
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the supramolecular chemistry of cyclopropeniums: halogen-bonding-induced electrostatic assembly of polymers†
Shiwen
Huang
 a,
Jianlin
Zheng
a,
Zihao
Jiang
a,
Jiaxiong
Liu
a and
Yiliu
Liu
*ab
a,
Jianlin
Zheng
a,
Zihao
Jiang
a,
Jiaxiong
Liu
a and
Yiliu
Liu
*ab
aSouth China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter, South China University of Technology, Guangzhou 510640, China. E-mail: liuyiliu@scut.edu.cn
bGuangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 24th August 2023
Abstract
Exploring new noncovalent synthons for supramolecular assembly is essential for material innovation. Accordingly, we herein report a unique type of cyclopropenium-based supramolecular motif and demonstrate its applications to polymer self-assembly. Because of the “ion pair strain” effect, trisaminocyclopropenium iodides complex strongly with fluoroiodobenzene derivatives, forming stable adducts. Crystal structure analysis reveals that halogen-bonding between the iodide anion and the iodo substituent of the fluoroiodobenzene is the driving force for the formation of these electrostatically complexed adducts. Such halogen-bonding-induced electrostatic interactions were further successfully applied to drive the assembly of polymers in solution, on surfaces, and in bulk, demonstrating their potential for constructing supramolecular polymeric materials.
Introduction
The development of noncovalent synthons is crucially important for expanding the applications of supramolecular polymeric materials.1–8 The dynamic and reversible nature of noncovalent synthons endows polymer systems with adaptive properties, such as self-healing, easy-processability, and recyclability.9–11 A renowned example is the ureidopyrimidinone (UPy)-based hydrogen bonding synthon reported by Meijer et al.12 Owing to its strong binding affinity and synthetic accessibility, the UPy motif has been widely employed in the construction of supramolecular materials, leading to applications ranging from recyclable plastics and biomaterials to soft electronics.13–15 As the demands for material innovation are constantly growing, the exploration of new noncovalent synthons remains a major focus of research in supramolecular chemistry and materials science.Triaminocyclopropeniums (TACs) are a unique type of 2-π-electron aromatic compound that exhibit high thermo-/chemo-stability and intriguing electronic features.16,17 In recent decades, TACs have found versatile applications as catalysts,18–20 ionic liquids,21,22 nitrogen-based ligands,23 redox-active materials,24 and fluorophores.25 For example, Campos and Lambert recently introduced TACs into polymeric structures, expanding their application potential to polyelectrolytes and biomedical materials.26,27 Nevertheless, the study of TACs from a supramolecular perspective remains rare, despite TACs showing interesting self-assembly behaviors. As far back as the 1990s, Weiss et al. reported several intriguing phenomena in which the counter-ions of TACs were involved in the formation of unusual strong non-covalent complexes. It has been revealed that there exists an electronic “ion pair strain” between TACs and their electron-rich counter-anions, leading to counter-anions with unparalleled coordination ability in the formation of robust noncovalent complexes.28,29 For example, TAC with iodide as the counter-anion (TAC-I) can form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts with iodoacetylene. It is revealed that the iodide counter-anion of TAC-I possesses exceptional coordinative ability to strongly bind with iodoacetylene through halogen bonding (XB), which drives the formation of electrostatically complexed adducts.29
:1 adducts with iodoacetylene. It is revealed that the iodide counter-anion of TAC-I possesses exceptional coordinative ability to strongly bind with iodoacetylene through halogen bonding (XB), which drives the formation of electrostatically complexed adducts.29
From a supramolecular point of view, this kind of adduct formation may be regarded as an electrostatic “forced marriage” between a salt and a neutral molecule via XB-induced electrostatic (XBIE) interactions. This inspired us to explore whether such interactions can also be applied to the construction of supramolecular polymeric systems. Accordingly, we herein report our recent efforts in exploring TACs as noncovalent synthons to drive the self-assembly of polymers. Demonstration examples, including the construction of electrostatically co-assembled micelles, layer-by-layer assembly, and supramolecular polymer blends, are showcased (Scheme 1).
