
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integration of plasma and electrocatalysis to synthesize cyclohexanone oxime under ambient conditions using air as a nitrogen source†
Shunhan
Jia
 ab,
Xingxing
Tan
a,
Limin
Wu
ab,
Xiaodong
Ma
a,
Libing
Zhang
ab,
Jiaqi
Feng
a,
Liang
Xu
ac,
Xinning
Song
ab,
Qinggong
Zhu
ab,
Xinchen
Kang
ab,
Xiaofu
Sun
*ab and
Buxing
Han
*abd
ab,
Xingxing
Tan
a,
Limin
Wu
ab,
Xiaodong
Ma
a,
Libing
Zhang
ab,
Jiaqi
Feng
a,
Liang
Xu
ac,
Xinning
Song
ab,
Qinggong
Zhu
ab,
Xinchen
Kang
ab,
Xiaofu
Sun
*ab and
Buxing
Han
*abd
aBeijing National Laboratory for Molecular Sciences, CAS Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: sunxiaofu@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China
dShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 30th October 2023
Abstract
Direct fixation of N2 to N-containing value-added chemicals is a promising pathway for sustainable chemical manufacturing. There is extensive demand for cyclohexanone oxime because it is the essential feedstock of Nylon 6. Currently, cyclohexanone oxime is synthesized under harsh conditions that consume a considerable amount of energy. Herein, we report a novel approach to synthesize cyclohexanone oxime by in situ NO3− generation from air under ambient conditions. This process was carried out through an integrated strategy including plasma-assisted air-to-NOx and co-electrolysis of NOx and cyclohexanone. A high rate of cyclohexanone oxime formation at 20.1 mg h−1 cm−2 and a corresponding faradaic efficiency (FE) of 51.4% was achieved over a Cu/TiO2 catalyst, and the selectivity of cyclohexanone oxime was >99.9% on the basis of cyclohexanone. The C–N bond formation mechanism was examined by in situ experiments and theoretical calculations, which showed that cyclohexanone oxime forms through the reaction between an NH2OH intermediate and cyclohexanone.
Introduction
Dinitrogen (N2) constitutes approximately 80% of the air, and is an inexhaustible nitrogen source that can be used to produce N-containing chemicals with a wide range of applications.1,2 However, because of the ultra-inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds, the kinetics are sluggish and the thermodynamics are limited for the conversion of N2.3–5 Over the past few decades, researchers were mainly focused on the fixation of N2 to NH3 as a feedstock that would be further processed into various organonitrogen chemicals through reductive amination, oxidative cyanation, and ammoxidation.6–8 Industrial NH3 production through the traditional Haber–Bosch process consumes large amounts of energy and emits high levels of CO2, and therefore cannot meet the demands for carbon neutrality or sustainability goals.9–11 As an alternative route, the direct utilization of N2 to construct C–N bonds under mild conditions is a promising strategy for the production of value-added chemicals and the decoupling of chemical manufacturing from fossil fuel energy, but it is a challenge.12–14
N triple bonds, the kinetics are sluggish and the thermodynamics are limited for the conversion of N2.3–5 Over the past few decades, researchers were mainly focused on the fixation of N2 to NH3 as a feedstock that would be further processed into various organonitrogen chemicals through reductive amination, oxidative cyanation, and ammoxidation.6–8 Industrial NH3 production through the traditional Haber–Bosch process consumes large amounts of energy and emits high levels of CO2, and therefore cannot meet the demands for carbon neutrality or sustainability goals.9–11 As an alternative route, the direct utilization of N2 to construct C–N bonds under mild conditions is a promising strategy for the production of value-added chemicals and the decoupling of chemical manufacturing from fossil fuel energy, but it is a challenge.12–14
There is great interest in the use of the electrochemical N2 reduction reaction (NRR) to produce NH3 because of the utilization of renewable electricity and protons directly from water.15–17 The electrochemical construction of the C–N bond via the coupling of CO2 and N2 has also been considered to produce some organonitrogen chemicals, such as urea.18,19 However, the reported reactions are very limited, and they are hindered by low reactivity and selectivity due to low N2 solubility, difficult N2 activation, and undesired hydrogen evolution reaction (HER).
Based on the occurrence of lightning in nature, air plasma oxidation offers an effective method for N2 activation. Air can be converted into reactive NOx or NOx−, which can be used as a N source in electrolysis.20,21 The C–N coupling would be achieved by the in situ generation of nucleophilic N-containing intermediates followed by their coupling with carbon substrates or intermediates as the electrophile.22,23
Cyclohexanone oxime is the essential feedstock of Nylon 6, with worldwide annual demand of approximately 10 million tons and an approximate global market size of USD 25 billion, and is currently produced through the coupling between cyclohexanone and NH2OH intermediates from unsustainable (NH3OH)(NH4)SO4 (Scheme 1).24–26 Therefore, optimization of N sources for NH2OH intermediates is urgently needed for cyclohexanone oxime production. Researchers have recently explored the synthesis of cyclohexanone oxime using in situ-generated H2O2 to produce NH2OH intermediates from NH3.8 Furthermore, the direct use of N2via air plasma oxidation integrated with electroreduction may result in the formation of cyclohexanone oxime under ambient conditions.
