
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDibenzannulated peri-acenoacenes from anthanthrene derivatives†
Frédéric
Lirette
a,
Ali
Darvish
a,
Zheng
Zhou
 b,
Zheng
Wei
b,
Lukas
Renn
cd,
Marina A.
Petrukhina
b,
R. Thomas
Weitz
cd and
Jean-François
Morin
*a
b,
Zheng
Wei
b,
Lukas
Renn
cd,
Marina A.
Petrukhina
b,
R. Thomas
Weitz
cd and
Jean-François
Morin
*a
aDépartement de chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), 1045 Ave de la Médecine, Université Laval, Québec, G1V 0A6, Canada. E-mail: jean-francois.morin@chm.ulaval.ca
bDepartment of Chemistry, University at Albany, State University of New York, 1400 Washington Avenue, Albany, New York 12222-0100, USA
c1st Institute of Physics, Faculty of Physics, Georg-August-University, Göttingen, Germany
dInternational Center for Advanced Studies of Energy Conversion (ICASEC), University of Göttingen, Göttingen, Germany
First published on 30th August 2023
Abstract
A series of dibenzannulated phenyl-annulated [4,2]peri-acenoacenes have been synthesized in three straightforward steps from 4,10-dibromoanthanthrone (vat orange 3). The phenyl bisannulation of [4,2]peri-acenoacene provides extra stability by increasing the overall aromatic character of the molecules, and allows for a 45–80% increase of the molar extinction coefficient (ε) compared to their [5,2]peri-acenoacene isomers. Depending on the substituents attached to the π-conjugated core, some derivatives exhibit strong aggregation in the solid state with association constant (Ka) up to 255 M−1, resulting in a significant broadening of the absorption spectrum and a substantial decrease of the bandgap value (more than 0.3 V) from solution to the solid state. One [4,2]peri-acenoacene derivative was doubly reduced using cesium and the crystal structure of the resulting salt has been obtained. Field-effect transistors showing a temperature-dependent hole mobility have been tested.
Introduction
Large polyaromatic hydrocarbons (PAHs) have been widely studied for their fundamental and optoelectronic properties.1–7 Their tunable bandgap, planar geometry and ease of functionalization make them compounds of choice for organic electronics applications such as organic light emitting diodes (OLEDs),8–10 organic photovoltaic devices (OPV),11–13 and organic field-effect transistors (OFETs).14–16 Recently, zigzag-edged Kekulé and non-Kekulé PAHs such as annulated acenes,17,18 triangulenes,19–21peri-acenes22 and peri-acenoacenes23–26 (Fig. 1) have attracted a great deal of attention as they often exhibit a low bandgap and a diradicaloid character owing to their small number of Clar sextets in the closed-shell state. [n,m]peri-Acenoacenes are particularly intriguing as they are the only class of Kekulé PAHs in which all the edges are zigzag-shaped yet exhibiting relatively good thermal and chemical stability. Although anthanthrene (or [3,2]peri-acenoacene, Fig. 1) has been well studied and used as a building block for various applications, the reports on higher peri-acenoacenes are rather scarce.27–30 In 2019, Wu and coworkers reported the synthesis of peri-tetracenotetracene and peri-pentacenopentacene using substituted pyrene and naphthalene as the building blocks.31 They showed that the addition of one or two rings in the longitudinal [n] axis allowed for a drastic decrease in the bandgap values from 2.47 eV for anthanthrene to 1.83 and 1.47 eV, respectively. In 2022, Frigoli and coworkers reported an alternative synthetic method for the synthesis of a TIPS-modified [5,2]peri-acenoacene.32 They demonstrated the use of this new derivative in different organic optoelectronic devices, such as OFET with hole mobility of up to 1.0 cm2 V−1 s−1 and OLED with external quantum efficiency of 0.6% at 800 nm, making this compound one of the few OLEDs active beyond 800 nm. However, pentacenopentacene derivatives have quite a high open-shell character (y = 0.6), which could be a problem for long-term stability for their use as semiconductors. Thus, the synthesis of an analogous molecule with an increase of stability is needed.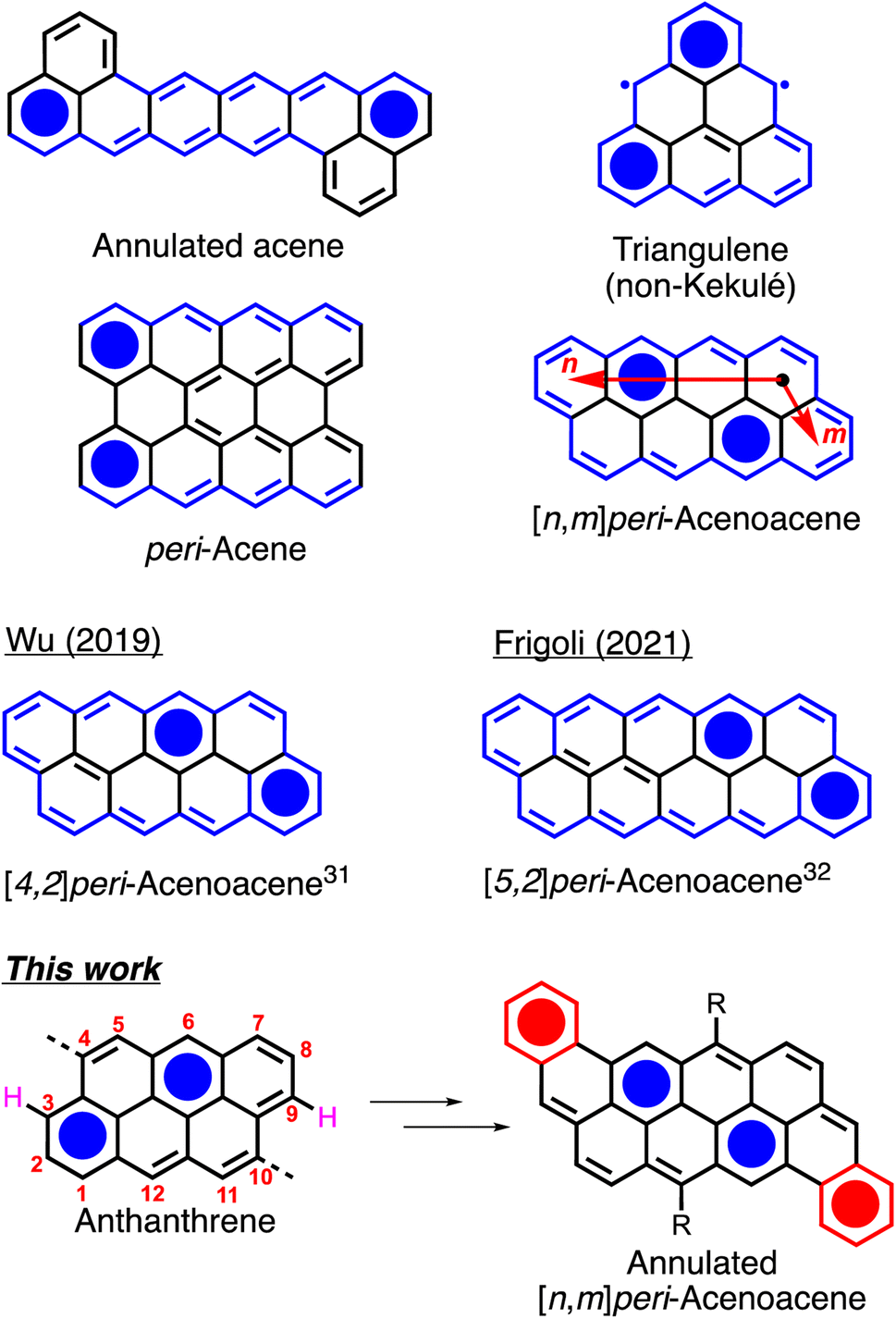 | ||
| Fig. 1 Examples of zigzag-edged polycyclic aromatic hydrocarbons. | ||
In recent years, our group has been using the commercially available 4,10-dibromoanthanthrone (vat orange 3) for the preparation of anthanthrene-based semiconductors for a full range of applications.33–36 One of the major drawbacks of this dye is the presence of protons in peri positions (see protons in pink, Fig. 1) relative to the 4 and 10 positions that yield to a relatively large dihedral angle between the anthanthrene core and the π-conjugated units attached at these positions, making the preparation of low bandgap molecules rather difficult. To overcome this issue, two options have been explored. The first is the use of alkyne spacers to increase the distance between the 4,10 protons and the coupled conjugated units.37 However, this method yields only to a slight decrease of the bandgap (<0.3 eV). The second option is to remove these protons through the formation of a sp3 carbon bridge on the anthanthrene unit, thus planarizing its conjugated core with its 4,10 aromatic substituents.38 Again, the effect on the bandgap is rather small with a decrease of only 0.14 eV. Thus, another strategy to further decrease the bandgap of the anthanthrene derivatives needs to be developed.
