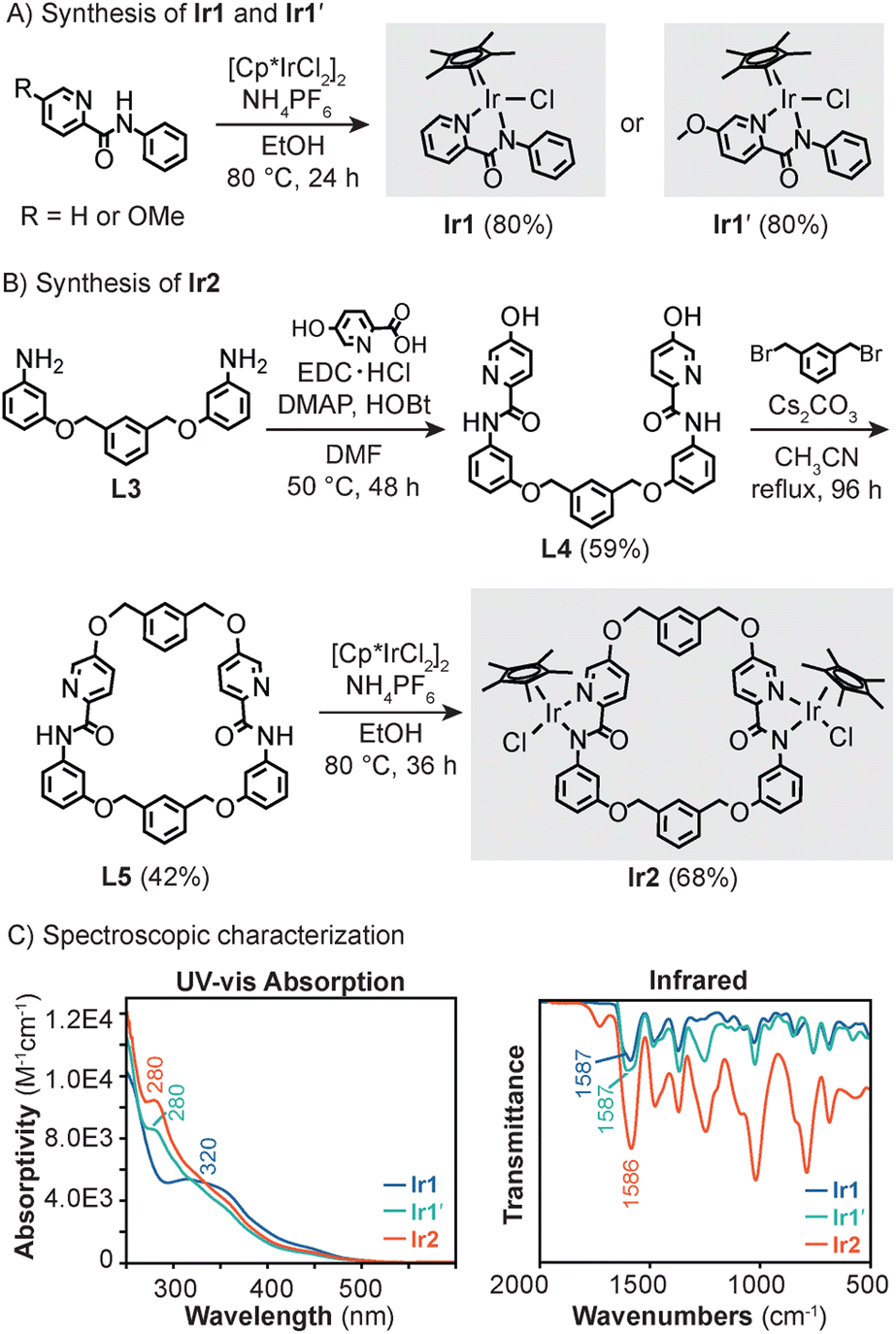 | ||
| Scheme 1 Synthesis of monoiridium Ir1 and Ir1′ (A) and diiridium Ir2 (B) and their UV-vis absorption (CH2Cl2, RT) and infrared spectra (C). | ||
Results and discussion
Because Ir1 lacks steric shielding on one hemisphere due to its half-sandwich structure, it can interact with external species of varying shapes and sizes. This feature is problematic because the catalyst can be readily poisoned by endogenous components and is unable to differentiate between functionally similar substrates. We reasoned that if two Ir1 units could be linked through several attachment points, the iridium centers would be spatially confined by restricting their rotational freedom. Based on this rationale, we synthesized the symmetric Ir2 complex, which has the formula [Cp*2Ir2(ligand L5)Cl2] (Scheme 1B). The ligand L5 was prepared starting from precursor L3, which contains 3-aminophenol units flanking a m-xylene spacer. Combining L3 with 5-hydroxypicolinic acid in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), 4-(dimethylamino)pyridine (DMAP), and N-hydroxybenzotriazole (HOBt) provided L4 in 59% yield. Template-assisted ring closing by treating L4 with aliquots of m-xylylene dibromide and Cs2CO3 over a period of 96 h under dilute conditions afforded L5 as a white solid (42% yield). Finally, Ir2 was obtained by stirring L5 with [Cp*IrCl2]2 and NH4PF6 at 80 °C for 36 h to furnish a yellow solid in 68% yield after purification by silica gel column chromatography. As catalyst standards, we also prepared Ir1 (ref. 58) and a variant bearing N-phenyl-5-methoxypicolinamidate (Ir1′) (Scheme 1A). The latter was designed to be a mononuclear analogue of Ir2, except that it lacks the ability to coordinate with Lewis acids as will be demonstrated in a later section.Spectroscopic characterization of the mono- and diiridium complexes revealed that they exhibit similar features (Scheme 1C). For example, the ultraviolet-visible (UV-vis) absorption spectra of Ir1′ and Ir2 were nearly identical, showing a prominent band centered at 280 nm. In contrast, Ir1 has an absorption maximum at 320 nm. All three complexes displayed CO vibrational stretches at ∼1590 cm−1, suggesting that the amidate groups donate electron density to their respective metal centers to the same extent. These results indicate that the Ir centers in Ir1, Ir1′, and Ir2 are electronically similar despite the latter having a more sterically crowded environment due to the macrocycle structure. Thus, the effects of spatial confinement on the catalyst behavior could be teased out by comparing the reactivity of Ir1/Ir1′vs.Ir2.
To test its transfer hydrogenation activity under biologically relevant conditions, Ir2 (0.5 mol% relative to substrate) was combined with benzaldehyde and excess sodium formate (HCOONa) in various mixtures at 37 °C for 24 h. About 10% v/v of DMSO was needed to sufficiently solubilize Ir2. Our results showed that >80% yield of benzyl alcohol was obtained in H2O/DMSO and phosphate buffered saline (PBS)/DMSO (Fig. 1A). The use of cell culture media such as Roswell Park Memorial Institute (RPMI) 1640 and Dulbecco's Modified Eagle Medium (DMEM), with or without fetal bovine serum (FBS), lowered the reaction yield to below 50%. The presence of glutamine (Gln), lysine (Lys), or arginine (Arg) as additives had minimal effects on Ir2 (∼70–80% yield). However, having high concentrations (5–25 mM) of glucose (Glc) reduced the reaction efficiency to ≤52% yield. In previous studies,46 we also observed that Ir1 was more active in PBS than in cell culture media, which was attributed to the inhibiting effects of sulfur- and nitrogen-containing species present in the latter.
