
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRearrangement of a carboxy-substituted spiro[4.4]nonatriene to annulated fulvenes through a Pd(II)-mediated 1,5-vinyl shift†
Karan
Goyal
a,
Garrett A.
Kukier
b,
Xiangyang
Chen
b,
Aneta
Turlik
b,
K. N.
Houk
 *b and
Richmond
Sarpong
*a
*b and
Richmond
Sarpong
*a
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: rsarpong@berkeley.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA. E-mail: houk@chem.ucla.edu
First published on 4th October 2023
Abstract
A novel synthesis of aryl-substituted, enantioenriched fulvenes from an oxidative Heck cascade and rearrangement of a carboxy-substituted spiro[4.4]nonatriene is disclosed. Mechanistic investigations with density functional theory (DFT) calculations and empirical results support the net transformation occurring through a novel Pd(II)-mediated 1,5-vinyl shift from a vinyl–palladium intermediate that terminates with protodepalladation. This spiro-to-fused bicycle conversion tolerates a range of electron-rich and deficient arylboronic acids to give a range of mono- and diaryl substituted annulated fulvenes in moderate to good yields and enantiomeric ratios. Overall, this work connects two classes of molecules with a rich history in physical organic chemistry.
Introduction
A subset of π-conjugated organic molecules possess p-orbitals in distinct spatial arrangements which give rise to interesting phenomena such as homoconjugation—the through-space interaction of non-contiguous orbitals or orbital systems.1 Among this class of molecules are the homotropylium cation,2 methylenenorbornadienes,3 and spiro[4.4]nonapolyenes.4 Spiro[4.4]nonatetraenes and the corresponding trienes have a rich history in physical organic chemistry because they exhibit spiroconjugation, the through-space interaction of two perpendicular π molecular orbitals (MOs) which splits degeneracy of the two orbital systems (Fig. 1A, left).5,6 This electronic phenomenon has been utilized as a design element for optoelectronic materials7 and dyes8 to manipulate the HOMO–LUMO gap in these systems (Fig. 1A, right). While there has been some investigation of how these orbital perturbations impact the reactivity of spirononenes, much of their reactivity remains to be explored. A report by Semmelhack and coworkers showed that these structures undergo skeletal rearrangements to indene systems when heated to high temperature (Fig. 1B).4 Peripheral rearrangements (e.g., acetate transposition) have also been demonstrated on related spirononadienes under milder, palladium-mediated conditions (Fig. 1C).9 Here, we demonstrate the use of palladium-mediated conditions to facilitate the enantioselective transformation of a carboxy-substituted spiro[4.4]nonatriene to annulated fulvenes (Fig. 2A).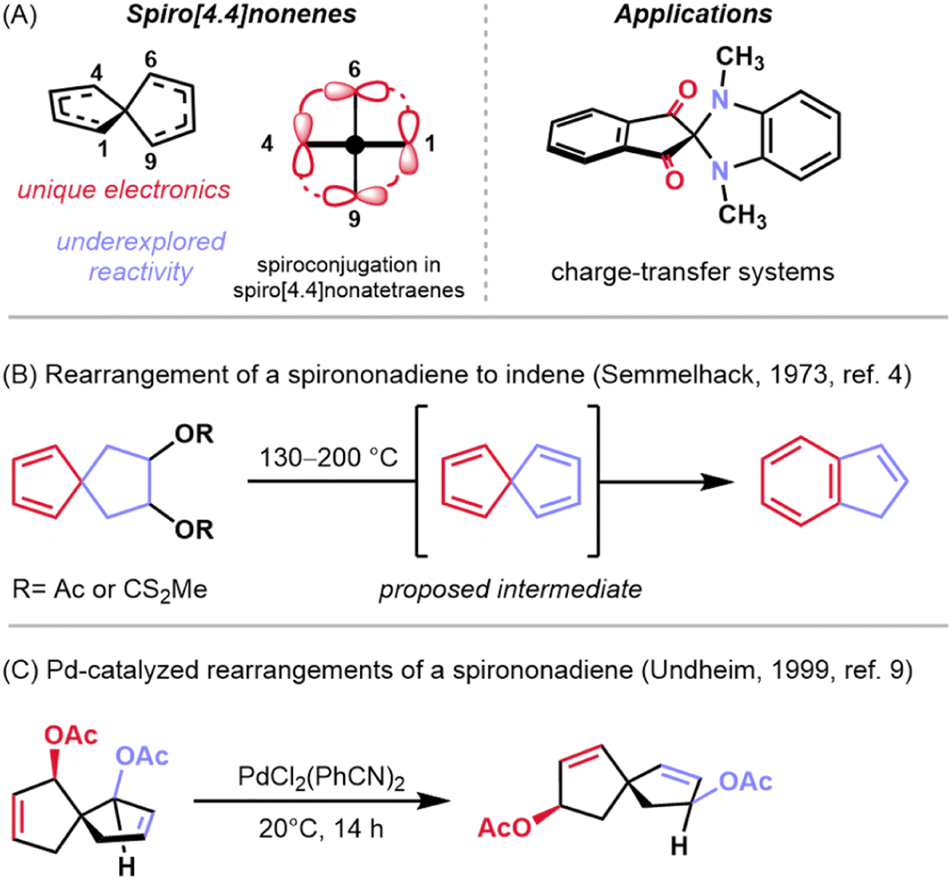 | ||
| Fig. 1 Introduction to the reactivity and applications of spiro[4.4]nonenes. | ||
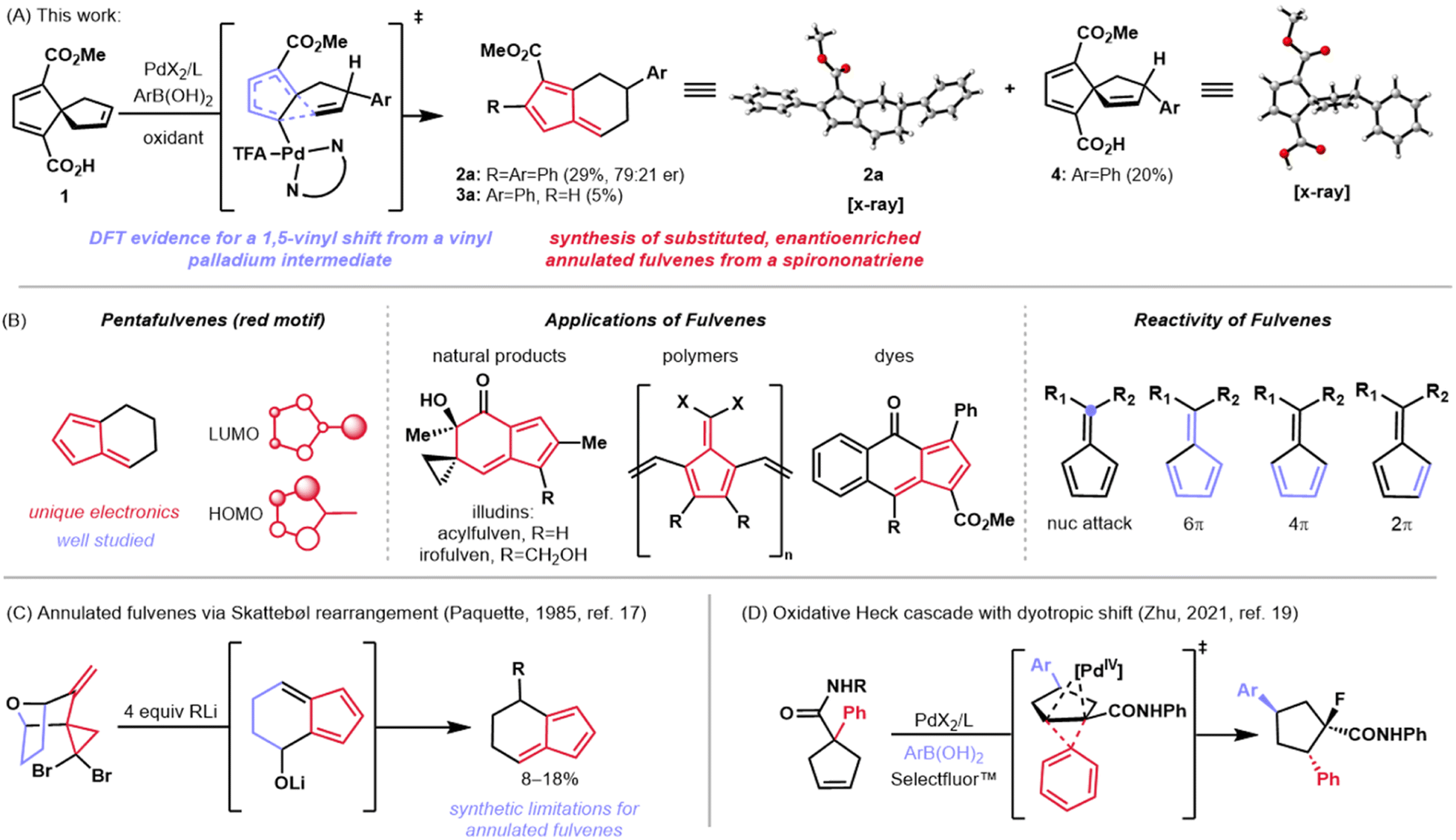 | ||
| Fig. 2 (A) This work: rearrangement of a spiro[4.4]nonatriene to annulated fulvenes through an interrupted oxidative Heck reaction. (B) Electronics, applications, and reactivity of pentafulvenes. (C) Synthesis of annulated fulvenes reported by Paquette. (D) Oxidative Heck reaction and dyotropic shift reported by Zhu. | ||
Fulvenes, such as the pentafulvenes, are another class of molecules that possess unique π orbital systems (Fig. 2B, left).10 The arrangement of p-orbitals in these systems confers some aromatic character, as well as versatile reactivity—both of which can be affected by substituent changes.11 Unlike spiro[4.4]nonenes, however, pentafulvenes have found much broader application in synthesis. These structural motifs are not only featured in polymers12 and dyes13,14 due to their tunable electronic properties, but are also found in some natural products such as the illudin family (e.g., see acylfulvene and irofulven in Fig. 2B, middle).15 Furthermore, the reactivity of fulvenes with nucleophiles and with other π systems as diverse cycloaddition partners has made them synthetically attractive (Fig. 2B, right), as demonstrated by Carreira and coworkers in the total synthesis of the pallambin natural products.16
