
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct decarboxylative Giese amidations: photocatalytic vs. metal- and light-free†
David M.
Kitcatt
a,
Katie A.
Scott
a,
Elena
Rongione
a,
Simon
Nicolle
b and
Ai-Lan
Lee
 *a
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: A.Lee@hw.ac.uk
bGlaxoSmithKline, Gunnels Wood Rd, Stevenage SG1 2NY, UK
First published on 24th August 2023
Abstract
A direct intermolecular decarboxylative Giese amidation reaction from bench stable, non-toxic and environmentally benign oxamic acids has been developed, which allows for easy access to 1,4-difunctionalised compounds which are not otherwise readily accessible. Crucially, a more general acceptor substrate scope is now possible, which renders the Giese amidation applicable to more complex substrates such as natural products and chiral building blocks. Two different photocatalytic methods (one via oxidative and the other via reductive quenching cycles) and one metal- and light-free method were developed and the flexibility provided by different conditions proved to be crucial for enabling a more general substrate scope.
Introduction
Giese radical conjugate addition reactions have re-emerged at the forefront of radical chemistry as a powerful method for forming C–C bonds which are not otherwise attainable via conventional nucleophilic protocols.1,2 The current popularity of the Giese reaction is largely due to the recent emergence of mild photocatalytic methodologies.2 The Giese alkylation, for example, has been exploited in a myriad of applications, including chemoselective bioconjugation of peptides,3 synthesis of unnatural amino acids,4 macrocyclisations,5 polymerisations,6 natural product7 and drug molecule synthesis.8 Within this context, Giese reactions that can proceed via direct decarboxylation from carboxylic acids (rather than via less atom economical activated radical precursors),2a are highly sought after since carboxylic acids are readily available, non-toxic, easy to handle, atom economical and the carboxy group can be expelled as traceless CO2 from the reaction.9Although direct decarboxylative Giese alkylations10 and acylations11 have been well established, there are currently very few examples of Giese amidation reactions and crucially, no direct decarboxylative methods from oxamic acids are known.2a,12 Only two Giese amidation reactions were reported when we commenced our work, both from activated carbamoyl precursors.13 The seminal report by Konev and Wangelin utilised activated Hantzsch ester derivatives 1 as radical precursors under organophotocatalytic conditions (Scheme 1A).13a Although 1 has the advantage of being activated, it however results in poor atom economy. The substrate scope of the acceptor is also limited to highly activated ones, usually with two strong electron-withdrawing groups (EWGs, 2).
 | ||
| Scheme 1 Intermolecular Giese amidations. | ||
Conversely, Melchiorre's pioneering procedure using carbamoyl chlorides 3 as radical precursors used acceptors 4 with only one EWG, although the exemplified substrate scope appeared restrictive (4a–c, Scheme 1B).13b The use of moisture sensitive carbamoyl chlorides 3, some of which are carcinogenic, can also be problematic, since they are often made from highly toxic triphosgene.13b
While the above two approaches are important as they constitute the first two examples of Giese amidation, it is also clear that two major limitations exist. Firstly, the ease of use, toxicity, atom economy and accessibility related to the identity of the carbamoyl radical precursor (1, 3) needs to be improved significantly for Giese amidations to be synthetically useful and more widely adopted by the synthetic community. The use of oxamic acids 7 as an environmentally benign precursor to carbamoyl radicals14 would solve this issue, but there are currently no reports of its use in Giese reactions. Secondly, the acceptor substrate scope needs to be substantially expanded beyond the current limitation of requiring either two activating EWGs (2), phenyl vinyl sulfone, acrylonitrile, or dimethyl maleate (4). During the preparation of this manuscript, Kerr disclosed an elegant Giese amidation procedure from metal oxamates 5 (Scheme 1C).15 Kerr's procedure partly addresses some of the Michael acceptor scope limitations, however, the amidation scope seems to be limited to tertiary amides. Metal oxamates 5 are a significant improvement on precursor 3 in terms of toxicity, but oxamates 5 are still hygroscopic. The key challenges of a direct reaction from oxamic acids 7 and a more general substrate scope are therefore still pertinent.