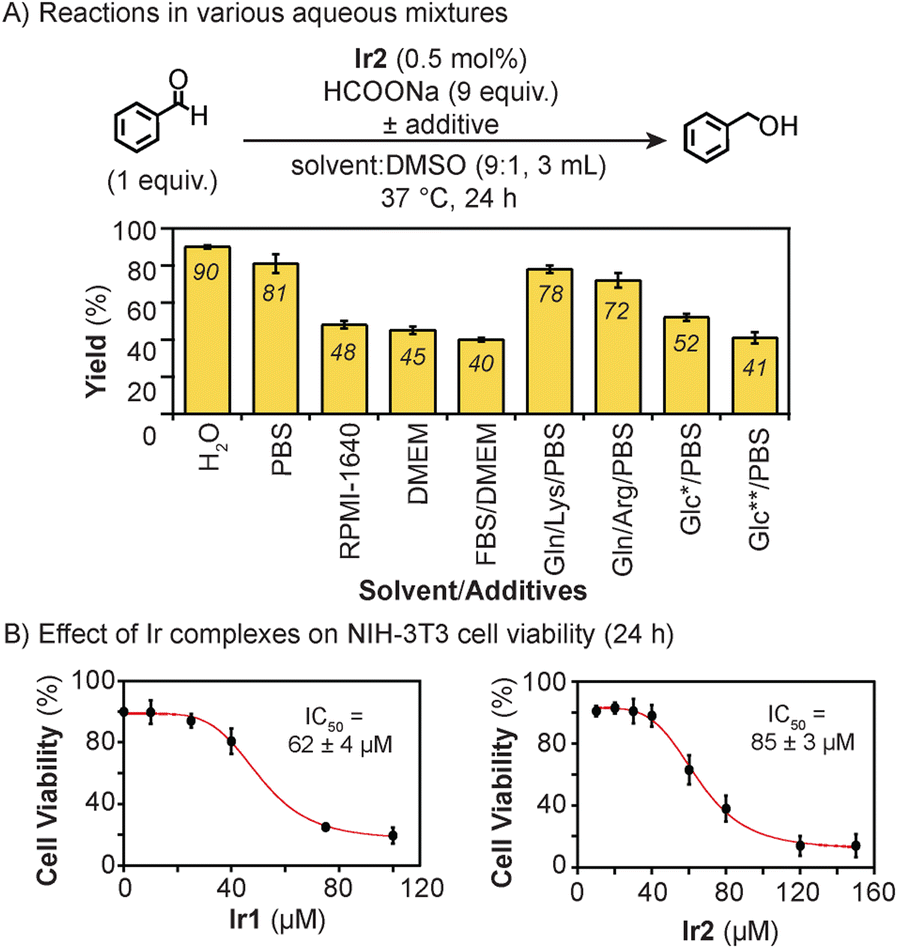 | ||
| Fig. 1 (A) Comparison of the transfer hydrogenation yield using Ir2 in various aqueous media. Abbreviations: PBS = phosphate buffered saline, RMPI = Roswell Park Memorial Institute, DMEM = Dulbecco's Modified Eagle Medium, FBS = fetal bovine serum, Gln = glutamine, Lys = lysine, Arg = arginine, Glc = glucose. Single asterisk (*) = 5 mM; double asterisk (**) = 25 mM. (B) Cell viability curves for Ir1 and Ir2 in NIH-3T3 cells after 24 h exposure. | ||
To assess the potential of Ir2 to be applied in living systems, we measured its effects on NIH-3T3 mouse fibroblast cells after treatment for 24 h (Fig. 1B). Our results showed that the half-maximal inhibition concentration (IC50) of Ir2 was 85 μM, which is higher than that for Ir1 (IC50 = 62 μM), suggesting that the former may be less cytotoxic than the latter. To determine whether the iridium complexes are cell permeable, we measured the iridium content of NIH-3T3 cells treated with either Ir1 or Ir2 for 24 h (Table S13 and Fig. S74†). Our inductively coupled plasma mass spectrometric (ICP-MS) results showed that the cells contained about 1.7× more Ir2 than Ir1 (1487 vs. 858 ng Ir/106 cells, respectively).57 Based on these cytotoxicity and cellular uptake data, Ir2 will likely be compatible with biological hosts at low catalyst loadings (i.e., below its IC50).
We evaluated the substrate scope of Ir2 under our standard transfer hydrogenation conditions (Fig. 2). In these experiments, an aldehyde was combined with Ir2 (0.5 mol% relative to substrate) and HCOONa as the hydride source in H2O/DMSO (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 37 °C under air for 24 h. Reactions of benzaldehyde or its derivatives featuring non-polar substituents (1a–1d) gave the corresponding benzyl alcohol (2a–2d) in excellent yields (>80%) (Fig. 2A). Benzaldehyde variants with electron-withdrawing 3-carbonyl (1h) or 4-nitro (1l) groups were also reduced efficiently (86 and 96% yield of 2h and 2l, respectively). In contrast, benzaldehyde substrates with electron-donating groups (e.g., 4-methoxy in 1e, 4-benzyloxy in 1f, 4-hydroxy in 1g, 4-hydroxy-3-methoxy in 1i, 4-acetylamino in 1j, and 4-dimethylamino in 1k) afforded yields of the alcohol products below 50%.
:1) at 37 °C under air for 24 h. Reactions of benzaldehyde or its derivatives featuring non-polar substituents (1a–1d) gave the corresponding benzyl alcohol (2a–2d) in excellent yields (>80%) (Fig. 2A). Benzaldehyde variants with electron-withdrawing 3-carbonyl (1h) or 4-nitro (1l) groups were also reduced efficiently (86 and 96% yield of 2h and 2l, respectively). In contrast, benzaldehyde substrates with electron-donating groups (e.g., 4-methoxy in 1e, 4-benzyloxy in 1f, 4-hydroxy in 1g, 4-hydroxy-3-methoxy in 1i, 4-acetylamino in 1j, and 4-dimethylamino in 1k) afforded yields of the alcohol products below 50%.