A subset of fulvenes, known as annulated fulvenes, were first synthesized by Paquette and coworkers in 1985 and used as intermediates toward conformationally restricted methylenenorbornadiene derivatives (Fig. 2C).17 While the Paquette approach yielded the desired molecules, it proceeded in low yield. Specifically, treatment of a gem-dibromocyclopropane with four equivalents of an organolithium reagent resulted in a Skattebøl rearrangement18 followed by nucleophilic attack to give the substituted annulated fulvenes in low yields, alongside indene and ring-opened aryl alkyne side products. Notably, this synthesis only allowed for substitution at a single position and afforded racemic products.
Herein, we report the development of a method which results in the synthesis of enantioenriched, aryl-substituted annulated fulvenes in moderate to good yields and enantiomeric ratios (er's) through an intercepted oxidative Heck reaction of a carboxy-substituted spiro[4.4]nonatriene (Fig. 2A). This discovery points to an intriguing mechanism in which a C–C cleavage occurs at low temperature and under weakly oxidative conditions. Recently, Zhu and coworkers demonstrated a cleavage of unstrained C–C bonds in related five-membered ring systems through an oxidative Heck reaction followed by a dyotropic shift (Fig. 2D).19 We reasoned that a similar pathway might be operative in this spirononatriene system. Our mechanistic investigations using a combination of DFT and empirical insights have led to a mechanistic hypothesis involving C–C cleavage from a key vinyl palladium species. The net coupling/rearrangement transformation of 1 to 2a (Fig. 2A) occurs with a range of commercially available arylboronic acids. Overall, this work expands the chemistry of spirononenes by showcasing their transformation to annulated fulvenes and provides evidence for a novel mechanism of C–C cleavage and migration—a Pd(II)-mediated 1,5 vinyl shift.
Results and discussion
Reaction discovery and optimization
In the course of an ongoing synthetic study, an oxidative Heck reaction of spirononatriene 1 to synthesize 4 was desired. Upon applying the oxidative Heck conditions reported by Zhu20 to this spirononatriene system, the desired Heck product (4) was indeed isolated in 20% yield. However, diaryl fulvene 2a was obtained as the major product in 29% yield, alongside a small amount of monoaryl fulvene 3a in 5% yield (Fig. 2A). Extensive optimization of different parameters led to reaction conditions which gave diaryl fulvene 2a as the major product in 59% isolated yield and 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 enantiomeric ratio (er) (Table 1, entry 1; see the ESI for the full optimization tables†). Deviation from these conditions, as illustrated in Table 1, inform our overall mechanistic understanding (vide infra). As described in the optimized conditions, pyridine oxazoline (PyrOx) and quinoline oxazoline (Quinox) ligands emerged as the best performers with (S)-isopropyl quinoline oxazoline ((S)-iPr-quinox) giving the highest yields and er's.
:13 enantiomeric ratio (er) (Table 1, entry 1; see the ESI for the full optimization tables†). Deviation from these conditions, as illustrated in Table 1, inform our overall mechanistic understanding (vide infra). As described in the optimized conditions, pyridine oxazoline (PyrOx) and quinoline oxazoline (Quinox) ligands emerged as the best performers with (S)-isopropyl quinoline oxazoline ((S)-iPr-quinox) giving the highest yields and er's.
