
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiexciton quintet state populations in a rigid pyrene-bridged parallel tetracene dimer†
Liang-Chun
Lin
a,
Tanner
Smith
b,
Qianxiang
Ai
b,
Brandon K.
Rugg
 c,
Chad
Risko
b,
John E.
Anthony
b,
Niels H.
Damrauer
ad and
Justin C.
Johnson
*cd
c,
Chad
Risko
b,
John E.
Anthony
b,
Niels H.
Damrauer
ad and
Justin C.
Johnson
*cd
aDepartment of Chemistry, University of Colorado Boulder, Boulder, CO 80309, USA
bDepartment of Chemistry & Center for Applied Energy Research, University of Kentucky, Lexington, Kentucky 40506-0055, USA
cNational Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401, USA. E-mail: justin.johnson@nrel.gov
dRenewable and Sustainable Energy Institute (RASEI), University of Colorado Boulder, Boulder, CO 80309, USA
First published on 2nd October 2023
Abstract
The multiexciton quintet state, 5TT, generated as a singlet fission intermediate in pairs of molecular chromophores, is a promising candidate as a qubit or qudit in future quantum information science schemes. In this work, we synthesize a pyrene-bridged parallel tetracene dimer, TPT, with an optimized interchromophore coupling strength to prevent the dissociation of 5TT to two decorrelated triplet (T1) states, which would contaminate the spin-state mixture. Long-lived and strongly spin-polarized pure 5TT state population is observed via transient absorption spectroscopy and transient/pulsed electron paramagnetic resonance spectroscopy, and its lifetime is estimated to be >35 µs, with the dephasing time (T2) for the 5TT-based qubit measured to be 726 ns at 10 K. Direct relaxation from 1TT to the ground state does diminish the overall excited state population, but the exclusive 5TT population at large enough persistent density for pulsed echo determination of spin coherence time is consistent with recent theoretical models that predict such behavior for strict parallel chromophore alignment and large exchange coupling.
Introduction
Singlet fission (SF) is a process in which the photoexcited singlet (S1) transforms into two low-energy triplets (T1), and it has been widely investigated to improve the photovoltaic conversion efficiency.1–5 The singlet-coupled triplet pair, 1TT, is a spin-entangled biexciton state that forms in every SF reaction via internal conversion from S1. For the past decade or so, researchers have been focusing on generating maximum decorrelated T1 through 1TT for solar cell applications.4,6 Here, in contrast, we prevent 1TT from decaying to T1 and drive the intersystem crossing from 1TT to 5TT manifolds in a highly state-selective fashion for quantum information science (QIS) applications by designing and synthesizing a parallel rigid organic dimer. The SF-born 5TT manifold is regarded as a novel platform to make a quantum bit, or qubit, due to its strong spin polarization via optical excitation and the ability to perform fast quantum gate operations with microwave pulses.7 As an S = 2 manifold, it also has the potential to operate in a qudit scheme. Furthermore, results from time-resolved electron paramagnetic resonance spectroscopy (TREPR), and pulsed EPR in both monomer thin films8–11 and organic dimers12–16 have verified 5TT formation and laid the groundwork for understanding the spin evolution.Considering the angular momentum coupling between the triplets in (TT) states, nine possible (TT) states can be constructed, including 1TT (one spin sublevel), 3TT (three spin sublevels) and 5TT (five spin sublevels). The state selectivity from 1TT to 5TT requires symmetry of the spin-exciton Hamiltonian, H = JSA·SB + HA + HB, where J represents isotropic exchange energy between the spin on each chromophore, and HA,B are the anisotropic terms for dipolar interaction within chromophore A and B, 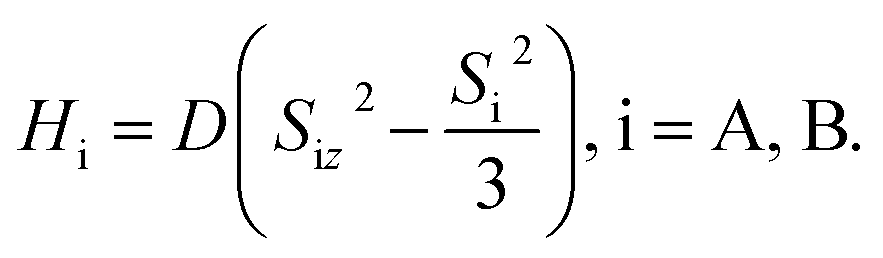 The fine–structure interaction, governing the interconversion between the nine states, perturbs the spin-exciton Hamiltonian and makes the eigenstates of the Hamiltonian not 2S+1TTM (−S≤M≤S) states but the combination of them. However, under conditions of large J, these interactions are typically negligible. The parallel JDE model7 of the spin-exciton Hamiltonian assumes the two chromophores to be identical and parallel to share the same coordinate axis. Under these conditions, the conversion from 1TT to 5TT while isolating 3TT is guaranteed by the Hamiltonian symmetry. Also, based on the parallel JDE model selection rules only the transition from 1TT to 5TT0 (ms = 0) and 5TT±2 (ms = ±2) are allowed when the external magnetic axis and the molecular axis are aligned. Further, the relative kinetics of the 1TT → 5TT0 and 5TT±2 transitions are proportional to the zero-field parameters D2 and E2, respectively, leaving the 5TT0 pathway as dominant because D ≥ 3E by definition. All accessible dimer geometries must be included in this analysis, and for flexible systems, particularly about the bridge, significant conformational diversity can result. This motivates the use of both chromophore and bridge with limited flexibility to simplify the spin evolution pathways.
The fine–structure interaction, governing the interconversion between the nine states, perturbs the spin-exciton Hamiltonian and makes the eigenstates of the Hamiltonian not 2S+1TTM (−S≤M≤S) states but the combination of them. However, under conditions of large J, these interactions are typically negligible. The parallel JDE model7 of the spin-exciton Hamiltonian assumes the two chromophores to be identical and parallel to share the same coordinate axis. Under these conditions, the conversion from 1TT to 5TT while isolating 3TT is guaranteed by the Hamiltonian symmetry. Also, based on the parallel JDE model selection rules only the transition from 1TT to 5TT0 (ms = 0) and 5TT±2 (ms = ±2) are allowed when the external magnetic axis and the molecular axis are aligned. Further, the relative kinetics of the 1TT → 5TT0 and 5TT±2 transitions are proportional to the zero-field parameters D2 and E2, respectively, leaving the 5TT0 pathway as dominant because D ≥ 3E by definition. All accessible dimer geometries must be included in this analysis, and for flexible systems, particularly about the bridge, significant conformational diversity can result. This motivates the use of both chromophore and bridge with limited flexibility to simplify the spin evolution pathways.