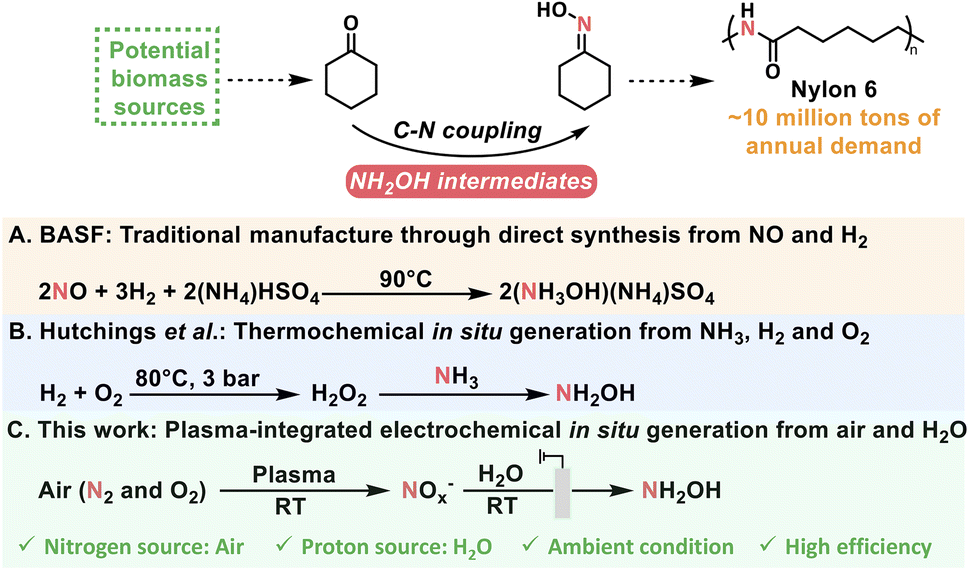 | ||
| Scheme 1 Schematic diagram of different production routes for cyclohexanone oxime. | ||
Herein, we report the first work to efficiently synthesize cyclohexanone oxime using air as the nitrogen source by combining plasma-assisted air-to-NOx and co-electrolysis of NOx and cyclohexanone. A faradaic efficiency (FE) of 51.4% was achieved for cyclohexanone oxime with a corresponding formation rate of 20.1 mg h−1 cm−2 and remarkable catalyst (Cu/TiO2) recyclability. The superior performance can be attributed to Cu sites supported on TiO2 with a stable oxidation state. In addition, the reaction pathway was studied based on in situ experiments and theoretical calculations.
Results and discussion
As shown in Fig. 1A, as a proof-of-concept, we synthesized cyclohexanone oxime through the coupling of plasma air activation and NOx electroreduction with cyclohexanone (Fig. S1†). A model air mixture of N2/O2 (v/v = 4/1) was used as the nitrogen source. The reaction between N2 and O2 activated by plasma produced NOx, which was absorbed by 1 mol L−1 NaOH aqueous solution, and the as-obtained solution was directly used as an electrolyte for further electrosynthesis (Fig. S2 and S3†). This process holds the potential to block the interference of trace H2O and CO2 from real air. The concentration of NO3− and NO2− was quantified by UV-Vis spectroscopy measurements (Fig. S4†). Fig. 1B shows that the yield of NOx linearly increased over the plasma activation time, in which the concentration of NO2− was approximately 2 times that of NO3−. A high total NOx concentration of approximately 1 mol L−1 was achieved after 400 min of plasma activation.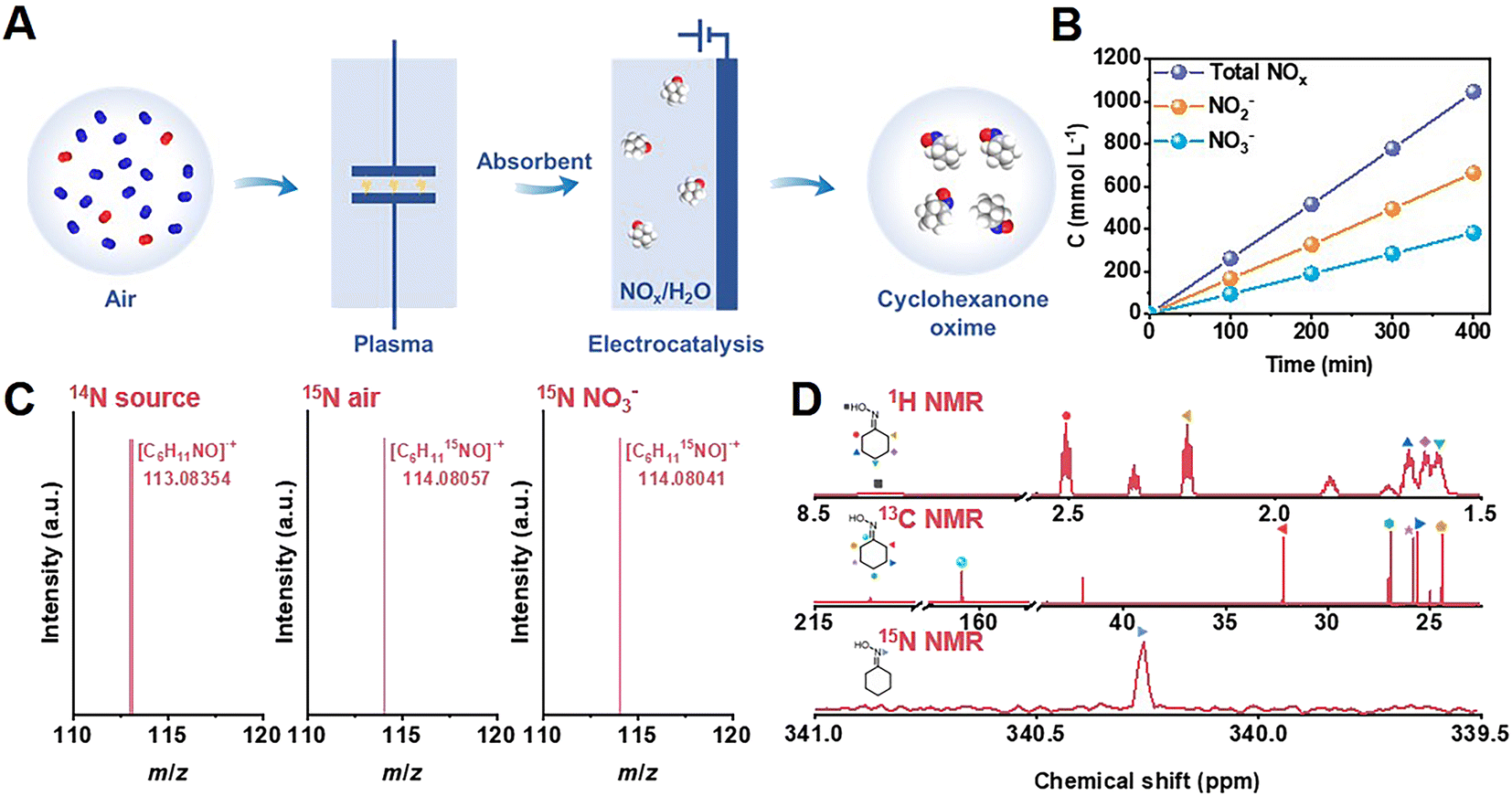 | ||
| Fig. 1 The integration system for cyclohexanone oxime synthesis. (A) Schematic illustration of the synthesis of cyclohexanone oxime with air as nitrogen source. (B) The concentration of NOx− species under air activation. (C) GC-MS and (D) 1H NMR, 13C NMR, and 15N NMR detection of cyclohexanone oxime. | ||
In the following electrosynthesis process, the electroreduction of NO3− with cyclohexanone was initially verified as a model reaction in a flow cell with 1 mol L−1 NaNO3 and 50 mmol L−1 cyclohexanone aqueous solution as the catholyte, and 1 mol L−1 NaOH as the anolyte. Copper-based catalysts were selected due to their exceptional performance in nitrate reduction, as described in recent reports.27–31 Cu/TiO2 catalysts loaded onto porous carbon paper served as the cathode, and Ni foam was used as the anode.