|
|
||
|---|---|---|
| Entry | Deviation | Resultsa |
| a Yields were determined by 1H NMR integration using pyrazine as an internal standard; isolated yields are shown in parentheses. | ||
| 1 | None | 76% (59%) 2a, 87:13 er |
| 2 | 5% AcOH (v/v) instead of base | 88% (46%) 2a, 84:16 er |
| 3 | No base | 65% (53%) 2a, 84:16 er |
| 4 | KOAc instead of Na2CO3 | 43% 2a, 44% 4 |
| 5 | Pd(OAc)2 instead of Pd(TFA)2; no base | 45% 2a, 34% 4 |
| 6 | 5% AcOH (v/v), no base; 0.4 equiv. Ag2O | 28% 2a, 24% 4, 21% 1 |
| 7 | BQ instead of 2,5-dimethyl BQ | 49% 2a, 39% 4 |
| 8 | 1 equiv. PhB(OH)2 | 46% 2a, 43% 1 |
| 9 | 0.1 M instead of 0.01 M | 47% 2a, 12% 4 |
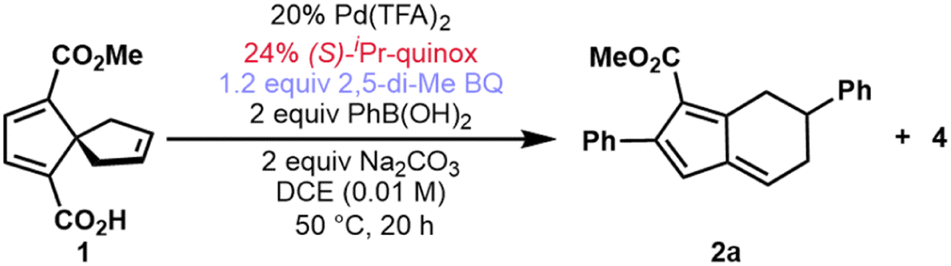
While the reaction was shown to work well under neutral, basic, or acidic conditions (Table 1, entries 1–3), basic conditions proved most suitable for a range of electron-rich and -deficient arylboronic acids (see ESI, Table S6†). Protodeboronation became more pronounced under more acidic conditions.21 Additionally, the presence of acetate, either from the added base or in the form of Pd(OAc)2, favored formation of Heck product 4 (Table 1, entries 4 and 5). The use of silver salts to promote decarboxylation22 and favor fulvene product 2a had no such effect and led to lower overall conversion and nearly equal amounts of the two products (Table 1, compare entries 2 and 6). The choice of oxidant was found to be crucial with 2,5-dimethyl-para-benzoquinone outperforming p-benzoquinone (Table 1, entry 7). The use of two equivalents of boronic acid was found to be necessary to obtain high yields. Using only one equivalent of the boronic acid coupling partner still led primarily to products that had incorporated two of the boronic acid units, albeit in low overall conversion (Table 1, entry 8). Finally, the reaction yield was highest at a concentration of 0.01 M (Table 1, entry 9).
Arylboronic acid substrate scope
With optimized conditions established, we first sought to evaluate the scope of arylboronic acids (Scheme 1). 4-Substituted arylboronic acids performed well regardless of the electronic nature of the substituents, giving fulvene products in 50–60% yield and approximately 85:15 er's. More electron-deficient arylboronic acids such 4-NO2, 4-CF3, and 4-CHO phenylboronic acids gave greater amounts of monoaryl fulvenes (3) as compared to the reaction profile of more electron-neutral or electron-rich arylboronic acids. The monoaryl fulvene products were generally formed in higher er's as compared to the corresponding diaryl fulvenes. 3-Substituted arylboronic acids performed analogously to give monoaryl fulvene and diaryl fulvene products in similar yields, er's, and ratios. 2-Substituted arylboronic acids, however, did not perform well, likely due to pronounced steric hindrance that could impede migratory insertion of the aryl-palladium intermediate across the spirononatriene olefin group.
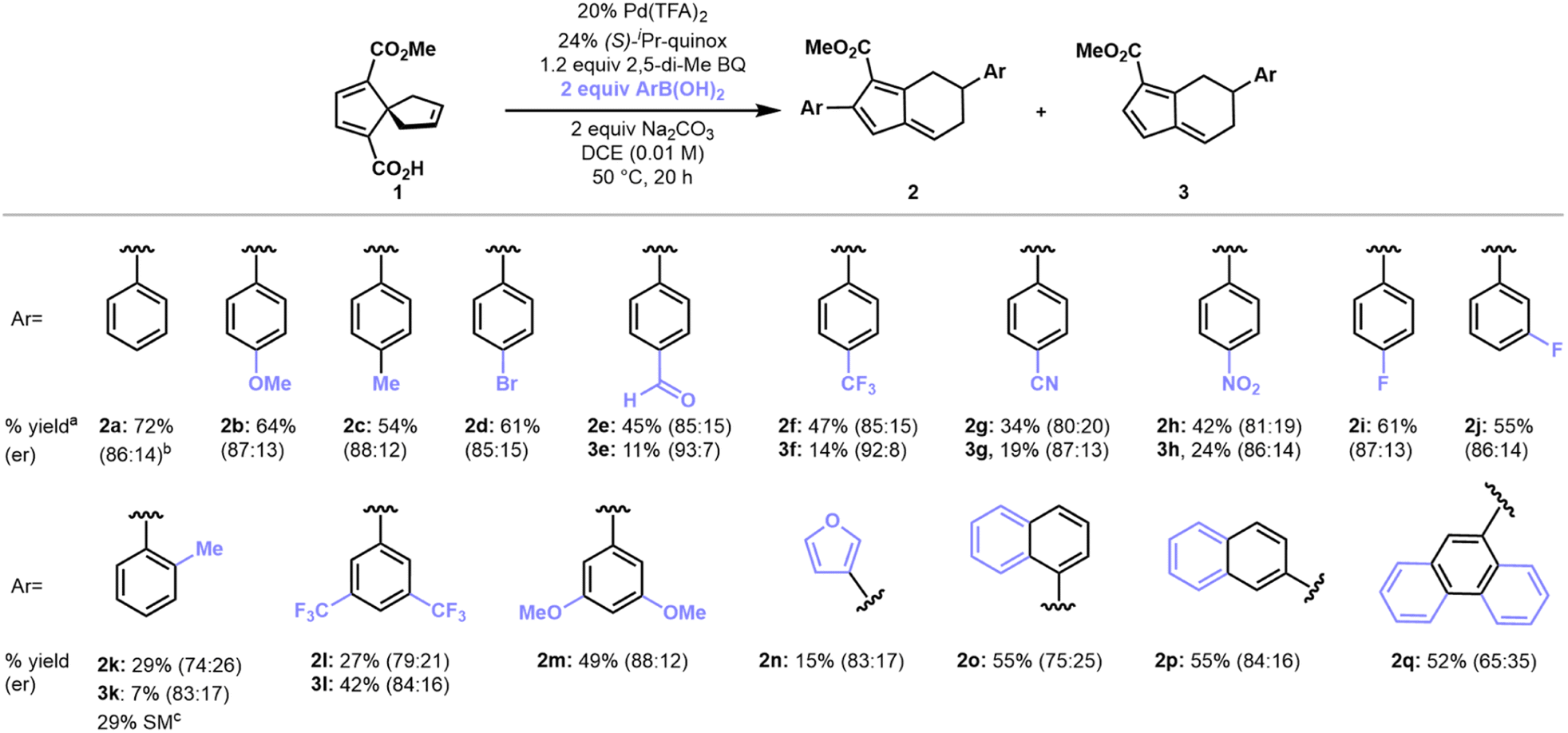 | ||
| Scheme 1 Substrate scope for arylboronic acids. aAll yields shown are isolated yields for reactions run with 0.05 mmol (11 mg) of 1. bReaction was run on 1.0 mmol scale. cRemaining starting material (SM) was determined by 1H NMR analysis of the crude reaction mixture with pyrazine as an internal standard. | ||
Arylboronic acids with extended aromatic systems also gave fulvene products such as 2o–2q in moderate yields, albeit with diminished enantiomeric ratios. Finally, heterocyclic arylboronic acids performed poorly as coupling partners under the optimal conditions that we have identified. Nonetheless, 3-furanyl-boronic acid gave a 15% yield isolated yield of fulvene product 2n. While low-yielding, this result suggests the potential for optimization of this method for heteroarylboronic acid coupling partners. Vinyl boronic acids and arylboronic pinacol esters did not participate in the coupling/rearrangement transformation, presumably due to inefficient transmetalation to the organo-palladium intermediate as well as competing decomposition of the vinyl boronic acid coupling partner (see ESI, Table S7†).23,24
Mechanistic hypotheses
A focal point of our mechanistic studies has been the C–C cleavage of the spirononatriene to afford the fused 5,6-bicycle. DFT calculations, in conjunction with experiments, were used to gain insight into this transformation. Three hypotheses were initially considered (Scheme 2). The first pathway that was considered was a 1,5-vinyl shift from isolable Heck product 4. Semmelhack and coworkers have proposed that simple, unsubstituted spirononatetraenes and trienes similar to 4 undergo rearrangement to 5,6-fused ring systems through such a mechanism but have high barriers that require temperatures of over 100 °C.25,26 However, it may be that the presence of substituents on 4 or coordination of the spirononatriene to a palladium complex may lead to lower barriers, giving rise to 5 (Scheme 2A).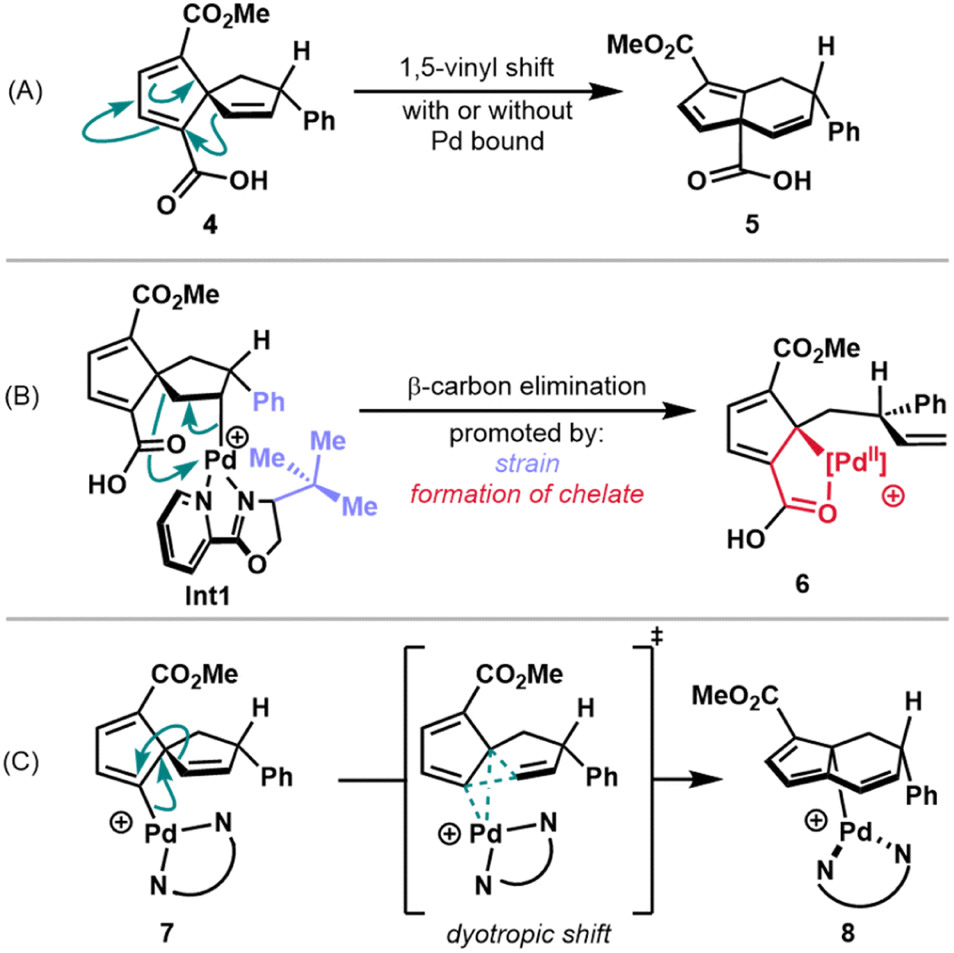 | ||