In this work, the rigid parallel organic dimer consists of two silylacetylene-substituted tetracene chromophores bridged with pyrene, TPT, and the structure can be seen in Fig. 2b. A similar dimer, APA, where two silylacetylene-substituted anthracenes are bridged with pyrene, has previously been characterized with UV-Vis and computational chemistry analysis.17 Electronic coupling between the two chromophores is weak, yet the interaction between the anthracenic moiety and the pyrene bridge is strong, making the S1 energy of the chromophore to be between that of silylacetylene-substituted anthracene and tetracene. For TPT, the properties of the tetracene moiety can also be tuned by the pyrene bridge to mimic a combination of silylacetylene-substituted tetracene and pentacene. We will demonstrate that the mixed nature of the chromophore excited state facilitates the formation and preservation of 5TT. This stands in contrast to other flexible-bridged pentacenic dimer systems wherein independent triplets are the primary long-lived product (whether directly from 5TT or via intersystem crossing from 3TT), rather than 5TT as the surviving species.13,14,18,19 Another rigid-bridged pentacene dimer was recently reported to almost exclusively form 5TT0, but the apparent yield was low.12 Our results reveal the first non-pentacene dimer to form a high yield of exclusively 5TT that persists to µs.
Results
Synthesis of TPT
Shown in Fig. 1 and S1,† the synthetic routes to TPT and PT involve intermediates 1 and 3, which have previously been reported.17 Reaction of 1 with 1,4-anthraquinone and KI yields the pyrenebis(tetracenequinone) 2. The addition of lithiated n-octyldiisopropylsilyl (NODIPS) acetylene followed by the addition of tin(II) chloride in aqueous HCl gives TPT. The synthesis of PT likewise consisted of the addition of 1,4-anthraquinone to 3 to yield pyrene-tetracenequinone 4, followed by addition of lithiated NODIPS-acetylene. Removal of Br from the diol intermediate 5 was initially attempted using a Pd catalyst at 100 °C, however these conditions resulted in apparent decomposition of the starting material and none of the target product was recovered. Debromination of 5 was ultimately achieved through metal–halogen exchange at low temperature, and subsequent addition of aq. HCl and tin(II) chloride to yield the aromatized product PT.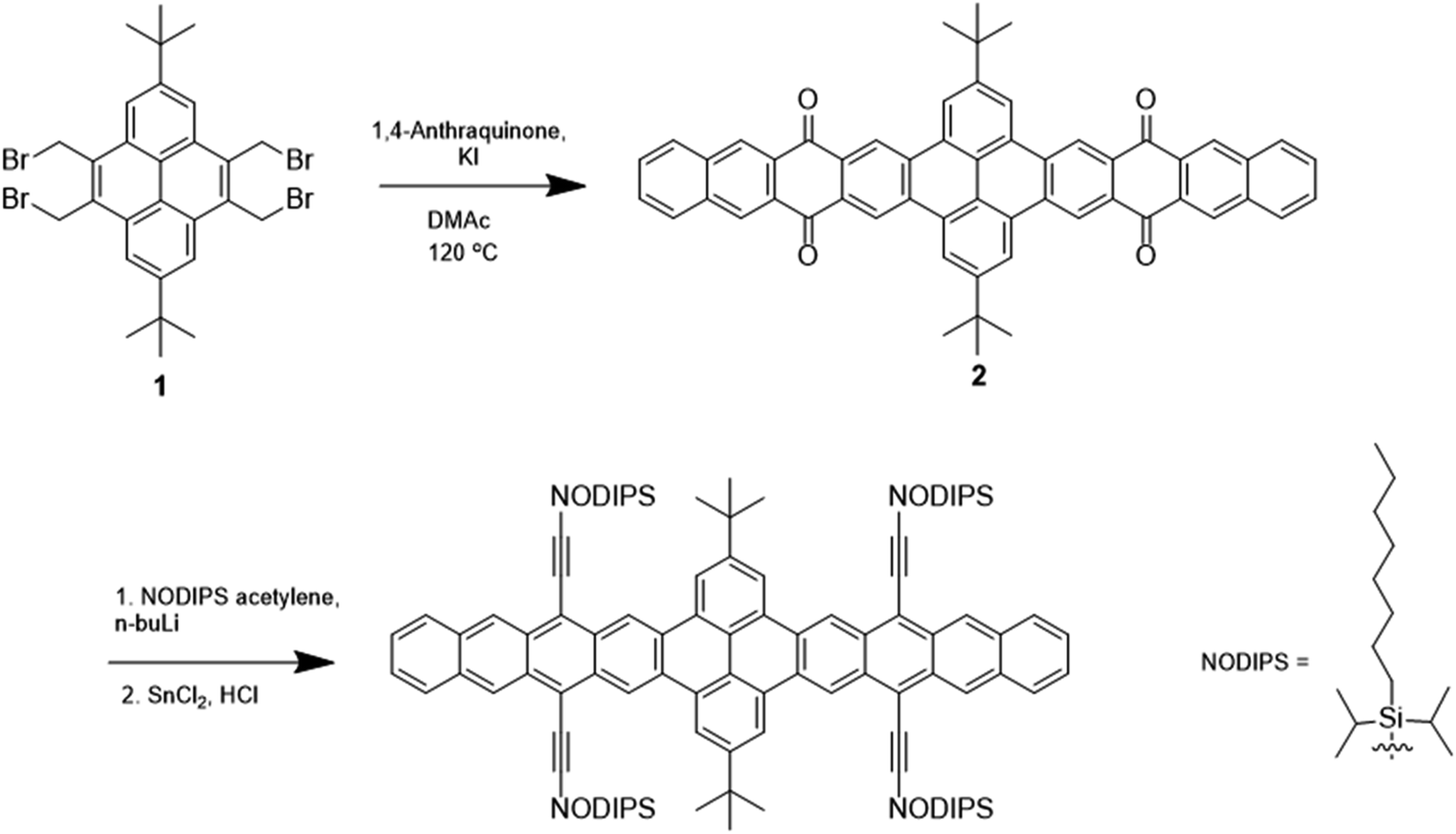 | ||
| Fig. 1 Synthetic scheme for TPT. | ||
Optical spectra and origin of transitions
Fig. 2a shows molar extinction spectra for PT and TPT. To the red, the vibronic peaks ranging from 500 nm to 625 nm are attributed to the short axis polarized S1 ← S0 transition of the tetracene moieties. The peak for the S1 ← S0 0–0 vibronic transition of PT is centered at 603 nm, located in between that of TIPS-tetracene and TIPS-pentacene,20,21 measured to be at 530 nm and 638 nm, respectively. The ∼70 nm redshift from TIPS-tetracene and ∼40 nm blueshift from TIPS-pentacene for PT (0.28 eV and 0.11 eV, respectively)20 result from the limited extension of the acene system.17 Comparing PT and TPT, as seen in Fig. 2a, for the S1 ← S0 transition, TPT has extinction coefficient ∼2 times that of PT. The fact that the extinction coefficient of a dimer is 2 times of the monomer suggests that the TPT is in a weakly coupled regime in terms of the interaction between the two chromophore units. This is further substantiated by the only ∼2 nm redshift for 0–0 transition from PT to TPT. Furthermore, the ratio of 0–0 to 0–1 absorption/emission intensity (I0–1/I0–0) for TPT and PT are both ∼1.4 as can be seen in Fig. S7.† However, for the long-axis S3 ← S0 transition of the chromophores around 315 nm, the extinction coefficient of TPT is less than two times of PT, suggesting some perturbation of higher-lying excited states. The extinction coefficient of the expected pyrene peaks in TPT is enhanced by roughly 5 times from the pyrene itself and from PT. | ||
| Fig. 2 Molar extinction spectra for both PT (blue line) and TPT (red line) in toluene. The molar extinction spectrum for pyrene in cyclohexane is in yellow. | ||
The vibronic peaks for PT and TPT within the 350 nm to 425 nm window, as indicated by TDDFT calculations, originate from a singlet excitation with contributions from both pyrene and tetracene segments. In PT, this excitation has two similarly weighted natural transition orbital (NTO) pairs that, as shown in Fig. S5a,† resemble tetracene HOMO → pyrene LUMO and pyrene HOMO → tetracene LUMO, respectively. As a result, the net charge transfer from pyrene to tetracene, compared to charge redistribution within these fragments, is negligible. This is confirmed by the two-fragment charge transfer matrix (Fig. S5b†) in which off-diagonal (diagonal) elements represent inter-fragment charge transfer (charge redistribution). The transition densities of this excitation are shown in Fig. S5c† with a transition dipole moment of 3.82 a.u. polarized along the long-axis (the directions along which the transition density switches its sign). This is in contrast to the vibronic peaks from 500 nm to 625 nm (S1 ← S0) that result from the local excitation of tetracene with a transition dipole moment polarized along the short-axis.15
Similar to PT, the excitation responsible for vibronic peaks from 350 nm to 425 nm in TPT is polarized along the long axis (Fig. S5f†). The NTOs (Fig. S5d†) also largely resemble frontier molecular orbitals of pyrene and tetracene, featuring bidirectional charge transfer from the pyrene segment to both tetracene segments. This is corroborated by the three-fragment charge transfer matrix (Fig. S5e†) in which one-direction charge transfer contributions between pyrene and tetracene fragments largely cancel each other, resulting in a small net charge transfer from pyrene to tetracene fragments. The addition of another tetracene segment leads to an increased transition dipole moment of 7.80 a.u. for this excitation, approximately double of that in PT. This is in good agreement with the experimentally observed extinction coefficient ratio (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) between TPT and PT.