The obtained catalysts contained 0.3, 0.6, and 0.9 wt% Cu, as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). We denoted these catalysts as 0.3% Cu/TiO2, 0.6% Cu/TiO2, and 0.9% Cu/TiO2 for clarity. H2 was the only gas product, and cyclohexanone oxime was the only organic product during electrolysis quantified by gas chromatography (GC). Colorimetric methods were used to study the aqueous byproducts, including NO2− and NH3 (Fig. S4 and S5†). We also identified cyclohexanone oxime via GC-mass spectrometry (GC-MS) and nuclear magnetic resonance (NMR) (Fig. S6–S10†). As shown in Fig. 1C, the molecular weight of 113.08354 provided by high-resolution GC-MS indicated that the molecular formula of the product is C6H11NO.
Using model air with 15N2 gas and Na15NO3 as the reactant, the molecular ion peak of the product at 114.08057 and 114.08041 matched the calculated weight of 114.08055 for C6H1115NO, which verified that the origin of nitrogen in cyclohexanone oxime was NO3−in situ generated from air. Moreover, all the peaks in the 1H NMR, 13C NMR, and 15N NMR (conducted with the 15N-labeled product) spectra were well matched with the standard samples (Fig. 1D and S9†). These results confirmed the formation of cyclohexanone oxime, with NO3− as the nitrogen source.
Fig. 2A displays the linear sweep voltammetry (LSV) results for 0.6% Cu/TiO2 in electrolyte with and without NaNO3 plus cyclohexanone substrates. After the addition of the substrates, the sharpness of the increase in the current density (j) was greater than that of the initial NaOH electrolyte, which demonstrated that the electrosynthesis of cyclohexanone was more kinetically favorable than the HER. Electrochemical performance tests were carried out in the range of −1.0 V to −2.0 V versus Ag/AgCl, and the electrochemical performance of cyclohexanone oxime formation on Cu/TiO2 is shown in Fig. 2B and C. The reactions under all the applied potentials presented a combined FE of approximately 100% for all products. A maximum cyclohexanone oxime FE of 50.0% and a corresponding formation rate of 20.1 mg h−1 cm−2 was achieved at −1.8 V over 0.6% Cu/TiO2.