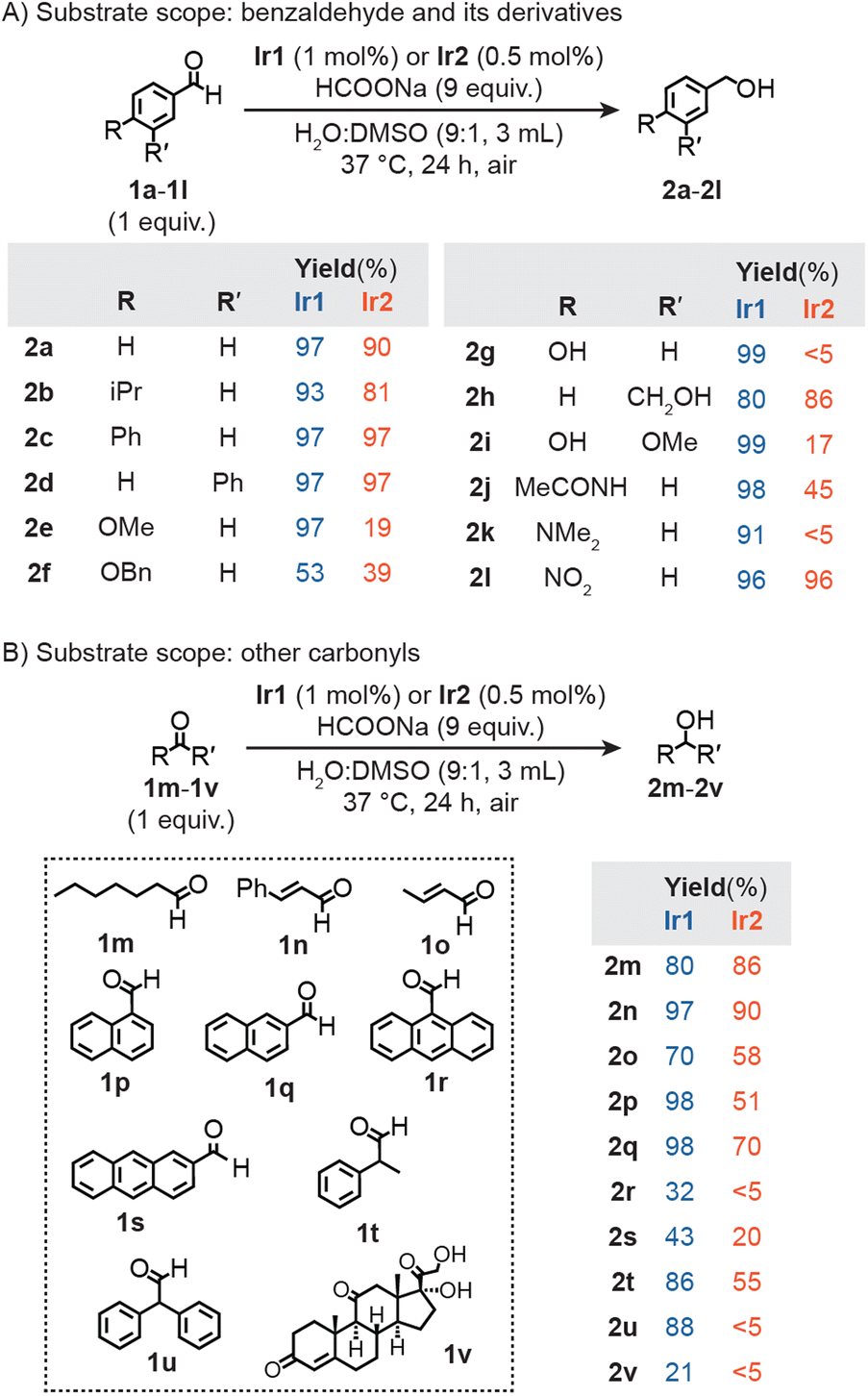 | ||
| Fig. 2 Transfer hydrogenation studies using Ir1 or Ir2 and HCOONa with benzaldehyde and its derivatives (A) or other carbonyl-containing (B) substrates. | ||
A similar set of reactions were performed using the monoiridium complexes, except that the catalyst loading was doubled to 1.0 mol% (relative to substrate) so that the same amount of iridium was used as in the Ir2 reactions. Our data showed that in the presence of HCOONa, Ir1 converted nearly all benzaldehydes to their corresponding benzyl alcohols with almost quantitative yields (Fig. 2A). The lower yield of 2f (53%) from 1f is presumably due to the steric hindrance of the benzyloxy (OBn) group. Since this substituent was not cleaved under our transfer hydrogenation conditions, it suggests that the OBn moiety in the Ir2 macrocycle is unaffected during catalysis. Substrates 1a, 1e, 1k, and 1l were also subjected to transfer hydrogenation reactions using Ir1′ (Table S3†). The results showed that Ir1′ was just as efficient as Ir1, giving ≥90% yields of 2a, 2e, 2k, and 2l, respectively. Neither Ir1 nor Ir2 with HCOONa were able to reduce 4-formylpyridine 1y (Fig. S34†), presumably due to pyridine coordination to Ir.59 Surprisingly, we observed significant yield differences between Ir1vs.Ir2 for products 2e (97 vs. 19%), 2g (99 vs. <5%), 2i (99 vs. 17%), and 2k (91 vs. <5%). Since the iridium centers in Ir1, Ir1′, and Ir2 are electronically similar according to their IR spectral data, the differences are not likely due to electronic effects. We can rule out steric effects because the bulky 4-isopropylbenzaldehyde (1b) and 4-phenylbenzaldehyde (1c) substrates were readily reduced by Ir2/HCOONa. Based on our catalyst structure studies (vide infra), we hypothesize that substrates containing nucleophilic groups (e.g., hydroxy, alkoxy, or amino) are capable of binding to the iridium center via their substituents and are slow to dissociate inside the supramolecular Ir2 + Na+ structure. In contrast, because Ir1 and Ir1′ are not spatially confined, reorientation of the substrates to favor hydride transfer to the electrophilic CO moiety is more facile.
We expanded our substrate scope studies to include other aldehydes (Fig. 2B). Using our typical reaction conditions, we screened aliphatic (1m), α,β-unsaturated (1n, 1o), aromatic (1p–1s), and acetaldehyde substrates (1t–1u). Both Ir1 and Ir2 with HCOONa gave similar yields of 2m, 2n (both CO and CC bonds were reduced), and 2o. For all other alcohol products, the monoiridium catalyst afforded significantly greater yields (>20%) than the diiridium catalyst, indicating that the former is more promiscuous toward substrates. The influence of spatial confinement in Ir2 is most apparent when comparing the reaction yields between chemically similar substrates. For example, Ir2 gave higher yields of 2q (70%) than its structural isomer 2p (51%) since the aldehyde group in 1q is less sterically shielded than that in 1p. Similarly, Ir2 produced 2t (55% yield) with much higher efficiency than 2u (<5% yield). The two α-phenyl groups in 1u likely make the substate too bulky to engage in hydride exchange with the Ir catalyst and formate. Overall, our results clearly showed that Ir2 favors substrates with less sterically accessible aldehyde groups. The ability to differentiate molecular species based on shape and size has not been achieved using conventional half-sandwich catalysts (e.g., Ir1 or Ir1′).