:1) between TPT and PT.
The associated peaks for pyrene in cyclohexane22 are measured to be from 300 nm to 350 nm. The peak centered ∼315 nm for PT and TPT corresponds to the S3 ← S0 transition of the tetracene moieties. NTOs of this excitation of PT are shown in Fig. S6a.† The two equally weighted NTO pairs resemble pentacene HOMO−2 → pentacene LUMO and pentacene HOMO → pentacene LUMO+2, respectively, as the extension of the acene system with the pyrene bridge mimics a pentacene unit. This is primarily a local excitation at the acene fragment, as indicated by the large diagonal element in its fragment charge transfer matrix (Fig. S6b†), with moderate bidirectional charge transfer yielding a small net charge transfer from tetracene to pyrene. Fig. S6c† shows the transition densities with a dipole moment of 4.45 a.u. along the long axis. Both NTOs and transition densities are similar to that of the 1Bb state of pentacene.23
The situation is more complicated for TPT. As shown in Fig. S6d,† another NTO pair (the third one), resembling pyrene HOMO → tetracene LUMO, appears with non-negligible contributions. Similar contributions were made to the hole and electron densities from the three fragments, leading to small differences among elements in the fragment charge transfer matrix (Fig.S6e†). Unlike the case in the previous section, where transition densities of TPT are roughly that of PT duplicated by a mirror plane along the 2, 7 positions of pyrene, transition densities of this excitation (Fig. S6f†) cannot be produced as such, likely due to the aforementioned additional NTO pair. The transition dipole moment is calculated to be 4.20 a.u. which is similar to that of PT, agreeing with the similar extinction coefficients for PT and TPT observed experimentally.
Davydov splitting, caused by interchromophore interaction of transition dipoles in a singlet fission dimer, should also be mentioned, especially for the rigidly bridged dimer. Davydov splitting for both S1 ← S0 and S3 ← S0 transitions have been observed from both norbornyl-bridged tetracene and pentacene dimers.20,21 For TPT, the two tetracene chromophores are held firmly at 180° from each other to form a planar dimer. Due to the flat structure of the molecule and the parallel nature of the two tetracene chromophores, the Davydov splitting for long-axis (S3 ← S0) and short-axis (S1 ← S0) transitions are not observed in Fig. 2a.
Photoluminescence
In Fig. S7 and S8,† S0 ← S1 0–0 emission peaks for TPT and PT are found to be 2.04 eV and 2.05 eV, respectively. Similar S1 energies for dimer and monomer suggests minimal electronic delocalization between the two tetracenic chromophores for TPT. However, the emission quantum yield Φemifor TPT is significantly smaller than PT, measured to be 6% and 100%, respectively. Photoluminescence kinetics for PT, obtained via time-correlated single photon counting (TCSPC), are displayed in Fig. S9b.† The relationship between Φemi and the kinetics of S1 can be described with the formula: where kr and knr represent radiative and nonradiative S1 decay coefficient, respectively. We obtain 1/kr to be 23 ± 0.02 ns from fitting the TCSPC measurement to a single exponential decay under conditions where Φemi is 100%. We show that 1/(kr + knr) is 22.6 ± 0.2 ns from TA (vida infra) in Fig. S10a and b,† consistent with the measured Φemi of 100%. For TCSPC traces of TPT, shown in Fig. S9c and d,† we can see an early decay from the fast fission process and a slow decay with a lifetime fitted to be 21.2 ± 0.02 ns, which is similar to the S1 lifetime from PT (Fig. S9a and b†). We assign the low emission quantum yield of TPT as due to the efficient singlet fission process, which will be verified below.
where kr and knr represent radiative and nonradiative S1 decay coefficient, respectively. We obtain 1/kr to be 23 ± 0.02 ns from fitting the TCSPC measurement to a single exponential decay under conditions where Φemi is 100%. We show that 1/(kr + knr) is 22.6 ± 0.2 ns from TA (vida infra) in Fig. S10a and b,† consistent with the measured Φemi of 100%. For TCSPC traces of TPT, shown in Fig. S9c and d,† we can see an early decay from the fast fission process and a slow decay with a lifetime fitted to be 21.2 ± 0.02 ns, which is similar to the S1 lifetime from PT (Fig. S9a and b†). We assign the low emission quantum yield of TPT as due to the efficient singlet fission process, which will be verified below.
 | ||
| Fig. 3 (a) Spectra at selected delay times for fsTA of TPT in 2-MeTHF at room temperature. Dots represent the raw data, and the solid lines represent the fit. (Inset) Spectra in visible region only. (b) Kinetic traces at selected wavelengths from (a). (c) Spectra at selected delay times for nsTA of TPT in 2-MeTHF at room temperature. (Inset) Spectra in visible region only. (d) Kinetic traces at selected wavelengths from (c). | ||
Applying the (A → B → 0) model based on the biexponential fitting result, we determined the species associated spectra (SAS) for S1 and 1TT, which can be seen in Fig. S11.† The S1 ESA from TPT shows a similar spectral shape compared to the S1 ESA from PT. In Fig. S11a† the redshift from S1(TPT) to S1(PT) at ∼465 nm and ∼1350 nm is apparent, suggesting Sn delocalization is facilitated by the bridge, to be discussed further below. Turning to the triplet, the spectral shape of the 1TT ESA features approximately aligns with those of the T1 measured through anthracene sensitization, as shown in Fig. S11b and S15.† However, a blueshift in the visible peak and a redshift in the NIR region (974 nm → 987 nm) is apparent from 1TT to T1.