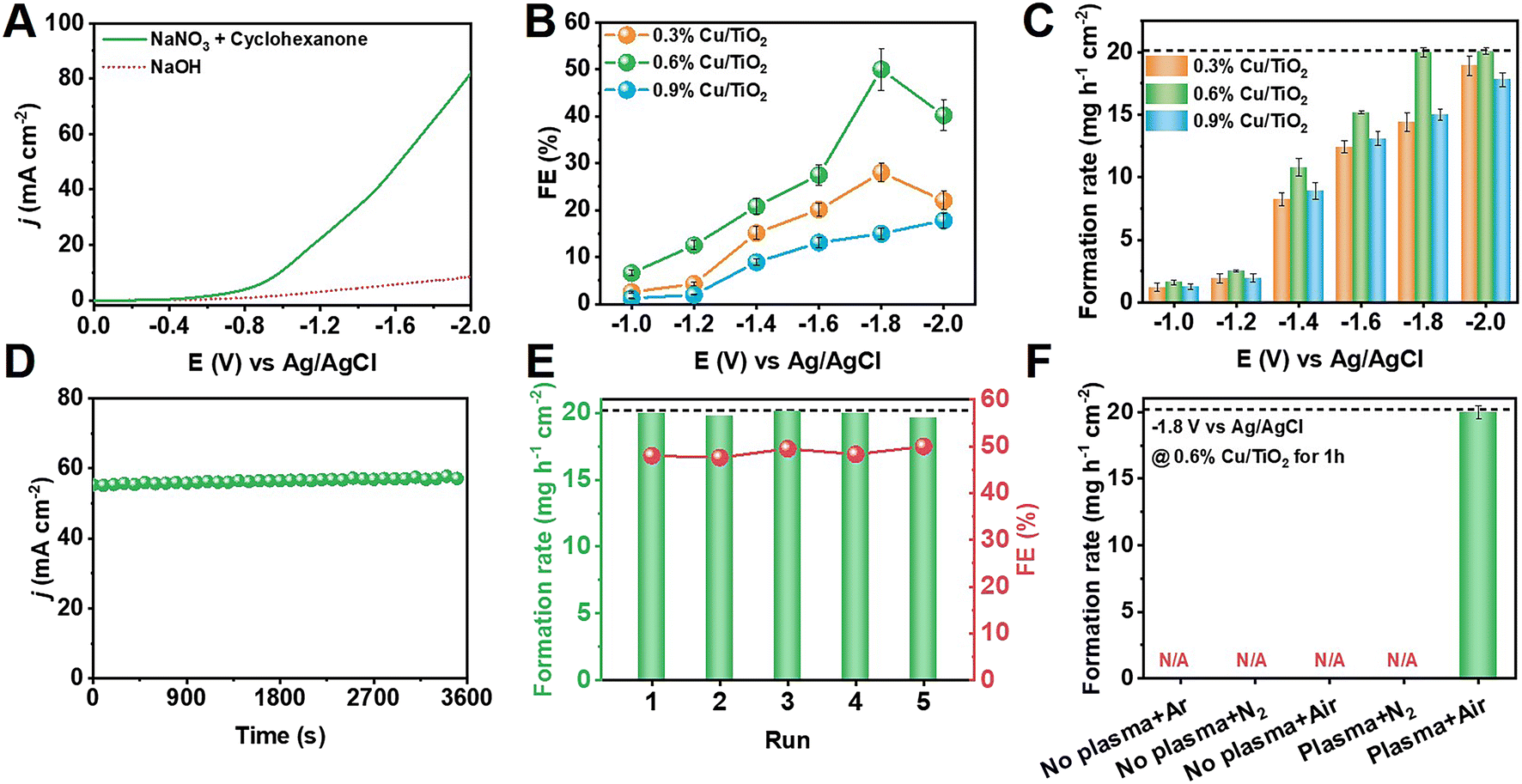 | ||
| Fig. 2 Electrochemistry study of cyclohexanone synthesis. (A) LSV curves of 0.6% Cu/TiO2 catalysts in different electrolyte. Potential-dependent (B) FE and (C) formation rate of cyclohexanone oxime production on different Cu-loaded TiO2 catalysts. (D) Chronoamperometry curves of 0.6% Cu/TiO2 under optimal conditions for cyclohexanone oxime synthesis. (E) Recyclability of 0.6% Cu/TiO2 electrocatalysts. (The dotted line shows the formation rate of cyclohexanone at 100% conversion.) (F) The formation rate of cyclohexanone oxime with different gas feed and plasma conditions. | ||
At low potentials, NO2− was the main byproduct, which could be attributed to the hindered reductive hydrogenation of NO3−. Once the potential increased so that it was more negative than −1.8 V, the enhancement of the HER and the further electroreduction of NH2OH intermediates led to a decrease in the selectivity for cyclohexanone oxime (Fig. S11†). Intriguingly, the byproducts generated during the electrolysis process, specifically NH3 and H2, hold significant value as chemicals with substantial potential for further utilization. Under optimal conditions, the FE of NH3 and H2 were 21.6% and 16.4%, respectively. Similar trends were found for the Cu/TiO2 catalysts with various Cu loadings. In addition, as shown in Fig. 2D, the corresponding chronoamperometry curve of 0.6% Cu/TiO2 catalysts at −0.8 V remained stable during electrosynthesis.