In the context of bioorthogonal chemistry, catalysts that exhibit controlled reactivity are desirable because they can target specific substrates without negatively impacting other biological components.1,2 As a representative example of an essential biomolecule, hydrocortisone (1v)60 was tested as a potential substrate in transfer hydrogenation (Fig. 2B). When Ir1 and HCOONa were combined with 1v in D2O/DMSO-d6 at 37 °C, reduction of the C3 carbonyl group to 2v was observed (Fig. S5†).61 Up to 21% yield of 2v was obtained after 24 h. In contrast, when Ir2 was used as the catalyst, conversion of 1v to 2v did not occur after the same time period (Fig. S7†).
Since many essential aldehydes (e.g., pyridoxal phosphate, retinal, glyceraldehyde-3-phosphate, etc.) are bulky whereas toxic aldehydes (e.g., acrolein, malondialdehyde, etc.) are non-bulky,62–64Ir2 could potentially be used as a selective detoxification agent.55–57 For example, our data showed that reduction of the natural aldehyde vanillin 1i by Ir2/HCOONa is relatively inefficient (17% yield, Fig. 2A). Because vanillin has more potent antioxidant effects than the corresponding vanillyl alcohol, it is desirable to avoid reducing 1i inside the cell.65–68 In contrast, we found that the cell-damaging aldehydes crotonaldehyde (1o, Fig. 2B), 4-hydroxynon-2-enal (Table S11,† entry 2), and hexa-2,4-dienal (Table S11,† entry 3) were converted to their corresponding non-toxic alcohol products by Ir2/HCOONa with high yields under our standard reaction conditions. Although further studies are needed to test the selectivity of Ir2 inside live cells, our results suggest that it exhibits a key feature of bioorthogonal catalysts by virtue of its ability to differentiate between beneficial vs. harmful substances.2
Based on previous studies,47,69 transfer hydrogenation reactions using Ir catalysts can proceed via several pathways (Fig. 3A). The productive route involves reaction of the Ir catalyst with HCOONa to generate an iridium–hydride (Ir–H) intermediate, which can react further with aldehydes to give alcohol products. Alternatively, the Ir–H species can reduce O2 to generate H2O2.69 When thiols are present, such as glutathione, cysteine (Cys), or homocysteine (Hsc), they can deactivate the Ir centers via coordination inhibition. The presence of H2O2 in the reaction mixture could lead to the conversion of thiols to disulfides, which are non-coordinating. Oxidation of aldehydes to carboxylic acids by H2O2 can also occur, but this process is slower than its reduction under our reaction conditions (Tables S5 and S9†).
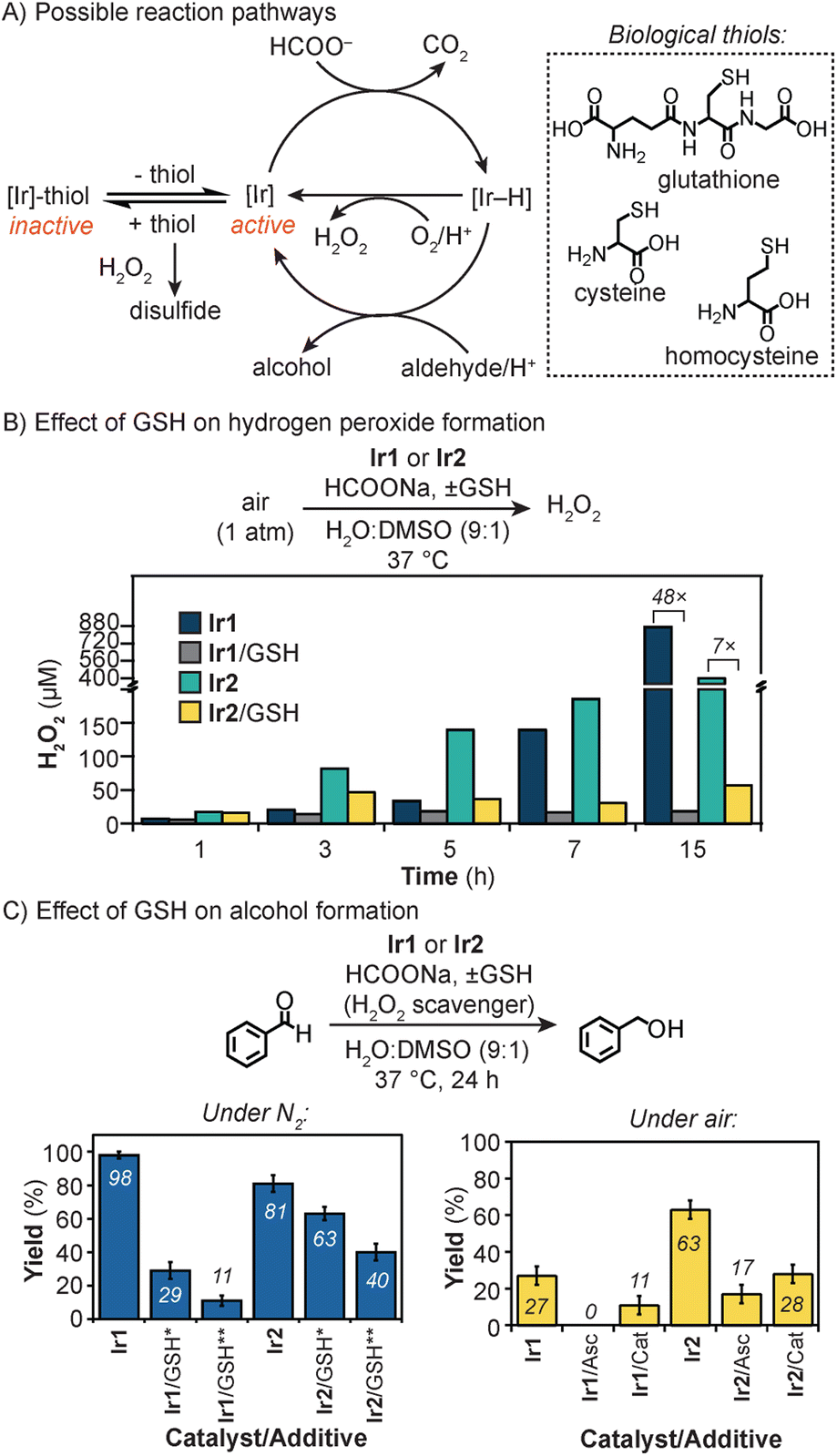 | ||
| Fig. 3 Possible reactions of iridium catalysts during the transfer hydrogenation process (A) and the effects of GSH on H2O2 (B) and alcohol product formation (C). Amounts used: Ir complex (0.15 μmol of Ir1, 0.075 μmol of Ir2), HCOONa (135 μmol), GSH (up to 3.0 μmol), 1a (15 μmol, if any), Asc (6 μmol, if any), Cat (1000 units per mL, if any) in H2O/DMSO (9:1, 3 mL) at 37 °C, 24 h. Single asterisk (*) = 1.5 μmol; double asterisk (**) = 3.0 μmol. Abbreviations: Asc = sodium ascorbate, Cat = catalase. | ||
To examine the thiol tolerance of the iridium complexes, we measured their ability to generate H2O2 in the presence and absence of glutathione (Fig. 3B). The iridium catalyst (50 μM of Ir1 or 25 μM of Ir2) was combined with HCOONa (45 mM) and GSH (50 μM) in H2O/DMSO (9:1) and stirred under air at 37 °C. The formation of hydrogen peroxide was monitored using commercial semi-quantitative H2O2 test strips (Fig. S14 and S15†).69 Our results showed that in all reactions, H2O2 concentrations increased with and without GSH over the course of 15 h. However, the addition of GSH to Ir1 led to a 48-fold inhibition in H2O2 formation (870 vs. 18 μM), whereas the addition of GSH to Ir2 led to only a 7-fold inhibition (401 vs. 57 μM). These observations indicate that Ir2 is less sensitive to thiols than Ir1. Since GSH is a relatively large small-molecule, it may not bind to Ir2 as readily as Ir1 due to its protected active sites.