Herein, we report the synthesis, properties and device performance of four new π-extended, fully conjugated anthanthrene derivatives that can be regarded as dibenzannulated phenyl-annulated [4,2]peri-acenoacenes. Compared to other reported peri-acenoacenes, these molecules possess two extra phenyl rings (in red in Fig. 1) fused on the zigzag edge of the aromatic core that ensure extra stabilization by lowering the diradical character. The optoelectronic properties, aggregation behavior and OFET performance were studied and compared to previously reported periacenoacenes. DFT calculations were also performed to study the aromaticity and the frontier orbitals distribution.
Results and discussion
The synthesis of the dibenzannulated phenyl-annulated [4,2]peri-acenoacene derivatives is shown in Scheme 1. A Suzuki–Miyaura cross-coupling reaction between 4,10-dibromo-6,12-bis(triisopropylsilylethynyl)anthanthrene39 and 2-formylbenzeneboronic acid or between 4,10-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6,12-bis(octyloxy)anthanthrene38 and 2-bromobenzaldehyde gives the corresponding substituted triisopropylsilylethynyl and or octyloxy di(2-formylphenyl)anthanthrenes dibenzaldehydeanthanthrene 1a and 1b in 61% and 83% yield, respectively. Then, a two-fold nucleophilic addition substitution with lithium TIPS-acetylide or 2-mesitylmagnesium bromide, followed by an intramolecular Friedel–Crafts cyclization using BF3·OEt2 lead to a mixture of protonated and the fully oxidized compounds 2a–d and their dehydro analogs. At the best of our knowledge, complete aromatization following the widely used BF3·OEt2 cyclization step without adding an oxidant (other than atmospheric oxygen during workup) has not been reported before. The resulting mixture was treated with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) to convert all the protonated analogs into their fully oxidized form to afford molecules 2a–d in low to moderate yields (19–32%). The rather low yields obtained for these reactions can be explained by the formation of an unidentified paramagnetic side product, especially in the case of compound 2a, that can only be removed after a very tedious purification step by column chromatography. However, once clean, fully oxidized compounds 2a–d are stable over time (at least 2 years) under ambient light and in aerobic conditions.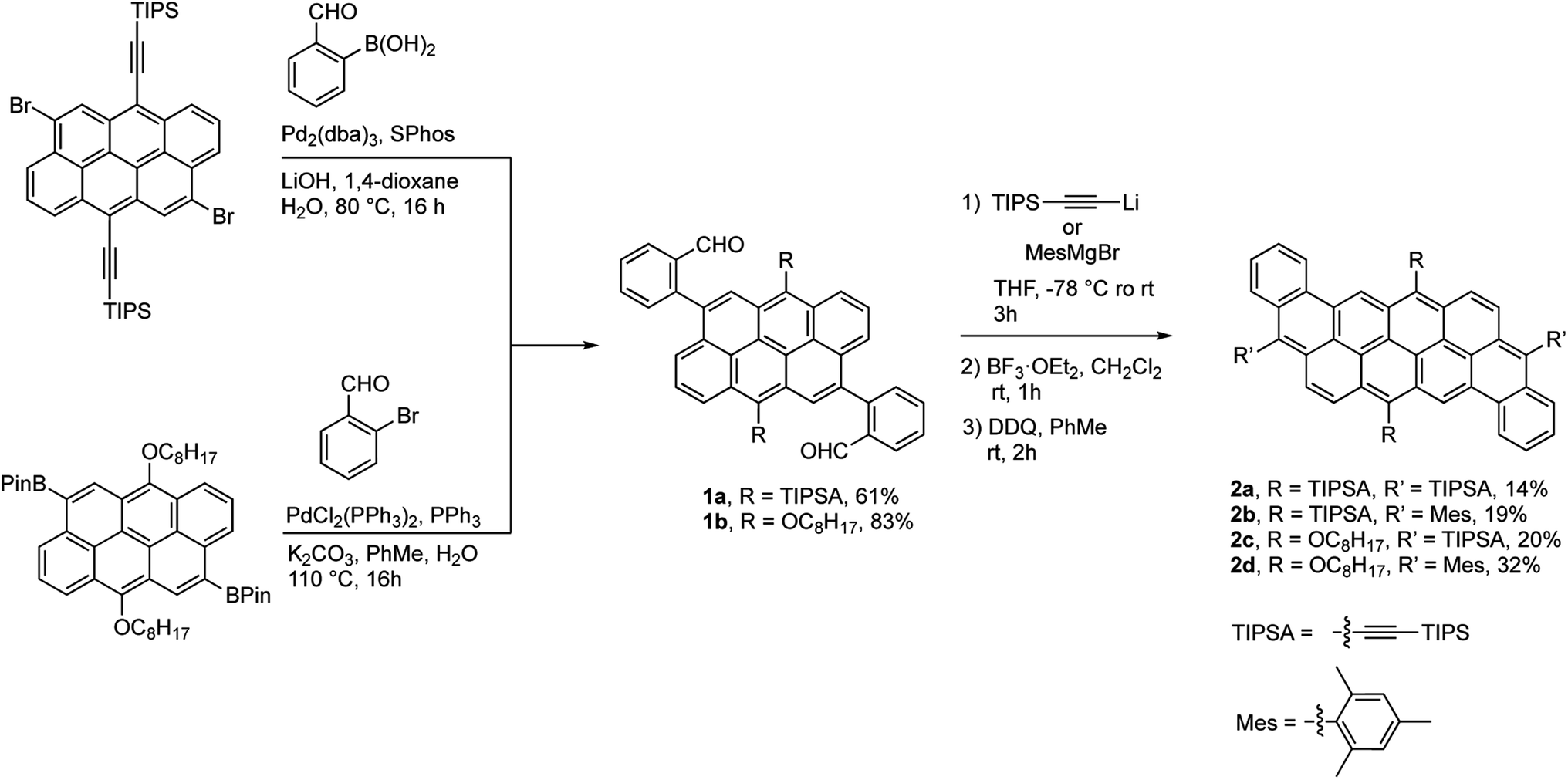 | ||
| Scheme 1 Synthesis of dibenzannulated phenyl-annulated [4,2]peri-acenoacenes 2a–d. | ||
All absorption spectra of compounds 2a–d were recorded in CHCl3 at 0.01 mM or in thin film by drop-casting and slow evaporation of a 0.4 mM solution (CHCl3) solution on a glass slide. The UV-visible and fluorescence spectra are presented in Fig. 2 and S14–S17.† All dibenzannulated phenyl-annulated [4,2]peri-acenoacene derivatives showed sharp transitions in the visible region with a λmax absorption ranging from 553 nm for compound 2d to 611 nm for compound 2a. Surprisingly, switching from a TIPS-acetylene to a mesityl (Mes) group at the 3 and 12 positions leads to a significant blueshift of 40 nm. In contrast, adding octyloxy chains instead of the TIPS-acetylene substituents has a weaker effect on the absorption, with a hypsochromic shift of 15 nm. Every compound showed good fluorescence properties, which mirrored the visible absorption region with Stokes shifts as low as 10 nm. Compared to the sp3 carbon-planarized anthanthrene derivatives reported previously,38 the absorption spectrum of compound 2d is red-shifted by 67 nm (486 nm to 553 nm). Moreover, the molar absorption coefficient (ε) of all dibenzannulated phenyl-annulated [4,2]peri-acenoacenes is more than twice the value reported for the sp3 carbon-bridged derivative. The absorption spectra of compounds 2a–d are significantly blue-shifted compared to the [5,2]peri-acenoacene reported by Frigoli and coworkers (λmax = 726 nm),32 due to the decrease of the acene character. In fact, dibenzannulated phenyl-annulated [4,2]peri-acenoacenes 2a–d are more aromatic (4 Clar sextets vs. 2 for [5,2]peri-acenoacene, vide infra) and their geometry allows for a much shorter delocalization on consecutive phenyl units. Interestingly, the ε values measured for compounds 2a–d are between 45% and 80% higher than that measured for [5,2]peri-acenoacene (see Table 1).