We herein report the first direct decarboxylative Giese amidation reaction from bench stable, non-toxic and user friendly oxamic acids 7,14 which benefits from having only traceless CO2 released from the radical precursor (Scheme 1D). Crucially, a more general acceptor substrate scope 8 is now possible for the Giese amidation, which renders the reaction applicable to more complex substrates such as natural products and chiral building blocks. Three different conditions were developed and compared to ascertain the most suitable methodology: photocatalytic reductive quenching cycle (conditions A), metal- and light-free (conditions B), and photocatalytic oxidative quenching cycle (conditions C). The complementarity and flexibility provided by different conditions will prove to be crucial for enabling a more general substrate scope.
Results and discussion
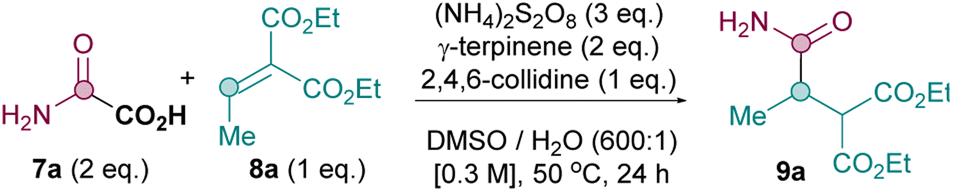
Our proposed mechanisms for the three sets of conditions are shown in Scheme 2. We initially adapted the conditions originally developed by Macmillan based on a reductive quenching cycle mechanism (conditions A),8 since this protocol has been used in a number of decarboxylative Giese alkylation and acylation reactions reported thereafter.2a In this reductive quenching cycle, the excited photocatalyst [e.g. *IrIII (E1/2*III/II = +1.21 V vs. SCE) for [Ir{dF(CF3)ppy}2(dtbpy)PF6]16 undergoes single electron transfer (SET) to yield the carboxylate radical from I (Eox = +1.17 V vs. SCE),17 which should then decarboxylate to form the carbamoyl radical II. Radical addition of II to 8 furnishes radical III. SET reduction by IrII (E1/2III/II = −1.37 V vs. SCE)16 to produce IV followed by protonation yields the Giese product 9. Unfortunately, it soon became apparent that adapting these Giese alkylation conditions for amidations was sub-optimal, yielding only a poor 34% of desired 9a with model substrates 7a and 8a (see later, Table 3). | ||
| Scheme 2 Proposed mechanisms. Conditions A: photocatalytic reductive quenching cycle. Conditions B: metal- and light-free thermal decarboxylation. Conditions C: photocatalytic oxidative quenching cycle. | ||
When reductive quenching cycle conditions failed to work in a key decarboxylative Giese alkylation step in Baran's synthesis of (−)-maximiscin, silver catalysis using oxidative Kochi conditions [Ag(I) and Na2S2O8] was ultimately utilised.7d For this reason, we decided to develop two oxidative methodologies in our effort to achieve the first efficient direct decarboxylative Giese amidations. Rather than using Kochi conditions, however, we set out to develop a metal- and light-free Giese method (conditions B), inspired by our recent success with metal- and light-free Minisci reactions.18 Using DMSO as the solvent allows for the breakdown of S2O82−V to the active SO4−·VI (Eox = +2.51–3.1 V vs. SHE)19 under mild conditions (40–50 °C), without the need for metal mediation or photolysis (Scheme 2).18,20 This could potentially be exploited in the Giese reaction, since SET between VI and carboxylate I (Eox = +1.17 V vs. SCE)17,21 can then occur to give radical VII,22 which should decarboxylate to give the carbamoyl radical II9a,23 for the Giese addition with 8. Unlike conditions A, radical III would presumably undergo hydrogen atom transfer (HAT) instead of SET/protonation to yield 9, due to the absence of an obvious reductant.
We also envisaged a related photocatalytic oxidative quenching cycle (conditions C, Scheme 2) where excited state *IrIII is generated from photoexcitation of the IrIII catalyst at 450 nm (E1/2Ir(III)*/Ir(IV) = −0.89 vs. SCE),24 which then undergoes SET with persulfate V (Eox = +1.75 V vs. SCE)20 to produce the oxidising species IrIV (E1/2Ir(IV)/Ir(III) = +1.69 vs. SCE).24 Subsequent SET with carboxylate I can either be induced by IrIV or the resulting sulfate radical anion VI (as in conditions B). Development of conditions C would allow the reaction to occur at ambient temperature as well as allow for a comparison between a photocatalytic oxidative (C) and reductive quenching cycle (A) for the Giese amidations.