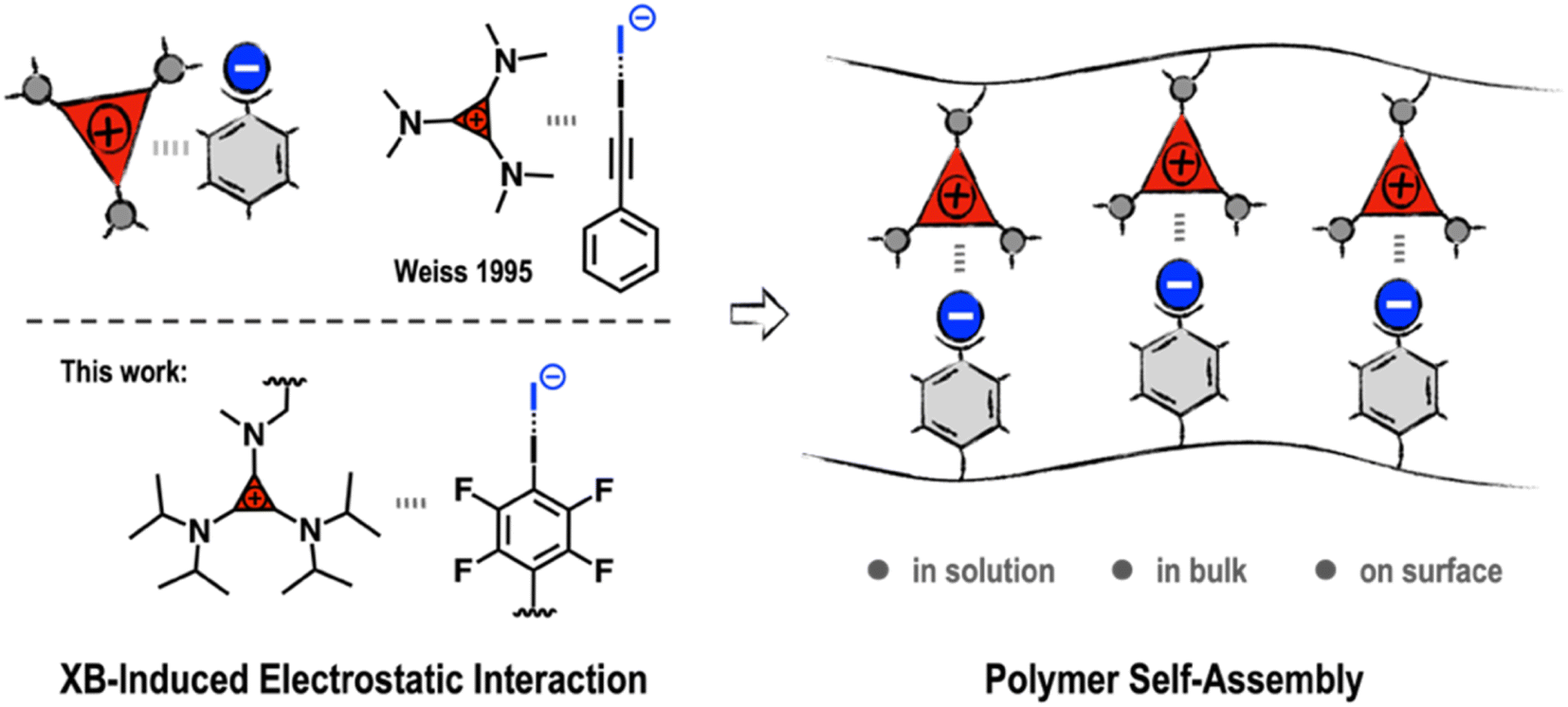 | ||
| Scheme 1 Halogen-bonding-induced electrostatic interactions for polymer self-assembly. | ||
Results and discussion
Instead of using iodoacetylene, fluoroiodobenzene derivatives were chosen as XB-donors for our study. Electron-withdrawing fluoro-substituents maximize the electron-deficiency of the iodine substituent, thus enhancing its XB-forming capacity.30 Model molecule TFAI, which contains an amide group at the para-position of the iodo-substituent, was synthesized from 2,3,5,6-tetrafluoro-4-iodobenzoic acid. Furthermore, TACE-I was chosen as a model TAC molecule to investigate the proposed XBIE interactions with TFAI. First, the complexation of TACE-I and TFAI was studied by 19F NMR using deuterated acetone as the solvent. As shown in Fig. 1a, upon titrating TACE-I into a solution of TFAI, the fluoride signals for TFAI gradually move up-field, suggesting the formation of XB between the iodide anion and TFAI. This was further supported by the results obtained from 13C NMR and infrared (IR) spectroscopy. As shown in Fig. S4,† the 13C NMR signal of the iodo-substituted carbon of TFAI shows a clear shift to the downfield upon mixing with TACE-I. Moreover, IR measurements reveal that the C–I stretching band of TFAI shifts to lower wavenumbers after complexing with TACE-I (Fig. S13a†). Fitting the titration curve obtained from 19F NMR affords an association constant Ka of 46.4 M−1. For comparison, the association constants between TFAI and other iodide salts, imidazolium iodide (DMIM-I) and sodium iodide (NaI), were also determined by the same method, revealing Ka values of 24.3 and 29.6 M−1, respectively. The higher Ka between TACE-I and TFAI confirms the effect of “ion pair strain” in promoting noncovalent interactions.31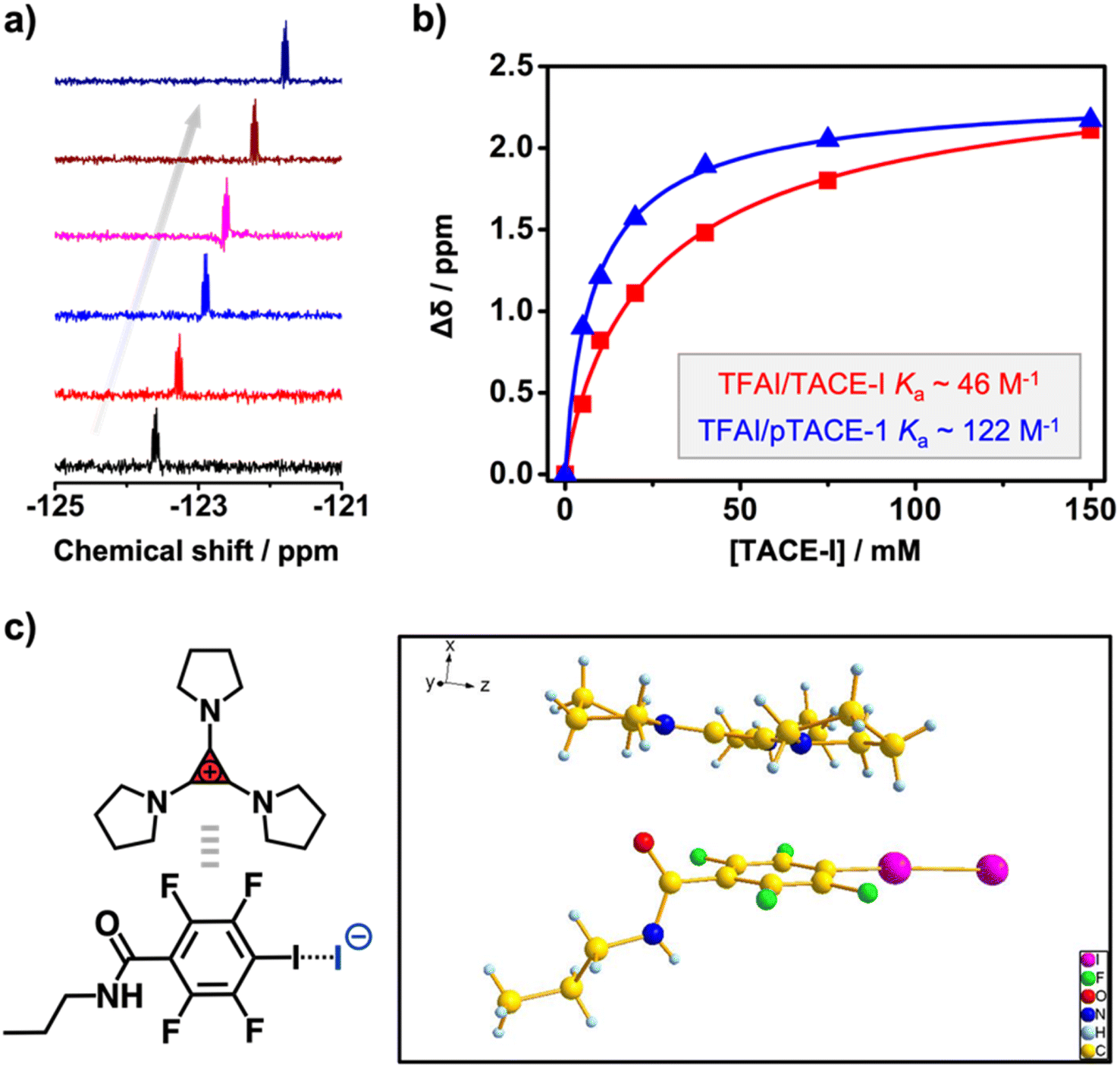 | ||
