
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free, photoinduced remote C(sp3)–H borylation†
Jiachen
He
and
Silas P.
Cook
 *
*
Department of Chemistry, Indiana University, 800 East Kirkwood Avenue, Bloomington, IN 47405-7102, USA. E-mail: sicook@iu.edu
First published on 22nd August 2023
Abstract
Here, we describe a protocol for the metal-free, photo-induced borylation of unactivated C(sp3)–H bonds distal to an O-oxalate hydroxamic ester functionality. The methodology requires only substrate and bis(catecholato)diboron under light irradiation to effect the desired transformation. A range of linear and cyclic tertiary and secondary borylation products are obtained in good yields and high site-selectivity enabling the late-stage C(sp3)–H borylation of natural product derivatives and drug-like compounds.
Alkyl boronic esters represent a class of powerful linchpin synthons for organic chemistry due to their ability to feed into a variety of functional group interconversions.1 While methods exist to prepare alkyl boronic esters, they often rely on multistep synthesis and prefunctionalization of the site of interest.2 Consequently, the development of complementary methods remains an important goal. Specifically, direct C(sp3)–H borylation offers both atom-economy and orthogonal reactivity to install boronic esters.3
Despite the progress in the borylation of C(sp2)–H bonds, the site-selective borylation of C(sp3)–H bonds is far less advanced.4 Transition-metal-catalyzed C–H borylation represents the most common strategy, and impressive progress has been achieved with various transition metals such as Ir,5 Rh,6 Co,7 Ru,8 and Pd9 (Scheme 1a). While synthetically attractive, these methods often target activated C–H bonds, rely on heteroatom directing groups, sometimes require harsh reaction conditions, and employ excess alkanes, thereby limiting scope.10 Moreover, the targeting of tertiary C(sp3)–H bonds is especially difficult due to both steric hindrance and the potential of β-hydride elimination with the late, noble transition metals.
 | ||
| Scheme 1 Borylation of unactivated C(sp3)–H bonds. B2cat2 = bis(catecholato)diboron. | ||
A less-explored approach is the borylation of carbon-based radicals generated by hydrogen atom transfer (HAT). In 2020, the Aggarwal group employed a Cl˙− assisted HAT strategy to achieve the primary-selective C(sp3)–H borylation of alkanes and silanes in the presence of B2cat2 and alkoxyphthalimide under mild conditions (Scheme 1b).11 Later, Xia, Dai, and Aggarwal groups reported an iron/copper-catalyzed C(sp3)–H borylation enabled by photoinduced ligand-to-metal charge transfer (LMCT).12 Gevorgyan group also achieved a metal-free radical α-C–H borylation of aliphatic amines (Scheme 1b).13 By employing iodobenzoyl radical translocation group, α-aminoalkyl radical intermediates are generated via 1,5-hydrogen atom transfer (1,5-HAT). This method allows activation of primary and secondary α-C–H sites of a broad range of acyclic and cyclic amines. Despite recent developments, metal-free borylation of tertiary C(sp3)–H bonds remains an unsolved problem. Therefore, a general approach that expands the scope, works with a common functional group, and site-selectively targets tertiary sites fills an important gap.
Recently, our group achieved the nickel-catalyzed reductive arylation of remote C(sp3)–H bonds with aryl electrophiles by using the O-oxalate hydroxamic ester (Oohe) moiety as the N-radical precursor.14 In 2020, the Gong group found, under light irradiation, B2cat2 can activate the carbonyl oxygen of tertiary alkyl methyl oxalates, facilitating the formation of tertiary alkyl radicals via C–O bond fragmentation.15 Encouraged by these results, we hypothesized that N-radicals could be generated by irradiating O-oxalate hydroxamic esters in the presence of B2cat2. Subsequently, carbon radicals at the γ site would be formed via 1,5-HAT, chemoselectively affording alkyl boronic esters through B2cat2 capture (Scheme 1c). During the preparation of this manuscript, Mo and co-workers disclosed a metal-free borylation of O-benzoyl hydroxamic ester with B2cat2,16 however, a photoredox catalyst and amine additive are necessary. An excited [EY2−]* intermediate is proposed as a powerful reducing reagent that undergoes single-electron transfer to O-benzoyl hydroxamic ester, leading to the homolytic cleavage of the N–O bond and formation of N-based radicals. The amine additive is proposed to regenerate the photoredox catalyst. Here, we demonstrate that by using an O-oxalate hydroxamic ester, the C(sp3)–H borylation reaction can proceed with visible light irradiation only—without the need for a photoredox catalyst or amine additive.