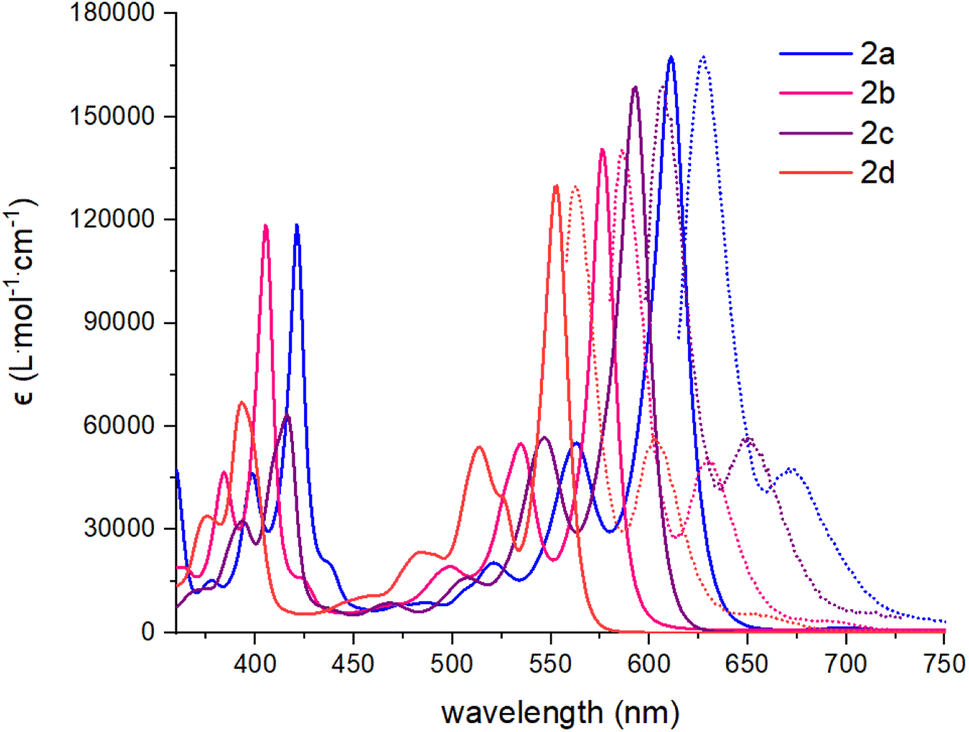 | ||
| Fig. 2 Absorption (CHCl3, solid lines) and fluorescence spectra (CHCl3, dashed lines) of compounds 2a–d. | ||
| Compound | λ solmax (nm) | ε (M−1 cm−1) | λ filmmax (nm) | E solg (eV) | E solidg (eV) | E theog (eV) | λ solem (nm) | Stokes shift (nm) |
|---|---|---|---|---|---|---|---|---|
| a [5,2]pAA = [5,2]peri-acenoacene.32 | ||||||||
| 2a | 611 | 168![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
620 | 1.97 | 1.65 | 1.85 | 627 | 16 |
| 2b | 576 | 141000 |
584 | 2.07 | 1.98 | 2.05 | 586 | 10 |
| 2c | 593 | 159000 |
633 | 2.01 | 1.70 | 1.95 | 606 | 13 |
| 2d | 553 | 130000 |
563 | 2.16 | 2.03 | 2.18 | 563 | 10 |
| [5,2]pAAa | 726 | 90000 |
N/A | N/A | N/A | N/A | 739 | 13 |
All dibenzannulated phenyl-annulated [4,2]peri-acenoacene derivatives exhibit a red shift absorption in the solid state compared to solution (Fig. 3 and S14–S16†). For the mesityl-substituted compounds 2b and 2d, the redshift is rather small while the 3,12-TIPS-acetylene-substituted derivatives (2a and 2c) show a significant broadening of the absorption band and are shifted by 9 nm and 40 nm, respectively. The bandgap values measured in the solid state for 2a and 2c are lowered by 0.32 eV, which is a good indication of strong π-aggregation. The different behavior between the mesityl- and TIPS-acetylene-substituted derivatives can be ascribed to the ability of the mesityl groups to prevent efficient π-stacking in the solid state.
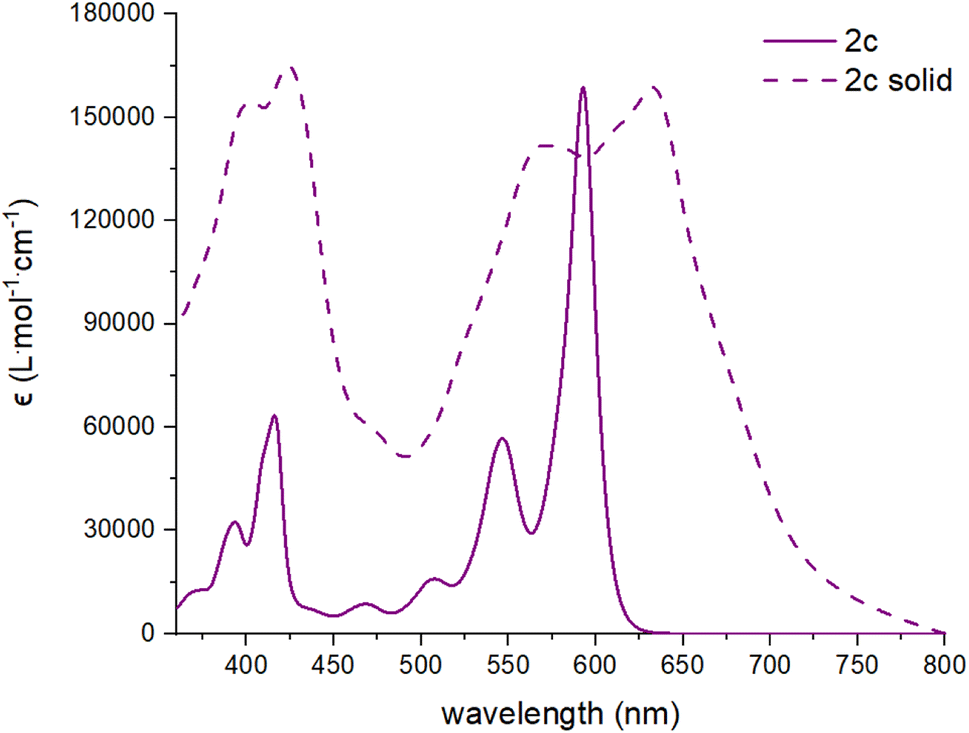 | ||
| Fig. 3 Absorption spectra in solution (CHCl3, solid line) and in solid state (dashed lines) of compound 2c. | ||
The significant broadening of the absorption spectrum in solid state for compounds 2a and 2c prompted us to study their aggregation in solution using 1H NMR spectroscopy in CHCl3 at variable concentrations. The dependence between chemical shifts associated to the protons of the aromatic core and the concentration (0.3 mM to 40 mM) are described in Tables S1 and S2.† Upon an increase in concentration, all protons are mildly to strongly shifted upfield, which can be attributed to face-to-face interactions (Fig. 4, S18 and S19†).40 It is noteworthy that no shift has been observed for compounds 2b and 2d, probably due to the presence of bulky mesityl moieties that prevent aggregation in solution. Association constant values (Ka) of 135 and 255 M−1 have been calculated for compounds 2a and 2c, respectively, assuming a monomer-dimer model.41–43 The difference between the two compounds could be explained by the better flexibility of the octyloxy chains, allowing additional intermolecular van der Waals interactions compared to the TIPS-containing derivatives.44,45 As presented in Table S1,† the aromatic protons Hb and Hc in compounds 2a do not shift significantly upon increasing the concentration (Δδ < 0.05 ppm) unlike all the other protons. In comparison, every aromatic protons of compound 2c significantly shifted upon increasing the concentration. In this case, Hb and Hc shifted by up to 0.22 ppm, showing a better stacking and stronger π-π intermolecular interactions for compound 2c than 2a, which correlates well with the behavior observed in the absorption spectra.