We further studied the influence of substrate concentrations on the electrochemical performance by varying the molar concentrations of cyclohexanone and NaNO3 in the electrolytes at the applied potential of −1.8 V vs. Ag/AgCl (Fig. S12†). This indicated that a higher cyclohexanone and NO3− concentration promoted the formation of cyclohexanone oxime. However, when the concentration of cyclohexanone was greater than 50 mmol L−1, there was a significant decrease in the cyclohexanone oxime FE and formation rate due to the phase separation in the catholyte. The increase in the NO3− concentration up to 2 mol L−1 strengthened the side reaction of NH3 formation, which was unsuitable for the formation of NH2OH intermediates and cyclohexanone oxime. Consequently, the optimized substrate concentration was found to be 50 mmol L−1 cyclohexanone and 1 mol L−1 NaNO3 for cyclohexanone oxime production.
The TiO2 support played a crucial role in the improvement of cyclohexanone oxime formation. Fig. S13† shows that the catalytic performance of the bulk TiO2 was very poor. For comparison, carbon black was also used as a support for Cu loading, but the as-prepared Cu/carbon black catalyst exhibited a maximum cyclohexanone oxime FE of only 8.4% with a formation rate of 2.5 mg h−1 cm−2. Therefore, the optimized catalytic activity could be ascribed to the cooperation between Cu and the TiO2 support.
The stability of the 0.6% Cu/TiO2 catalyst was also studied at −1.8 V. Fig. 2E shows that there was no notable change in the cyclohexanone oxime FE or formation rate during 5 successive cycles, which was indicative of prominent recyclability and is important for practical applications.
After establishing the baseline of NO3− electrolysis activity, NOx obtained from plasma air activation was fed together with cyclohexanone in the electrosynthesis process. Fig. 2F shows that the integrated system provided cyclohexanone oxime at a formation rate of 20.1 mg h−1 cm−2 using air as the nitrogen source. No cyclohexanone oxime was detected in the absence of either plasma, N2, or O2, indicating the necessity of air as the nitrogen source under plasma oxidation for cyclohexanone oxime production. It was therefore concluded that the two-step integration of plasma air activation with electrocatalytic reduction is practically feasible for highly efficient and selective nitrogen synthesis of cyclohexanone oxime under ambient conditions.
Structural and morphological measurements clarified that Cu nanoparticles were uniformly loaded on TiO2 in the Cu/TiO2 catalysts (Fig. S14–S22†). In situ X-ray absorption near-edge spectroscopy (XANES) was conducted to further investigate the electronic structure of the catalysts during cyclohexanone oxime synthesis. Fig. 3A shows that an initial 0.6% Cu/TiO2 catalyst exhibited an absorption edge between that of Cu foil and Cu2O samples, indicating that the oxidation state of Cu was between 0 and +1. We further acquired the Cu average oxidation state as a function of the Cu K-edge energy shift, and the value was 0.42 during the in situ measurements (Fig. 3B). The value did not change over electrolysis time, indicating the excellent stability of the Cu active sites during electrolysis. The results indicated that a stable slightly positive oxidation state for Cu was beneficial for the reaction. In Fig. S23 and S24,† no obvious changes were observed in the structure or morphology of the catalyst after electrolysis, further indicating the stability of the catalyst.