| Fig. 1 (a) 19F NMR spectra recorded upon titrating TACE-I into TFAI (signals from the fluoro-substituents neighboring the iodo-substituent; ratio of TACE-I increases from bottom to top); (b) titration curves obtained from titrating TFAI with TACE-I (red) and pTACE-1 (blue); (c) crystal structure of the TFAI/TACP-I adduct (C: yellow; O: red; N: blue; F: green; I: purple). | ||
Crystallographic analysis was then performed to reveal more structural information. Crystal growth of TFAI/TACE-I was not successful. Instead, we obtained colorless needle-like crystals of the adduct between TFAI and TACP-I (an analog of TACE-I; Scheme 2). The resolved crystal structure is shown in Fig. 1c. The iodide anion of TACP-I is located in the vicinity of the iodo substituent of TFAI instead of that of the cyclopropenium ring. The I⋯I distance is 325.8 pm, which is significantly shorter than the sum of their van der Waals radii (396 pm), confirming the formation of strong halogen bonding. Besides the XB, the cyclopropenium ring of TACP-I stacked above/below the phenyl ring of TACP-I, suggesting an extra aromatic interaction between the two counterparts. Noteworthily, the adduct can be easily prepared by mixing TFAI and TACP-I in dichloromethane which then precipitates in diethyl ether. Considering that TFAI itself is highly soluble in diethyl ether, the co-precipitation confirms a high affinity between the two counterparts.