Next, we investigated the effects of thiols on the reduction of benzaldehyde by the Ir catalysts and HCOONa. The first set of experiments was carried out under N2 to suppress the formation of H2O2 (Fig. 3C and Table S4†). When GSH (1 mM) was added to Ir1 (50 μM) and HCOONa (45 mM) in H2O/DMSO (9:1), the conversion of 1a to 2a dropped by 8.9-fold, relative to the standard reaction without GSH (11 vs. 98% yield, respectively). In comparison, the addition of GSH (1 mM) to Ir2 (25 μM) only lowered the activity by about 2.0-fold with respect to the control (40 vs. 81% yield, respectively).
A second set of experiments was conducted in air to mimic the aerobic environment of mammalian cells. We found that in the presence of 0.5 mM of GSH, reduction of 1a by Ir1 (50 μM) or Ir2 (25 μM) and HCOONa (45 mM) gave 27 and 63% yield of 2a, respectively (Fig. 3C). To minimize oxidation of GSH (Fig. S8†), the reactions were treated with either the anti-oxidant sodium ascorbate (Asc) or H2O2-disproportionating enzyme catalase (Cat) (Table S5†). As expected, the yields of benzyl alcohol decreased due to the availability of more GSH to deactivate the Ir catalysts. For example, the addition of Cat to Ir1 or Ir2 gave 2a in 11 and 28% yield, respectively. Once again, however, reactions with Ir2 generated more transfer hydrogenation products than those with Ir1, demonstrating that the sterically protected Ir2 exhibit better thiol tolerance. The catalyst inhibiting effects of GSH could be reversed by introducing thiol scavengers such as Michael acceptors or chemical oxidants (Table S6†).24,70
Under similar reaction conditions, the use of Cys or Hcy instead of GSH completely deactivated the iridium catalysts (Fig. S12†). Since Cys and Hcy are single amino acids rather than a tripeptide like GSH, their smaller size may allow them to bind Ir2 more readily. Finally, thioethers (e.g., methionine) and disulfides (e.g., cystine and GSH disulfide) have minimal effects on the Ir catalysts.
Because the macrocyclic backbone in Ir2 features multiple amide donors, we wondered if they could chelate cations with different affinities.71,72 This interaction could tune simultaneously the steric and electronic properties of the Ir centers, leading to corresponding changes in their substrate reactivity. We found that when Ir2 and 1p were treated with HCOOLi, HCOONa, HCOOK, or HCOOCs under our standard transfer hydrogenation conditions, 2p (indicated as ** in Fig. 4) was produced in 17, 38, 34, and 41% yield, respectively. However, switching the formate salt to either HCOONMe4 or HCOONBu4 gave significantly larger quantities of 2p (>70% yield). It appears that the behavior of Ir2 is strongly dependent on the identity of the cation used. Because 1p is sensitive to the Ir environment due to its sterically crowded CO group, these results suggest that the catalyst structure is more confined in the reaction with alkali ions than with tetraalkylammonium ions. For some substrates, the amount of product generated was independent of X in HCOOX. For example, the reduction of 1a and 1q to the corresponding alcohol 2a (*) and 2q (***), respectively, proceeded with ≥75% yield for all formate salts tested. Because the aldehyde moiety in 1a and 1q is unhindered, these substrates can be readily reduced regardless of Ir2's spatial arrangement.
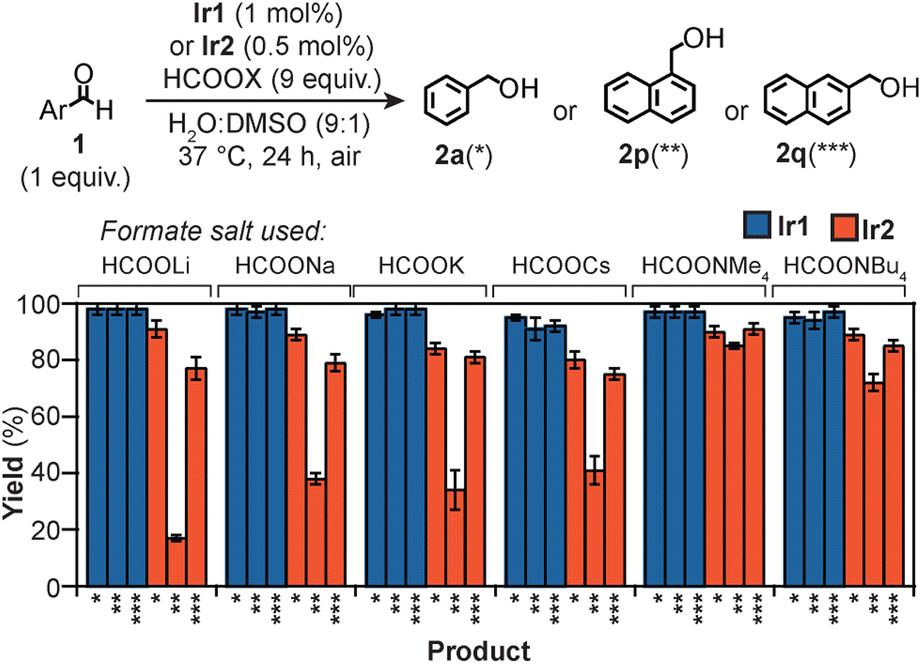 | ||
| Fig. 4 Transfer hydrogenation reactions using Ir catalysts and various formate salts. Reaction conditions used: substrate (15 μmol), formate salt (135 μmol), Ir complex (0.15 μmol of Ir1, 0.075 μmol or Ir2), 37 °C, 24 h. See Table S7† for specific yields. | ||
For comparison, we performed similar reactions using the monoiridium catalysts. Our results showed that Ir1 gave quantitative yields of 2a, 2p, and 2q with all HCOOX salts (Fig. 4), suggesting that the identity of X had no impact on the catalyst activity.