At 77 K, the biexciton formation time increases to 8.3 ± 0.1 ps (Fig. S12†). The spectral shape of the triplet shows only a minor redshift at 77 K compared to room temperature, shown in Fig. 4a. The slowing of the fission process at 77 K suggests that singlet fission of TPT is thermally activated; selected kinetic traces for both temperatures in VIS and NIR regions are shown in Fig. S13a and b.† As a thermally activated yet reversible singlet fission process, the equilibrium between S1 and 1TT is also temperature dependent. The S1 ESA remaining on ns timescales in the normalized fsTA time trace at room temperature and at 77 K (1345 nm and 1356 nm in Fig. 3b and S12b,† respectively) are measured to be ∼0.065 and ∼0.042, respectively, which are the signal portions relative to maximum S1 signal at early times. The 6.5% remaining S1 ESA amplitude is consistent with the emission quantum yield measurement mentioned above (∼6%).
 | ||
| Fig. 4 (a) Normalized 1TT spectra at different temperatures. (b) Arrhenius plot for the 1TT decay rate coefficient at two different concentrations. | ||
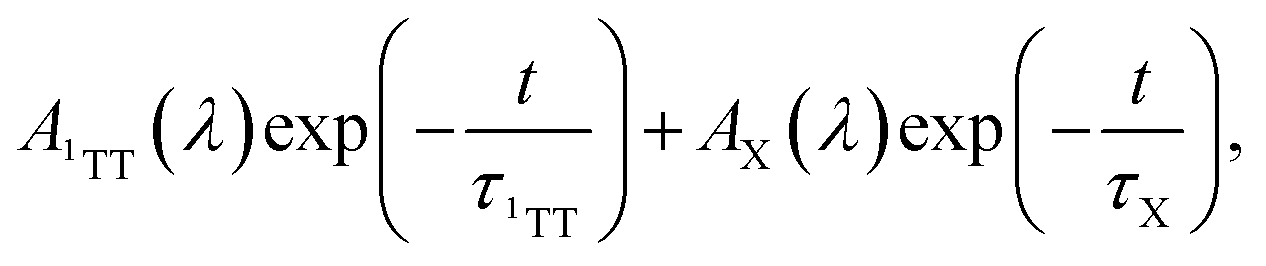 to represent both 1TT and the other species initially labeled X. In Fig. 3c, the characteristic triplet peak at 540 nm and the shoulder at 502 nm decay to the long-lasting X, with peaks at ∼520 nm and a shoulder at ∼556 nm. τ1TT obtained from the global fit is 197.4 ± 1.5 ns. However, τX from the fit is inaccurate due to the relatively small signal, which is <5% of 1TT. Therefore, we report only the lower limit of τX to be ∼35 µs. While a lower limit, the lifetime is markedly below that determined for T1 using sensitization experiments where we observe >150 µs. At 77 K, the 1TT ESA shows similar spectral features as the ESA in room temperature, except with a ∼10 nm redshift. The X ESA observed at room temperature can also be seen in the later time at 77 K in Fig. S12c.† Therefore, we again fit the nsTA at 77 K with the bi-exponential function. The τ1TT extracted from the global fit is 286.2 ± 5.3 ns, which is longer than 197 ns measured at room temperature.
to represent both 1TT and the other species initially labeled X. In Fig. 3c, the characteristic triplet peak at 540 nm and the shoulder at 502 nm decay to the long-lasting X, with peaks at ∼520 nm and a shoulder at ∼556 nm. τ1TT obtained from the global fit is 197.4 ± 1.5 ns. However, τX from the fit is inaccurate due to the relatively small signal, which is <5% of 1TT. Therefore, we report only the lower limit of τX to be ∼35 µs. While a lower limit, the lifetime is markedly below that determined for T1 using sensitization experiments where we observe >150 µs. At 77 K, the 1TT ESA shows similar spectral features as the ESA in room temperature, except with a ∼10 nm redshift. The X ESA observed at room temperature can also be seen in the later time at 77 K in Fig. S12c.† Therefore, we again fit the nsTA at 77 K with the bi-exponential function. The τ1TT extracted from the global fit is 286.2 ± 5.3 ns, which is longer than 197 ns measured at room temperature.
We now turn to the temperature dependent kinetics of 1TT and X based on the biexponential fit introduced above. From the global fit of nsTA, we normalize the ESA of 1TT to the maximum of the triplet peak, which is shown in Fig. 4a. The triplet feature redshifts by 10 nm from 296 K to 77 K. Fig. 4b is the Arrhenius plot for the decay rate coefficient of 1TT, 1/τ1TT, measured at two different concentrations: 2.2 µM and 22 µM. The agreement between τ1TT and the nearly constant spectral shapes at the two concentrations (cf., Fig. S17 and S18† for concentration and temperature dependence) indicates that the environment for molecules in the solutions is similar, and that aggregation is not playing a significant role in the photophysics. However, the data from 130 K to 77 K show a stark change in slope compared with temperatures higher than 130 K, indicating a violation of the expected Arrhenius trend. The change occurs very near the glass transition of 2-MeTHF at ∼137 K. Despite this abrupt change in temperature-dependent decay kinetics of 1TT, the spectral amplitude of the long-lived X does not show much temperature dependence from 77 K to 296 K, as seen in Fig. S14b.†
 | ||
| Fig. 5 (a) Normalized TREPR 2D plot for TPT in 2-MeTHF at 77 K. (b) Normalized EPR spectra from (a) at the selected delay times. | ||
To support our assignment for the EPR signal mentioned above, pulsed EPR experiments were conducted on TPT in 2-MeTHF at 10 K. Transient nutation traces were obtained with the pulse sequence shown in Fig. 6a at both 308 mT and 338 mT, and the data are shown in Fig. 6c. The Fourier transformed results (Fig. 6d) indicate extracted frequencies that are centered at 32.5 MHz and 40 MHz for the 308 mT and 338 mT nutation experiments, respectively. The ratio of these frequencies (0.81:1) is a close match to the ratio 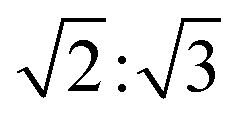 (i.e., 0.816:1). Based on the equation
(i.e., 0.816:1). Based on the equation  where B1 is the microwave field strength and ms represents spin quantum number, the EPR signal at 308 mT can be assigned to a transition between ms = ±2 and ms = ±1, while the signal at 338 mT is associated with the transition between ms = ±1 and ms = 0, all within the 5TT manifold.