We therefore commenced our optimisation of the metal- and light-free conditions B using model substrates 7a and 8a (Table 1). To our delight, the Giese amidation works very well as long as an efficient HAT source is present (entries 1 vs. entries 2–6),25 with 2 equiv. of γ-terpinene identified as optimal (entry 3). The presence of a base is required for good yields (entries 7–10), with 2,4,6-collidine providing the best results (entry 3). Persulfate is crucial for reactivity (entry 15), with (NH4)2S2O8 outperforming Na2S2O8 and K2S2O8 (entries 11–12), likely due to the former's superior solubility in DMSO. A solvent screen shows that the reaction requires DMSO for appreciable conversion (entries 3 vs. entries 16–19). The yield drops at lower temperature (35 °C, entry 20) and under air (entry 23). A control reaction in the dark proves that the reaction under conditions B is not light mediated (entry 22) and the reaction is inhibited in the presence of TEMPO (entry 24), consistent with a radical mechanistic pathway.
|
|
||
|---|---|---|
| Entry | Deviations from standard conditions | Yieldb (%) |
| a Reactions performed on a 0.12 mmol scale of 8a under Ar atmosphere. b Yields estimated by 1H NMR analysis of the crude mixture using dibromomethane as the internal standard. 1,4-CHD: 1,4-cyclohexadiene. N.d.: not detected. See ESI† for full optimisation studies. | ||
| 1 | No γ-terpinene | 36 |
| 2 | 1,4-CHD instead of γ-terpinene | 65 |
| 3 | None | 96 |
| 4 | 1 eq. of γ-terpinene | 79 |
| 5 | 3 eq. of γ-terpinene | 73 |
| 6 | Hantzsch ester instead of γ-terpinene | 83 |
| 7 | Cs2CO3 instead of 2,4,6-collidine | 65 |
| 8 | K2HPO4 instead of 2,4,6-collidine | 86 |
| 9 | 2,6-Lutidine instead of 2,4,6-collidine | 84 |
| 10 | No 2,4,6-collidine | 55 |
| 11 | Na2S2O8 instead of (NH4)2S2O8 | 14 |
| 12 | K2S2O8 instead of (NH4)2S2O8 | 23 |
| 13 | 1 eq. of (NH4)2S2O8 | 72 |
| 14 | 5 eq. of (NH4)2S2O8 | 62 |
| 15 | No (NH4)2S2O8 | n.d. |
| 16 | H2O used as solvent | 26 |
| 17 | Acetone used as solvent | n.d. |
| 18 | DMF used as solvent | 9 |
| 19 | MeCN used as solvent | 14 |
| 20 | At 35 °C | 38 |
| 21 | At 80 °C | 99 |
| 22 | In the dark | 97 |
| 23 | Under air | 79 |
| 24 | With 3 eq. TEMPO | n.d. |
For the photocatalytic oxidative quenching cycle conditions C, optimisation studies showed that [Ir{dF(CF3)ppy}2(dtbpy)PF6] catalyst at 1 mol% loading yielded the best results (Table 2, entry 1, see ESI† for full optimisation studies). The Fukuzumi organophotocatalyst 9-mesityl-10-methylacridinium perchlorate26 gave a slightly lower yield (entry 2) but is a good alternative to the Ir catalyst should cost, toxicity and sustainability of the Ir catalyst be an issue. Control experiments prove that a HAT source (entry 3), persulfate (entry 4), photocatalyst (entry 5), base (entry 6) and light (entry 7) are all required for good reactivity under photocatalytic conditions C.