Next, further studies were performed to interrogate the nature of the Ir2–Lewis acid interaction. First, we studied the binding of Na+ to the iridium complexes in H2O/DMSO (9:1) using UV-vis absorption spectroscopy (Fig. 5A). When NaCl was added to a solution containing Ir1, no spectral changes were observed, indicating that Na+ does not bind to the Ir complex. In contrast, when Ir2 was treated with NaCl, the mixture became cloudy and its UV-vis absorption baseline increased. This effect was also observed when NaCl was replaced with NaPF6 (Fig. S20†). Titration of Ir2 with NMe4Cl, which contains a non-coordinating ammonium cation, resulted in no spectral changes (Fig. S24†). These observations indicate that Ir2 binds preferentially to Lewis acidic cations, whereas Ir1 does not.
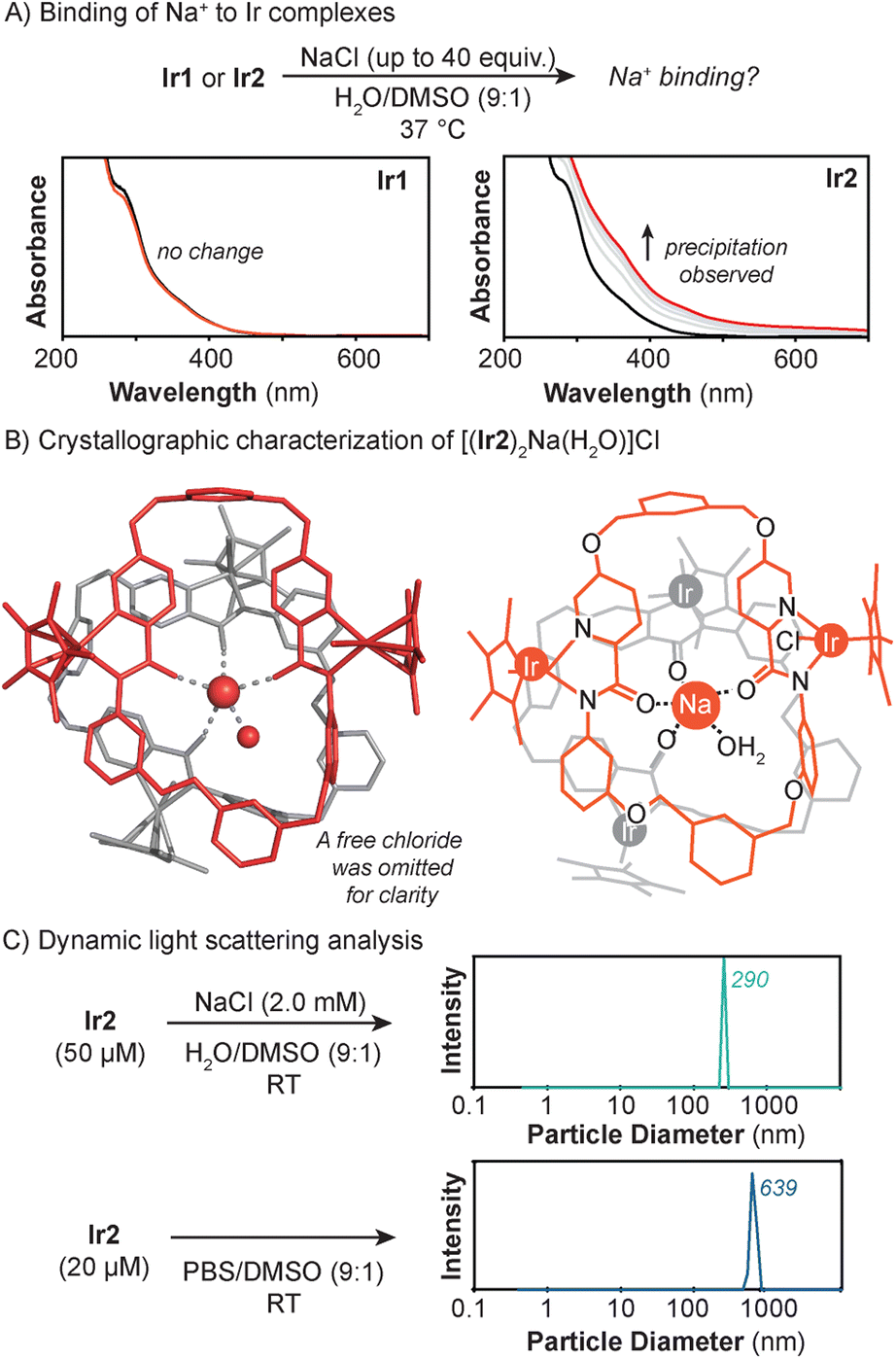 | ||
| Fig. 5 Characterization of the Ir2 + Lewis acid species using UV-vis absorption spectroscopy (A), single crystal X-ray crystallography (B), and dynamic light scattering (C). In part B, the X-ray structure (left) and cartoon representation (right) of [(Ir2)2Na(H2O)]Cl are shown in gray and red-orange to highlight the presence of two different Ir2 units. | ||
Second, to obtain structural characterization of the Ir2 complex in the presence of Lewis acids, X-ray crystallography was used. Single crystals were grown by layering pentane over a dichloromethane solution of Ir2 containing trace amounts of NaCl. Crystallographic analysis showed the presence of a multi-nuclear species with the formula [(Ir2)2Na(H2O)]Cl (Ir2–Na) (Fig. 5B). This complex features two Ir2 units coordinated to a central Na+ ion via the amide groups of their ligand backbone. The iridium centers adopt the expected half-sandwich structure and the sodium center is five-coordinate due to binding with an additional water molecule. A chloride counteranion was also located within the crystallographic asymmetric unit.