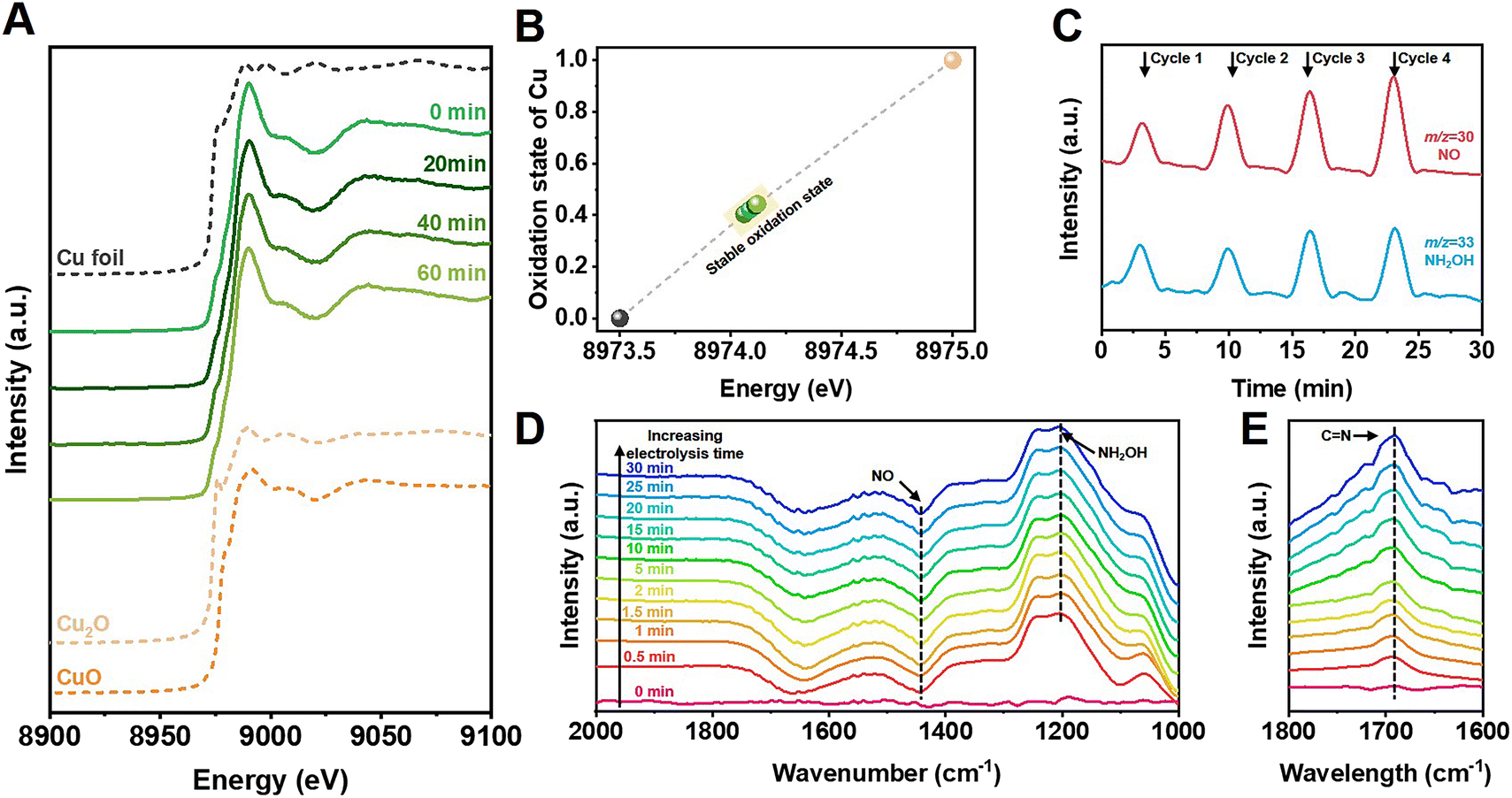 | ||
| Fig. 3 In situ study. (A) XANES spectra at Cu K-edge and (B) Cu oxidation state of 0.6% Cu/TiO2 measured at different electrolysis times during cyclohexanone oxime synthesis. (C) Online DEMS graph of NO and NH2OH intermediates. In situ FTIR spectra of electrochemical cyclohexanone synthesis conducted at prolonged electrolysis time on 0.6% Cu/TiO2 in (D) H2O and (E) D2O solvent. | ||
Further study was conducted to reveal the possible reaction pathway. Online differential electrochemical mass spectrometry (DEMS, Fig. S25†) was used to detect the m/z signals of 30 and 33, as shown in Fig. 3C, which corresponded to the NO and NH2OH intermediates, respectively, during NO3− reduction over 0.6% Cu/TiO2 catalyst. The weak signal of NH2OH indicated that the generated NH2OH spontaneously coupled with cyclohexanone to produce oxime products and could not remain in the bulk electrolyte. Signals of other byproducts were also detected, as shown in Fig. S26.†In situ Fourier transform infrared (FTIR) spectroscopy (Fig. S27†) also confirmed the possible involvement of NO and NH2OH signals at 1442 and 1204 cm−1 in Fig. 3D on the surface of the 0.6% Cu/TiO2 catalyst during electrosynthesis.32–34 Additionally, when using D2O as the electrolyte solvent, an obvious peak at 1689 cm−1 was observed, as shown in Fig. 3E, indicating the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in cyclohexanone oxime molecules that was not observed in H2O solvent because of the interference of O–H bending (1645 cm−1).
N bonds in cyclohexanone oxime molecules that was not observed in H2O solvent because of the interference of O–H bending (1645 cm−1).
Systematic control experiments were performed to study the reaction pathways of the electrosynthesis of cyclohexanone oxime (Table S1†). No cyclohexanone oxime was detected when there was no cyclohexanone or NO3− in the electrolyte (entries 1–3). We also used NO2−, NO, and NH2OH as the feedstock, which were formerly identified as the intermediates (entries 4–6). All three entries produced cyclohexanone oxime products. In addition, the C–N coupling between cyclohexanone and NH2OH was proved to be spontaneous, and occurred without electrolysis (entries 6–7).
The discernible fluctuations in the FE, notably when feeding with NO, were ascribable to the limited solubility of NO gas in aqueous solutions, leading to heightened hydrogen evolution side reactions. Notwithstanding these disparities in the FE, the rate of cyclohexanone oxime formation continued to exhibit a noteworthy level of consistency across all nitrogenous substrates. Therefore, the above results present the possible reaction pathways of NO3− to NH2OH intermediates through NO3− → NO2− → NO → NH2OH progression, followed by the nonelectrochemical spontaneous condensation of NH2OH and cyclohexanone to produce cyclohexanone oxime.