| Scheme 2 Proposed pathways for C–C cleavage: (A) 1,5-vinyl shift from 4 (B) β-carbon elimination from alkyl palladium species Int1 (C) dyotropic shift from vinyl palladium species 7. | ||
A second proposed pathway involves a β-carbon elimination from the intermediate alkyl palladium species generated after migratory insertion into the spirononatriene olefin (see Int1, Scheme 2B). β-Carbon eliminations in relatively unstrained systems of this type are rare due to the higher kinetic barriers and thermodynamic considerations.27 However, Lautens and coworkers have reported a Heck cascade in which a β-carbon elimination is driven by the relief of strain arising from bulky ortho-aryl substituents.28 We hypothesized that in our system, analogous relief of developing strain between the newly introduced phenyl group and bulky substituent on the ligand may serve as a driving force (see Int1, Scheme 2B). Additionally, enthalpic gain through formation of a chelate with the pendant carboxylic acid could further facilitate this rearrangement to 6 (Scheme 2B).
Finally, a dyotropic shift pathway could be operative. Here, concerted migration of two bonds across a stationary scaffold could occur from vinyl palladium species 7 (Scheme 2C).29 In this case, such a rearrangement would furnish the observed 5,6-fused ring system of the annulated fulvene. The dyotropic shifts that have been invoked for transition metal intermediates generally involve high oxidation state metal centers such as putative Cu(III) or Pd(IV) intermediates.18,30 In a related report, a possible dyotropic rearrangement in a Heck reaction with Pd(II) is proposed, albeit without sufficient supporting data.31 Under our reaction conditions, accessing a Pd(IV) intermediate appears unlikely and so a Pd(II)-mediated dyotropic shift was considered.
Mechanistic investigations
Initial mechanistic insight was obtained through a series of experiments as summarized in Table 2. Proposed Pathway A was examined by subjecting Heck product 4 to a variety of reaction conditions that may facilitate rearrangement to the 5,6-fused bicycle and checking for formation of annulated fulvenes 2a or 3a. Simply heating 4 in a DCE/AcOH mixture only returned the starting spirononatriene (Table 2, entry 1). While no reports of Pd-mediated 1,5-vinyl shifts are known, it has been shown that coordination of Pd(II) to 1,5-hexadienes can catalyze [3,3]-sigmatropic rearrangements.32 To test for a possible effect of Pd(II) or Pd(0)-coordination on the vinyl-shift, 4 was added to pre-stirred mixtures of either a Pd(0) or Pd(II) precatalyst and ligand (Table 2, entries 2 and 3). Only starting material (4) was recovered in both of these cases as well. Additionally, re-subjecting 4 to the optimized reaction conditions only gave trace amounts of annulated fulvene products as determined by 1H NMR of the crude product mixture (Table 2, entry 4). This observation indicates that 4 itself may not rearrange to the fulvene products but rather, rearrangement likely occurs through an intermediate related to 4 along the oxidative Heck pathway.|
|
||
|---|---|---|
| Entry | Conditions | Results |
| a Fulvene products (2a and 3a) do not readily form from Heck product (4). | ||
| 1 | 5% AcOH/DCE, 50 °C | No reaction |
| 2 | 5% AcOH/DCE, 50 °C, Pd2(dba)3/(S)-tBu-quinox | No reaction |
| 3 | 5% AcOH/DCE, 50 °C, Pd(TFA)2/(S)-tBu-quinox | No reaction |
| 4 | Optimized reaction conditions | Only trace 2a/3a, largely SM |
These empirical observations are supported by DFT calculations. For all calculations involving Pd, alkyl palladium species Int1 was used as the starting point (i.e., ΔG = 0 kcal mol−1). The energies of all cationic structures include the energy of a trifluoroacetate (TFA) anion at an infinite distance from the Pd-complex. Pathway A was initially considered with coordination of palladium (Fig. 3A). In this scenario, 9, which may be accessed through vinyl shift of the Pd-bound alkene (Int2), was determined to be uphill by 15.9 kcal mol−1 (ΔG) and has a transition state (TS) energy, ΔG‡, of 44.8 kcal mol−1 (see the ESI for the structure of TS1). This value is unlikely for a transformation that occurs at 50 °C (approximate maximum of 28 kcal mol−1 accessible).33 In considering Pathway A without the involvement of Pd(II), 4 was used as the thermodynamic starting point (Fig. 3B). Overall, while this transformation was found to be more kinetically and thermodynamically accessible, it was still unlikely to occur under the reaction conditions that had been employed. This reaction was endergonic by 1.0 kcal mol−1 with an activation barrier of 29.4 kcal mol−1 (Fig. 3B, see the ESI† for the structure of TS2). Altogether, these computational and experimental results do not support Pathway A as a viable reaction path for the formation of the annulated fulvene products from the precursor spirononatriene.