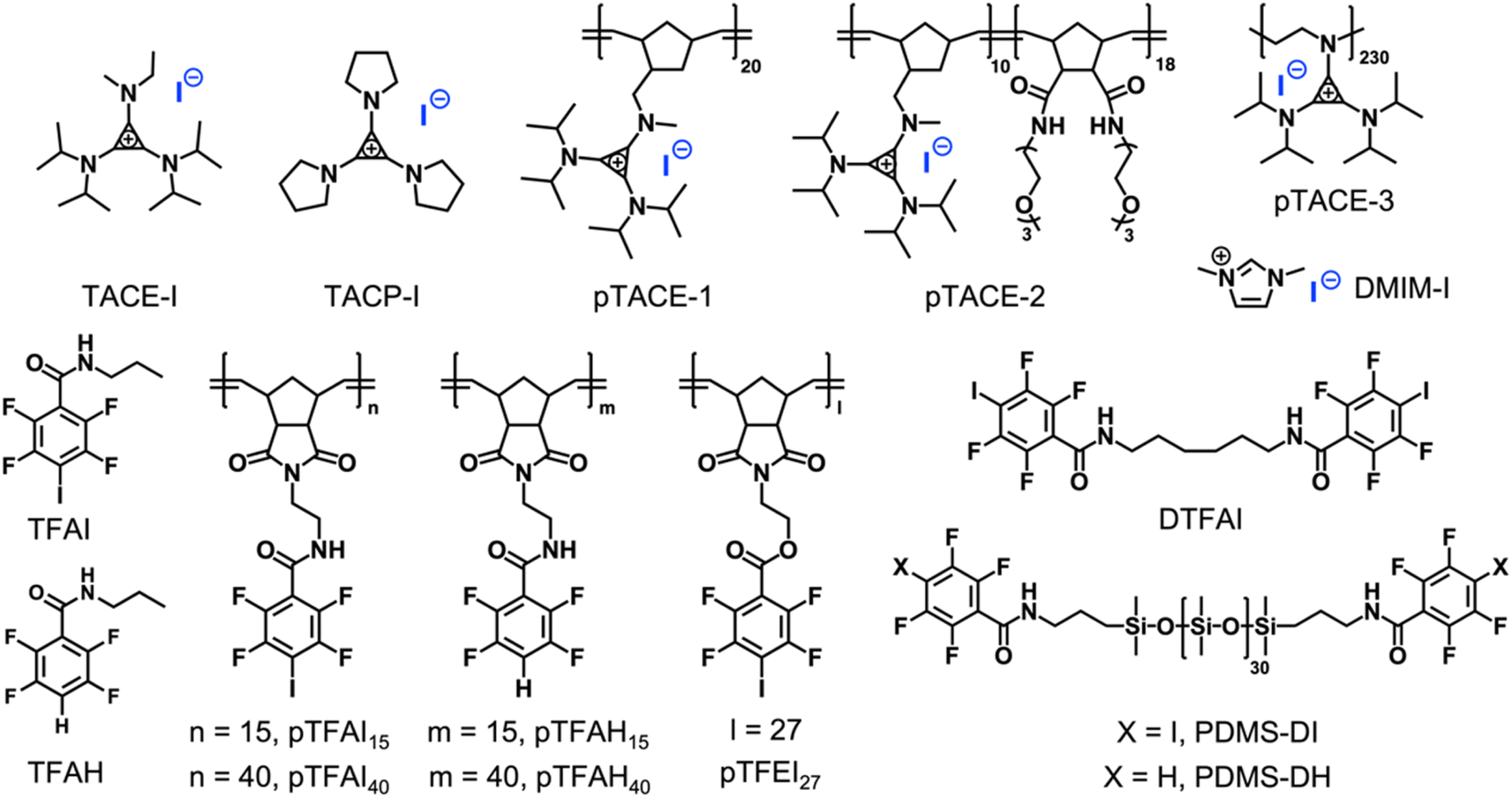 | ||
| Scheme 2 Chemical structures of the model molecules and polymers involved in the study. | ||
Single-set noncovalent interaction is normally not strong enough to drive the formation of stable supramolecular polymeric systems. A potent strategy to overcome this is the introduction of multivalent interactions.32,33 To study how such multivalency can improve complexation capacity, pTACE-1, which contains multiple TACE-I pendant groups, was designed and synthesized (Scheme 2; see the ESI† for details). It is worthy of mention that, to our knowledge, our synthesis of pTACE-1 is the first example of using ring-opening metathesis polymerization (ROMP) to make cyclopropenium-containing polymers. The association constant for the XB between pTACE-1 and TFAI was determined to be 122.9 M−1 (per TACE unit), which is significantly higher than that of the “monovalent” system. The substantial increase in Ka can be attributed to the polymer structure, which creates a favorable microenvironment with a higher local concentration of iodide anions and a lower dielectric constant.34 More importantly, when the TFAI-based dimer (DTFAI) or polymer (pTFAI15) was mixed with pTACE-1, precipitates formed immediately, indicating strong complexation (Fig. S8 and S9†). It is reported that the iodide anions can also form hydrogen-bonding (HB) with the proton of the amide moiety.35 Several control experiments were conducted to confirm the importance of XBIE interaction in polymer complexation. Initially, the association constant of the hydrogen bond between iodide anions and the amide group was studied through 1H NMR titration experiments by using TACE-1 and TFAH (without an iodo-substituent) as model systems (Scheme 2 and Fig. S5†). The Ka value measured was below 10 M−1 in acetone, indicating a relatively weak association. Subsequently, a mixture of the control polymers pTFAH15 and pTACE-1 was prepared in acetone. This resulted in a clear solution without any precipitate formation (Fig. S10†), confirming the necessity of XBIE interaction for intermolecular complexation. Additionally, a different polymer, pTFEI27, was designed and synthesized, where ester bonds were used instead of amide bonds (Scheme 2). When pTACE-1 was mixed with pTFEI27, immediate precipitation occurred, further supporting the fact that XBIE interaction is the primary driving force behind polymer precipitation. Overall, these results demonstrate the feasibility of XBIE interactions in intermolecular complexation, which were subsequently utilized in the self-assembly of polymers.