We began our investigation by studying the reaction between Oohe 1 and B2cat2 in N,N-dimethylfomamide (DMF) under blue LEDs irradiation (Table 1). Pleasingly, the conditions provided targeted alkyl pinacol boronic ester 11 in 98% isolated yield after treating the crude reaction with pinacol and triethylamine. Increasing the concentration led to lower yields, likely because higher concentrations favored N–B bond formation over 1,5-HAT resulting in reduced product formation. The N-Bcat product can undergo hydrolysis to form the N–H during work-up to produce the formal hydrogenation product (Table 1, entries 2–4). Evaluation of the solvent revealed that amide-based solvents were uniquely effective for the transformation, likely due to the stabilization of the boryl radical involved in the radical chain process (see ESI†).17 The crucial effect of light was confirmed by the absence of desired product 11 when the reaction was performed in the dark (Table 1, entry 6). The conditions previously reported14 by our group provided 11 in only 6% yield (Table 1, entry 7). Compared to blue light, white light and green light provided the product in lower yields, 42% and 23%, respectively. The important role of the catechol ligand on the diboron reagent was highlighted by no reactions with bis(pinacolato)diboron (B2pin2) and B2(OH)4 (Table 1, entries 10 and 11). Replacing B2cat2 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of B2(OH)4 and catechol also gave the desired product in moderate yield (Table 1, entry 13). Reducing the amount of B2cat2 to 1.5 equiv. resulted in a decrease in the reaction yield (Table 1, entry 13), from 98% to 60%. A series of amidyl-radical precursors 2–8 were also tested, the reaction did not work under the developed conditions. Interestingly, 61% yield could be achieved when O-benzoyl hydroxamic ester 9 (ref. 16 and 18) was used as starting material, without the use of a photocatalyst.
:1 mixture of B2(OH)4 and catechol also gave the desired product in moderate yield (Table 1, entry 13). Reducing the amount of B2cat2 to 1.5 equiv. resulted in a decrease in the reaction yield (Table 1, entry 13), from 98% to 60%. A series of amidyl-radical precursors 2–8 were also tested, the reaction did not work under the developed conditions. Interestingly, 61% yield could be achieved when O-benzoyl hydroxamic ester 9 (ref. 16 and 18) was used as starting material, without the use of a photocatalyst.
|
|
||
|---|---|---|
| Entry | Deviation | Yieldb (%) |
| a Performed on 0.1 mmol scale (0.1 mmol 1) in 0.05 M solvent. b Yields were determined using 1H NMR analysis with 1,3,5-trimethoxybenzene (Ar–H) as internal standard. c Performed with 0.1 mmol 1, 15 mol% Ni(hfacac)2·xH2O, 0.8 equiv. pyridine, 1.5 equiv. Zn and 1.2 equiv. MgCl2, in 0.2 M DMA/THF (3/1), 16 h.13 | ||
| 1 | None | 98 |
| 2 | 0.4 M DMF | 26 |
| 3 | 0.25 M DMF | 65 |
| 4 | 0.1 M DMF | 96 |
| 5 | 0.1 M DMA | 45 |
| 6 | No light | Trace |
| 7 | Cook's conditionsc | 6 |
| 8 | White light | 42 |
| 9 | Green light | 23 |
| 10 | 3 equiv. B2Pin2 | Trace |
| 11 | 3 equiv. B2(OH)4 | Trace |
| 12 | 3 equiv. B2(OH)4 + 3 equiv. catechol | 63 |
| 13 | 1.5 equiv. B2Cat2 | 60 |

|
||
With the optimized reaction conditions in hand, we next evaluated a range of substrates with tertiary and secondary C(sp3)–H bonds (Table 2). Various methine C–H borylation worked successfully with excellent yields including cyclic congeners and sterically hindered adamantyl, which is typically difficult to access. For all cases, only a single regioisomer was observed. Notably, the borylation occurred site-selectively for substrates that contain more than one tertiary C–H bond (18 and 21), demonstrating the advantage of the intramolecular HAT-strategy. Various functional groups, such as halides (22 and 30), ketone (23), and alkene (33), are all amenable to the reaction conditions. Substates containing oxygen atom adjacent to the reaction site also afforded desired products 24, 25 and 34.