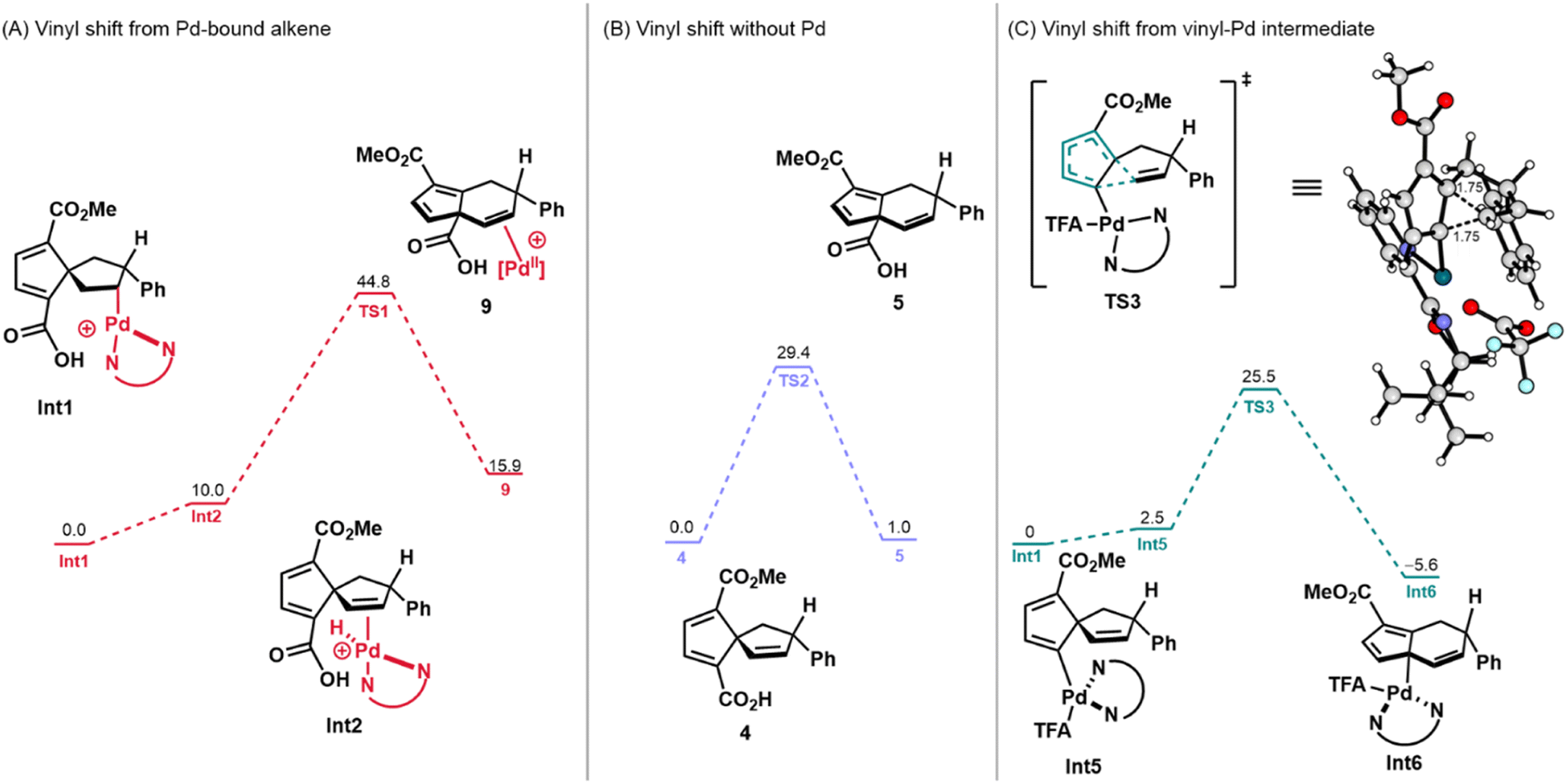 | ||
| Fig. 3 Calculated energies (in kcal mol−1) of transition states and intermediates for pathways involving vinyl shifts. A trifluoroacetate (TFA) counter-anion at infinite distance is implied for all cationic species. | ||
Next, the β-carbon elimination pathway (Scheme 2B) was investigated using DFT calculations. A transition state for β-carbon elimination was found with a TS energy of 38.7 kcal mol−1 (see ESI, Scheme S1†). The reaction itself was also found to be endergonic, giving a product that lies 11.6 kcal mol−1 higher. On the basis of these initial results, we concluded that the β-carbon elimination pathway was unlikely and additional mechanistic studies and calculations to support or refute this possibility were not pursued.
Finally, Pathway C (Scheme 2C), which involves a dyotropic shift, was investigated computationally. Following an extensive search, a dyotropic shift transition state was not found. However, searches for this transition state converged to one where the C–C bond migrates but the C–Pd bond remains stationary, constituting a 1,5-vinyl shift from vinyl–Pd species Int5 (Fig. 3C). Since transition state optimizations of geometries constituting a dyotropic rearrangement converged to this Pd(II)-mediated 1,5-vinyl shift transition state, we conclude that this pathway must be lower in energy than the proposed dyotropic rearrangement. Initial searches furnished a 1,5-vinyl shift transition state involving a cationic Pd(II) intermediate. It was then found that coordination of TFA reduces the energy of this transition state, resulting in a sufficiently low transition state energy of 25.5 kcal mol−1 and an exergonic reaction which places Int6 at −5.6 kcal mol−1 (Fig. 3C). On the basis of these calculations, the 1,5-vinyl shift pathway from vinyl palladium species Int5 was then deemed as a plausible pathway and the energies of intermediates leading to Int5 were calculated (Scheme 3).