First, the solution self-assembly of polymers driven by XBIE interactions was investigated. The block copolymer pTACE-2, which contains a block bearing TACE-I pendant groups and a block bearing oligo-ethylene glycol (OEG) sidechains, was synthesized by ROMP (Scheme 2). Both pTACE-2 and pTFAI15 are highly soluble in acetone, forming solutions containing molecularly dissolved polymers. The solution of pTFAI15 was titrated into the solution of pTACE-2, and the resulting mixtures were investigated by dynamic light scattering (DLS). As shown in Fig. 2a and S14,† aggregates with hydrodynamic radii (Rh) around 25 nm formed in all the mixed solutions. Conversely, the control sample prepared by mixing pTFAH15 and pTACE-2 showed no aggregation, confirming that the driving force for the polymer assembly is XBIE interaction. The aggregates formed in the pTFAI15/pTACE-2 mixture were investigated by transmission electron microscopy (TEM), and spherical micelles with sizes consistent with the DLS results were observed (Fig. 2a, inset).
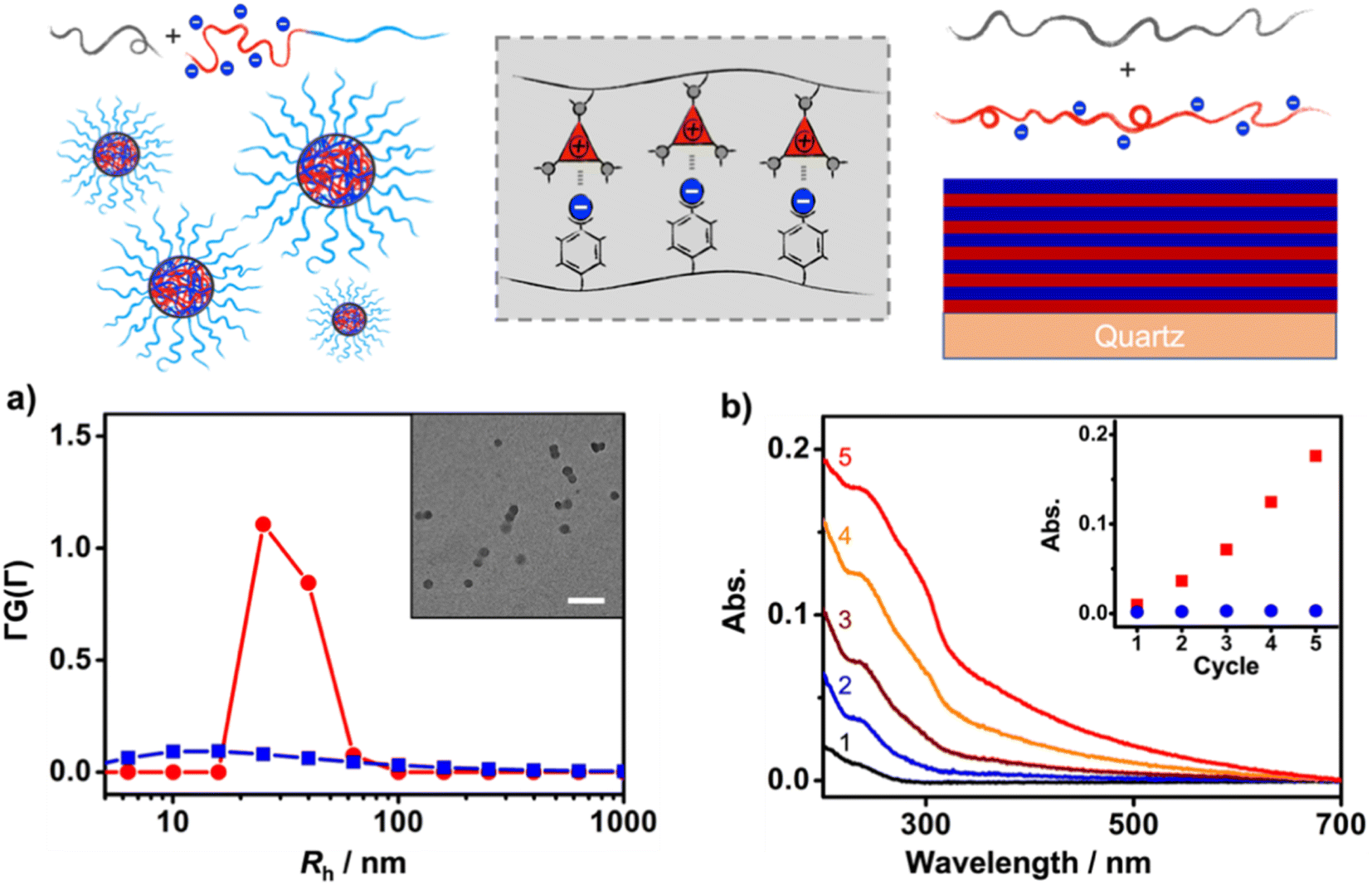 | ||
| Fig. 2 Application of XBIE interactions to polymer assembly in solution and on a surface; (a) DLS results for pTACE-2/pTFAI15 (red) and pTACE-2/pTFAH15 (blue) (inset: the TEM image of a pTACE-2/pTFAI15 solution); (b) UV-vis spectra from LBL assembly of pTACE-3/pTFAI40 (inset: abs. at 237 nm as a function of cycle number; pTACE-3/pTFAI40 (red) and pTACE-3/pTFAH40 (blue)). | ||
The layer-by-layer (LBL) deposition technique is a versatile approach to assembling polymers on surfaces.36 Although LBL driven by electrostatic interactions or XB has been reported,37,38 XBIE-driven LBL has yet to be explored. Here, pTACE-3 and pTFAI40 were used as the two components for LBL study. First, a clean hydroxylated quartz plate was immersed in an acetone solution of pTACE-3 for 10 min and then rinsed with acetone three times to eliminate unattached polymers. After drying under ambient conditions, the quartz plate was immersed in an acetone solution of pTFAI40 for another 10 min and then rinsed with acetone and dried. This process was repeated several times, and the quartz plate was characterized by UV-vis spectroscopy between each cycle. Concurrently, LBL deposition was also performed with pTACE-3 and pTFAH40 (without iodo-based XB donors) for comparison. In the pTACE-3/pTFAI40 LBL system, the absorbance increases linearly with cycle repetitions, confirming the success of LBL assembly with similar amounts of polymer being adsorbed in each cycle. In significant contrast, LBL assembly of pTACE-3 and pTFAH40 results in almost no increase in absorbance, most likely because of the lack of XBIE interactions between the two polymers (Fig. S16†).