where B1 is the microwave field strength and ms represents spin quantum number, the EPR signal at 308 mT can be assigned to a transition between ms = ±2 and ms = ±1, while the signal at 338 mT is associated with the transition between ms = ±1 and ms = 0, all within the 5TT manifold.
 | ||
| Fig. 6 (a) Pulse sequence for the transient nutation measurement, where T is 300 ns, τ is 148 ns, the π/2 pulse is 6 ns, π pulse is 12 ns, and the delay after flash (DAF) is t0 + dt, where dt = 30 ns. (b) Pulse sequence for the T2 measurement. (c) The nutation traces at 308 mT and 338 mT for TPT in 2-MeTHF at 10 K. (d) Fourier transformed data from (c). (e) Net spin magnetization decay trace at 338 mT for TPT in 2-MeTHF at 10 K using the pulse sequence in (b). | ||
To measure the spin dephasing lifetime (T2), we operate on the spin states corresponding to the EPR signal at 338 mT with the Hahn echo pulse sequence shown in Fig. 6a. The transient net spin magnetization decay trace is displayed in Fig. 6d, obtained by modifying the relaxation time between the 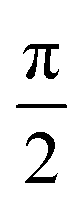 pulse, π pulse, and the echo. The decay trace is fit with a single exponential function leading to a determination of T2 = 726 ns at 10 K.
pulse, π pulse, and the echo. The decay trace is fit with a single exponential function leading to a determination of T2 = 726 ns at 10 K.
Discussion
Despite evidence of some spectral shifts and transition dipole enhancement for the electronic transitions involving the pyrene bridge caused by interaction with the tetracenic chromophores, the overall picture still implicates weak coupling in the TPT dimer. Interaction between the tetracene and pyrene could be large, yet the coupling between the two tetracene chromophores through the pyrene bridge is relatively low based on the only weakly perturbed S1 ← S0 transition. Upon excitation from S0 to S1, TT is formed with a risetime of 4.5 ps at room temperature and 9.3 ps at 77 K. Due to the fast observed rate, internal conversion from S1 to 1TT is the most likely reaction pathway.26 The temperature dependence suggests that singlet fission of TPT is a thermally activated process, which could in principle be an indication of a slightly endothermic energy balance between S1 and 1TT. However, the apparent equilibrium between S1 and 1TT, which has been observed in tetracenic systems,20,25,27–29 strongly favors 1TT in TPT, suggesting instead the overall reaction is exoergic. Because we observe no other species besides S1 and 1TT on the ns timescale, the equilibrium indicates that the singlet fission yield is about 94%, as the S1 emission quantum yield is measured to be 6%. The singlet fission yield for pentacene dimers in most cases is close to 100%,13,14,20,30–39 and is typically higher than tetracene dimers, which is lower than 50% in general.16,20,40,41 For tetracene dimers bridged with a norbonyl ring studied by Gilligan et al.,20 TIPS-BT1 and TIPS-BT1′, an equilibrium constant between S1 and 1TT was found to be 0.1 and 1, respectively. The small equilibrium constant of the tetracene dimers is due to the similar energetics of S1 and 1TT. On the other hand, the pentacene dimer, TIPS-BP1, has an equilibrium constant of 102–105 and the emission quantum yield is <0.01, indicating strong exothermicity from S1 to 1TT. From this perspective, TPT behaves closer to pentacene than tetracene, commensurate with its S0–S1 energy red-shift.Given the slight exothermicity, the relatively fast fission rate for TPT compared with other weakly coupled tetracenes is not surprising. The rate falls below that of the fastest pentacene dimer systems with sub-ps singlet fission rates, and in those cases strong electronic coupling between chromophores can be implicated for accelerating the rate. In TPT however, the bridge does not evidently lead to strong chromophore–chromophore coupling, as judged by the unperturbed and only slightly shifted S1 ← S0 absorption bands. Only higher-lying absorption bands show evidence of mixed-state behavior involving charge redistribution, but these are not directly excited nor involved in singlet fission during our measurements. Nevertheless, models of singlet fission originally developed by Ratner, Michl, et al.42 clearly show a role for virtual CT states in the SF process through the mediated mechanism. Modulating the importance of CT state involvement using solvent polarizability or polarity could be insightful, although here poor solubility in many solvents prevents a systematic study.
Now we turn to the eventual fate of 1TT. From nsTA data, the decay lifetimes of 1TT are 197 ns and 276 ns at 296 K and 77 K, respectively, indicating the decay of the 1TT state is also a thermally activated process. One of the decay pathways for 1TT is internal conversion to S0, which can be seen from the dominant ground state bleach recovery on the ns timescale (Fig. 3d at 560 nm and Fig. S12d† at 568 nm, yellow traces). The other decay pathway could be from 1TT to 5TT, and we suggest here that nsTA data are consistent with the EPR data in this regard. In addition to the distinct 5TT peak pattern with primary separation of D/3, the TREPR signals are further characterized to be from the pure 5TT state using nutation frequencies from pulsed EPR at 10 K (Fig. 6). Importantly, these data lack any clear contribution from T1. This then suggests that the longer-lived species identified in nsTA experiments – what was referred to earlier as species X – is due to 5TT. Because we can now verify the long-lived species common to EPR and TA is 5TT, we suggest that 5TT has unique spectral features compared with 1TT, which can be seen in Fig. S14a.† Distinct ESA features for 1TT and 5TT were also observed by Pace et al.43 A more definitive method for connecting 5TT photoinduced absorption and spin signatures involving optically detected magnetic resonance44,45 is planned.