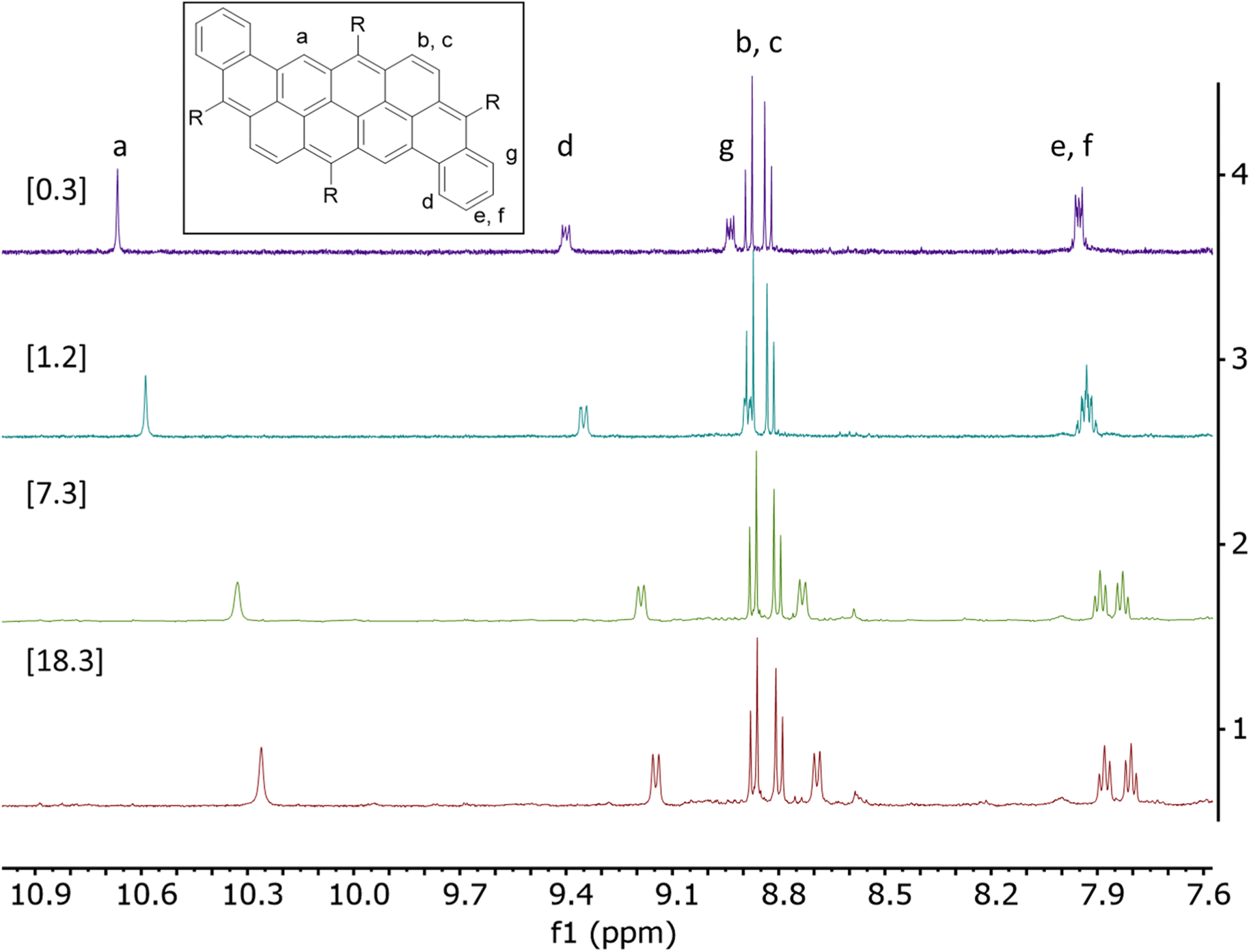 | ||
| Fig. 4 1H NMR of compound 2a between 7.5 ppm and 11.0 ppm at different concentrations [0.3–18.3 mM] in CDCl3 at room temperature. | ||
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were used to determine the oxidation/reduction potentials and the energy level of the frontier orbitals of compounds 2a–d. All curves are shown in Fig. S20–S23,† and experimental data are summarized in Table 2. All compounds show at least two reversible oxidation processes, with compounds 2c and 2d having their first oxidation potential 0.2 V below the TIPS-acetylene-substituted 2a and 2c, showing the effect of the electro-donating octyloxy side chains that increased the energy level of the HOMO. One clear reversible redox process can be observed for compounds 2a and 2b. Half-wave potentials of the first oxidation/reduction peaks are at 0.21/−1.42 V and 0.22/−1.65 V, respectively. The HOMO and LUMO energy levels were found to be −5.01/−3.38 eV and −5.02/−3.15 eV, giving bandgap values of 1.63 eV and 1.86 for 2a and 2b, respectively. No reversible redox process has been found for the octyloxy-bearing compounds, with a first oxidation process at 0.03 V for 2c and 0.08 V for 2d, giving HOMO energy levels of −4.83 eV and −4.88 eV, respectively.
Spectroelectrochemistry analysis has been performed on compounds 2a–d and the results are shown in Fig. S24–S29.† All compounds have the same behavior for both oxidation processes. During the first oxidation, all absorption bands become broader and less intense, with two new peaks appearing in the NIR region at ∼1000 nm and ∼1400 nm. The transition with the lowest energy follows the same trend as the λmax of the neutral species, 2a > 2c > 2b > 2d, ranging from 1465 nm to 1324 nm. Absorption of 2x2+ in the NIR region vanishes comparatively to 2x˙+, with a single peak rising in the visible region between 700 nm and 800 nm. For the reduced species, 2a˙− and 2b˙− are showing a loss of intensity, with broad absorption covering all the visible region up to 950 nm. No spectrum of the reduced form of compounds 2c–d was recorded as only degradation was observed. Compound 2d precipitated in the quartz cell during reduction.
Density functional theory (DFT) calculations were performed on compounds 2a–d to assess the frontier orbitals distribution and energy levels using 6-31G(d,p) as the basis set and B3LYP as the functional. The results are summarized in Fig. 5a. For all derivatives, the HOMO and LUMO orbitals are delocalized over the entire molecule, while the LUMO orbital is located mainly on the edge of the peri-acenoacene core with a weak contribution on the annulated phenyl benzene. Interestingly, the four derivatives exhibit different orbital energy levels, despite their similarity. For example, the LUMO energy level of compound 2b, which differentiates from 2a only by the presence of mesityl groups instead of TIPS-acetylene at the 3 and 12 positions, is 0.25 eV higher in energy while the HOMO energy level increased slightly (+0.05 eV). Similar shifts are observed from compounds 2c to 2d. Surprisingly, replacing a TIPS-acetylene at the 9 and 18 positions by electron-donating octyloxy groups did not yield to a significant increase of the HOMO energy level, although the LUMO energy level increased significantly. Overall, the substitution pattern at the 3,12 and 9,18 positions provided a bandgap variation from Eg = 1.85 eV for 2a (lowest) to Eg = 2.18 eV for 2d (highest). These results correlate well with the electronic spectra (see above).