Furthermore, density functional theory (DFT) calculations were conducted to study the reaction pathway (Fig. 4A and S28†). According to high-resolution transmission electron microscopy (HRTEM) images and X-ray diffraction (XRD) patterns, Cu(111) and Cu(111)-Cu2O(110) heterostructures loaded on anatase phase TiO2(101) were used as the catalyst models. A free energy diagram of NO3−-to-NH2OH is presented in Fig. 4B. NO3− was chemically absorbed on the catalyst to initially form 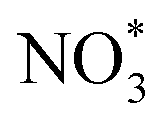 with a decrease in free energy. Then, the deoxygenation and hydrogenation steps were continuously proceeded by H+/e− transfer to form
with a decrease in free energy. Then, the deoxygenation and hydrogenation steps were continuously proceeded by H+/e− transfer to form  and NO*. The conversion of
and NO*. The conversion of 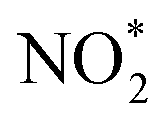 to NOOH* was endergonic and acted as the rate-determining step (RDS), which was consistent with the detection of NO2− byproducts during electrolysis. Subsequently, the NO* intermediate was gradually converted to NOH*, NHOH*, and finally to NH2OH* under hydrogenation.
to NOOH* was endergonic and acted as the rate-determining step (RDS), which was consistent with the detection of NO2− byproducts during electrolysis. Subsequently, the NO* intermediate was gradually converted to NOH*, NHOH*, and finally to NH2OH* under hydrogenation.
 | ||
| Fig. 4 Theoretical calculations. (A) Catalytic pathway from NO3− to NH2OH on Cu/TiO2 catalyst according to the optimized configurations with adsorbed intermediates. Cyan, blue, purple, red, and pink balls denote Ti, Cu, N, O, and H, respectively. (B) Gibbs free energy diagrams of NH2OH generation from NO3− on different models. (C) Calculated free energy diagram for (C) cyclohexanone* and (D) cyclohexanone oxime* adsorption on different TiO2-loaded Cu catalytic sites. (E) The proposed pathway of NH2OH generation and cyclohexanone oxime production. | ||
The high selectivity of >99.9% is an obvious advantage of cyclohexanone oxime production through an electrocatalysis strategy.8,35,36 Interestingly, the reduction of cyclohexanone and cyclohexanone oxime were not detected as side reactions because no GC-MS peaks of cyclohexanol or cyclohexylamine appeared in Fig. S6A.† The reduction of excess NOx substrates as well as a coherent base environment (pH 13.4) of the electrolyte hindered the reduction of organic molecules.12 Additionally, DFT study revealed the limitation of reduction of cyclohexanone (Fig. 4C) and cyclohexanone oxime (Fig. 4D). The calculated free energy of the absorption of cyclohexanone and cyclohexanone oxime on Cu(111)-Cu2O(110) models was found to be lower than that on the Cu(111) structure, which implies that the side reactions were hindered during efficient production of cyclohexanone oxime on Cu/TiO2 catalysts. As discussed above, the pathway of cyclohexanone is presented in Fig. 4E. NOx substrates from air oxidation produced NH2OH through electrocatalysis, followed by its non-electrochemical coupling with cyclohexanone to produce oxime.
The universality of our method was further confirmed by successfully converting a range of aldehydes and ketones in addition to cyclohexanone (e.g., benzaldehyde, acetone, cyclopentanone, and diethyl ketone) into oximes with high FE and selectivity under optimal conditions (Table S2†). This highlights the broad applicability of the electrocatalytic strategy to synthesize oxime, and affirms our proposed reaction pathway.
Conclusions
We propose a sustainable strategy to efficiently synthesize value-added cyclohexanone oxime using air as a nitrogen source, which includes plasma-assisted air-to-NOx and co-electrolysis of NOx and cyclohexanone. The highest performance was provided by 0.6% Cu/TiO2, which produced cyclohexanone at a yield rate of 20.1 mg h−1 cm−2 and an FE of 51.4%. The selectivity of cyclohexanone oxime was >99.9% on the basis of cyclohexanone. Detailed study indicated that the optimized catalytic activity was ascribed to Cu sites supported on TiO2 with a stable oxidation state.Further in situ characterizations combined with DFT calculations revealed the possible pathway of NO3− → NO2− → NO → NH2OH → cyclohexanone oxime. Together with transformation routes reported by our group, including the production of phenol using lignin and selective phenol-to-cyclohexanone conversion, this work provides a rational pathway to the synthesis of cyclohexanone oxime with biomass, an air nitrogen source, and green solvent.37–39 It will also inspire innovative routes to the carbon-natural chemical manufacturing of industrially important fine C–N chemicals from renewable carbon sources and nitrogen using clean energy.
Data availability
All experimental data is available in the ESI.†Author contributions
S. H. J., X. F. S., and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. S. H. J. performed all the experiments. X. X. T., L. M. W., and X. D. M. performed the analysis of the experimental data. L. B. Z., J. Q. F., L. X., and X. N. S. conducted portions of the characterization study. Q. G. Z. and X. C. K. participated in discussions. X. F. S. and B. X. H. supervised the entire project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22002172, 21890761, and 22121002), Beijing Natural Science Foundation (J210020), CAS Project for Young Scientists in Basic Research (Grant No. YSBR-050), and the Photon Science Center for Carbon Neutrality. The X-ray absorption spectroscopy measurements were performed at Beamline 4B9A at the Beijing Synchrotron Radiation Facility (BSRF). We also thank Dr Jiang Yan, Dr Ying Wang, Ms. Ziwei Zhao, and Ms. Jun He for their support in the GC-MS measurements. The authors thank the staff at the Centre for Physiochemical Analysis & Measurement of ICCAS for material characterizations.References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS
.