| a Performed on 0.1 mmol scale with 3 equiv. of B2Cat2, in 0.05 M DMF. The reaction time was 24 h. Isolated yields are shown. b Performed in 0.025 M DMF. c Replacing O-oxalate hydroxamic esters with O-benzoyl hydroxamic ester, 48 h. d dr was calculated by 1H NMR analysis of the crude reaction mixture. |
|---|

|
Compared to the methine borylation, yields for the less activated secondary C–H bonds were slightly lower. The reaction provided exclusive γ-borylation even in the presence of tertiary C(sp3)–H bonds (28). While O-oxalate hydroxamic ester failed to target benzylic sites, we turned to O-benzoyl hydroxamic ester, which allowed the formation of benzylic boronates, as exemplified by 29–31. Aryl rings substituted with methoxy and bromo groups also proved to be suitable substrates. The methodology failed to produce primary boronic esters due to unidentifiable side product formation. Encouraged by the broad scope of our method, we next targeted the site-selective C–H borylation of drug derivatives and biologically relevant compounds. Stearic acid (32), oleic acid (33) and dihydrochlolesterol (34) substrates could be successfully borylated, thereby demonstrating the value of this method for the late-stage functionalization of complex alkanes.
While the primary goal of this work targeted N-directed C–H functionalization, we became interested in whether our strategy can apply to δ-C–H bond borylation of amine derivatives (Table 3). The starting materials were readily prepared from the corresponding amines (see ESI†). While tert-butyl O-hydroxamic ester 35 only provided trace yields, the adamantoyl-based O-hydroxamic ester 36 produced the desired boronic esters in 58% yield. Additionally, we observed the desired borylation of benzyol-based O-hydroxamic ester substrates bearing either electron-withdrawing or electron-donating groups on the aromatic ring, providing good yields of the target boronic esters (37–41). Notably, borylation of the aliphatic C–H bond predominated over that of the benzylic C–H bond in substrates having two possible 1,5-HAT pathways (42). Product 43 was obtained in lower yield compared to product 38, likely due to the Thorpe–Ingold effect.19 Again, the method provided a broad substrate scope with excellent regioselectivity.
| a Performed on 0.1 mmol scale with 3 equiv. of B2Cat2, in 0.05 M DMF. The reaction time was 24 h. Isolated yields are shown. b Performed in 0.025 M DMF. c dr was calculated by 1H NMR analysis of the crude reaction mixture. d Due to signal overlap in the 1H NMR, unambiguous determination of the selectivity failed. |
|---|

|
Compounds 44–47 were formed as mixture of the two diastereoisomers. Transannular borylation occurred with complete regioselectivity to produce 47 as the sole product. The borylation protocol was successfully applied to an amine-derived from gemfibrozil, affording product 48 in a synthetically useful yield.
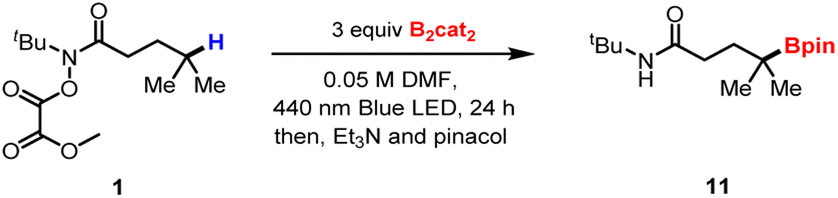
Performing the reaction on 1.0 mmol also produced the desired product in good yield, demonstrating the practicality of this method (Scheme 2a). As expected, the boronic ester products can be easily transformed into other synthetically useful motifs (Scheme 2b). For example, the transition-metal-free cross-coupling20 between 11 and furan furnished the γ-arylated derivative 49. Mild oxidation and Zweifel olefination21 of 11 provided the corresponding products, 50 and 51, respectively. Additionally, Matteson homologation22 of the obtained product provided boronate, which was further transformed into alcohol derivate 52.