|
|
||||
|---|---|---|---|---|
| a Reactions performed on a 0.12 mmol scale of 8a under Ar atmosphere in a Penn PhD M2 Photoreactor, 450 nm at 50% light intensity. b Yields estimated by 1H NMR analysis of the crude mixture using 1,3,5-trimethoxybenzene as the internal standard. [Ir] = [Ir{dF(CF3)ppy}2(dtbpy)PF6]. See ESI† for full optimisation studies. | ||||
| Entry | Photocat. | Mol% | Deviations | Yieldb (%) |
| 1 | [Ir] | 1 | — | 92 |
| 2 | [Mes–Acr]+[ClO4]− | 1.5 | — | 86 |
| 3 | [Ir] | 2 | 100% intensity; no γ-terpinene | 13 |
| 4 | [Ir] | 1 | No (NH4)2S2O8 | 22 |
| 5 | None | 0 | No photocat. | 17 |
| 6 | [Ir] | 1 | No collidine | 51 |
| 7 | [Ir] | 1 | In dark | 15 |

In addition, the average quantum yield27 (Φ) was found to be 2.83 × 10−3 (std. dev. = 0.69 × 10−3) for conditions A and 11.5 × 10−3 (std. dev. = 8.8 × 10−3) for conditions C (see ESI†), thus ruling out the presence of any chain reactions under these conditions.
With optimal conditions in hand, an oxamic acid 7 substrate scope study was carried out next (Table 3). When comparing conditions A, B and C for amidation with 7a to form primary amide 9a, the metal- and light-free conditions B were superior to both photocatalytic methods A and C (A: 34%, B: 75%, C: 63%).
| a Reactions performed on a 0.12 mmol scale of 8 under argon atmosphere and isolated yields reported unless otherwise stated. Conditions A and C were carried out in a Penn PhD M2 Photoreactor, 450 nm at 50% light intensity. [Ir-cat] = [Ir{dF(CF3)ppy}2(dtbpy)PF6]. b Yield determine 1H NMR using dibromomethane as internal standard. c 72% yield at 1 mmol scale and 55% yield at 5.4 mmol scale. d Reaction performed on a 0.20 mmol scale. e Used 3 eq. of 7, 4 eq. of (NH4)2S2O8, 3 eq. of 2,4,6-collidine at 75 °C. f Reaction performed on a 0.24 mmol scale. g Used 4 eq. of 7 and 2.4 eq. of K2HPO4. Yield was 38% under standard conditions. h Reacted for 48 h at 40 °C. |
|---|

|
A similar pattern was observed for installing secondary amides: oxidative conditions (either B or C) generally outperformed reductive quenching cycle conditions A for 9c (A: 67%, B: 85%, C: 71%), 9g (A: 87%, B: 88%, C: 68%), 9k (A: 47%, B: 79%, C: 57%) and 9n (A: 50%, B: 74%, C: 82%). For this reason, only conditions B and C were investigated for the formation of the rest of the secondary amides shown in Table 3. Various aliphatic substituents on the nitrogen were tolerated well (9b–j, 63–88%), including primary alkyl (9b–d), secondary alkyl (9e–h) and tertiary alkyl (9i–j) substituents. Pleasingly, these include cyclic N-alkyl substituents (9f–h, 9j) as well as alkyl substituents with CF3 (9c 85%) and benzyls (9d 81%, 9e 76%). N-Aryl substituents were also tolerated (9k–o), with electron-rich aryls (9l–n, 69–82%) performing better than electron-poor ones (9o, 49%).28 This trend reflects the lower nucleophilicity of the resulting carbamoyl radical with electron-withdrawing substituents.
In general, for the synthesis of secondary amides, oxidative conditions B and C both performed well. The inferior yields under conditions A in these cases are likely due to significant formation of unwanted formamide (RR′NCHO 11) side products compared to conditions B (e.g.1H NMR analysis of the crude mixture for 9c shows ∼10% formamide 11 under conditions A vs. >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 9:11 under conditions B).
:1 9:11 under conditions B).