To gain a full kinetic picture, kinetic simulations are performed based on the scheme shown in Fig. 7. Comparison of the relative state populations as a function of time are shown in Fig. S21.† The long-lived 5TT appears in nsTA throughout the temperature range with no obvious temperature dependence. For 1TT, we assume that the competing pathways (1TT → S0, 1TT →5TT) could both play some role in dictating the temperature dependence of TT deactivation. One possible mechanism for reduced 1TT → S0 decay at low temperature involves the constraint of the solvent environment that severely restricts the molecular motion of TPT upon freezing, decelerating relaxation from the 1TT state to the S0 state. The exact dynamics involved in the transition is unclear, as is the role that the aforementioned CT states might play, but the lower polarizability of the frozen solution may modulate their involvement. While 1TT → S0 is delayed as the solvent is gradually frozen to solid, 1TT → 5TT may also be slowed. The exact mechanism responsible here is again unknown, but it may be that large J fluctuations required to promote the transition7 and caused by thermally induced motions are suppressed, reducing the observed rate of 1TT → 5TT population flow.
 | ||
| Fig. 7 (a) The kinetic scheme for TPT in 2-MeTHF upon excitation centered at 600 nm. (b) Resulting rate coefficients from a fit of the kinetic scheme to TA data. kfiss was determined from fsTA data, and k5TT was given an upper bound based on estimated decay on a µs timescale. Φemi was fixed based on fluorescence quantum yield. kSQ and k1TT were varied in the fit of nsTA to match the 1TT and 5TT populations. Further details of the fit are in the ESI.† | ||
Despite the coincidence of long-lived signals assignable to 5TT in TA and EPR, there is a lifetime mismatch between the long-living ESA from nsTA (τ > 35 µs) and the 5TT signal from TREPR that decays in ∼3 µs. The EPR signal arises from spin polarization, proportional to the population difference between the magnetic spin states.46 Consequently, the decay of the EPR signal does not signify the disappearance of population from the state, but instead the decrease of the population difference between the corresponding states. Namely, although the 5TT signal from TREPR decays in ∼3 µs due to spin-lattice relaxation, the 5TT population lifetime can still be much longer. We fit the kinetic trace for TREPR spectra at 338 mT with a set of ordinary differential equations based on the scheme in Fig. S19c.† The normalized population for 1TT and 5TT states is shown in Fig. S19a.† The population difference between 5TT(0) and 5TT(1), representing the spin polarization between the two states, follows the kinetic trace for TREPR spectra at 338 mT, including the sign flipping at ∼2500 ns. Furthermore, the fitted lifetimes of 1TT and 5TT roughly correlate with the lifetimes obtained from nsTA. From the ODE fit, τ11, τ10, τSLR, τ1, and τ0 are 106.6 ns, 446.3 ns, 2 µs, 19.1 µs, and 38.4 µs, respectively. From nsTA, τ1TT is 276 ns (similar to the average of τ11 and τ10, the two 1TT decay pathways), and τ5TT is >35 µs, consistent with the average of τ1 and τ0, the 5TT decay pathways from different spin sublevels.
As for the EPR spectral shape of the 5TT state, according to the literature,7,12 it depends on the molecular geometry and the alignment between molecular axis and magnetic axis. For the organic dimers with the two chromophores aligned in parallel as in TPT, the population within the 5TT state should be symmetric regarding ms = 0, that is, P(ms = −1) = P(ms = 1) and P(ms = −2) = P(ms = 2), where P represents population. Because of the symmetric population distribution for the 5TT manifold, the arrangement of features in the EPR spectrum should be approximately symmetric, accordingly. However, in Fig. 5, the spectrum throughout the magnetic field range is mostly positive and is asymmetric with respect to B0 (334 mT). Absorptive spin polarization for 5TT has been observed10,14–16 and discussed in other systems. The transition from 1TT to 5TT is mediated by the fluctuation of exchange energy, in films or aggregates caused by triplet migration. This process is discussed by Kobori et al.10,47 in disordered TIPS-pentacene thin films and aggregated TIPS-pentacene solutions. In their chemically induced dynamic electron polarization (CIDEP) model, SQ0 mixing takes place when the coupling of the triplet pair is weakened via triplet migration. Meanwhile, SQ−2 and SQ−1 become populated via level crossing at the corresponding distances between the triplet pair, and through zero-field splitting, population is trapped in 5TT0, 5TT−2, and 5TT−1, resulting in absorptive spin polarization of 5TT. No triplet migration is possible in TPT, nor are large-scale nuclear motions, so we disfavor this mechanism. Instead, we suggest that large and rare fluctuations in J produce 5TT populations. We compare TPT behavior with predictions for planar but flexible pentacene dimers studied by MacDonald et al.,48 who interpret net-absorptive quintet peaks in CW EPR and symmetric 5TT spectra in pulsed-EPR to arise from SQ-mixing (i.e., low J, fast decoherence)49 and stochastic processes (high J, slower decoherence),50 respectively. These regimes may be simultaneously available in their ensemble due to conformational flexibility, absent in TPT. The fact that cw-EPR and echo-detected X-band spectra for TPT roughly match (Fig. 5b and S20a,† both are net-absorptive with similar shapes), confirms that quintets are not likely forming via low-J SQ mixing because such net-absorptive quintets should be strongly suppressed by the echo detection process. Therefore, net-absorptive X-band spectra seemingly arise in the stochastic regime due to asymmetrically-populated quintets not subject to fast decoherence, whereas the population becomes more symmetric at Q-band (Fig. S20b†). This type of field-dependent mechanism requires further theoretical investigation, and we note that the full collection of cw- and pulsed-EPR at different field strengths are needed to develop a complete picture.51
One of the primary goals for QIS applications with the 5TT state is to increase the dephasing time, T2, and the population of purely spin-polarized 5TT sublevels. In this study, abundant 5TT is produced such that the 5TT state can be monitored by both nsTA and EPR. Higher 5TT yield has been reported in monomer thin films8–10,43,51 or weakly coupled dimers,13–16,18,19 but it inevitably comes with isolated T1 because the interchromophore interaction can become weak enough to enable the spin mixing and decorrelation process to T1.14,19 The 1TT decay pathways for these cases are proposed to be 1TT → 5TT ⇌ T1 + T1 or through intersystem crossing from 5TT to 3TT, generating T1 in two steps. However, TPT is an example of a weakly coupled dimer from the pure electronic coupling standpoint (and as judged by steady-state spectroscopy), where there is no evidence for isolated triplets generated through 5TT. The features that could give rise to this behavior are the rigid bridge, the co-planar orientation, and the near-resonance of mixed chromophore-bridge electronic states. The outcome of these characteristics is an unusually large J at the equilibrium geometry, which is one of the keys to generate spin-polarized 5TT without coexistence of T1, stated in the parallel JDE model.7 For the weakly coupled dimers and the monomer thin films that generate T1, J is estimated to be <100 GHz,10,13,15,16,43,51 and sometimes close to 0 GHz, indicating the energies of 1TT, 5TT and 2*T1 are about the same. For TPT, from broken symmetry DFT calculation as described in detail in the ESI,†J is calculated to be −512 GHz, where the absolute value is higher than the weakly coupled chromophores mentioned above, and the negative sign suggests 5TT to be lower in energy than 1TT. We thus hypothesize that the exchange energy of TPT is strong enough to slow the formation of uncorrelated T1, given that there are limited intramolecular degrees of freedom available to promote the 5TT → (T1 + T1) transition, unlike dimers with flexible bridges.
The dephasing time, T2, of the 5TT manifolds is measured to be 726 ns, and it is long enough for us to demonstrate more than 4 Rabi oscillating cycles with the pulsed microwave setting mentioned previously. At 338 mT, for example, the EPR signal corresponds to the transition between ms = 0 and ms = 1 in 5TT, and with pulsed microwaves, we can rotate the population between the two magnetic spin states, therefore demonstrating this two-level system as a qubit.