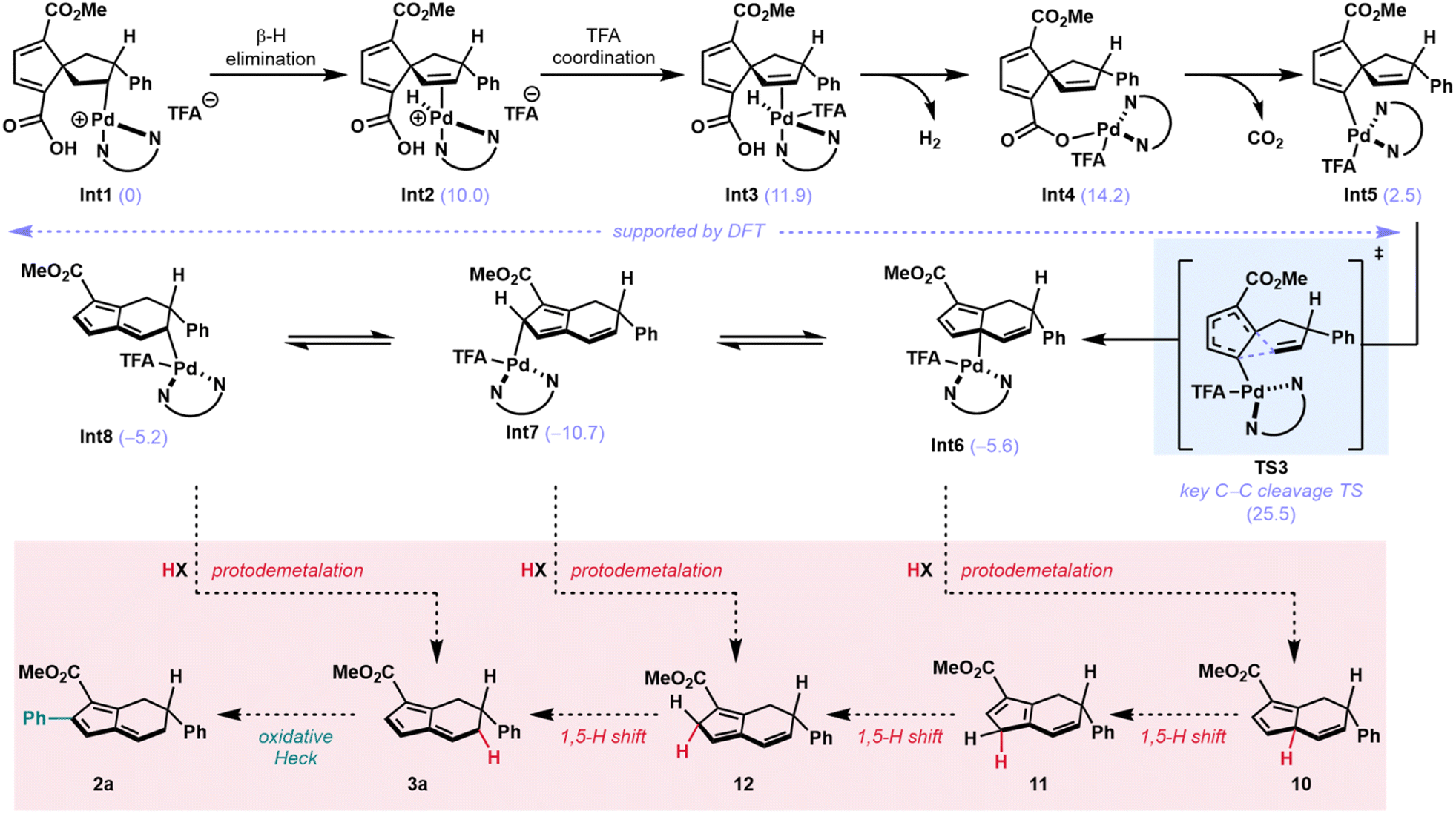 | ||
| Scheme 3 Intermediates en route to and following key TS3 are shown above. Calculated energies for intermediates are shown in parentheses in units of kcal mol−1. The remaining hypothesized steps for this pathway are shown in a red box and further discussed below. | ||
The immediate step following formation of alkyl Pd-species Int1 (Scheme 3) is expected to be a β-hydride elimination to give the spirononatriene-bound Pd–H species Int2 at 10.0 kcal mol−1.
Subsequently, a TFA anion coordinates to the cationic Pd(II)-complex to give a neutral trigonal bipyramidal complex (Int3 at 11.9 kcal mol−1). In Int3, the Pd–H is positioned such that the hydride is proximal to the pendant carboxylic acid. At this stage, we propose a deprotonation-palladation sequence which results in the loss of H2 and the formation of a palladated carboxylate. Pd(II)-hydrides have been reported to be basic enough to deprotonate phenols.34 As such, we posit that the deprotonation of a more acidic carboxylic acid should be possible under these conditions. Calculations show this transformation from Int3 to Int4 to be thermodynamically feasible with the palladated carboxylate at 14.2 kcal mol−1. Furthermore, it has been shown in the context of organolithium systems that complex-induced proximity effects (CIPE) can be leveraged for selective deprotonation.35 Due to the observation that 4 does not form any rearranged products when subjected to the reaction conditions (Table 2, entry 4), we hypothesize that CIPE may be operative in this intramolecular deprotonation to form Int4, and that Int4 does not form from intermolecular palladation of 4. This direct palladation of 4 is likely impeded by steric hindrance between the newly installed aryl group and bulky palladium complex.
From Int4, we propose that a Pd(II)-mediated decarboxylation,36–38 which is calculated to be exergonic by 12.3 kcal mol−1, furnishes an initial vinyl Pd species at 5.8 kcal mol−1 (see ESI, Int-S1).39 A slight conformational change to pre-reaction complex Int5 at 2.5 kcal mol−1 then leads to C–C cleavage through a kinetically feasible and exergonic Pd(II)-mediated 1,5-vinyl shift (with a TS energy of 25.5 kcal mol−1) to give cyclopentadienyl Pd-complex Int6 at −5.6 kcal mol−1. The η-1 bound palladium isomers Int7 and Int8 were calculated to have energies of −10.7 and −5.2 kcal mol−1, respectively (Scheme 3). At this stage, the catalytic cycle could terminate in protodemetalation from any of these intermediates (Int6–Int8). Direct protodemetalation from Int8, or protodemetalation from Int6 and Int7 followed by 1,5-hydride shifts, could furnish monoaryl fulvene 3a (Scheme 3, red box). A subsequent oxidative Heck reaction would then yield diaryl fulvene 2a.
Our hypotheses regarding protodemetalation and second oxidative Heck reaction were both investigated experimentally. The protodemetalation step was tested through a series of deuterium-labeling studies (Fig. 4). First, spirononatriene 1 was subjected to the standard reaction conditions in the absence of base with an added 5% (v/v) of deuterated acetic acid (AcOD). For reactions with both 4-NO2 and 4-CF3 phenylboronic acids, the mono- and diaryl fulvene products were obtained with partial deuterium incorporation at multiple positions (Fig. 4A). This outcome not only provides evidence for protodemetalation but the observation of deuteration at multiple sites provides additional mechanistic insight. Specifically, it is likely that alkyl palladium intermediates that result from the Pd(II)-mediated vinyl shift (e.g., Int6–Int8, Scheme 3) equilibrate prior to protodemetalation. Suprafacial 1,5-hydride shifts may ultimately give the fulvenyl π system. This rationale is in line with Semmelhack's proposal of rapid 1,5-hydride shifts following the vinyl shift in spiro[4.4]nonenes to the 5,6-fused bicycles under high temperature conditions.25 To support the proposal that deuteration results from protodemetallation en route to the fulvene, and not after fulvene formation, diaryl fulvene 2h was subjected to the conditions with added deuterated acetic acid (Fig. 4B). In this case, deuterium incorporation was not observed, supporting our initial hypothesis.