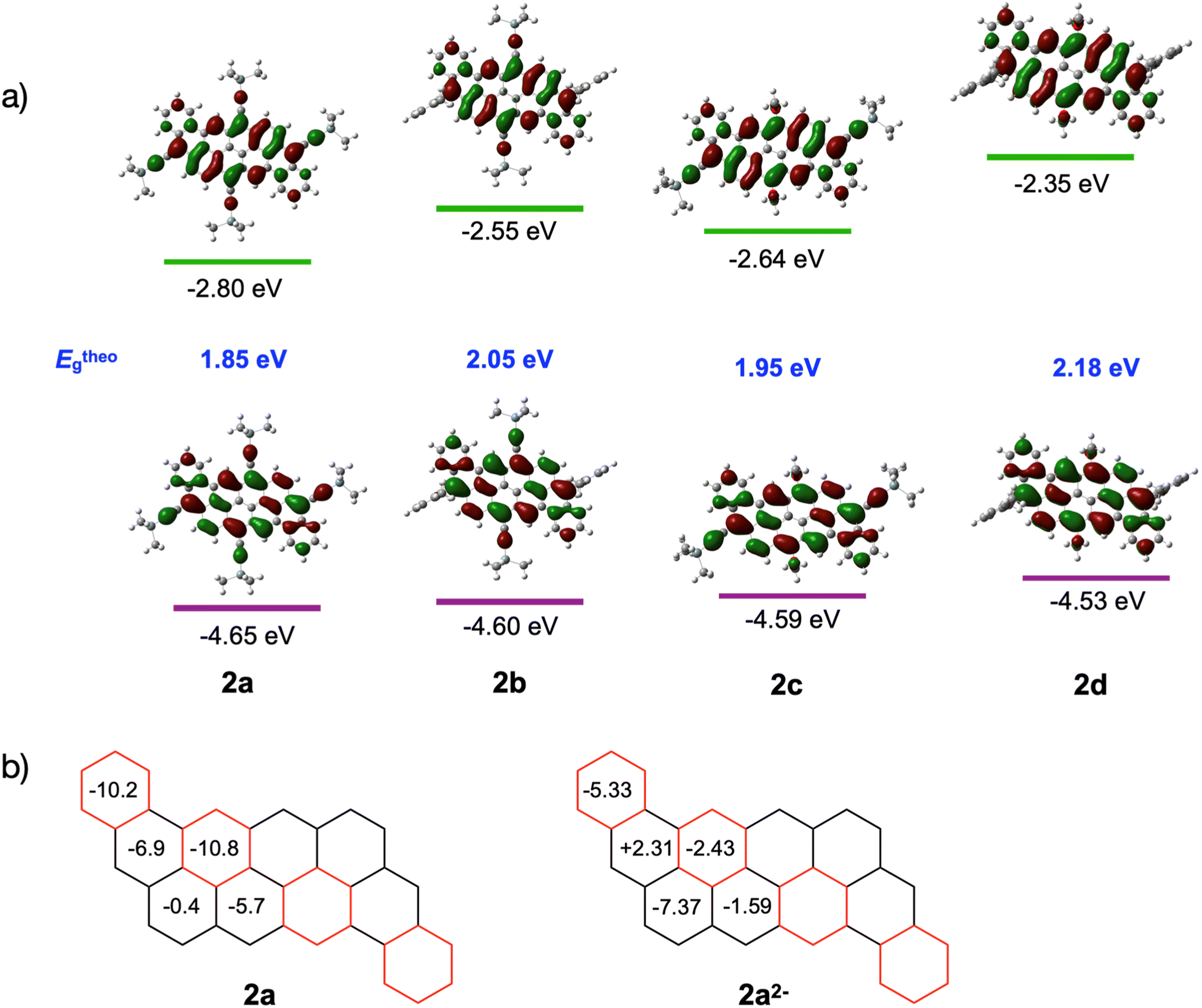 | ||
| Fig. 5 (a) Kohn–Sham molecular orbitals of model compounds of 2a–d based on calculations at the B3LYP/6-31G(d,p) level of theory. (b) NICS(0) values (in ppm) for the different rings in dibenzannulated phenyl-annulated [4,2]peri-acenoacene 2a and 2a2−. The values have been calculated for compound 2a. The TIPS-acetylene groups have been omitted for clarity. | ||
The aromaticity of dibenzannulated phenyl-annulated [4,2]peri-acenoacene was evaluated using computed Nuclear Independent Chemical Shift (NICS) values. As shown in Fig. 5b, the rings colored in red and forming a diagonal within the molecule show the strongest aromatic character at δ = −10.2 and −10.8 ppm. The ring adjacent to this diagonal exhibits a much weaker aromatic character at δ = −6.9, −5.7 and −0.4 ppm. These NICS values correlate well with the electronic structure possessing the maximum number of Clar sextets (4), as shown in Fig. 1.
To get a better understanding of the redox behavior of dibenzannulated phenyl-annulated [4,2]peri-acenoacenes, the chemical reduction of 2a was investigated with Cs metal in hexanes at room temperature. The reaction mixture went through a purple color (monoanion) and deepened to dark brown in ca. 4 hours, which is indicative of the formation of the doubly-reduced product. After 24 hours, the dark-brown precipitate was removed from solution and dissolved in THF (see the ESI for more details†). Dark-brown needle-shaped crystals were isolated by slow diffusion of hexanes into THF solution in the presence of 18-crown-6 ether. The X-ray diffraction analysis confirmed the formation of the [{Cs+(18-crown-6)2}2(2a2−)] product (crystallized with one interstitial THF molecule).
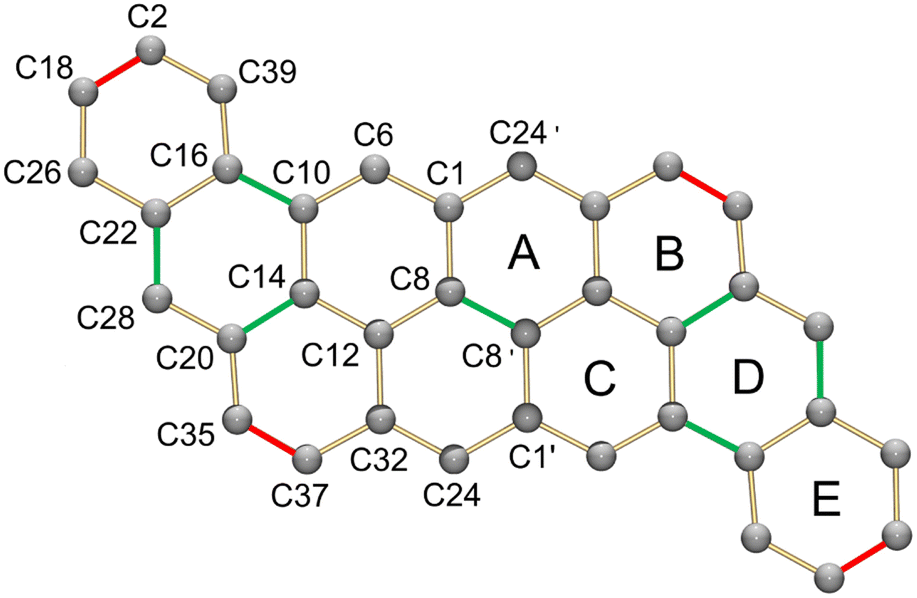
In the crystal structure (Fig. 6), two crystallographically independent Cs+ ions are fully wrapped by two 18-crown-6 ether ligands each (Cs–O: 3.243(16)–3.629(13) Å) leaving a “naked” dianion, thus allowing evaluation of the 2a2− core structure without any metal binding influence. The C–C bond length distances and planarity calculation along with the dihedral angles are shown in Tables 3 and 4, respectively. In comparison to the structure of neutral peri-acenoacene having a close core,46 the internal C–C bonds in 2a2− are overall longer than regular aromatic C–C bond length (1.4 Å). In particular, the bond length of C8–C8′, C10–C16, C14–C20, and C22–C28 fall in the range of 1.447(12)–1.460(12) Å (highlighted in green, Table 3 scheme). As a result, the aromaticity reduction can be expected in rings A and D, which is consistent with the NICS(0) values calculated for 2a2− (Fig. 5b). In contrast, the external C–C bonds, especially C2–C18 and C35–C37, are much shorter (1.345(14)/1.355(13) Å, highlighted in red) than those in neutral peri-acenoacene. Moreover, all six-membered rings remain almost planar upon reduction, but the dihedral angle increase from A/B (1.8°) to A/E (7.9°) may be indicative of an S-distortion along the core of the dianion.