The Giese amidation reaction could also be scaled up using conditions B to yield appreciable amounts of 9d, albeit with a slight drop in yield with each 5 to 8-fold increase. Product 9d was successfully formed in 72% yield at 1 mmol scale and a still synthetically useful 55% at gram (5.4 mmol) scale.29
Next, the synthesis of tertiary amides was investigated. As shown in Scheme 3A, standard oxidative conditions B and C using oxamic acid 7b surprisingly gave the dealkylated product 9b instead of the desired tertiary amide 9p as the major product. Subjecting product 9p to reaction conditions B did not result in 9b, thus ruling out dealkylation from the desired Giese products (see ESI†).30 Instead, we postulated that upon the conjugate addition of II to 8 to form IIIa (Scheme 2), 1,5-HAT31 could occur to give VIII (Scheme 3B). SET of VIII and hydrolysis of the corresponding iminium32IX could yield the dealkylated product 9b (Scheme 3B).33
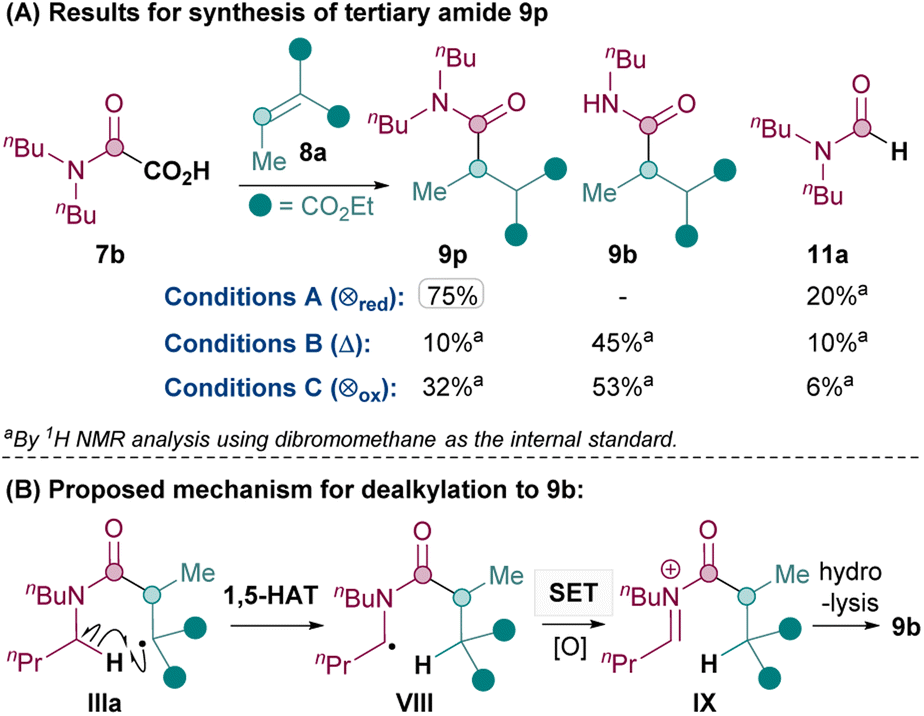 | ||
| Scheme 3 Dealkylation observed with conditions B and C for synthesis of tertiary amide. | ||
Since the formation of undesired 9b requires an oxidation (VIII to IX), it was thought that exploiting the reductive quenching catalytic cycle (conditions A) should prevent the formation of 9b. Pleasingly, this hypothesis proved to be correct and conditions A successfully yielded 9p in 75% yield (Scheme 3A). It should be noted that the dealkylated side products such as 9b under oxidative conditions are only observed with tertiary amides and not secondary amides. The formation of the formamide side product 11 (from radical II) though, is generally much more prevalent with conditions A (e.g. 20% 11a and also 31% of the corresponding formamide was isolated along with 9q) than with standard oxidative conditions B and C (e.g. 10% 11a).