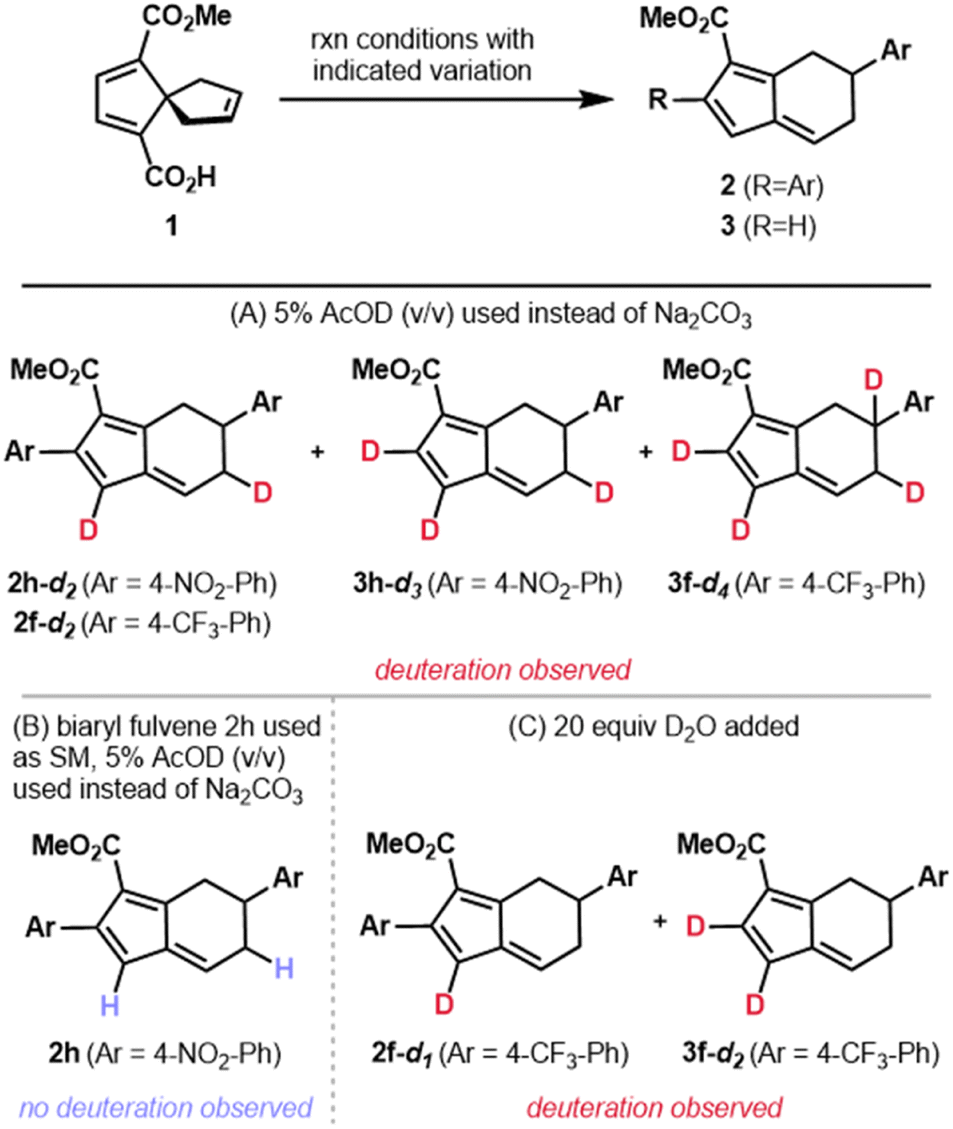 | ||
| Fig. 4 The results of investigating protodemetalation are shown above. | ||
Because the standard reaction conditions use base, we sought to determine the source of the proton that terminates the transformation. We hypothesized that adventitious water might be the source of the proton. Addition of D2O to the reaction conditions did, indeed, lead to partial deuterium incorporation (Fig. 4C). Additionally, on the basis of the pKa's of aryl boronic acids (pKa = 4.2–9.0),40 which tend to be lower than the pKa of H2O (pKa = 14),41 the proton involved in protodemetalation might arise from the boronic acid coupling partner or a by-product of boronic acid transmetalation. These observations provide strong support for a terminating protodemetalation and 1,5-hydride shifts in the formation of the initial monoaryl fulvene product (3a). On the basis of these results, we hypothesize that 1,5-hydride shifts of the benzylic hydrogen in intermediates similar to 12 (Scheme 3) may also be responsible for the lower er's observed for diaryl fulvenes as compared to the corresponding monoaryl fulvenes (i.e.2hvs.3h).
Subjecting 4-NO2 monoaryl fulvene 3h to the reaction conditions cleanly gives diaryl fulvene 2h in 73% yield, consistent with the second aryl group arising through a second oxidative Heck reaction.42 Measuring the product distribution of 2hvs.3h over the course of the reaction provides further support for this second oxidative Heck reaction. An initial increase in monoaryl fulvene 3h is observed, followed by a decrease in 3h as a simultaneous increase in the amount of diarylfulvene 2h is observed (see the ESI, Section 3.5†). Overall, these experiments provide support for the hypothesis for conversion of alkyl palladium intermediates Int6–8 to fulvenes 2a and 3a (Scheme 3, red box).
Collectively, these findings support the mechanistic picture illustrated in Scheme 3, which is consistent with our empirical observations and calculations. Additional insight may be provided by some observations made during the process of reaction optimization. DFT calculations indicate that the key transition state (TS3) bearing acetate instead of trifluoroacetate as the X-type ligand is lower in energy (18.2 kcal mol−1, see ESI, Scheme S2†) despite the observation that addition of acetate results in reduction in the amount of fulvene product (2a) and greater amounts of the Heck product (4; see ESI, Table S8†). In this case, it may be that acetate competitively binds the Pd(II) center, displacing arylated spirononatriene 4 from either the Pd(II)-bound alkene (Int3) or palladated carboxylate (Int4). Silver salts might serve an analogous role due to their ability to bind carboxylates.43
Conclusions
In conclusion, we have developed a new oxidative Heck cascade reaction which results in the rearrangement of 1-carboxy-4-methyl-ester-spiro[4.4]nonatriene to enantioenriched, aryl-substituted annulated fulvenes. The products are obtained in moderate to good yields and enantiomeric ratios. Overall, this transformation expands the reactivity of spirononatrienes and provides a complementary synthesis of annulated fulvenes. In this way, the work reported here connects these two classes of π-conjugated molecules which have a rich history in physical organic chemistry. Our mechanistic investigations, bolstered by DFT calculations, support a pathway where cleavage of a relatively unstrained C–C bond occurs through a novel palladium-mediated 1,5-vinyl shift. This work adds to the growing number of reactions wherein palladium intermediates can accelerate sigmatropic rearrangements but provides a distinct contrast to existing work as it involves rearrangement from a covalently bonded organopalladium intermediate as opposed to an organopalladium coordination complex.44,45Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
K. G. and R. S. made the original discovery, conceived the project, and formulated the initial mechanistic hypotheses. K. G. performed all experimental work under the guidance of R. S. G. A. K., A. T., and X. C. performed all computational work under the guidance of K. N. H. K. G. and R. S. wrote the manuscript with input from G. A. K., A. T., X. C., and K. N. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R.S. is grateful to the ACS–PRF New Directions (PRF# 56509-ND1) for seed funding and the NIGMS (R35 GM130345) for funding. K.N.H. is grateful to the National Science Foundation (CHE-2153972) for funding. A. T. is grateful for fellowship support provided by the National Institutes of Health (NIH) (F32GM134709). We thank Dr Hasan Celik and UC Berkeley's NMR facility in the College of Chemistry for spectroscopic assistance. Instruments in the CoC NMR facility are funded in part by NIH S10OD024998. We thank Dr Ulla Andersen and Dr Zhongrui Zhou at the UC Berkeley QB3 Mass Spectrometry Facility for HRMS analysis. We also thank Dr Nicholas Settineri for single-crystal X-ray diffraction studies. Instruments in the UC Berkeley X-ray facility are supported by NIH Shared Instrument Grant S10-RR027172. Additionally, we thank Dr Kathy Durkin and Dr Dave Small who run UC Berkeley's Molecular Graphics and Computational Facility (MGCF). Calculations were performed at the MGCF, which is supported by NIH grant S10OD023532, and on the Hoffman2 cluster at UCLA.Notes and references
- P. Bischof and E. Heilbronner, Helv. Chim. Acta, 1970, 53, 1425–1434 CrossRef CAS.