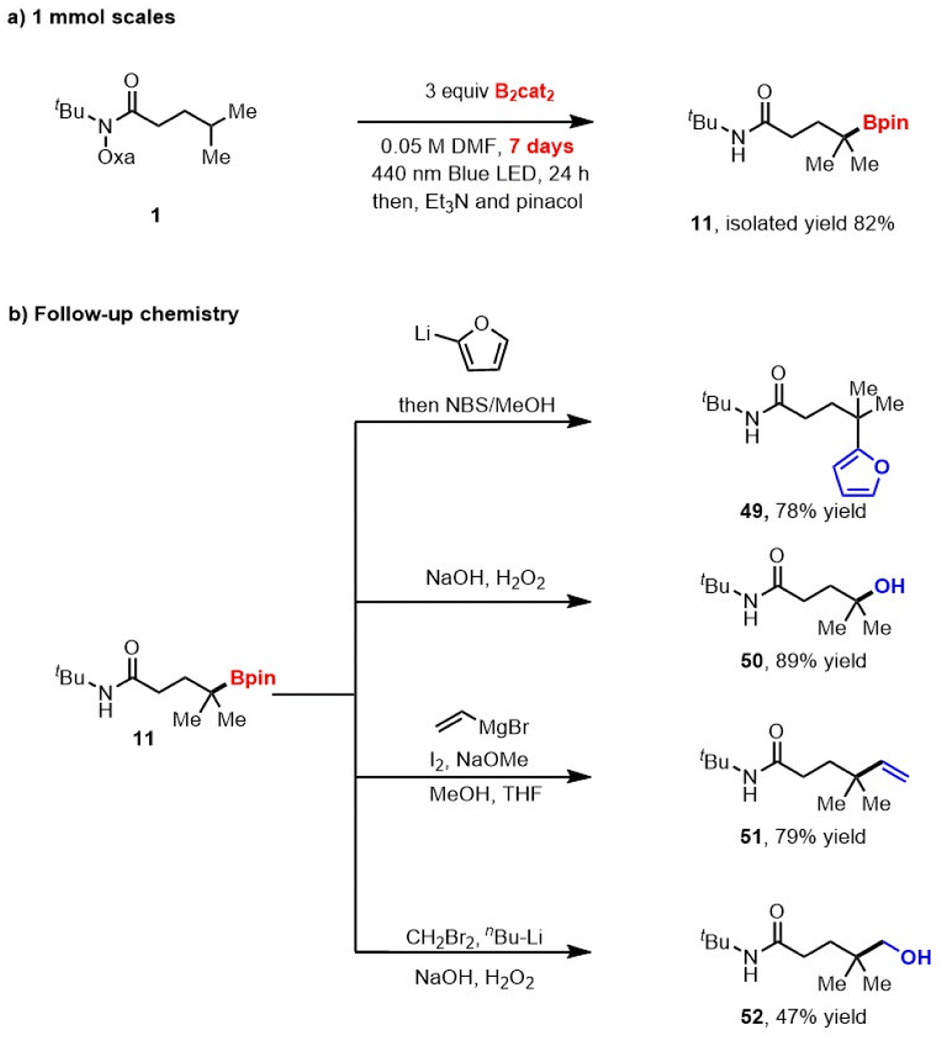 | ||
| Scheme 2 The method offers value in both laboratory scalability and synthetic utility. | ||
To gain insight into the reaction mechanism, several control experiments were performed (Scheme 3). Only trace of the desired product was detected when the reaction was performed in the presence of TEMPO. Other radical scavengers such as BHT also significantly diminish the yield (Scheme 3a). The reaction of unsaturated the O-oxalate hydroxamic ester 53a as a radical probe afforded the cyclized product 53 (Scheme 3b).23 Additionally, a substrate containing cyclopropyl methyl moiety undergoes ring-opening to result primary borylated product 54 (Scheme 3c).24 Together, the results in Scheme 3 suggest that the reaction proceeds via a radical H-atom abstraction mechanism. Additionally, we investigated the photoinitiation process by measuring absorption spectra of the reaction components. The lack of change in the UV/Vis spectrum suggested that an electron donor–acceptor complex between O-oxalate hydroxamic ester and B2cat2 does not occur, is fast on the UV/Vis timescale, or is generated in small amounts (Fig. S1†).25 A weak absorption at ∼430 nm suggests the formation of a DMF·B2(cat)2 adduct.15 Likely, photoexcitation of the DMF·B2(cat)2 complex generates a DMF-stabilized boryl radical, which can further initiate a radical chain process.15,26 Interestingly, a bathochromic shift in the absorption spectrum was observed when O-benzoyl hydroxamic ester 9 was treated with B2cat2 in DMF (Fig. S3†), suggesting the formation of an electron–donor–acceptor (EDA) complex.
 | ||
| Scheme 3 Mechanistic studies. (a) Reaction with radical scavengers; (b) radical-mediated cyclization; (c) radial-mediated cyclopropane ring-opening. | ||
Based on the combination of mechanistic results and literature reports, a proposed mechanism is shown in Scheme 4. Under light irradiation, the formation of a (DMF)2–B2cat2A will give a DMF-stabilized Bcat radical B to activate the oxalate. Then, radical fragmentation of the N–O bond generates the corresponding amidyl radical C, followed by 1,5-HAT to give a C-centered alkyl radical D. Radical D is then trapped by diboron B2cat2A to yield the adduct E, with subsequent B–B bond cleavage facilitated by complexation with DMF to form F. The weak B–B bond homolyzes to give the borylated product G along with DMF-Bcat radical B.