Thus, the Giese amidation formed tertiary amides 9p, 9q and 9r successfully in 75%, 53% and 51% respectively using reductive quenching cycle conditions A (Table 3). The amidation seemed sensitive to sterics, with 9s and 9t being formed in a moderate 38% and 41% yield respectively, although the yield of 9t was successfully improved to 55% upon more forcing conditions. Cyclic tertiary amides were produced in only moderate yields with both conditions A and B (35% and 31% 9u).34
Next, the Michael acceptor scope was investigated (Table 4). Since the model oxamic acid 7 chosen gave significantly better yields under conditions B in Table 3 (9k), a result that is further confirmed by direct comparison of conditions A, B and C for producing 9z, 9al and 9am, conditions B were therefore utilised for the rest of the Michael acceptor scope. Activated Michael acceptors with two electron-withdrawing groups performed well, as expected, to give 9k, 9v–x in 56–79% yields. The production of 9x from a coumarin derivative was an exception where conditions A performed better, due to competitive oxidative rearomatisation under conditions B (see ESI†). To our delight, Michael acceptors with only one electron-withdrawing group were also suitable substrates, including cyclic acceptors such as cyclopentenone (9y, 64%), cyclohexanone (9z, 75%), cycloheptanone (9aa, 74%), butenolide (9ab, 57%), pentenolide (9ac, 47%) and α,β-unsaturated amide (9ad, 54%). A substituent in the α-position of cyclohexanone was also tolerated (9ae, 59%), although the lower yield for 9af (31%) indicates that the reaction was sensitive to the alkene moiety in carvone. Cyclic Michael acceptors with the electron-withdrawing group exo to the ring can also be utilised (9ag, 58% and 9ah, 55%). Other acyclic acceptors reacted smoothly including diethyl maleate and diethyl fumarate, giving product 9ai in good yields (89% and 77% respectively). The EWGs need not be carbonyls, for example, diethyl vinylphosphonate and vinyl sulfones were also good substrates, furnishing 9aj, 9ak and 9al in 64%, 76% and 61% yields respectively. Nevertheless, a current limitation is that acyclic ketones such as 9am seem to react with more moderate yields (34%).35

The Michael acceptor substrate scope has thus been significantly expanded compared to previous methods (2 and 4, Scheme 1). In particular, the ease of reaction with many endocyclic acceptors (e.g.9x–9ah) for the first time renders the Giese amidation applicable to various natural products and building blocks with such motifs.
Thus, the Giese amidation was successfully applied to amino acids, natural products and chiral building blocks (Table 5).36 Oxamic acids of alanine and valine reacted smoothly to give 9an and 9ao in 79% and 64% yields respectively, with conservation of enantiopurity for 9ao (see ESI†). More complex amines such as the natural product leelamine can also be introduced via the Giese amidation in good yield (9ap, 65%). The reaction can also be applied to amidate Michael acceptor natural product cryptone (9aq, 67%) and a common chiral building block37 (9ar, 51%). Finally, in order to challenge the system further, an attempt was made to combine a complex amine with a complex Michael acceptor. Despite the challenge, 9as was successfully formed in 35% yield.
| a Reactions performed on a 0.12 mmol scale of 7 under argon atmosphere unless otherwise stated. b Reaction performed on a 0.24 mmol scale.; no racemisation of stereogenic centre observed by CSP-HPLC. c Reaction performed on a 0.11 mmol scale. d Yield determined by 1H NMR analysis using dibromomethane as internal standard. ⊗ox = Cond. C. e 3 eq. of 8, 3 eq. of 2,4,6-collidine and 4 eq. (NH4)2S2O8, 24 h at 50 °C and 24 h at 75 °C. Yield was 25% under standard conditions. |
|---|

|
Conclusions
We have successfully developed the first direct Giese amidation reaction from oxamic acids 7, which benefits from having a significantly better substrate scope compared to previously reported Giese amidation methods. Crucially, the ability to use the bench stable, non-toxic and environmentally benign oxamic acids 7 as the carbamoyl precursor directly for the first time greatly improves the practicality of the Giese amidation. The significantly expanded Michael acceptor substrate scope, especially the applicability of endocyclic Michael acceptors for the first time, now renders the Giese amidation applicable to natural products and chiral building blocks.Three different conditions were developed and compared: photocatalytic reductive quenching cycle (conditions A), metal- and light-free (conditions B) and photocatalytic oxidative quenching cycle (conditions C). The methods were found to be complementary, with the flexibility provided by different conditions allowing for a more general substrate scope.
Data availability
RAW NMR data, HRMS and IR spectra available at: DOI: 10.17861/50eb4ef7-ce19-4ac4-952d-76b7c96386c3.Author contributions
DMK performed the bulk of the experiments and analysed the data. KAS and ER both assisted with optimisation studies and/or part of the substrate scope studies. A-LL conceived and supervised the research. SN co-supervised the research. A-LL and DMK co-wrote the manuscript, with input from SN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council and GSK for financial support [Industrial CASE PhD studentship to DMK; Grant code: EP/V519522/1] and Heriot-Watt University for an ICS undergraduate summer project bursary for ER. We thank undergraduate project student Lennox Stewart for experimental assistance with oxamic acids for 9an–9ao and Georgina M. Rosair for X-ray crystallography.References
- For recent review on radical chemistry, see: M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- For recent reviews on Giese reactions, see: (a) D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC; (b) A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed.