- P. Warner, D. L. Harris, C. H. Bradley and S. Winstein, Tetrahedron Lett., 1970, 4013–4016 CrossRef CAS.
- L. A. Paquette, T. M. Kravetz and P. Charumilind, Tetrahedron, 1986, 42, 1789–1795 CrossRef CAS.
- M. F. Semmelhack, J. S. Foos and S. Katz, J. Am. Chem. Soc., 1973, 95, 7325–7336 CrossRef CAS.
- H. E. Simmons and T. Fukunaga, J. Am. Chem. Soc., 1967, 89, 5208–5215 CrossRef CAS.
- C. Batich, E. Heilbronner, E. Rommel, M. F. Semmelhack and J. S. Foos, J. Am. Chem. Soc., 1974, 96, 7662–7668 CrossRef CAS.
- W.-J. Son, S.-Y. Kwak, H. Koo, B. Choi, S. Kim, H. S. Lee, M. H. Whangbo and H. Choi, ChemPhysChem, 2018, 19, 1711–1715 CrossRef CAS PubMed.
- P. Maslak, Adv. Mater., 1994, 6, 405–407 CrossRef CAS.
- D. Sirbu, L. Falck-Pedersen, C. Remming and I. Undheim, Tetrahedron, 1999, 55, 6703–6712 CrossRef CAS.
- K. N. Houk, J. K. George and R. E. Duke, Tetrahedron, 1974, 30, 523–533 CrossRef CAS.
- P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930–3989 CrossRef CAS PubMed.
- C. Dahlstrand, B. O. Jahn, A. Grigoriev, S. Villaume, R. Ahuja and H. Ottosson, J. Phys. Chem. C, 2015, 119, 25726–25737 CrossRef CAS.
- E. Shurdha, B. K. Repasy, H. A. Miller, K. Dees, S. T. Iacono, D. W. Ball and G. J. Balaich, RSC Adv., 2014, 4, 41989–41992 RSC.
- A. Lücht, S. Sobottka, L. J. Patalag, P. G. Jones, H. U. Reissig, B. Sarkar and D. B. Werz, Chem. - Eur. J., 2019, 25, 10359–10365 CrossRef PubMed.
- D. S. Siegel, G. Piizzi, G. Piersanti and M. Movassaghi, J. Org. Chem., 2009, 74, 9292–9304 CrossRef CAS PubMed.
- C. Ebner and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 11227–11230 CrossRef CAS PubMed.
- T. M. Kravetz and L. A. Paquette, J. Am. Chem. Soc., 1985, 107, 6400–6402 CrossRef CAS.
- H. K. Holm and L. A. Skattebøl, J. Am. Chem. Soc., 1977, 99, 5480–5481 CrossRef.
- J. Cao, H. Wu, Q. Wang and J. Zhu, Nat. Chem., 2021, 13, 671–676 CrossRef CAS PubMed.
- G. Chen, J. Cao, Q. Wang and J. Zhu, Org. Lett., 2020, 22, 322–325 CrossRef CAS PubMed.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–433 RSC.
- D. V. Partyka, Chem. Rev., 2011, 111, 1529–1595 CrossRef CAS PubMed.
- M. F. Semmelhack and H. N. Weller, J. Org. Chem., 1978, 43, 3791–3792 CrossRef CAS.
- M. F. Semmelhack, H. N. Weller and J. S. Foos, J. Am. Chem. Soc., 1977, 99, 292–294 CrossRef CAS.
- M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed.
- J. Ye, Z. Shi, T. Sperger, Y. Yukusawa, C. Kingston, F. Schoenebeck and M. Lautens, Nat. Chem., 2017, 9, 361–368 CrossRef CAS PubMed.
- I. Fernández, F. P. Cossío and M. A. Sierra, Chem. Rev., 2009, 109, 6687–6711 CrossRef PubMed.
- M. F. Croisant, R. van Hoveln and J. M. Schomaker, Eur. J. Org Chem., 2015, 5897–5907 CrossRef CAS.
- B. M. M. Wheatley and B. A. Kay, J. Org. Chem., 2007, 72, 7253–7259 CrossRef CAS PubMed.
- L. E. Overman and F. M. Knoll, J. Am. Chem. Soc., 1980, 102, 865–867 CrossRef CAS.
- P. H. Scudder, Electron Flow in Organic Chemistry, Jon Wiley & Sons, 2013, p. 49 Search PubMed.
- E. S. Osipova, N. V. Belkova, L. M. Epstein, O. A. Filippov, V. A. Kirkina, E. M. Titova, A. Rossin, M. Peruzzini and E. S. Shubina, Eur. J. Inorg. Chem., 2016, 1415–1424 CrossRef CAS.
- M. C. Whisley, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206–2225 CrossRef PubMed.
- J. S. Dickestein, C. A. Mulrooney, E. M. O'Brien, B. J. Morgan and M. C. Kozlowski, Org. Lett., 2007, 9, 2441–2444 CrossRef PubMed.
- P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898–11899 CrossRef CAS PubMed.
- M. Li, C. Wang and H. Ge, Org. Lett., 2011, 13, 2062–2064 CrossRef CAS PubMed.
- Due to the difficulty in finding a feasible transition state for the decarboxylation using DFT calculations, it is possible that the decarboxylation may be concerted with the 1,5-vinyl shift (see TS3). See the ESI† for more details.
- J. Yan, G. Springsteen and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS.
- On the basis of the observation that monoaryl fulvenes are only formed in isolable quantities when electron-deficient boronic acids are used (e.g. 4-CN, 4-NO2, 4-CN arylboronic acids), the rate of the migratory insertion for more electron-deficient aryl palladium intermediates across the fulvene alkene is likely slower than for more electron-rich arylboronic acids.
- T. P. Silverstein and S. T. Heller, J. Chem. Educ., 2017, 94, 690–695 CrossRef CAS.
- A. Baur, K. A. Bustin, E. Aguilera, J. L. Petersen and J. M. Hoover, Org. Chem. Front., 2017, 4, 519–524 RSC.
- E. E. Lee and R. A. Batey, Angew. Chem., Int. Ed., 2004, 43, 1865–1868 CrossRef CAS PubMed.
- O. Gutierrez, C. E. Hendrick and M. C. Kozlowski, Org. Lett., 2018, 20, 6539–6543 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2245692 (for 1), 2245684 (for 2a), and 2245677 (for 4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03222a |
| This journal is © The Royal Society of Chemistry 2023 |