Finally, XBIE interactions were applied to the self-assembly of incompatible polymers in bulk.39 The telechelic polydimethylsiloxanes PDMS-DI and PDMS-DH, which contain either TFAI or TFAH as end-groups, were employed (Scheme 2). First, these polymers were respectively mixed with pTACE-3 in dichloromethane. Evaporation of the solvent led to two types of polymer blends, namely PDMS-DI/pTACE-3 and PDMS-DH/pTACE-3. PDMS is incompatible with most polar species, and macroscale phase separation occurs when there are no specific interactions between PDMS and other components in the blend. Photographs of the two blends are shown in Fig. 3a. In the PDMS-DH/pTACE-3 blend, pTACE-3 precipitates as a solid and PDMS-DH remains as a flowable liquid. Conversely, PDMS-DI/pTACE-3 forms a homogeneous gel-like blend, suggesting that XBIE interactions between TACE-I and TFAI endow the two polymers with compatibility. This was confirmed by solid-phase 19F NMR measurements (Fig. S23†). When pTACE-3 is mixed with PDMS-DI, the fluoride signals from PDMS-DI exhibit noticeable shifts towards higher fields. These shifts are consistent with the halogen-bonding formation observed in the TACE-I & TFAI model systems in solution, indicating a similar phenomenon in the solid state.
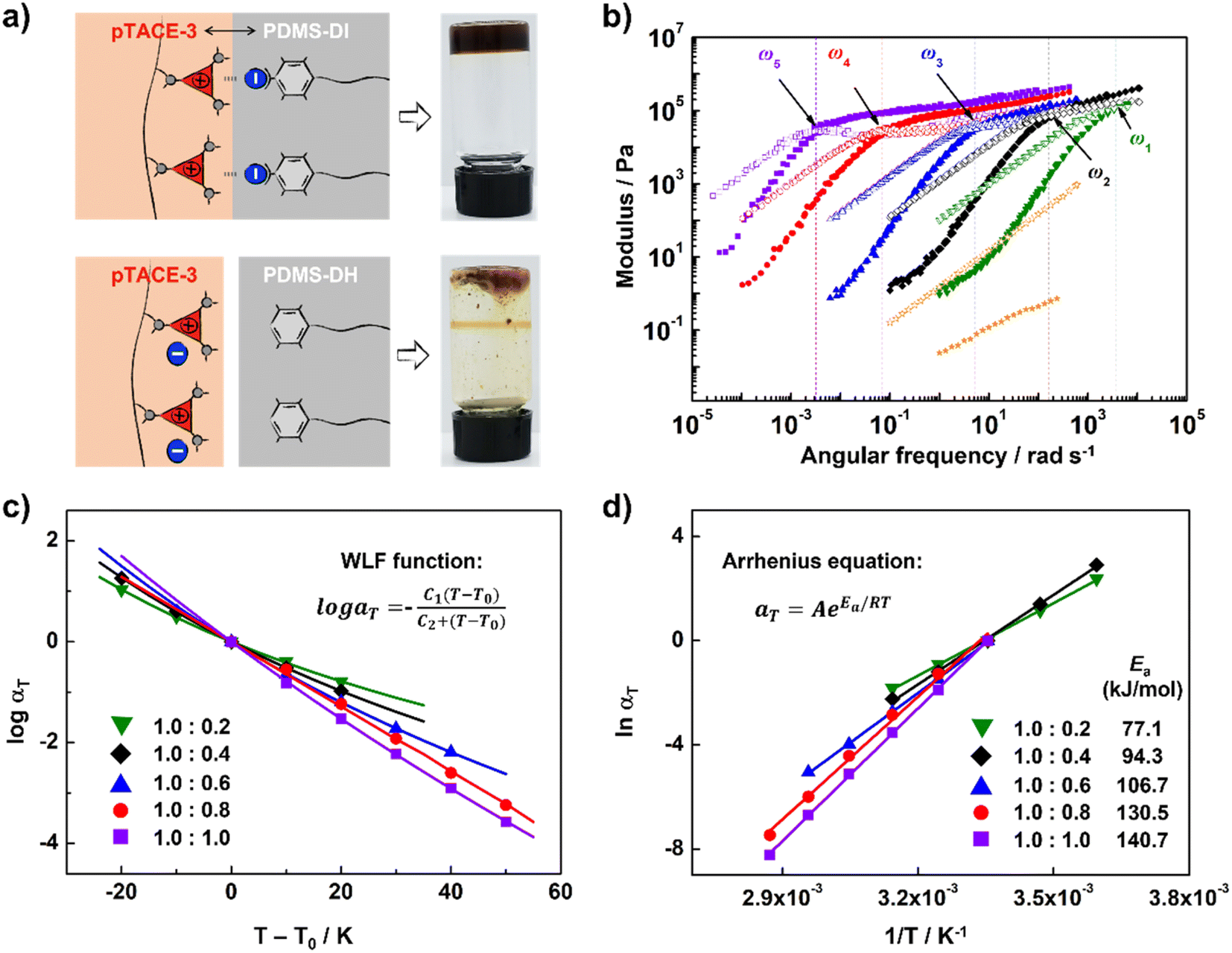 | ||
| Fig. 3 Application of XBIE interactions to constructing supramolecular polymer blends. (a) Schematics and pictures of PDMS-DI/pTACE-3 and PDMS-DH/pTACE-3 blends; (b) master curves for supramolecular polymer blends with [PDMS-DI]/[pTACE-3] = 1.0:0.2 (green), 1.0:0.4 (black), 1.0:0.6 (blue), 1.0:0.8 (red), 1.0:1.0 (purple), and pure PDMS-DI (orange) (G′ (solid) and G′′ (hollow)); (c) WLF fitting of aT as a function of temperature. (d) Determination of Ea using Arrhenius plots. | ||
To gain a deeper understanding of the dynamic viscoelastic properties of the supramolecular polymer blend, samples with different ratios of PDMS-DI and pTACE-3 were prepared and characterized by frequency-sweep analysis. As shown in Fig. 3b, master curves for the storage moduli (G′) and loss moduli (G′′) for PDMS-DI/pTACE-3 blends (1.0/0.2–0.8) at a reference temperature of 25 °C are well constructed according to the time–temperature superposition principle. For each sample, the G′ and G′′ cross at a specific frequency (ω), and the G′ approaches a plateau value in the high-frequency (low-temperature) region, demonstrating responses of elastic solids.40 The terminal slopes at low frequency show G′′ ∝ ω and G′ ∝ ω2 behavior, which indicates that PDMS-DI and pTACE-3 are homogeneously mixed in these supramolecular polymeric blends.41 This was further corroborated by the analysis conducted using differential scanning calorimetry (DSC). The results revealed a singular, yet notably higher glass transition temperature (Tg) in the supramolecular polymeric blend compared to that observed in PDMS-DI (Fig. S25†). Meanwhile, the horizontal shift factor aT follows the Williams–Landel–Ferry (WLF) function well, and the vertical shift factor bT is close