- (a) K. Merkens, F. J. Aguilar Troyano, K. Anwar and A. Gómez-Suárez, J. Org. Chem., 2021, 86, 8448–8456 CrossRef CAS PubMed; (b) O. Zhang and J. W. Schubert, J. Org. Chem., 2020, 85, 6225–6232 CrossRef CAS PubMed; (c) P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557–1562 CrossRef CAS PubMed; (d) A. A. Shah, M. J. Kelly, III and J. J. Perkins, Org. Lett., 2020, 22, 2196–2200 CrossRef CAS PubMed.
- S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed.
- M. Yamawaki, A. Ukai, Y. Kamiya, S. Sugihara, M. Sakai and Y. Yoshimi, ACS Macro Lett., 2017, 6, 381–385 CrossRef CAS PubMed.
- (a) S. Inuki, K. Sato, T. Fukuyama, I. Ryu and Y. Fujimoto, J. Org. Chem., 2017, 82, 1248–1253 CrossRef CAS PubMed; (b) J. C. DeForest, R. A. Samame, G. Suryn, A. Burtea and S. D. Rychnovsky, J. Org. Chem., 2018, 83, 8914–8925 CrossRef CAS PubMed; (c) K. Minagawa, D. Kamakura, K. Hagiwara and M. Inoue, Tetrahedron, 2020, 76, 131385 CrossRef CAS; (d) K. S. McClymont, F.-Y. Wang, A. Minakar and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 8608–8613 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- (a) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (b) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (c) L. Li, Y. Yao and N. Fu, Eur. J. Org Chem., 2023, 26, e202300166 CrossRef CAS.
- For representative examples, see ref. 4–8 and: (a) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854–7856 RSC; (b) D. W. Manley, R. T. McBurney, P. Miller, J. C. Walton, A. Mills and C. O'Rourke, J. Org. Chem., 2014, 79, 1386–1398 CrossRef CAS PubMed; (c) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed; (d) J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 RSC; (e) A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464–13468 CrossRef CAS PubMed; (f) N. P. Ramirez and J. C. Gonzalez-Gomez, Eur. J. Org Chem., 2017, 2017, 2154–2163 CrossRef CAS; (g) A. Gualandi, E. Matteucci, F. Monti, A. Baschieri, N. Armaroli, L. Sambri and P. G. Cozzi, Chem. Sci., 2017, 8, 1613–1620 RSC; (h) S. Zhang, Z. Tan, H. Zhang, J. Liu, W. Xu and K. Xu, Chem. Commun., 2017, 53, 11642–11645 RSC; (i) J.-Q. Chen, R. Chang, Y.-L. Wei, J.-N. Mo, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2018, 83, 253–259 CrossRef CAS PubMed; (j) Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS PubMed; (k) T. Guo, L. Zhang, Y. Fang, X. Jin, Y. Li, R. Li, X. Li, W. Cen, X. Liu and Z. Tian, Adv. Synth. Catal., 2018, 360, 1352–1357 CrossRef CAS; (l) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed; (m) F. El-Hage, C. Schöll and J. Pospech, J. Org. Chem., 2020, 85, 13853–13867 CrossRef CAS PubMed; (n) L. Gingipalli, J. Boerth, D. Emmons, T. Grebe, H. Hatoum-Mokdad, B. Peng, L. Sha, S. Tentarelli, H. Wang, Y. Wu, X. Zheng, S. Edmondson and A. Gopalsamy, Org. Lett., 2020, 22, 3418–3422 CrossRef CAS PubMed; (o) X. Chen, X. Luo, X. Peng, J. Guo, J. Zai and P. Wang, Chem.–Eur. J., 2020, 26, 3226–3230 CrossRef CAS PubMed; (p) H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 CrossRef CAS PubMed; (q) Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- For representative examples, see: (a) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed; (b) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed; (c) J.-J. Zhao, H.-H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed; (d) D.-L. Zhu, Q. Wu, D. J. Young, H. Wang, Z.-G. Ren and H.-X. Li, Org. Lett., 2020, 22, 6832–6837 CrossRef CAS PubMed.