Conclusion
We design a parallel pyrene-bridged tetracene dimer, TPT, with coupled triplet spins for purposes of demonstrating pure spin polarization production relevant to QIS applications. Excited states are calculated to involve both tetracene and pyrene, yet the lowest singlet transitions are only weakly perturbed from the PT to TPT, suggesting minimal interchromophore communication across the bridge. The interaction between tetracene chromophores is optimized to allow the singlet-quintet mixing and trap the population in the 5TT state while preventing the production of T1. We have observed 5TT with optical transient absorption spectra, which is rarely reported, through kinetic and temperature-dependent measurements that correlate with observations of 5TT using EPR. We characterize the qubits in 5TT manifolds with TREPR and pulsed EPR, and the dephasing time approaches the µs timescale. The pure 5TT state produced in large population is a highlight of this work; however, to generate high-quality qubits for QIS applications, we not only need a large population solely in the 5TT state, but also pure spin-polarized 5TT state. Toward this end, we suggest aiming future efforts to align all TPT dimers with their principal magnetic axis along an applied magnetic field direction.Data availability
Crystallographic data for TPT has been deposited at the Cambridge Structural Database under deposition number 2299672. TDDFT log files have been uploaded as part of the ESI.†Author contributions
LL performed all spectroscopic experiments and analysis, supervised by JCJ and NHD, and wrote the experimental portions of the manuscript. JCJ assisted with spectroscopic experiments and advised on the data analysis and manuscript writing with NHD. JEA designed the target molecules and the experimental approach, and JEA and TS devised the appropriate chromophores for singlet fission studies. TS performed and optimized the synthesis and conducted appropriate structural characterization. QA contributed conceptualization and investigation of calculation methodology and wrote portions of the manuscript. CR supervised and conceptualized computational efforts and contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work on transient absorption and time-resolved electron paramagnetic resonance spectroscopy was supported by the Solar Photochemistry Program of the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences, and Geosciences. We acknowledge funding for steady-state spectroscopy and aggregation studies from the National Science Foundation (CHE-2102713). Q. A. and C. R. acknowledge funding from the National Science Foundation through the Designing Materials to Revolutionize and Engineer our Future (NSF DMREF) program under award number DMR 1627428, and thank the University of Kentucky Center for Computational Sciences and Information Technology Services Research Computing for their fantastic support and collaboration and use of the Lipscomb Compute Cluster and associated research computing resources. We thank Sean Parkin, University of Kentucky X-ray crystallography facility, for obtaining the TPT crystal structure. This work was authored by Alliance for Sustainable Energy, Limited Liability Company, the manager and operator of the National Renewable Energy Laboratory under Contract No. DE-AC36-08GO28308. The views expressed in the article do not necessarily represent the views of the Department of Energy or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid‐up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- M. J. Y. Tayebjee, D. R. McCamey and T. W. Schmidt, Beyond Shockley–Queisser: Molecular Approaches to High-Efficiency Photovoltaics, J. Phys. Chem. Lett., 2015, 6, 2367–2378 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Recent Advances in Singlet Fission, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Singlet Fission, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. C. Hanna and A. J. Nozik, Solar conversion efficiency of photovoltaic and photoelectrolysis cells with carrier multiplication absorbers, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- M. J. Y. Tayebjee, A. A. Gray-Weale and T. W. Schmidt, Thermodynamic Limit of Exciton Fission Solar Cell Efficiency, J. Phys. Chem. Lett., 2012, 3, 2749–2754 CrossRef CAS.
- I. Paci, et al. , Singlet Fission for Dye-Sensitized Solar Cells: Can a Suitable Sensitizer Be Found?, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef CAS PubMed.
- K. E. Smyser and J. D. Eaves, Singlet fission for quantum information and quantum computing: the parallel JDE model, Sci. Rep., 2020, 10, 18480 CrossRef CAS PubMed.
- D. Lubert-Perquel, et al. , Identifying triplet pathways in dilute pentacene films, Nat. Commun., 2018, 9, 4222 CrossRef PubMed.
- R. D. Pensack, et al. , Observation of Two Triplet-Pair Intermediates in Singlet Exciton Fission, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed.
- H. Nagashima, et al. , Singlet-Fission-Born Quintet State: Sublevel Selections and Trapping by Multiexciton Thermodynamics, J. Phys. Chem. Lett., 2018, 9, 5855–5861 CrossRef CAS PubMed.
- L. R. Weiss, et al. , Strongly exchange-coupled triplet pairs in an organic semiconductor, Nat. Phys., 2017, 13, 176–181 Search PubMed.
- R. D. Dill, K. E. Smyser, N. H. Damrauer and J. D. Eaves Entangled, Spin-polarized Excitons from Singlet Fission in a Rigid Dimer, Nat. Commun., 2023, 14, 1180 Search PubMed.
- M. J. Y. Tayebjee, et al. , Quintet multiexciton dynamics in singlet fission, Nat. Phys., 2017, 13, 182–188 Search PubMed.
- B. S. Basel, et al. , Unified model for singlet fission within a non-conjugated covalent pentacene dimer, Nat. Commun., 2017, 8, 15171 Search PubMed.
- S. Nakamura, et al. , Synergetic Role of Conformational Flexibility and Electronic Coupling for Quantitative Intramolecular Singlet Fission, J. Phys. Chem. C, 2021, 125, 18287–18296 CrossRef CAS.
- Y. Matsui, et al. , Exergonic Intramolecular Singlet Fission of an Adamantane-Linked Tetracene Dyad via Twin Quintet Multiexcitons, J. Phys. Chem. C, 2019, 123, 18813–18823 CrossRef CAS.
- Q. Ai, et al. , Nanoribbons or weakly connected acenes? The influence of pyrene insertion on linearly extended ring systems, J. Mater. Chem. C, 2021, 9, 16929–16934 RSC.
- B. S. Basel, et al. , Evidence for Charge-Transfer Mediation in the Primary Events of Singlet Fission in a Weakly Coupled Pentacene Dimer, Chem, 2018, 4, 1092–1111 CAS.
- A. B. Pun, et al. , Ultra-fast intramolecular singlet fission to persistent multiexcitons by molecular design, Nat. Chem., 2019, 11, 821–828 CrossRef CAS PubMed.
- A. T. Gilligan, E. G. Miller, T. Sammakia and N. H. Damrauer, Using Structurally Well-Defined Norbornyl-Bridged Acene Dimers to Map a Mechanistic Landscape for Correlated Triplet Formation in Singlet Fission, J. Am. Chem. Soc., 2019, 141, 5961–5971 CrossRef CAS PubMed.
- J. D. Cook, T. J. Carey and N. H. Damrauer, Solution-Phase Singlet Fission in a Structurally Well-Defined Norbornyl-Bridged Tetracene Dimer, J. Phys. Chem. A, 2016, 120, 4473–4481 CrossRef CAS PubMed.