to 1, confirming the thermorheologically simple behavior of these supramolecular polymeric blends (Fig. 3c and S21†).40 The tanδ curve of PDMS-DI/pTACE-3 (1:1) blends shows a slight stacking deviation in the low temperature region, which may result from the phase separation caused by excessive pTACE-3 (Fig. S20†). Using the Arrhenius equation, the apparent activation energy (Ea) of these supramolecular polymeric blends was determined (Fig. 3d, Table S1†).42 For the blends studied here, the obtained Ea ranges from 77.1 to 140.7 kJ mol−1, increasing with the addition of pTACE-3, indicating that the supramolecular polymeric blends are inherently enhanced by introducing XBIE interactions.
Conclusions
In summary, we have developed triaminocyclopropenium iodides and fluoroiodobenzene derivatives as a new type of noncovalent synthon, which were applied, for the first time, to drive the self-assembly of polymers. The construction of electrostatically co-assembled micelles in solution, LBL deposition of polymers on a surface, and preparation of supramolecular polymer blends in bulk were demonstrated as showcases, which demonstrate the feasibility of this noncovalent synthon in constructing supramolecular polymeric systems. Considering the unique electronic features of TACs, such as their redox activity and conductivity, we envision that TAC-based noncovalent synthons can act as functional components while also being structural motifs. Accordingly, this line of research presents opportunities for developing supramolecular electronic materials. Furthermore, when compared with hydrogen bonding, XB offers several advantages, including high directivity, adjustable strength, and greater resistance to polar solvents. It has found extensive applications across various fields such as mesomorphic materials, organic catalysis, and drug delivery. We expect that the newly coined term “halogen-bonding-induced electrostatic interaction” will stimulate new research on TAC- and/or XB-based molecular systems.Data availability
All experimental data are available within the article and the ESI.†Author contributions
S. Huang and Y. Liu conceived the study. S. Huang, J. Zheng, Z. Jiang, and J. Liu performed the experiments. All authors contributed to the finalization of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We gratefully acknowledge the financial support from the National Key R&D Program of China (grant no. 2021YFA1501600), the Recruitment Program of Guangdong (2016ZT06C322), the Natural Science Foundation of Guangdong Province (2023A1515011561), and the Fundamental Research Funds for the Central Universities (2022ZYGXZR038). We also thank Zhiwei Yan and Prof. Rongchun Zhang for solid-phase NMR measurements and Prof. Taolin Sun for helpful discussion and assistance in the dynamic mechanical analysis of polymer blends.References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, 1907247 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- K. Liu, Y. Jiang, Z. Bao and X. Yan, CCS Chem., 2019, 1, 431–447 CrossRef CAS.