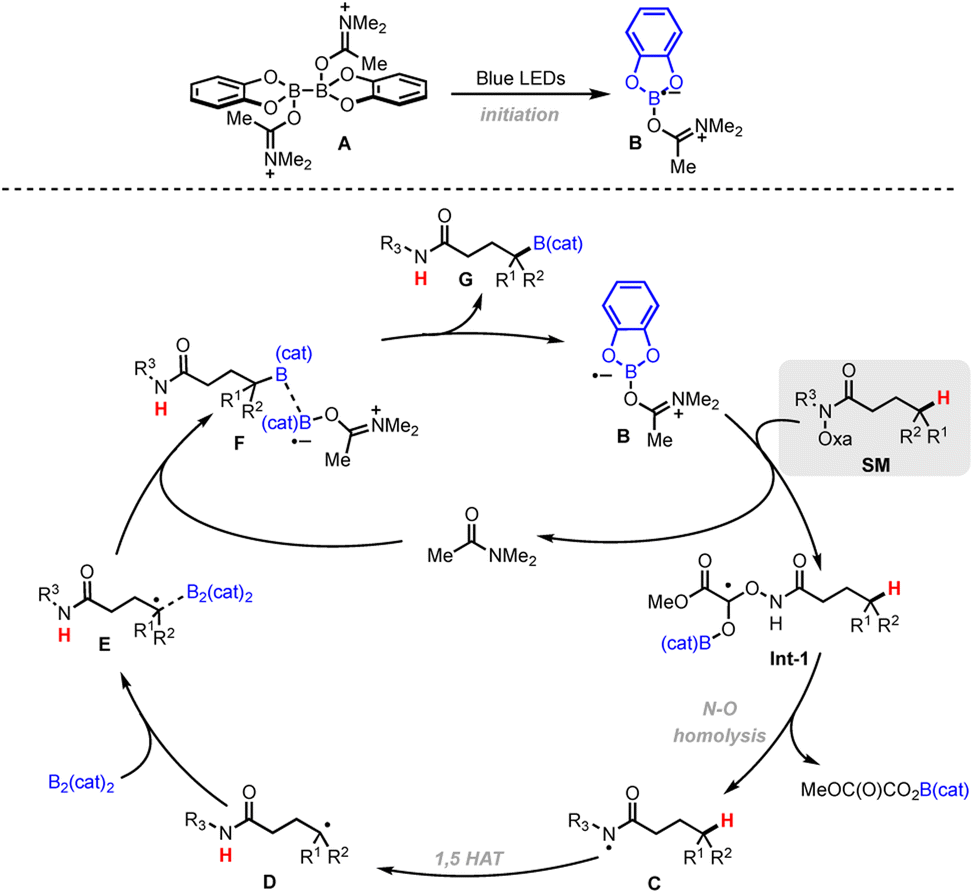 | ||
| Scheme 4 Proposed reaction mechanism. | ||
In conclusion, we have developed a photoinduced remote C(sp3)–H bond borylation of O-oxalate hydroxamic esters. The reaction does not require a photocatalyst, transition metal catalyst, or additional radical initiator. Remarkably, this method enables the targeting of an impressive array of aliphatic C–H bonds (2° and 3°) with broad functional group tolerance and highly site-selectivity and can be used in the late-stage borylation of bioactive molecules. Key to the success of the reaction was the use of O-oxalate hydroxamic esters, which efficiently reacted with B2(cat)2 in a photoinitiated radical chain mechanism.
Author contributions
J. H. and S. P. C. designed the experiments. J. H. performed the experiments. J. H. and S. P. C. wrote the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge Indiana University for partial support. We also acknowledge the NIH (GM121668), We acknowledge funds from Indiana University in partial support of this work. We also gratefully acknowledge the NIH (R01GM121668). Eli Lilly & Co. and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award. IU mass spectrometry for HRMS (NSF Grant CHE1726633).Notes and references
- (a) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed; (b) Y.-M. Tian, X.-N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed; (c) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- (a) S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed; (b) F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503–8518 RSC.