Third, to determine whether the structure of Ir2–Na is representative of the species in bulk solution, we used dynamic light scattering (DLS) to measure the Ir2 + NaCl particles in various aqueous mixtures (Fig. 5C). DLS analysis of a H2O/DMSO (9:1) solution containing 50 μM of Ir2 and 2 mM of NaCl showed the presence of particles with a diameter of ∼290 nm. Since biological media contains significant amounts of Lewis acids, we also prepared samples of Ir2 in PBS/DMSO (9:1) (Fig. S23†). The commercial PBS used is composed of KCl (2.7 mM), NaCl (137 mM), Na2HPO4 (10 mM), and KH2PO4 (1.8 mM). Analysis of this heterogeneous solution by DLS showed that the particles formed have an average size of ∼639 nm. Similarly, large nanoassemblies (>750 nm) were observed when Ir2 was measured in the cell culture media Dulbecco's Modified Eagle Medium (DMEM) (Fig. S27 and S28†), which also contains high salt concentrations. Based on the molecular structure of Ir2–Na, the longest length of a single Ir2 complex is ∼1.8 nm. Thus, each Ir2 + NaCl particle must contain hundreds of Ir2 species. However, it is unclear whether the Ir2 + Lewis acid particles comprise aggregates of discrete clusters or a continuous metal–organic network. Although our cell uptake data showed that Ir2 is internalized inside cells, it is uncertain whether the complex is taken up as a discrete molecular species or nanoassembly.52,73–75
Our investigations led to several key findings. First, the macrocyclic effect is critical in enabling Ir2 to form adducts with Lewis acids, since Ir1 and Ir1′ lack this ability despite also possessing backbone amides. Second, the Ir2–Lewis acid nanoassemblies are likely porous because some substrates were reduced with high efficiency (e.g., 1a–1d, 1h, 1l, 1m, and 1n). Presumably, the supramolecular motif allows small molecules to diffuse in and out of the active sites but at the same time, restricts access to species bearing nucleophilic substituents (e.g., 1e, 1g, 1i, 1j, 1k, and 1y) or sterically hindered CO groups (e.g., 1r, 1u, and 1v). Our results also suggest that the confined active sites in the Ir2–Lewis acid nanoassemblies disfavor binding by GSH, which allows the free thiols to scavenge H2O2 and further increase the Ir catalyst's reaction efficiency. Third, substrate selectivity may be further tuned by judicious selection of the Lewis acid. For example, using Li+ gave lower yields of 2p than using Na+, K+, or Cs+ (Fig. 4). A comparison of the IR spectra of Ir2 with and without NaCl did not reveal any significant differences (Fig. S31†). However, further studies are needed to determine how the binding of different cations changes the structural and electronic properties of the Ir2 supramolecular motif. Although Korendovych and coworkers have reported an elegant example using Cp*Ir nanoassemblies for enantioselective transfer hydrogenation reactions,76 their vesicle-like structures were formed via hydrophobic interactions rather than metal–ligand coordination. Lastly, although the intrinsic activity of Ir1 is higher than Ir2 + Lewis acid because the former has a more sterically accessible active site (Fig. 3C, left), Ir2 + Lewis acid showed higher activity when reactions were conducted in the presence of GSH and air (Fig. 3C, right). Thus, when developing catalysts for bioorthogonal chemistry applications, it is important to consider how they might perform under cell-like rather than controlled synthetic environments.
Conclusions
In summary, we present a new strategy to design bioorthogonal catalysts by leveraging Lewis acid-driven coordination self-assembly. Our macrocyclic construct Ir2 was demonstrated to be a highly active catalyst for transfer hydrogenation reactions under physiologically relevant conditions. In the presence of Lewis acids, Ir2 tolerates up to 1.0 mM of GSH and reacts preferentially with small aldehydes. Their ability to discriminate substrates based on shape and size could have useful biological applications, such as the selective detoxification of toxic aldehydes over essential aldehydes. Our studies suggest that the unique reactivity of the Ir2–Lewis acid nanoassemblies stems from the spatial confinement of the Ir centers in a supramolecular framework. Although the exact nature of this extended structure is currently under investigation, the preorganized amide donors in Ir2 are likely responsible for its ability to form stable adducts with secondary cations. Our MIC confinement approach is advantageous over other methods because it can be applied in situ by exploiting naturally occurring salts in biological media (e.g., Li+, Na+, K+, etc.). Based on preliminary cell studies, Ir2 is non-toxic when used at low concentrations (i.e., below its IC50 value of 85 μM in NIH-3T3 cells). However, the extent to which its reactivity in the flask is replicated inside living cells and its cellular uptake mechanism need to be investigated. Finally, we are also exploring advanced applications, such as exploiting the heterogeneous cellular environment to achieve dynamic or switchable self-assembly.49 Importantly, this work expands the options available for developing metal-based intracellular catalysts, which will likely provide new opportunities for bioorthogonal chemistry discovery.Data availability
Crystallographic data for Ir2-Na has been deposited at the CCDC under 2267222. The data supporting this article have been uploaded as part of the ESI.†Author contributions
H. D. N.: conceptualization, investigation, analysis, writing. R. D. J.: investigation, editing. D. T. C.: investigation. T. V. T.: conceptualization, editing. L. H. D.: conceptualization, analysis, writing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Welch Foundation (Grant No. E-1894 to L. H. D.) and the National Institutes of Health (Grant No. R01GM129276 to L. H. D.) for funding this work. We thank Mina Omidiyan, Jong Moon Lee, and Prof. T. Randall Lee for assisting and allowing us to access their dynamic light scattering instrument.References
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- M. M. A. Mitry, F. Greco and H. M. I. Osborn, Chem.–Eur. J., 2023, 29, e202203942 CrossRef CAS PubMed.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogoub and G. Gasser, Chem. Sci., 2023, 14, 409–442 RSC.
- A. Seoane and J. L. Mascareñas, Eur. J. Org Chem., 2022, e202200118 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- A. H. Ngo, S. Bose and L. H. Do, Chem.–Eur. J., 2018, 24, 10584–10594 CrossRef CAS PubMed.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Chem. Sci., 2022, 13, 6478–6495 RSC.
- N. Li, R. K. V. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- S. N. W. Toussaint, R. T. Calkins, S. Lee and B. W. Michel, J. Am. Chem. Soc., 2018, 140, 13151–13155 CrossRef CAS PubMed.
- N. S. Schunck and S. Mecking, Angew. Chem., Int. Ed., 2022, 61, e202211285 CrossRef CAS PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- N. Singh, A. Gupta, P. Prasad, P. Mahawar, S. Gupta and P. K. Sasmal, Inorg. Chem., 2021, 60, 12644–12650 CrossRef CAS PubMed.
- S. Bose, A. H. Ngo and L. H. Do, J. Am. Chem. Soc., 2017, 139, 8792–8795 CrossRef CAS PubMed.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 22017–22025 CrossRef PubMed.
- A. Gutierrez-Gonzalez, D. Marcos-Atanes, L. G. Cool, F. Lopez and J. L. Mascareñas, Chem. Sci., 2023, 14, 6408–6413 RSC.
- C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascareñas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS PubMed.
- Y. M. Wilson, M. Dürrenberger, E. S. Nogueira and T. R. Ward, J. Am. Chem. Soc., 2014, 136, 8928–8932 CrossRef CAS PubMed.
- H. D. Nguyen and L. H. Do, Curr. Opin. Chem. Biol., 2022, 71, 102213 CrossRef CAS PubMed.
- A. Sathyan, L. Deng, T. Loman and A. R. A. Palmans, Catal. Today, 2023, 418, 114116 CrossRef CAS.
- D. Diao, A. J. Simaan, A. Martinez and C. Colomban, Chem. Commun., 2023, 59, 4288–4299 RSC.
- Y. Liu, K. L. Lai and K. Vong, Eur. J. Inorg. Chem., 2022, e202200215 CrossRef CAS.