- H. Du, R. A. Fuh, J. Li, L. A. Corkan and J. S. Lindsey, PhotochemCAD++: A Computer-Aided Design and Research Tool in Photochemsitry, Photochem. Photobiol., 1998, 62, 141–142 Search PubMed.
- H. F. Bettinger, C. Tönshoff, M. Doerr and E. Sanchez-Garcia, Electronically Excited States of Higher Acenes up to Nonacene: A Density Functional Theory/Multireference Configuration Interaction Study, J. Chem. Theory Comput., 2016, 12, 305–312 CrossRef CAS PubMed.
- J. C. Dean, et al. , Photophysical characterization and time-resolved spectroscopy of a anthradithiophene dimer: exploring the role of conformation in singlet fission, Phys. Chem. Chem. Phys., 2017, 19, 23162–23175 RSC.
- K. Miyata, et al. , Coherent singlet fission activated by symmetry breaking, Nat. Chem., 2017, 9, 983–989 CrossRef CAS PubMed.
- N. V. Korovina, N. F. Pompetti and J. C. Johnson, Lessons from intramolecular singlet fission with covalently bound chromophores, J. Chem. Phys., 2020, 152, 040904 CrossRef CAS PubMed.
- N. R. Monahan, et al. , Dynamics of the triplet-pair state reveals the likely coexistence of coherent and incoherent singlet fission in crystalline hexacene, Nat. Chem., 2017, 9, 341–346 CrossRef CAS PubMed.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Triplet Pair States in Singlet Fission, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- M. W. B. Wilson, et al. , Temperature-Independent Singlet Exciton Fission in Tetracene, J. Am. Chem. Soc., 2013, 135, 16680–16688 CrossRef CAS PubMed.
- J. Zirzlmeier, et al. , Singlet fission in pentacene dimers, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- C. Hetzer, et al. , Chromophore Multiplication To Enable Exciton Delocalization and Triplet Diffusion Following Singlet Fission in Tetrameric Pentacene, Angew. Chem., Int. Ed., 2019, 58, 15263–15267 CrossRef CAS PubMed.
- J. Zirzlmeier, et al. , Solution-based intramolecular singlet fission in cross-conjugated pentacene dimers, Nanoscale, 2016, 8, 10113–10123 RSC.
- S. N. Sanders, et al. , Quantitative Intramolecular Singlet Fission in Bipentacenes, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed.
- K. C. Krishnapriya, et al. , Spin density encodes intramolecular singlet exciton fission in pentacene dimers, Nat. Commun., 2019, 10, 33 CrossRef CAS PubMed.
- H. Sakai, et al. , Multiexciton Dynamics Depending on Intramolecular Orientations in Pentacene Dimers: Recombination and Dissociation of Correlated Triplet Pairs, J. Phys. Chem. Lett., 2018, 9, 3354–3360 CrossRef CAS PubMed.
- T. Sakuma, et al. , Long-Lived Triplet Excited States of Bent-Shaped Pentacene Dimers by Intramolecular Singlet Fission, J. Phys. Chem. A, 2016, 120, 1867–1875 CrossRef CAS PubMed.
- E. Kumarasamy, et al. , Tuning Singlet Fission in π-Bridge-π Chromophores, J. Am. Chem. Soc., 2017, 139, 12488–12494 CrossRef CAS PubMed.
- T. Yamakado, Conformational planarization vs. singlet fission: Distinct excited-state dynamics of cyclooctatetraene-fused acene dimers, Angew. Chem., Int. Ed., 2018, 57, 5438 CrossRef CAS PubMed.
- S. Lukman, et al. , Tuneable Singlet Exciton Fission and Triplet–Triplet Annihilation in an Orthogonal Pentacene Dimer, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- A. M. Müller, Y. S. Avlasevich, K. Müllen and C. J. Bardeen, Evidence for exciton fission and fusion in a covalently linked tetracene dimer, Chem. Phys. Lett., 2006, 421, 518–522 CrossRef.
- N. V. Korovina, et al. , Linker-Dependent Singlet Fission in Tetracene Dimers, J. Am. Chem. Soc., 2018, 140, 10179–10190 CrossRef CAS PubMed.
- E. C. Greyson, J. Vura-Weis, J. Michl and M. A. Ratner, Maximizing Singlet Fission in Organic Dimers: Theoretical Investigation of Triplet Yield in the Regime of Localized Excitation and Fast Coherent Electron Transfer, J. Phys. Chem. B, 2010, 114, 14168–14177 CrossRef CAS PubMed.
- N. A. Pace, et al. , Conversion between triplet pair states is controlled by molecular coupling in pentadithiophene thin films, Chem. Sci., 2020, 11, 7226–7238 RSC.
- G. Joshi, et al. , Optical readout of singlet fission biexcitons in a heteroacene with photoluminescence detected magnetic resonance, J. Chem. Phys., 2022, 157, 164702 CrossRef CAS PubMed.
- R. D. Dill, et al. , Near-Infrared Absorption Features of Triplet-Pair States Assigned by Photoinduced-Absorption-Detected Magnetic Resonance, J. Phys. Chem. Lett., 2023, 14, 2387–2394 CrossRef CAS PubMed.
- C. Hintze, U. E. Steiner and M. Drescher, Photoexcited Triplet State Kinetics Studied by Electron Paramagnetic Resonance Spectroscopy, ChemPhysChem, 2017, 18, 6–16 CrossRef CAS PubMed.
- S. Matsuda, S. Oyama and Y. Kobori, Electron spin polarization generated by transport of singlet and quintet multiexcitons to spin-correlated triplet pairs during singlet fissions, Chem. Sci., 2020, 11, 2934–2942 RSC.
- T. S. C. MacDonald, M. J. Y. Tayebjee, M. I. Collins, E. Kumarasamy, S. N. Sanders, M. Y. Sfeir, L. M. Campos and D. R. McCamey, Anisotropic Multiexciton Quintet and Triplet Dynamics in Singlet Fission via Pulsed Electron Spin Resonance, J. Am. Chem. Soc., 2023, 145(28), 15275–15283 CrossRef CAS PubMed.
- M. I. Collins, M. R. McCamey and M. J. Y. Tayebjee, Fluctuating exchange interactions enable quintet multiexciton formation in singlet fission, J. Chem. Phys., 2019, 151, 164104 CrossRef PubMed.
- M. I. Collins, F. Campaioli, M. J. Y. Tayebjee, J. H. Cole and D. R. McCamey, Quintet formation, exchange fluctuations, and the role of stochastic resonance in singlet fission, Commun. Phys., 2023, 6, 65 CrossRef.
- B. K. Rugg, et al., Triplet-pair spin signatures from macroscopically aligned heteroacenes in an oriented single crystal, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201879119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details and characterization, additional steady-state and transient optical and EPR spectroscopy, TDDFT and exchange energy calculations, details of the kinetic models and associated fits. See DOI: https://doi.org/10.1039/d3sc03153e |
| This journal is © The Royal Society of Chemistry 2023 |