- H. Wang, X. Ji, Z. Li and F. Huang, Adv. Mater., 2017, 29, 1606117 CrossRef.
- G. M. Ter Huurne, A. R. A. Palmans and E. W. Meijer, CCS Chem., 2019, 1, 64–82 CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- J. Zhu, G. Y. Chen, L. Yu, H. Xu, X. Liu and J. Sun, CCS Chem., 2020, 2, 280–292 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- B. Qin, S. Zhang, P. Sun, B. Tang, Z. Yin, X. Cao, Q. Chen, J. Xu and X. Zhang, Adv. Mater., 2020, 32, 2000096 CrossRef CAS.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33, 2008111 CrossRef CAS PubMed.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef PubMed.
- Z. Yoshida and Y. Tawara, J. Am. Chem. Soc., 1971, 93, 2573–2574 CrossRef CAS.
- J. S. Bandar and T. H. Lambert, Synthesis, 2013, 45, 2485–2498 CrossRef CAS.
- R. M. Wilson and T. H. Lambert, Acc. Chem. Res., 2022, 55, 3057–3069 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- S. Huang, X. Su, Y. Wu, X. Xiong and Y. Liu, Chem. Sci., 2022, 13, 11352–11359 RSC.
- O. J. Curnow, D. R. MacFarlane and K. J. Walst, Chem. Commun., 2011, 47, 10248–10250 RSC.
- J. Litterscheidt, J. S. Bandar, M. Ebert, R. Forschner, K. Bader, T. Lambert, W. Frey, A. Buhlmeyer, M. Brandle, F. Schulz and S. Laschat, Angew. Chem., Int. Ed., 2020, 59, 10557–10565 CrossRef CAS.
- H. Bruns, M. Patil, J. Carreras, A. Vazquez, W. Thiel, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2010, 49, 3680–3683 CrossRef CAS PubMed.
- S. S. Sevov, S. K. Samaroo and M. S. Sanford, Adv. Energy Mater., 2017, 7, 1602027 CrossRef.
- M. Guest, R. Mir, G. Foran, B. Hickson, A. Necakov and T. Dudding, J. Org. Chem., 2020, 85, 13997–14011 CrossRef CAS PubMed.
- Y. Jiang, J. L. Freyer, P. Cotanda, S. D. Brucks, K. L. Killops, J. S. Bandar, C. Torsitano, N. P. Balsara, T. H. Lambert and L. M. Campos, Nat. Commun., 2015, 6, 5950 CrossRef CAS PubMed.
- P. J. Griffin, J. L. Freyer, N. Han, N. Geller, X. Yin, C. D. Gheewala, T. H. Lambert, L. M. Campos and K. I. Winey, Macromolecules, 2018, 51, 1681–1687 CrossRef CAS.
- R. Weiss, T. Brenner, F. Hampel and A. Wolski, Angew. Chem., Int. Ed., 1995, 34, 439–441 CrossRef CAS.
- R. Weiss, M. Rechinger, F. Hampel and A. Wolski, Angew. Chem., Int. Ed., 1995, 34, 441–443 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- J. R. Butchard, O. J. Curnow, D. J. Garrett and R. G. A. R. Maclagan, Angew. Chem., Int. Ed., 2006, 45, 7550–7553 CrossRef CAS PubMed.
- J. Xu, L. Chen and X. Zhang, Chem. –Eur. J., 2015, 21, 11938–11946 CrossRef CAS PubMed.
- Y. Li, Y. Ding, B. Yang, T. Cao, J. Xu, Y. Dong, Q. Chen, L. Xu and D. Liu, CCS Chem., 2022, 5, 434–444 CrossRef.
- K. M. Bqk, S. C. Patrick, X. Li, P. D. Beer and J. J. Davis, Angew. Chem., Int. Ed., 2023, 62, e202300867 CrossRef PubMed.
- M. Giese, M. Albrecht, T. Krappitz, M. Peters, V. Gossen, G. Raabe, A. Valkonen and K. Rissanen, Chem. Commun., 2012, 48, 9983–9985 RSC.
- X. Zhang, H. Chen and H. Zhang, Chem. Commun., 2007, 1395–1405 RSC.
- Y. Lvov, G. Decher and H. Moehwald, Langmuir, 1993, 9, 481–486 CrossRef CAS.
- F. Wang, N. Ma, Q. Chen, W. Wang and L. Wang, Langmuir, 2007, 23, 9540–9542 CrossRef CAS PubMed.
- L. M. Pitet, A. H. M. van Loon, E. J. Kramer, C. J. Hawker and E. W. Meijer, ACS Macro Lett., 2013, 2, 1006–1010 CrossRef CAS PubMed.
- T. L. Sun, F. Luo, W. Hong, K. Cui, Y. Huang, H. J. Zhang, D. R. King, T. Kurokawa, T. Nakajima and J. P. Gong, Macromolecules, 2017, 50, 2923–2931 CrossRef CAS.
- K. J. Henderson and K. R. Shull, Macromolecules, 2012, 45, 1631–1635 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, X. Li, H. Guo, Y. Huang, H. Zhang and J. P. Gong, Polymer, 2017, 116, 487–497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2236596. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03086e |
| This journal is © The Royal Society of Chemistry 2023 |