- B. Maity, M. Taher, S. Mazumdar and T. Ueno, Coord. Chem. Rev., 2022, 469, 214593 CrossRef CAS.
- S. Liu, P. Du, H. Sun, H.-Y. Yu and Z.-G. Wang, ACS Catal., 2020, 10, 14937–14958 CrossRef CAS.
- Y. Okamoto and T. R. Ward, Angew. Chem., Int. Ed., 2017, 56, 10156–10160 CrossRef CAS PubMed.
- M. Basauri-Molina, C. F. Riemersma, M. A. Würdemann, H. Kleijn and R. J. M. Klein Gebbink, Chem. Commun., 2015, 51, 6792–6795 RSC.
- M. Taher, B. Maity, T. Nakane, S. Abe, T. Ueno and S. Mazumdar, Angew. Chem., Int. Ed., 2022, 61, e202116623 CrossRef CAS PubMed.
- S. C. Bete and M. Otte, Angew. Chem., Int. Ed., 2021, 60, 18582–18586 CrossRef CAS PubMed.
- G. Izzet, J. Zeitouny, H. Akdas-Killig, Y. Frapart, S. Ménage, B. Douziech, I. Jabin, Y. Le Mest and O. Reinaud, J. Am. Chem. Soc., 2008, 130, 9514–9523 CrossRef CAS PubMed.
- D. Zhang, K. Jamieson, L. Guy, G. Gao, J.-P. Dutasta and A. Martinez, Chem. Sci., 2017, 8, 789–794 RSC.
- J. G. Rebelein, Y. Cotelle, B. Garabedian and T. R. Ward, ACS Catal., 2019, 9, 4173–4178 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Chen, E. S. Garcia and S. C. Zimmerman, Acc. Chem. Res., 2020, 53, 1244–1256 CrossRef CAS PubMed.
- G. Qiu, P. Nava, A. Martinez and C. Colomban, Chem. Commun., 2021, 57, 2281–2284 RSC.
- C.-M. Hirschbiegel, X. Zhang, R. Huang, Y. A. Cicek, S. Fedeli and V. M. Rotello, Adv. Drug Delivery Rev., 2023, 195, 114730 CrossRef CAS PubMed.
- Y. Fan, S. Liu, Y. Yi, H. Rong and J. Zhang, ACS Nano, 2021, 15, 2005–2037 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- L. A. Khawli and S. Prabhu, Mol. Pharmaceutics, 2013, 10, 1471–1472 CrossRef CAS PubMed.
- A. H. Ngo, M. Ibañez and L. H. Do, ACS Catal., 2016, 6, 2637–2641 CrossRef CAS.
- A. H. Ngo and L. H. Do, Inorg. Chem. Front., 2020, 7, 583–591 RSC.
- W. A. Catterall, M. J. Lenaeus and T. M. Gamal El-Din, Annu. Rev. Pharmacol. Toxicol., 2020, 60, 133–154 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Chem. Rev., 2014, 114, 538–556 CrossRef CAS PubMed.
- G. Moreno-Alcántar and A. Casini, FEBS Lett., 2023, 597, 191–202 CrossRef PubMed.
- S. V. Dummert, H. Saini, M. Z. Hussain, K. Yadava, K. Jayaramulu, A. Casini and R. A. Fischer, Chem. Soc. Rev., 2022, 51, 5175–5213 RSC.
- G. Moreno-Alcántar, Eur. J. Inorg. Chem., 2023, 26, e202200788 CrossRef.
- S. Lo, C. Z.-J. Ren, P. Solís-Muñana and J. L.-Y. Chen, in Supramolecular Nanotechnology, 2023, pp. 469–493 Search PubMed.
- C. C. James, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2023, e202306645 Search PubMed.
- Z. Xie, S. P. Baba, B. R. Sweeney and O. A. Barski, Chem.-Biol. Interact., 2013, 202, 288–297 CrossRef CAS PubMed.
- L. T. Beringer, S. Li, G. Gilmore, J. Lister and S. Averick, Mol. Pharmaceutics, 2015, 12, 3776–3781 CrossRef CAS PubMed.
- R. D. Jana, A. H. Ngo, S. Bose and L. H. Do, Chem.–Eur. J., 2023, e202300842 CrossRef CAS PubMed.
- Z. Almodares, S. J. Lucas, B. D. Crossley, A. M. Basri, C. M. Pask, A. J. Hebden, R. M. Phillips and P. C. McGowan, Inorg. Chem., 2014, 53, 727–736 CrossRef CAS PubMed.
- A. C. Carrasco, V. Rodríguez-Fanjul and A. M. Pizarro, Inorg. Chem., 2020, 59, 16454–16466 CrossRef CAS PubMed.
- L. M. Mongioì, R. A. Condorelli, F. Barbagallo, S. La Vignera and A. E. Calogero, Endocrine, 2020, 67, 507–515 CrossRef PubMed.
- A. H. Ngo, M. J. Adams and L. H. Do, Organometallics, 2014, 33, 6742–6745 CrossRef CAS.
- R. M. LoPachin and T. Gavin, Chem. Res. Toxicol., 2014, 27, 1081–1091 Search PubMed.
- S. Dalleau, M. Baradat, F. Guéraud and L. Huc, Cell Death Differ., 2013, 20, 1615–1630 CrossRef CAS PubMed.
- D. T. Nam, M. Arseneault, V. Murthy and C. Ramassamy, Curr. Mol. Pharmacol., 2010, 3, 66–78 CrossRef CAS.
- D. P. Bezerra, A. K. N. Soares and D. P. de Sousa, Oxid. Med. Cell. Longevity, 2016, 2016, 9734816 Search PubMed.
- D. Zhao, J. Sun, B. Sun, M. Zhao, F. Zheng, M. Huang, X. Sun and H. Li, RSC Adv., 2017, 7, 46395–46405 RSC.
- A. Tai, T. Sawano, F. Yazama and H. Ito, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 170–177 CrossRef CAS PubMed.
- A. Tai, T. Sawano and F. Yazama, Biosci., Biotechnol., Biochem., 2011, 75, 2346–2350 CrossRef CAS PubMed.
- H. T. H. Nguyen and L. H. Do, Chem. Commun., 2020, 56, 13381–13384 RSC.
- S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Chem. Sci., 2017, 8, 2107–2114 RSC.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- A. J. M. Miller, Dalton Trans., 2017, 46, 11987–12000 RSC.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- K. Kettler, K. Veltman, D. van de Meent, A. van Wezel and A. J. Hendriks, Environ. Toxicol. Chem., 2014, 33, 481–492 CrossRef CAS PubMed.
- M. A. Dolan, P. N. Basa, O. Zozulia, Z. Lengyel, R. Lebl, E. M. Kohn, S. Bhattacharya and I. V. Korendovych, ACS Nano, 2019, 13, 9292–9297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2267222. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02836d |
| This journal is © The Royal Society of Chemistry 2023 |