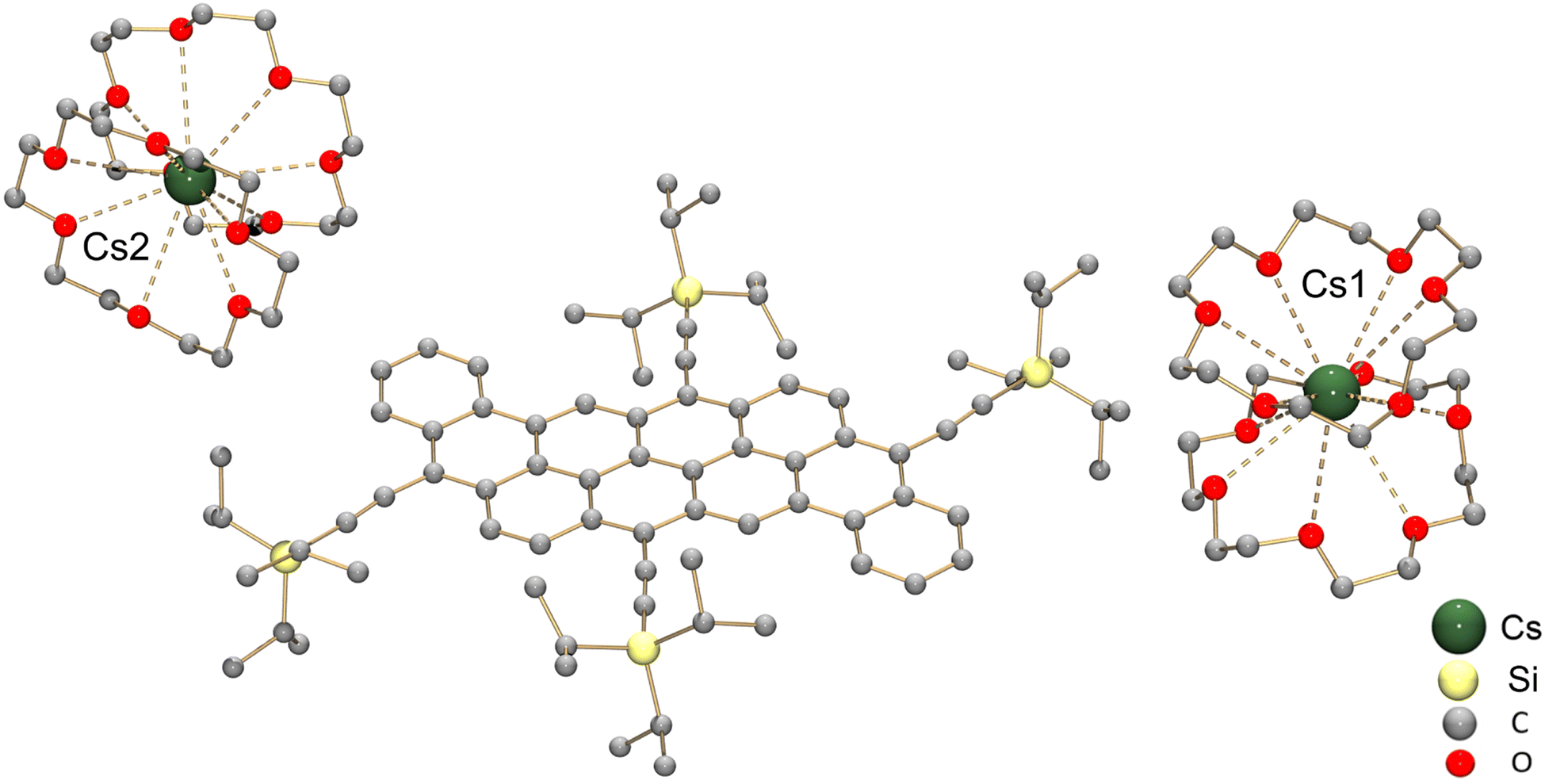 | ||
| Fig. 6 Crystal structure of [{Cs+(18-crown-6)2}2(2a2−)] in ball-and-stick model, H-atoms are omitted for clarity. | ||
|
|
|||||
|---|---|---|---|---|---|
| Bond | Distance | Bond | Distance | Bond | Distance |
| C1–C6 | 1.408(12) | C10–C14 | 1.436(11) | C20–C28 | 1.432(12) |
| C1–C8 | 1.424(11) | C10–C16 | 1.460(12) | C20–C35 | 1.375(12) |
| C1–C24′ | 1.421(11) | C12–C14 | 1.427(11) | C22–C26 | 1.436(13) |
| C2–C18 | 1.345(14) | C12–C32 | 1.439(11) | C22–C28 | 1.447(12) |
| C2–C39 | 1.399(13) | C14–C20 | 1.447(11) | C24–C32 | 1.411(12) |
| C6–C10 | 1.377(11) | C16–C22 | 1.425(12) | C32–C37 | 1.413(12) |
| C8–C8′ | 1.459(16) | C16–C39 | 1.383(12) | C35–C37 | 1.355(13) |
| C8–C12 | 1.406(11) | C18–C26 | 1.384(14) | ||
| RMSD/A | ||||
|---|---|---|---|---|
| A | B | C | D | E |
| 0.012 | 0.017 | 0.011 | 0.012 | 0.008 |
| Dihedral angle | |||
|---|---|---|---|
| A/B | A/C | A/D | A/E |
| 1.8 | 1.3 | 4.2 | 7.9 |
In the solid-state structure of [{Cs+(18-crown-6)2}2(2a2−)], the 2a2− dianions are stacked into a one-dimensional column with the insertion of two {Cs+(18-crown-6)2} cations in between (Fig. 7). The stacks are held by C–H⋯π interactions ranging over 2.423(12)–2.848(12) Å.
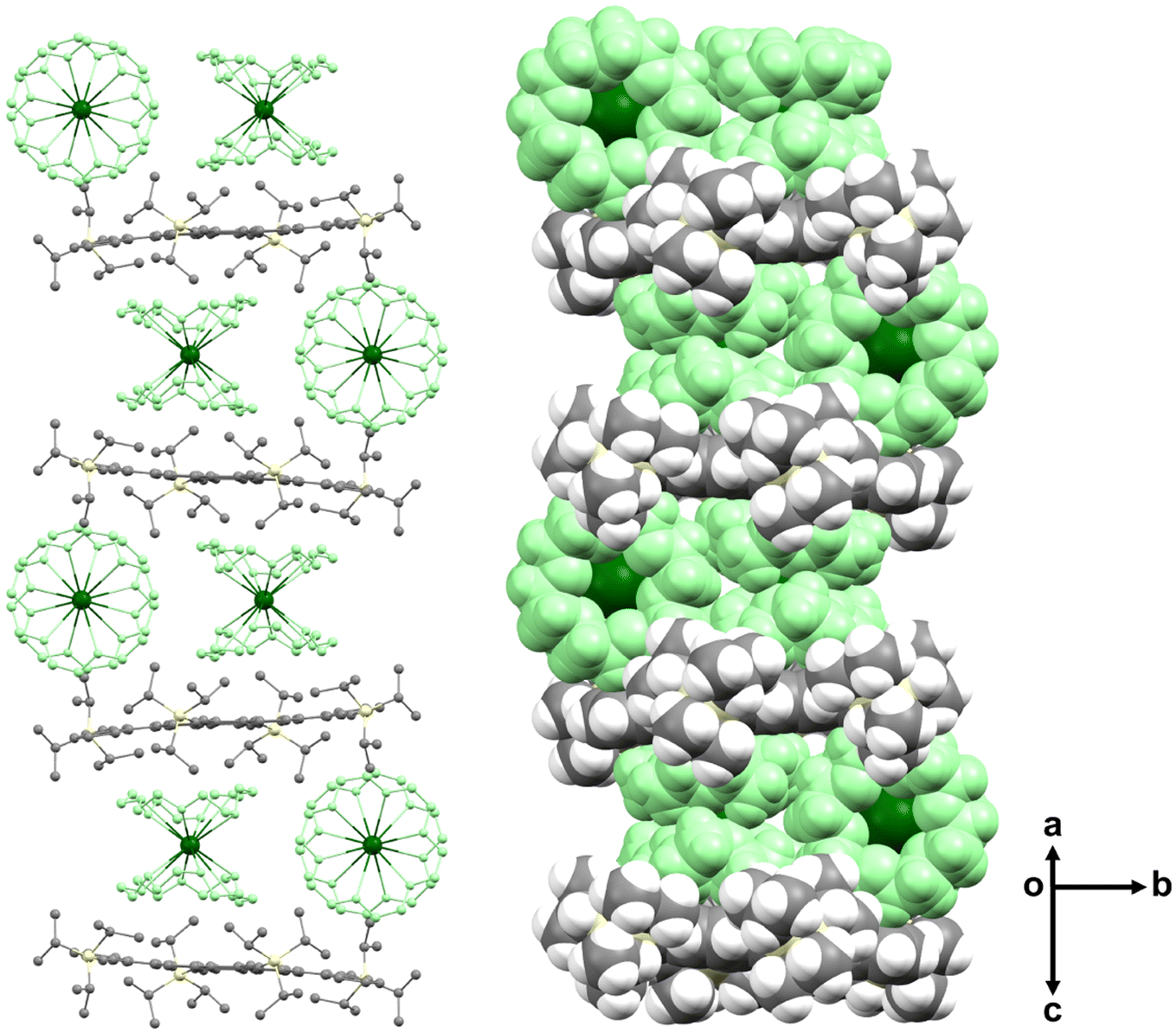 | ||
| Fig. 7 Solid-state packing of [{Cs+(18-crown-6)2}2(2a2−)] in ball-and-stick (no H-atoms, left) and space-filling (right) models. The {Cs+(18-crown-6)2} moieties are shown in green. | ||
The charge transport properties of molecule 2a and 2c were investigated in organic field-effect transistors (OFETs) in a bottom-gate top-contact configuration. The best performing OFETs were obtained when compound 2a was evaporated onto Si/SiO2 wafers, which were passivated with an n-octadecytrichlorosilane self-assembled monolayer (ODTS SAM) prior to the evaporation, and when compound 2c was spin-coated from a chloroform solution directly onto Si/SiO2 wafers. The thin film morphology of both compounds is shown in Fig. S33 in the ESI section.†
The output curve (Fig. 8a) measured in a vacuum (to avoid degradation due to the ambient)47 at room temperature with an OFET based on compound 2a displays close to ideal p-type charge transport with small injection barrier at the contacts48 consistent with the expected good energetic alignment between the gold contacts and the HOMO of compound 2a (see Table 2). The corresponding linear-regime transfer curve (Fig. 8b) exhibits an onset voltage of Von = −25 V and was used to determine the charge carrier mobility μlin ≈ 6.5 × 10−6 cm2 V−1 s−1, which is shown in Fig. 8c. Charge transport measurements conducted on compound 2c under the same conditions (see ESI Fig. S34†) display very similar results with a slightly higher mobility of μlin ≈ 8 × 10−6 cm2 V−1 s−1.
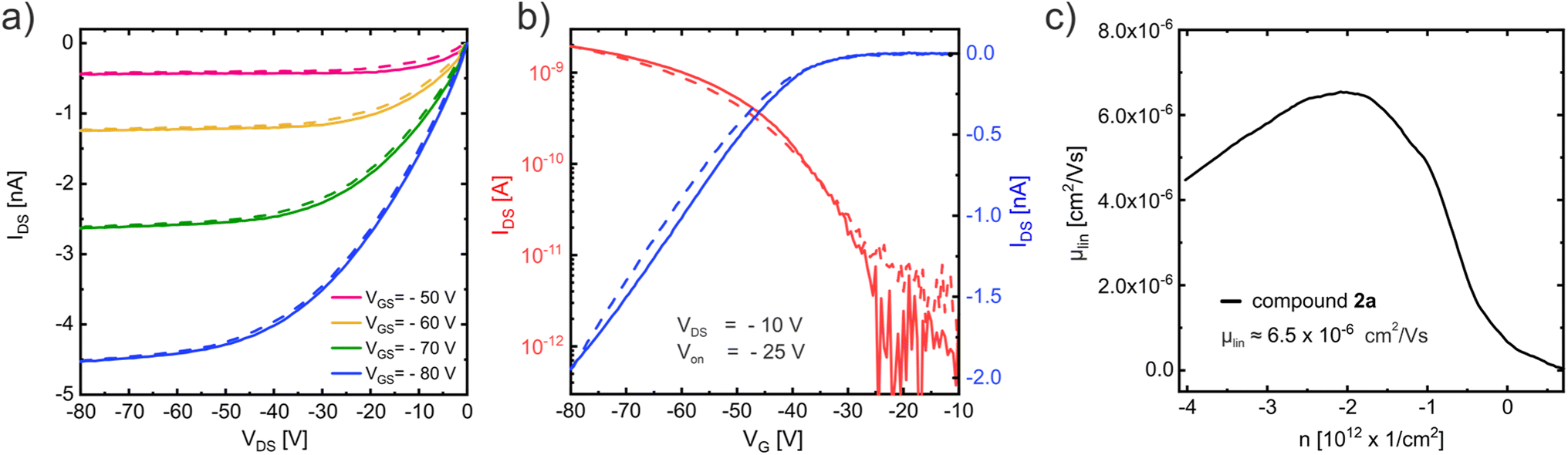 | ||
| Fig. 8 Charge transport measurements of compound 2a conducted in a vacuum at room temperature. (a) The output curve displays close to ideal p-type transport. (b) Linear-regime transfer curve and (c) the corresponding charge carrier mobility. | ||