- M. A. Tarselli, Nat. Chem., 2012, 4, 686 CrossRef CAS PubMed
- J. N. Galloway, A. R. Townsend, J. W. Erisman, M. Bekunda, Z. Cai, J. R. Freney, L. A. Martinelli, S. P. Seitzinger and M. A. Sutton, Science, 2008, 320, 889–892 CrossRef CAS PubMed
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef
- X. Chen, S. Song, H. Li, G. Gözaydın and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed
- M. Hua, J. Song, X. Huang, H. Liu, H. Fan, W. Wang, Z. He, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21479–21485 CrossRef CAS PubMed
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed
- R. Xia, S. Overa and F. Jiao, JACS Au, 2022, 2, 1054–1070 CrossRef CAS PubMed
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202112835 CrossRef CAS PubMed
- Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Natl. Sci. Rev., 2020, 7, 1564–1583 CrossRef CAS PubMed
- R. Xu, D.-H. Si, S.-S. Zhao, Q.-J. Wu, X.-S. Wang, T.-F. Liu, H. Zhao, R. Cao and Y.-B. Huang, J. Am. Chem. Soc., 2023, 145, 8261–8270 CrossRef CAS PubMed
- D.-L. Meng, M.-D. Zhang, D.-H. Si, M.-J. Mao, Y. Hou, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 25485–25492 CrossRef CAS PubMed
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 CrossRef CAS PubMed
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed
- S. Jia, X. Ma, X. Sun and B. Han, CCS Chem., 2022, 4, 3213–3229 CrossRef CAS
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 14131–14137 CrossRef CAS PubMed
- Y. Ren, C. Yu, L. Wang, X. Tan, Z. Wang, Q. Wei, Y. Zhang and J. Qiu, J. Am. Chem. Soc., 2022, 144, 10193–10200 CrossRef CAS PubMed
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem, 2022, 6, 303–319 CrossRef CAS PubMed
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev., 2001, 43, 381–441 CrossRef CAS
- R. Mokaya and M. Poliakoff, Nature, 2005, 437, 1243–1244 CrossRef CAS PubMed
- J. M. Thomas and R. Raja, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13732–13736 CrossRef CAS PubMed
- Y. Fu, S. Wang, Y. Wang, P. Wei, J. Shao, T. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2023, 62, e202303327 CrossRef CAS PubMed
- L. Wu, J. Feng, L. Zhang, S. Jia, X. Song, Q. Zhu, X. Kang, X. Xing, X. Sun and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202307952 CrossRef CAS PubMed
- Q. Gao, B. Yao, H. S. Pillai, W. Zang, X. Han, Y. Liu, S.-W. Yu, Z. Yan, B. Min, S. Zhang, H. Zhou, L. Ma, H. Xin, Q. He and H. Zhu, Nat. Synth., 2023, 2, 624–634 CrossRef
- S. Zhang, J. Wu, M. Zheng, X. Jin, Z. Shen, Z. Li, Y. Wang, Q. Wang, X. Wang, H. Wei, J. Zhang, P. Wang, S. Zhang, L. Yu, L. Dong, Q. Zhu, H. Zhang and J. Lu, Nat. Commun., 2023, 14, 3634 CrossRef CAS PubMed
- X. Li, Y. Chen, X. Zhan, Y. Xu, L. Hao, L. Xu, X. Li, M. Umer, X. Tan, B. Han, A. W. Robertson and Z. Sun, The Innovation Materials, 2023, 1, 100014 CrossRef
- M. Duca, M. C. Figueiredo, V. Climent, P. Rodriguez, J. M. Feliu and M. T. M. Koper, J. Am. Chem. Soc., 2011, 133, 10928–10939 CrossRef CAS PubMed
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrochim. Acta, 2017, 227, 77–84 CrossRef
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed
- Y. Wu, J. Zhao, C. Wang, T. Li, B.-H. Zhao, Z. Song, C. Liu and B. Zhang, Nat. Commun., 2023, 14, 3057 CrossRef CAS PubMed
- Y. Wu, W. Chen, Y. Jiang, Y. Xu, B. Zhou, L. Xu, C. Xie, M. Yang, M. Qiu, D. Wang, Q. Liu, Q. Liu, S. Wang and Y. Zou, Angew. Chem., Int. Ed., 2023, 62, e202305491 CrossRef CAS PubMed
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed
- R. Wu, Q. Meng, J. Yan, H. Liu, Q. Zhu, L. Zheng, J. Zhang and B. Han, J. Am. Chem. Soc., 2022, 144, 1556–1571 CrossRef CAS PubMed
- J. Yan, Q. Meng, X. Shen, B. Chen, Y. Sun, J. Xiang, H. Liu and B. Han, Sci. Adv., 2020, 6, eabd1951 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02871b |
| This journal is © The Royal Society of Chemistry 2023 |