- (a) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC.
- (a) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 12422–12425 CrossRef CAS PubMed; (b) Q. Li, C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755–8765 CrossRef CAS PubMed; (c) M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633–8643 CrossRef CAS PubMed; (d) R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Science, 2020, 368, 736–741 CrossRef CAS.
- (a) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS; (b) J. D. Lawrence, M. Takahashi, C. Bae and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 15334–15335 CrossRef CAS PubMed; (c) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995–1997 CrossRef CAS PubMed.
- W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766–769 CrossRef CAS.
- (a) J. M. Murphy, J. D. Lawrence, K. Kawamura, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 13684–13685 CrossRef CAS; (b) R.-L. Zhong and S. Sakaki, J. Am. Chem. Soc., 2020, 142, 16732–16747 CrossRef CAS PubMed.
- (a) L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899–3903 CrossRef CAS PubMed; (b) J. He, H. Jiang, R. Takise, R.-Y. Zhu, G. Chen, H.-X. Dai, T. G. M. Dhar, J. Shi, H. Zhang, P. T. W. Cheng and J.-Q. Yu, Angew. Chem., 2016, 128, 795–799 CrossRef; (c) H. B. Chandrashekar, P. Dolui, B. Li, A. Mandal, H. Liu, S. Guin, H. Ge and D. Maiti, Angew. Chem., 2021, 133, 18342–18348 CrossRef.
- J. Hu, J. Lv and Z. Shi, Trends Chem., 2022, 4, 685–698 CrossRef CAS.
- C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714–719 CrossRef CAS PubMed.
- (a) J.-L. Tu, A.-M. Hu, L. Guo and W.-J. Xia, J. Am. Chem. Soc., 2023, 145(13), 7600–7611 CrossRef CAS PubMed; (b) W. Fang, H.-Q. Wang, W. Zhou, Z.-W. Luo and J.-J. Dai, Chem. Commun., 2023, 59, 7108–7111 RSC; (c) R.-C. Sang, W.-Y. Han, H.-W. Zhang, C. M. Saunders, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2023, 145, 15207–15217 CrossRef CAS PubMed.
- S. Sarkar, S. Wagulde, X. Jia and V. Gevorgyan, Chem, 2022, 8, 3096–3108 CAS.
- Z.-Y. Liu and S. P. Cook, Org. Lett., 2022, 24, 3313–3318 CrossRef CAS.
- G. Ma, C. Chen, S. Talukdar, X. Zhao, C. Lei and H. Gong, Chem. Commun., 2020, 56, 10219–10222 RSC.
- B. Sun, W. Li, Q. Liu, G. Zhang and F. Mo, Commun. Chem., 2023, 6, 156 CrossRef CAS.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- (a) W. Jin and S. Yu, Org. Lett., 2021, 23, 6931–6935 CrossRef CAS PubMed; (b) H. Chen, L. Guo and S. Yu, Org. Lett., 2018, 20, 6255–6259 CrossRef CAS PubMed; (c) H. Chen, W. Fan, X.-A. Yuan and S. Yu, Nat. Commun., 2019, 10, 4743 CrossRef.
- (a) H. Sun, B. Cui, L. Duan and Y.-M. Li, Org. Lett., 2017, 19, 1520–1523 CrossRef CAS; (b) Y. Xue, H. S. Park, C. Jiang and J.-Q. Yu, ACS Catal., 2021, 11, 14188–14193 CrossRef CAS; (c) R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed.
- M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed.
- (a) R. J. Armstrong, W. Niwetmarin and V. K. Aggarwal, Org. Lett., 2017, 19, 2762–2765 CrossRef CAS PubMed; (b) R. Armstrong and V. Aggarwal, Synthesis, 2017, 49, 3323–3336 CrossRef CAS.
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- D. Zheng, K. Jana, F. A. Alasmary, C. G. Daniliuc and A. Studer, Org. Lett., 2021, 23, 7688–7692 CrossRef CAS PubMed.
- A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405–18410 CrossRef CAS PubMed.
- (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (b) J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS PubMed.
- (a) C. Shu, R. Madhavachary, A. Noble and V. K. Aggarwal, Org. Lett., 2020, 22, 7213–7218 CrossRef CAS PubMed; (b) D. Wei, T.-M. Liu, B. Zhou and B. Han, Org. Lett., 2020, 22, 234–238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03048b |
| This journal is © The Royal Society of Chemistry 2023 |