Temperature dependent measurements with compound 2a are shown in the ESI section (Fig. S35†) and revealed a strongly enhanced charge transport at higher temperatures with a maximum mobility of μlin ≈ 2.6 × 10−5 cm2 V−1 s−1 at 400 K and a carrier freeze out at temperatures below 250 K, which suggests a thermally activated hopping transport. An Arrhenius approach was used to determine the activation energy to a value of EA = 220 meV (see Fig. S35d†). Temperature dependent measurements with compound 2c showed similar results, as displayed in Fig. S36.†
Conclusion
In conclusion, four dibenzannulated phenyl-annulated [4,2]peri-acenoacene derivatives have been prepared in a few synthetic steps from the inexpensive vat orange 3. As expected, phenyl annulation provides extra stability compared to other peri-acenoacenes by increasing the overall aromatic character of the molecules. Yet, these molecules absorb light in the visible region with molar extinction coefficient up to 168000 M−1 cm−1 with bandgap values of ca. 2.0 eV. The large, flat π-conjugated core allows for strong intermolecular interactions, resulting in a significant broadening of the absorption spectrum, especially for the octyloxy-containing derivatives. The accessibility of the doubly-reduced state is illustrated by chemical reduction of 2a with Cs metal coupled with crystallographic characterization of the resulting product. Field-effect transistors using these dibenzannulated phenyl-annulated [4,2]peri-acenoacene derivatives as semiconductors exhibit hole mobility of up to 2.6 × 10−5 cm2 V−1 s−1 and show a temperature-dependent behavior, suggesting a thermally activated hopping transport mechanism.
Data availability
The ESI contains detailed description for the synthetic method, spectroscopic and crystallographic data (CCDC 2266823), computational method and device fabrications.†Author contributions
J.-F. M. supervised the project. F. L. and A. D. performed the synthetic experiments. L. R. performed device fabrication and characterization. J.-F. M. performed theoretical calculations. Z. Z., Z. W and M. P. performed the crystallographic analysis. All authors analyzed the data, discussed the results, and contributed to the manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC through a Discovery Grant. Frédéric Lirette thanks NSERC for a PhD scholarship. Financial support of this work from the U.S. National Science Foundation, CHE-2003411, is gratefully acknowledged by M. A. P. NSF's ChemMatCARS Sector 15 at the Advanced Phonon Source (APS), Argonne National Laboratory (ANL) is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. This research used the resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- N. Baig, S. Shetty, R. Tiwari, S. K. Pramanik and B. Alameddine, ACS Omega, 2022, 7, 45732–45739 CrossRef CAS PubMed.
- M. Feofanov, V. Akhmetov and K. Amsharov, Chem. - Eur. J., 2021, 27, 17322–17325 CrossRef CAS PubMed.
- S. Kumar, K. Yoshida, Y. Hattori, T. Higashino, H. Imahori and S. Seki, Chem. Sci., 2022, 13, 1594–1599 RSC.
- R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Org. Lett., 2019, 21, 8652–8656 CrossRef CAS PubMed.
- J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schütt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547–5558 RSC.