- For selected “interrupted Giese” reactions whereby radical VI participates in other pathways instead of HAT or SET/protonation, see: (a) Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed; (b) G. Chen, C. Li, J. Peng, Z. Yuan, P. Liu and X. Liu, Org. Biomol. Chem., 2019, 17, 8527–8532 RSC; (c) Z. Zhang, C. Jia, X. Kong, M. Hussain, Z. Liu, W. Liang, L. Jiang, H. Jiang and J. Ma, ACS Sustain. Chem. Eng., 2020, 8, 16463–16468 CrossRef CAS.
- (a) L. Cardinale, M. O. Konev and A. Jacobi von Wangelin, Chem.–Eur. J., 2020, 26, 8239–8243 CrossRef CAS PubMed; (b) E. de Pedro Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Chem. Sci., 2020, 11, 6312–6324 RSC.
- For a recent review, see: I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 7593–7607 RSC.
- J. D. Williams, S. G. Leach and W. J. Kerr, Chem.–Eur. J., 2023, 29, e202300403 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- (a) X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed ; see also:; (b) I. M. Ogbu, J. Lusseau, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2020, 56, 12226–12229 RSC.
- (a) D. R. Sutherland, M. Veguillas, C. L. Oates and A.-L. Lee, Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed; (b) M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Org. Lett., 2019, 21, 7119–7123 CrossRef CAS PubMed; (c) D. T. Mooney, B. D. T. Donkin, N. Demirel, P. R. Moore and A.-L. Lee, J. Org. Chem., 2021, 86, 17282–17293 CrossRef CAS PubMed; (d) D. T. Mooney, P. R. Moore and A.-L. Lee, Org. Lett., 2022, 24, 8008–8013 CrossRef CAS PubMed.
- C. Liang, I. L. Lee, I. Y. Hsu, C.-P. Liang and Y.-L. Lin, Chemosphere, 2008, 70, 426–435 CrossRef CAS PubMed.
- C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425–4431 CrossRef CAS PubMed.
- (a) G. Liu, S. You, Y. Tan and N. Ren, Environ. Sci. Technol., 2017, 51, 2339–2346 CrossRef CAS PubMed; (b) R. C. Thompson, Inorg. Chem., 1981, 20, 1005–1010 CrossRef CAS.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Green Chem., 2019, 21, 748–764 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- We have previously shown that the base 2,4,6-collidine can act as a HAT source in the absence of γ-terpinene or 1,4-CHD, albeit a less efficient one. See: E. B. McLean, D. T. Mooney, D. J. Burns and A.-L. Lee, Org. Lett., 2022, 24, 686–691 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- The lower yields for 9o is due to poorer conversions..
- The decrease in yield reflects a decrease in conversion, the remaining mass return is recovered starting material..
- An alternative mechanism involving direct HAT of 7b or 9p (at the position α-to N) by SO4−˙ (for example, see ref. 30a and b) was thus ruled out by this control experiment. (a) J. Kaur, A. Shahin and J. P. Barham, Org. Lett., 2021, 23, 2002–2006 CrossRef CAS PubMed; (b) J. Zhou, Q. Ren, N. Xu, C. Wang, S. Song, Z. Chen and J. Li, Green Chem., 2021, 23, 5753–5758 RSC.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974 RSC.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- In an attempt to increase the desired intermolecular HAT of IIIavs. the undesired intramolecular 1,5-HAT of IIIa, the equivalents of external HAT source γ-terpinene were increased and different HAT sources were evaluated, but the yields for 9p were still poor (see ESI†).
- The moderate yields for 9s–9u are due to poor conversions..
- More reactive acyclic acceptors such as acrylonitrile and methyl acrylate formed a complex mixture of products with <20% desired product.
- Since compounds in Table 5 are secondary amides, we opted for oxidative conditions, as we had observed from Table 3 that these were generally higher yielding.
- Y. H. Jin, P. Liu, J. Wang, R. Baker, J. Huggins and C. K. Chu, J. Org. Chem., 2003, 68, 9012–9018 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full optimisation studies, mechanistic studies, characterisation data and copies of NMR spectra of new compounds. CCDC 2258061. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03143h |
| This journal is © The Royal Society of Chemistry 2023 |