- G. M. Paternò, Goudappagouda, Q. Chen, G. Lanzani, F. Scotognella and A. Narita, Adv. Opt. Mater., 2021, 9, 2100508 CrossRef.
- O. Matuszewska, T. Battisti, R. R. Ferreira, N. Biot, N. Demitri, C. Mézière, M. Allain, M. Sallé, S. Mañas-Valero, E. Coronado, E. Fresta, R. D. Costa and D. Bonifazi, Chem. - Eur. J., 2023, 29, e202203115 CrossRef CAS PubMed.
- J. Wagner, P. Zimmermann Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed.
- D. Zhang and L. Duan, J. Phys. Chem. Lett., 2019, 10, 2528–2537 CrossRef CAS PubMed.
- G. Meng, D. Zhang, J. Wei, Y. Zhang, T. Huang, Z. Liu, C. Yin, X. Hong, X. Wang, X. Zeng, D. Yang, D. Ma, G. Li and L. Duan, Chem. Sci., 2022, 13, 5622–5630 RSC.
- Y. Li, X. Huang, K. Ding, H. K. M. Sheriff, L. Ye, H. Liu, C.-Z. Li, H. Ade and S. R. Forrest, Nat. Commun., 2021, 12, 5419 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Jr Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- J. B. Park, J.-W. Ha, I. H. Jung and D.-H. Hwang, ACS Appl. Energy Mater., 2019, 2, 5692–5697 CrossRef CAS.
- Z. Wang, Q. Peng, X. Huang, Q. Ma, J. Shao and Q. Shen, Dyes Pigm., 2021, 185, 108877 CrossRef CAS.
- B.-L. Hu and Q. Zhang, Chem. Rec., 2021, 21, 116–132 CrossRef CAS PubMed.
- C.-T. Hsieh, C.-Y. Chen, H.-Y. Lin, C.-J. Yang, T.-J. Chen, K.-Y. Wu and C.-L. Wang, J. Phys. Chem. C, 2018, 122, 16242–16248 CrossRef CAS.
- P. Hu and J. Wu, Can. J. Chem., 2017, 95, 223–233 CrossRef CAS.
- C. Zong, X. Zhu, Z. Xu, L. Zhang, J. Xu, J. Guo, Q. Xiang, Z. Zeng, W. Hu, J. Wu, R. Li and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 16230–16236 CrossRef CAS PubMed.
- S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS PubMed.
- C. Chen, J. Lu, Y. Lv, Y. Yan, Q. Sun, A. Narita, K. Müllen and X.-Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212594 CrossRef CAS PubMed.
- S. Mishra, K. Xu, K. Eimre, H. Komber, J. Ma, C. A. Pignedoli, R. Fasel, X. Feng and P. Ruffieux, Nanoscale, 2021, 13, 1624–1628 RSC.
- M. R. Ajayakumar, J. Ma and X. Feng, Eur. J. Org Chem., 2022, 2022, e202101428 CrossRef CAS.
- D. Lungerich, O. Papaianina, M. Feofanov, J. Liu, M. Devarajulu, S. I. Troyanov, S. Maier and K. Amsharov, Nat. Commun., 2018, 9, 4756 CrossRef PubMed.
- M. Franceschini, M. Crosta, R. R. Ferreira, D. Poletto, N. Demitri, J. P. Zobel, L. González and D. Bonifazi, J. Am. Chem. Soc., 2022, 144, 21470–21484 CrossRef CAS PubMed.
- Y. Gu, R. Muñoz-Mármol, W. Fan, Y. Han, S. Wu, Z. Li, V. Bonal, J. M. Villalvilla, J. A. Quintana, P. G. Boj, M. A. Díaz-García and J. Wu, Adv. Opt. Mater., 2022, 10, 2102782 CrossRef CAS.
- A. Omist, G. Ricci, A. Derradji, A. J. Pérez-Jiménez, E. San-Fabián, Y. Olivier and J. C. Sancho-García, Phys. Chem. Chem. Phys., 2021, 23, 24016–24028 RSC.
- M. Desroches and J.-F. Morin, Org. Lett., 2018, 20, 2797–2801 CrossRef CAS PubMed.
- Q. Liu, Y. Wang, L. Arunagiri, M. Khatib, S. Manzhos, K. Feron, S. E. Bottle, H. Haick, H. Yan, T. Michinobu and P. Sonar, Mater. Adv., 2020, 1, 3428–3438 RSC.
- H. D. Pham, T. T. Do, J. Kim, C. Charbonneau, S. Manzhos, K. Feron, W. C. Tsoi, J. R. Durrant, S. M. Jain and P. Sonar, Adv. Energy Mater., 2018, 8, 1703007 CrossRef.
- C. Aumaitre, D. Fong, A. Adronov and J.-F. Morin, Polym. Chem., 2019, 10, 6440–6446 RSC.
- Y. Gu, Y. G. Tullimilli, J. Feng, H. Phan, W. Zeng and J. Wu, Chem. Commun., 2019, 55, 5567–5570 RSC.
- T. Jousselin-Oba, M. Mamada, K. Wright, J. Marrot, C. Adachi, A. Yassar and M. Frigoli, Angew. Chem., Int. Ed., 2022, 61, e202112794 CrossRef CAS PubMed.
- M. Desroches and J.-F. Morin, Chem. - Eur. J., 2018, 24, 2858–2862 CrossRef CAS PubMed.
- J.-B. Giguère, Q. Verolet and J.-F. Morin, Chem. - Eur. J., 2013, 19, 372–381 CrossRef PubMed.
- M. Desroches, P. Mayorga Burrezo, J. Boismenu-Lavoie, M. Peña Álvarez, C. J. Gómez-García, J. M. Matxain, D. Casanova, J.-F. Morin and J. Casado, Angew. Chem., Int. Ed., 2017, 56, 16212–16217 CrossRef CAS PubMed.
- Y. Du, H. B. Lovell, F. Lirette, J.-F. Morin and K. N. Plunkett, J. Org. Chem., 2021, 86, 1456–1461 CrossRef CAS PubMed.
- J.-B. Giguère, J. Boismenu-Lavoie and J.-F. Morin, J. Org. Chem., 2014, 79, 2404–2418 CrossRef PubMed.
- F. Lirette, C. Aumaitre, C.-É. Fecteau, P. A. Johnson and J.-F. Morin, ACS Omega, 2019, 4, 14742–14749 CrossRef CAS PubMed.
- A. Lafleur-Lambert, J.-B. Giguère and J.-F. Morin, Macromolecules, 2015, 48, 8376–8381 CrossRef CAS.
- C. Giessner-Prettre, B. Pullman, P. N. Borer, L.-S. Kan and P. O. P. Ts’O, Biopolymers, 1976, 15, 2277–2286 CrossRef CAS PubMed.
- Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S. Araki, M. Sonoda, K. Hirose and K. Naemura, J. Am. Chem. Soc., 2002, 124, 5350–5364 CrossRef CAS PubMed.
- R. Kaur, M. Ramesh, P. V. Bharatam and R. Kishore, J. Phys. Chem. B, 2014, 118, 9199–9208 CrossRef CAS PubMed.
- W. Tang, H. Mo, M. Zhang, S. Parkin, J. Gong, J. Wang and T. Li, J. Phys. Chem. B, 2017, 121, 10118–10124 CrossRef CAS PubMed.
- K. S. Mali and S. De Feyter, Philos. Trans. R. Soc., A, 2013, 371, 20120304 CrossRef PubMed.
- Y. Ai, Y. Li, M. H.-Y. Chan, G. Xiao, B. Zou and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 10659–10667 CrossRef CAS PubMed.
- L. Zhang, A. Fonari, Y. Zhang, G. Zhao, V. Coropceanu, W. Hu, S. Parkin, J.-L. Brédas and A. L. Briseno, Chem. - Eur. J., 2013, 19, 17907–17916 CrossRef CAS PubMed.
- U. Zschieschang, K. Amsharov, M. Jansen, K. Kern, H. Klauk and R. T. Weitz, Org. Electron., 2015, 26, 340–344 CrossRef CAS.
- J. W. Borchert, R. T. Weitz, S. Ludwigs and H. Klauk, Adv. Mater., 2022, 34, 2104075 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2266823. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02898d |
| This journal is © The Royal Society of Chemistry 2023 |